

Kufor‐Rakeb syndrome (KRS) (Mendelian Inheritance in Man no. 606693) is an autosomal recessive, juvenile parkinsonism caused by mutations in the ATP13A2 (adenosine 5′‐triphosphatase [ATPase] type 13A2) gene. The clinical features comprise akinetic rigid parkinsonism, dementia, psychosis, supranuclear gaze palsy, oculogyric dystonic spasms, facial‐faucial‐finger minimyoclonus, and spasticity.1 The disease is rare, and only a few patients have been reported to date. Whole exome sequencing is an emerging tool in the screening for mutations in several diseases in a timely and cost‐effective manner. We describe a patient with KRS in whom we used exome sequencing and homozygosity mapping to identify a novel, homozygous missense mutation in ATP13A2.
Case Report
A man aged 32 years born of second‐degree, consanguineous parentage presented with insidious‐onset, progressive slowness in activities of daily living, change in speech, forgetfulness, and falls since the age of 21 years. He had psychiatric features including excessive fear, formed visual and auditory hallucinations from the age of 27 years, and had behavioral disturbances such as hyper sexuality in the form of excessive sexual thoughts, inappropriately touching himself, and masturbation since age 29 years. He was started on sustained‐release carbidopa/levodopa (l‐dopa) (50/200mg tablets; one‐half tablet twice daily) at age 22 years. His slowness improved, but he developed l‐dopa–induced dyskinesia 3 years later. Hence, l‐dopa was stopped, and ropinirole (12 mg daily) and trihexyphenidyl (6 mg daily) were started. The patient had received risperidone, olanzapine, and quetiapine for the psychosis. He had been on levosulpiride for 3 years 8 years after the onset of illness. There was no family history of parkinsonism. His elder sister died at the age 15 years from a respiratory illness (Fig. S1; see online supporting information).
His gait, speech, and cognition worsened over next 10 years, and he developed urinary and bowel incontinence. He presented to us 10 years later. On evaluation, cognition was impaired (Mini‐Mental State Examination [MMSE] score, 8 of 30), with the patient having deficits in orientation, calculation, and language functions on the MMSE. Cranial nerve examination showed impaired upward gaze and absent horizontal and vertical saccades. Spastic dysarthria was present. He had torticollis to the left and jaw‐opening dystonia while performing rapid alternate hand movements. Motor examination revealed mild, bilateral dorsiflexor weakness of the feet, generalized spasticity with brisk deep tendon reflexes and plantars were flexor. Truncal myoclonus was noted while standing with support. Resting tremor was observed in both upper limbs with prominent intention and postural component along with myoclonic jerks. Bradykinesia and rigidity were seen on the left side more than the right. His off‐medication score on the Unified Parkinson's Disease Rating Scale motor part was 71, and it improved to 55 with carbidopa/l‐dopa (25/100 mg) (video S1). There were no cerebellar signs. There was no orthostatic hypotension on tilt test.
He was diagnosed with early onset, atypical parkinsonism and was evaluated for the same with imaging and blood tests, as shown in Table 1. In view of his consanguineous parentage, cognitive decline, psychosis, pyramidal and extrapyramidal features, a diagnosis of KRS was considered, and whole exome sequencing was used to make a molecular diagnosis.
Table 1.
Diagnostic Patient Investigations
| Investigation | Result |
|---|---|
| 1. Peripheral smear for acanthocytes | Negative |
| 2. Genetic analysis | |
| Huntington's disease | Negative |
| Dentatorubro‐pallidoluysian atrophy | Negative |
| 3. Nerve conduction study | Normal |
| 4. Electroencephalogram | Normal |
| 5. Imaging | |
| Computed tomography: Brain | Diffuse cerebral atrophy, no calcification |
| Magnetic resonance imaging | |
| Braina | Diffuse cerebral and brainstem atrophy, no basal ganglia mineralization |
| Cervical spine | Normal |
| 6. Axillary skin biopsy | Negative for adult polyglucosan disease and neuronal ceroid lipofuscinosis |
See Figure 1.
Figure 1.
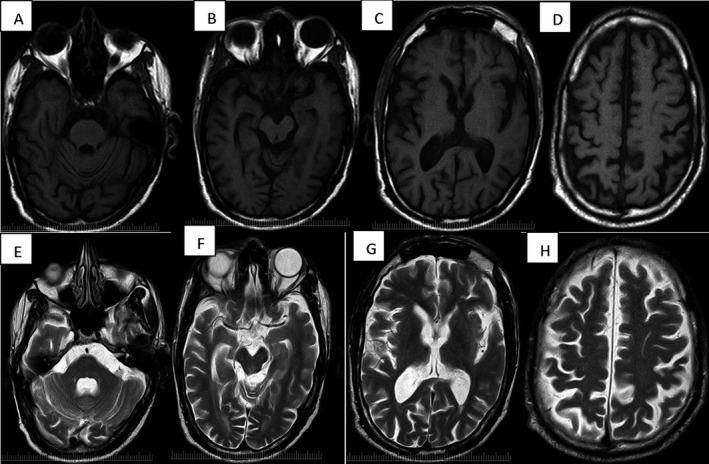
A‐D:MRI T1 axial sequence showing atrophy of pons, midbrain, basal ganglia and cortex. Similar changes noted on T2 axial weighted images(E‐H).
Genomic DNA extracted from whole blood was available for both parents and the index case. Regions of homozygosity greater than 0.5 megabases in the index case were identified using an Omni Express SNP chip (Illumina, Inc., San Diego, CA). Whole exome sequencing was performed using Illumina's TruSeq exome‐enrichment kit and HighSeq sequencing system (Illumina, Inc.) in the proband. This generated 43,413,599 unique reads, translating to a total variant count of 21,308. Using the collaborative consensus coding sequence (CCDS) hg19 definition of the exome (The CCDS Collaboration: National Center for Biotechnology Information, National Library of Medicine, Bethesda, MD; European Bioinformatics Institute, European Molecular Biology Laboratory, Hinxton, UK; USCS Genome Browser, Santa Cruz, CA; Wellcome Trust Sanger Institute, Hinxton, UK), 94% and 85% of the exome was covered by at least 2 reads and 10 reads, respectively. The mean read depth across the exome was 33 reads. Only variants within regions of homozygosity were used for filtering. We filtered out synonymous variants and any variant that was present at a global minor allele frequency of ≥0.001% in a range of publically available databases of sequence variation (1000 Genomes Project2; Complete Genomics [Shenzhen, China] 69 database3; and the National Heart, Lung, and Blood Institute [Bethesda, MD] Exome Sequencing Project database4) as well variants found in our in‐house exomes from individuals with unrelated diseases (n = 200). This left 40 homozygous variants. A missense mutation c.T2525C:p.L842P in ATP13A2 (Ensemble transcript ID ENST00000341676) was of immediate interest because of its association with KRS. The variant was validated by Sanger sequencing using the Big Dye Terminator v3.1 Cycle Sequencing Kit and an Applied Biosystems 3130XL Genetic Analyzer (Life Technologies, Carlsbad, CA) and confirmed that both parents were heterozygous carriers.
The residue at which the mutation occurs is highly conserved, with a GERP (Genomic Evolutionary Rate Profiling) score of 3.46 and is predicted with a high probability to be deleterious using in‐silico prediction software (a SIFT [Sorting Intolerant From Tolerant] score of 0 and a PolyPhen2 [Polymorphism Phenotyping v2] score of 1).
Review of other genes that cause autosomal recessive parkinsonian syndromes revealed good coverage. No mutations in these genes were detected on exome sequencing (Fig. S2; see online supporting information).
We report a novel homozygous missense mutation in ATP13A2 (c.T2525C:p.L842P) located in the hydrolase domain of the protein, a domain in which other pathogenic frameshift and missense mutations have previously been described. To date, only 3 other homozygous missense variants have been reported in association with KRS,5, 6, 7 and most are frameshift and splice‐site mutations. Currently, the mechanism(s) through which missense mutations cause KRS are not fully understood; however, a recent study in indicates that another missense mutation in the hydrolase domain (p.G877R) impairs protein stability and enhances degradation by the proteasome. Other missense mutations resulted in mislocalization of ATP13A2 to the endoplasmic reticulum.8
The patient was managed with gradually increasing dose of carbidopa/l‐dopa (25/100 mg; 2 tablets 3 times daily), clonazepam (2 mg daily), and clozapine (25 mg daily). Ropinirole, trihexyphenidyl, and quetiapine were stopped. He has noticed mild improvement in slowness, and his hallucinations have reduced. However, he still has truncal myoclonus, severe intentional tremor, and hypersexuality. He is completely dependent for all his activities of daily living.
There is a single case report of KRS from India in a patient who had a nonsense mutation in exon 22 of the ATP13A2 gene (chromosome 1, 17316187; G>A) detected by whole exome sequencing.9 The clinical phenotype in our patient is similar to that described in the literature, with early onset parkinsonism, cognitive decline, psychosis, gaze palsy, pyramidal features, and l‐dopa response with early dyskinesias. Interestingly, our patient had truncal myoclonus in addition to myoclonic jerks in both hands. The hypersexuality seen in our patient might be due to the disease or the dopaminergic therapy.
The current case demonstrates the utility of exome sequencing in diagnostically challenging patients. It allows the investigation of mutations in several genes simultaneously in a timely and cost‐effective manner. However, there are several technical limitations to using whole exome sequencing for diagnostic purposes. Selectively sequencing the exome (which, to our knowledge, is the most likely region of the genome to contain pathogenic mutations) also excludes noncoding and intronic regions. The contribution of mutations in noncoding regions to Mendelian disease has yet to be determined. Mutations in microRNAs, promotors, and ultraconserved elements may be associated with disease; however, these are not fully covered in current exome capture kits. Furthermore, mutations may be missed if the region is poorly covered. Whole exome sequencing is unable to detect structural variants or chromosomal rearrangements; therefore, other technologies need to be used to exclude these, e.g., multiplex ligation‐dependent probe amplification or quantitative polymerase chain reaction.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
M.M.A: 1B, 1C, 2A, 2B, 2C, 3A, 3B
S.T.G: 1B, 1C, 2B, 3B
U.M.S: 1B, 1C, 2A, 2B, 3A
K.P.B: 1A, 1B, 2A, 2C, 3 B
U.B.M: 1A, 1B, 2A, 2C, 3B
Disclosures
Funding Sources and Conflicts of Interest: The authors report no sources of funding and no conflicts of interest.
Financial Disclosures for the previous 12 months: The authors declare that there are no disclosures to report.
Supporting information
A video accompanying this article is available in the supporting information here.
Figure S1. Pedigree: Squares represent men and circles women. Black square represent the proband and black dots within represent the mutation in heterozygous state.
Figure S2. Average exome sequencing reads per exon for a range of autosomal recessive young‐onset Parkinsonism genes.
Video S1: Off period: The patient has features of Parkinsonism with bradykinesia and resting tremor with myoclonic jerks in outstretched hands. Horizontal and vertical saccades are absent. Truncal myoclonus is noted on standing. On period: patient has orofacial dyskinesias and prominent postural and intention tremor with myoclonic jerks. Bradykinesia has improved. Spastic gait is present while walking.
Supporting information may be found in the online version of this article.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. al‐Din Najim AS, Wriekat A, Mubaidin A, Dasouki M, Hiari M. Pallido‐pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor‐Rakeb syndrome. Acta Neurol Scand 1994;89:347–352. [DOI] [PubMed] [Google Scholar]
- 2. The International Genome Sample Resource (IGSR) and the 1000 Genomes Project . Global reference of human genetic variation. Available at: http://www.1000genomes.org/accessed July 2015.
- 3. Complete Genomics . Complete Genomics data. Shenzhen, China: Complete Genomics Inc.; 2015. Available at: http://www.completegenomics.com/public-data/69-Genomes/accessed July 2015. [Google Scholar]
- 4. National Heart, Lung, and Blood Institute (NHLBI) . NHLBI Exome Sequencing Project (ESP) database. Bethesda, MD: National Institutes of Health, US Department of Health and Human Services; 2015. Available at: http://evs.gs.washington.edu/EVS accessed July 2015.
- 5. Ning YP, Kanai K, Tomiyama H, et al. PARK9‐linked parkinsonism in eastern Asia: mutation detection in ATP13A2 and clinical phenotype. Neurology 2008;70:1491–1493. [DOI] [PubMed] [Google Scholar]
- 6. Di Fonzo A, Chien HF, Socal M, et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 2007;68:1557–1562. [DOI] [PubMed] [Google Scholar]
- 7. Santoro L, Breedveld GJ, Manganelli F, et al. Novel ATP13A2 (PARK9) homozygous mutation in a family with marked phenotype variability. Neurogenetics 2011;12:33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Podhajska A, Musso A, Trancikova A, et al. Common pathogenic effects of missense mutations in the P‐type ATPase ATP13A2 (PARK9) associated with early‐onset parkinsonism [serial online]. PLoS ONE 2012;7:e39942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Prashanth LK, Murugan S, Kamath V, et al. First report of Kufor‐Rakeb syndrome (PARK9) from India, and a novel nonsense mutation in ATP13A2 gene [letter]. Mov Disord Clin Pract 2015;2:326–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A video accompanying this article is available in the supporting information here.
Figure S1. Pedigree: Squares represent men and circles women. Black square represent the proband and black dots within represent the mutation in heterozygous state.
Figure S2. Average exome sequencing reads per exon for a range of autosomal recessive young‐onset Parkinsonism genes.
Video S1: Off period: The patient has features of Parkinsonism with bradykinesia and resting tremor with myoclonic jerks in outstretched hands. Horizontal and vertical saccades are absent. Truncal myoclonus is noted on standing. On period: patient has orofacial dyskinesias and prominent postural and intention tremor with myoclonic jerks. Bradykinesia has improved. Spastic gait is present while walking.