

Autosomal‐dominant progressive external ophthalmoplegia type 1 (adPEO1) is characterized by slowly progressive ophthalmoplegia. It can be caused by mutations in different genes, including the mitochondrial DNA polymerase γ (POLG), which results in heterogeneous clinical phenotypes associated with progressive external ophthalmoplegia, including myoclonic epilepsy,1 parkinsonism,2 and ataxia3 (Table 1). Other additional features may include premature ovarian failure and hypogonadism.2 POLG mutations causing adPEO1 can have both autosomal‐dominant or ‐recessive inheritance traits.4 Dystonia has been observed in patients with POLG mutations,5, 6, 7 however, to our knowledge, not in adPEO1. Here, we describe the first adPEO1 patient attributed to a POLG mutation showing dystonia as the presenting and core clinical feature.
Table 1.
Main disorders and clinical features caused by POLG mutations
| Disorder | Clinical Features |
|---|---|
| Autosomal‐dominant progressive external ophthalmoplegia | Ophthalmoplegia, myopathy, hearing loss, neuropathy, ataxia, parkinsonism, depression, hypogonadism, ptosis, dysphagia, dysarthria, and cataracts |
| Autosomal‐recessive progressive external ophthalmoplegia | Ophthalmoplegia, ptosis, myopathy, pes cavus, dysarthria, neuropathy, and mitral valve dysfunction |
| Myoclonic epilepsy, myopathy, sensory ataxia | Myoclonic epilepsy, neuropathy, and ataxia |
| Mitochondrial DNA depletion syndrome 4A (Alpers‐Huttenlocher syndrome) | Encephalopathy, failure to thrive, intractable epilepsy, visual disturbances, ataxia, vomiting, and hepatic failure |
| Mitochondrial DNA depletion syndrome 4B (neurogastrointestinal encephalopathy) | Gastrointestinal dysmotility, intestinal pseudo‐obstruction, abdominal pain, cachexia, ophthalmoplegia, ataxia, neuropathy, myopathy, hypotonia, seizures, and developmental delay |
| Childhood myocerebrohepatopathy spectrum | Developmental delay, myopathy, lactic acidosis, hearing loss, cyclic vomiting, liver failure, renal tubular acidosis, and pancreatitis |
| Myoclonic epilepsy myopathy sensory ataxia | Epilepsy, myopathy, and ataxia |
| Ataxia neuropathy spectrum (MIRAS and SANDO) | Ataxia, neuropathy, dysarthria, ophthalmoplegia, epilepsy, hearing loss, ptosis, myopathy, cognitive impairment, and depression |
| Other pure POLG disorders | Myoclonus, dystonia (torticollis, focal eyelid dystonia, limb dystonia), parkinsonism, and chorea |
MIRAS, mitochondrial recessive ataxia syndromes; SANDO, sensory ataxia, neuropathy, dysarthria, and ophthalmoplegia.
Case Presentation
A 22‐year‐old Caucasian female with Italian ancestry and medical history of secondary amenorrhea, with normal motor and cognitive development until 15 years of age, when she began to experience progressive abnormal twisted and sustained posture of the right arm, hand, and foot, is presented. In the next 6 months, she developed diplopia, dysarthria, and dysphagia. Four months later, she was progressively unable to feed, dress or toilet herself, and unable to walk without assistance because of the progression of dystonia and the development of axial ataxia. The full clinical picture developed in a 1‐year period. Her family history was remarkable for scoliosis in her younger sister, maternal aunt, and maternal grandmother, who had scoliosis for more than 10 years without showing walking problems or dystonic features. The patient's mother had a slight strabismus. The maternal grandfather and great‐grandfather both presented a dystonic syndrome involving the legs and neck as well as difficulty in swallowing in their fifties. A family tree is offered as Supporting Information.
On physical examination, the patient revealed cerebellar dysarthria, severe horizontal and vertical external ophthalmoparesis, esotropia, slight left ptosis, and asymmetric facial muscle weakness. Hemidystonia on the right side, predominantly in the upper limb, was noted with right plantar extensor response, clonus, and hyper‐reflexia. Tandem walk was impossible and assistance was needed for walking because of severe trunk and gait ataxia. Fundoscopy was unremarkable and visual acuity was normal. Cognitive and psychiatric status were normal. She had scoliosis; however, no cataracts, pes cavus, hearing impairment, parkinsonism, sensory loss, muscle atrophy, or visceromegaly were present. Brain MRI revealed slight cerebellar atrophy and decreased signal in the globus pallidus, SN, dentate nucleus, and red nuclei on fluid‐attenuated inversion recovery (FLAIR) and T2‐weighted sequences (Fig. S2). Initial lab workup showed normal creatine kinase, lactate, alpha‐fetoprotein, vitamin E, organic acids, thyroid hormones and antibodies, ceruloplasmin levels, and urinary copper. Polymerase chain reaction in cerebrospinal fluid for Tropheryma whipplei was negative. Hexosaminidase A, sphingomyelinase, and beta‐glucosidase were negative. Niemann‐Pick type C (NPC) was suspected after Filipin stain performed on skin fibroblasts from the patient rendered positive results. However, further studies ruled out this possibility given that oxysterols levels and NPC1 and NPC2 gene mutations were negative. Genetic testing for DYT1, SCA1, SCA2, SCA3, and mitochondrial DNA (mtDNA) point mutations and deletions for mitochondrial disorders (blood sample) proved negative.
A mitochondrial encephalopathy was suspected, and coenzyme Q10, flavonoids, creatine, and vitamins C and E were added, without clinical improvement. At the age of 16, trihexyphenidyl was tried to treat dystonia, without symptomatic relief after 3 months of treatment. Then, trihexyphenidyl was stopped and levodopa therapy was started and escalated up to 1,000 mg/day with partial improvement of limb and axial dystonia and gait. Physical activities in daily life and functional capacity improvements after l‐dopa introduction have been unchanged and sustained for the last 6 years of follow‐up.
Whole‐genome sequencing (WGS), followed by Sanger sequencing, was performed for the index patient at 22 years of age and her parents. A heterozygous variant c.3436C>T in exon 21 of the POLG gene, which results in a missense p.Arg1146Cys change, was detected. This mutation is consistent with a genetic diagnosis of adPEO1.8 The same variant was also detected in a heterozygous state in the patient's mother. Complete WGS analysis data are depicted in Table S1.
Discussion
The clinical phenotype of our patient with dystonia as the presenting and core clinical feature expands the clinical spectrum of adPEO1 associated with POLG mutations. Torticollis, focal eyelid dystonia, and limb dystonia have been previously described in patients with other POLG mutations5, 6, 7; however, it was not reported in adPEO1 patients harboring POLG gene mutations. Table 1 shows the main disorders and clinical features caused by POLG mutations. Dystonia in our patient showed favorable and sustained long‐term response to l‐dopa treatment, as was reported in parkinsonism2 associated with POLG mutations. However, improvement of dystonia with l‐dopa was partial even at a high dose, but allowed better functional capacity and walking without assistance. Unfortunately, functional dopaminergic imaging to evaluate nigrostriatal pre‐ or postsynaptic integrity was not available, which might have clarified whether an altered dopaminergic nigrostriatal pathway might explain some improvement with l‐dopa. However, these studies are usually normal in patients with dopa‐responsive dystonia.9
Noteworthy is the oligo‐symptomatic state of the patient's mother, who, although carrying the same heterozygous variant as our index case, only expresses slight strabismus. This might be explained because heterozygous mutations of the nuclear gene, POLG, can produce an inactive form of the enzyme that competes with wild‐type pol‐γ. As a result, mtDNA replication is impaired and error prone, allowing the introduction of point mutations and/or deletions of mtDNA molecules or the progressive depletion of mtDNA copy number. The interplay between mtDNA variants and mutations in nuclear genes may, in part, explain the extremely high variation in mitochondrial phenotypes. This phenomenon has been described for other nuclear POLG mutations in which the phenotype is strongly related to the downstream mtDNA effects in an individual patient, so that mtDNA depletion results in an early‐onset severe phenotype, whereas deletions are associated with a later‐onset disease.10 Although in the index case the presence of mutations in the mtDNA obtained from peripheral blood has been ruled out, the evaluation of mtDNA mutations in muscle is still pending.
It should be pointed out that the Filipin stain conducted to rule out NPC presented a positive pattern, in that the stain was present, but its subcellular distribution was very particular and different from the one observed in individuals affected by this pathology. Usually, patients with NPC show an intense perinuclear staining corresponding to the location of the late endosomes and lysosomes (Fig. 1C). The intense stain observed in the patient presented a subcellular localization suggesting a location in the Golgi apparatus (Fig. 1B). This finding, previously unreported in patients with nuclear POLG mutations, could be because cholesterol transport is affected by mitochondrial dysfunction.11
Figure 1.
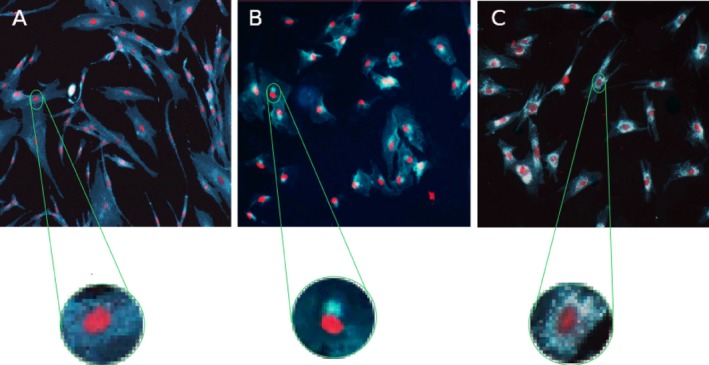
Filipin stain. Filipin stain was performed on skin fibroblasts from the patient, after culture in an low‐density lipoprotein–enriched medium. All Filipin staining analyses included both positive controls (a cell line from an affected patient with NPC confirmed by NPC1 gene mutation analysis) and negative controls (a cell line of wild‐type fibroblasts). Cholesterol detection was conducted by using Filipin dye (blue‐green color), and cell nuclei were stained with propidium iodide. (A) Individual negative for Filipin staining. (B) Patient with nuclear gene POLG mutation, showing an intense polarized staining in the region corresponding to the Golgi apparatus. (C) Patient with an NPC disease diagnosis, showing an intense staining in a perinuclear localization corresponding to late endosomes and lysosomes.
This case illustrates the phenotypic variability of POLG mutations and poses the possibility that adPEO1 attributed to mutations in the POLG gene may be considered within the crescent list of disorders inducing dystonia.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
M.R.: 1A, 1B, 1C, 2A, 2B, 2C, 3A, 3B
A.M.E.: 1A, 1B, 1C, 2A, 2B, 2C, 3A, 3B
D.C.: 1A, 1B, 1C, 2A, 2B, 2C, 3A, 3B
M.R.: 1A, 1B, 1C, 2A, 2B, 2C, 3A, 3B
S.T.: 1A, 1B, 1C, 2A, 2B, 2C, 3A, 3B
C.P.: 1A, 1B, 1C, 2A, 2B, 2C, 3A, 3B
M.M.: 1A, 1B, 1C, 2A, 2B, 2C, 3A, 3B
Disclosures
Ethical Compliance Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: The authors report no sources of funding and no conflicts of interest.
Financial Disclosures for previous 12 months: The authors declare that there are no disclosures to report.
Supporting information
A video accompanying this article is available in the supporting information here.
Figure S1. Pedigree of the reported patient. Pedigree of family exhibiting a heterozygous variant c.3436C>T in exon 21 of the POLG gene, which results in a missense p.Arg1146Cys change. Affected individuals are indicated in black, whereas subjects with symptoms who have not been genetically tested are shaded in grayscale: in dark gray those with a dystonic syndrome (I:1 and II:4) and in light gray those with scoliosis (II:5, III:4 and IV:2), which does not seem to be attributed to dystonia. The index patient (IV:1) is indicated by an arrow.
Figure S2. Brain MRI. Brain MRI evidence of the slight superior vermis atrophy (A) and the decreased signal in globus pallidus, SN, red nuclei (B, C, E, and F), and dentate nucleus (D) on fluid‐attenuated inversion recovery (FLAIR) and T2‐weighted sequences, that might be normal, given that these structures usually show high levels of iron.
Table S1. Whole‐genome sequencing data
Video 1. The video was taken at age 22 years, while on l‐dopa therapy for the last 6 years. Patient denied stopping l‐dopa for videotaping in the OFF condition. Video shows patient's severe horizontal and vertical external ophthalmoparesis, esotropia, facial muscle weakness, trunk and right hemidystonia, and marked gait ataxia.
Acknowledgments
Full consent was obtained from the patient and the parents before publication.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Van Goethem G, Mercelis R, Lofgren A, et al. Patient homozygous for a recessive POLG mutation presents with features of MERRF. Neurology 2003;61:1811–1813. [DOI] [PubMed] [Google Scholar]
- 2. Luoma P, Melberg A, Rinne JO, et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet 2004;364:875–882. [DOI] [PubMed] [Google Scholar]
- 3. Van Goethem G, Luoma P, Rantamaki M, et al. POLG mutations in neurodegenerative disorders with ataxia but no muscle involvement. Neurology 2004;63:1251–1257. [DOI] [PubMed] [Google Scholar]
- 4. Lamantea E, Tiranti V, Bordoni A, et al. Mutations of mitochondrial DNA polymerase gammaA are a frequent cause of autosomal dominant or recessive progressive external ophthalmoplegia. Ann Neurol 2002;52:211–219. [DOI] [PubMed] [Google Scholar]
- 5. Hinnell C, Haider S, Delamont S, Clough C, Hadzic N, Samuel M. Dystonia in mitochondrial spinocerebellar ataxia and epilepsy syndrome associated with novel recessive POLG mutations. Mov Disord 2012;27:162–163. [DOI] [PubMed] [Google Scholar]
- 6. Synofzik M, Schule R, Schulte C, et al. Complex hyperkinetic movement disorders associated with POLG mutations. Mov Disord 2010;25:2472–2475. [DOI] [PubMed] [Google Scholar]
- 7. Paus S, Zsurka G, Baron M, et al. Apraxia of lid opening mimicking ptosis in compound heterozygosity for A467T and W748S POLG1 mutations. Mov Disord 2008;23:1286–1288. [DOI] [PubMed] [Google Scholar]
- 8. Gonzalez‐Vioque E, Blazquez A, Fernandez‐Moreira D, et al. Association of novel POLG mutations and multiple mitochondrial DNA deletions with variable clinical phenotypes in a Spanish population. Arch Neurol 2006;63:107–111. [DOI] [PubMed] [Google Scholar]
- 9. Wijemanne S, Jankovic J. Dopa‐responsive dystonia—clinical and genetic heterogeneity. Nat Rev Neurol 2015;11:414–424. [DOI] [PubMed] [Google Scholar]
- 10. Rajakulendran S, Pitceathly RD, Taanman JW, et al. A clinical, neuropathological and genetic study of homozygous A467T POLG‐related mitochondrial disease. PLoS One 2016;11:e0145500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miller WL. Steroidogenic acute regulatory protein (StAR), a novel mitochondrial cholesterol transporter. Biochim Biophys Acta 2007;1771:663–676. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A video accompanying this article is available in the supporting information here.
Figure S1. Pedigree of the reported patient. Pedigree of family exhibiting a heterozygous variant c.3436C>T in exon 21 of the POLG gene, which results in a missense p.Arg1146Cys change. Affected individuals are indicated in black, whereas subjects with symptoms who have not been genetically tested are shaded in grayscale: in dark gray those with a dystonic syndrome (I:1 and II:4) and in light gray those with scoliosis (II:5, III:4 and IV:2), which does not seem to be attributed to dystonia. The index patient (IV:1) is indicated by an arrow.
Figure S2. Brain MRI. Brain MRI evidence of the slight superior vermis atrophy (A) and the decreased signal in globus pallidus, SN, red nuclei (B, C, E, and F), and dentate nucleus (D) on fluid‐attenuated inversion recovery (FLAIR) and T2‐weighted sequences, that might be normal, given that these structures usually show high levels of iron.
Table S1. Whole‐genome sequencing data
Video 1. The video was taken at age 22 years, while on l‐dopa therapy for the last 6 years. Patient denied stopping l‐dopa for videotaping in the OFF condition. Video shows patient's severe horizontal and vertical external ophthalmoparesis, esotropia, facial muscle weakness, trunk and right hemidystonia, and marked gait ataxia.