

Abstract
Autosomal‐dominant episodic ataxias (EAs) represent a clinically and genetically heterogeneous group of disorders characterized by recurrent episodes of cerebellar ataxia (CA). Ataxia episodes are usually of short duration and often triggered by specific stimuli. There are currently seven classified subtypes of EA. EA types 1 and 2 have the highest prevalence and are therefore the clinically most relevant. Between attacks, EA 1 is associated with myokymia. In EA 2, often an interictal downbeat nystagmus with other cerebellar ocular dysfunctions is present; patients with EA 2 may display slowly progessive ataxia and vermian atrophy. EA 1 and 2 are both channelopathies, affecting the potassium channel gene, KCNA1, in EA 1 and the PQ calcium channel‐encoding gene, CACNA1A, in EA 2. The types EA 3 to 7 are very rare and have to be further elucidated. Here, we review the historical, clinical, and genetic aspects of autosomal‐dominant EAs and their current treatment, focusing on EA 1 and 2.
Keywords: episodic ataxia, channelopathy, CACNA1A, KCNA1, CACNB4, SLC1A3
In 1986, Gancher and Nutt, from Portland, Oregon, described three distinct syndromes of autosomal‐dominant episodic cerebellar ataxias (CAs) in the first volume of Movement Disorders.1 Retrospectively, two of these syndromes correspond to episodic ataxia (EA) type 1 (EA 1) and type 2 (EA 2). The first published reports on the EA disease spectrum date back to 1946 by H.L. Parker from the Mayo Clinic.2 In 1978, acetazolamide (ACT) treatment was introduced in a subset of EA patients by R. Griggs from Rochester, New York.3 Missense mutations were first found in a potassium channel gene of EA 1 patients and in a calcium channel gene in patients with EA 2. Thus, EA 1 and 2 are both channelopathies, affecting the potassium channel gene, KCNA1, in EA 1 and the PQ calcium channel‐encoding gene, CACNA1A, in EA 2, respectively. Subsequently, autosomal‐dominant EAs were categorized on a clinical and genetic basis in seven subtypes, the most prevalent and clinically relevant being EA 1 and 2.4 Between ataxia attacks, EA 1 is associated with myokymia. In EA 2, often an interictal downbeat nystagmus with other cerebellar ocular dysfunctions is present; patients with EA 2 may display slowly progessive ataxia and vermian atrophy. Besides ataxia, the various types of EAs are variably associated with a broad spectrum of paroxysmal neurological symptoms, including epilepsy, movement disorders, hemiplegic migraine, and myasthenic syndrome. There is a high variability of these symptoms even among subjects with the same mutations or concordant twins, suggesting that nongenetic factors might modulate the phenotype.5 There is no such high variability in interictal EA features.
The Clinical Spectrum of EAs
Mutations of the potassium channel‐encoding gene (KCNA1) result in EA 1, which is characterized by brief attacks of midline cerebellar dysfunction, including limb ataxia, nystagmus, dysarthria, and tremor.1 The world‐wide prevalence is estimated at 1 in 500,000.6 Onset of EA 1 is typically in early childhood. Attacks consist of ataxia and sometimes are associated with dysarthria and tremor.7, 8 Vertigo is usually absent.9 Typically attacks are of brief duration (<15 minutes) and may occur up to 30 times per day. Atypical variants with longer duration of episodes have been reported.7, 10 Attacks occur spontaneously or may be triggered by shock, anxiety, exercise, kinesiogenic stimuli, emotional stress, alcohol intake, intercurrent illness, and hunger.7 Usually, there are no cerebellar deficits between attacks, but ataxia, hearing impairment, and postural tremor of the head and hands have been described.7, 8, 11 Approximately 20% of EA 1 individuals will accumlate persistent cerebellar symptoms and signs over time.12 Interictal myokymia is pathognomonic, but not specific for EA 1. It has been speculated that myokymia results from ectopic impulse generators within peripheral nerves.7, 13, 14 Other neuromuscular findings include painful stiffness and distal weakness, moderate muscle hypertrophy, and generalized increase in muscle tone associated with hypercontracted posture.6, 8 The fact that some EA 1 patients display cognitive dysfunction, delayed motor development, choreoathetosis, carpal spasms, shortening of the Achilles' tendon, clenching of the fists, and isolated myokymia argues against a pure cerebellar syndrome.7, 8, 15, 16, 17 Furthermore, skeletal deformities, a higher incidence of tonic‐clonic seizures, intermittent head turning and eye deviation to the same side, and breathing disorders during ataxic episodes have rarely been reported in EA 1.8, 15, 18, 19
EA 2 was mapped to the CACNA1A gene on chromosome 19p13, which encodes a calcium channel.4 EA 2 usually presents with recurrent episodes of vertigo and ataxia lasting for hours to days. The consortium for Clinical Investigation of Neurological Channelopathies estimated the prevalence of EA 2 as less than 1:100,000.20 In EA 2, attacks usually last longer than 10 minutes and consist of midline CA. The frequency of the attacks ranges from four times per week to once per year. Typical triggers of ataxic episodes are emotional stress and exercise.21 Other triggers include alcohol or caffeine intake, fever, and phenytoin treatment.20, 22 Clinical onset is typically in the first two decades of life, but late‐onset cases have been reported.23 Vertigo and dizziness (at least half of the patients), nausea, and vomiting are the most common associated symptoms (see Video 1).21 During attacks, patients may clinically exhibit severe ataxia, dysarthria, and primary position downbeat or gaze‐evoked nystagmus.24 Other attack features include diplopia, dysarthria, dystonia, and weakness. Additionally, there may be symptoms suggestive of brain stem and cortex involvement.24, 25, 26 Approximately 90% of EA 2 subjects show an interictal nystagmus, mainly gaze‐evoked or primary position downbeat nystagmus.27 Other neuro‐ophthalmological findings include impairment of visual tracking, optokinetic nystagmus, and suppression of the vestibulo‐ocular reflex (see Video 2). Saccade dysmetria and slowing of mean saccade velocity have been reported.22, 24, 28 Furthermore, patients with EA 2 display an increased epilepsy risk, compared to the background population.29 Gradual progressive baseline ataxia occurs in up to 50% of EA 2 patients.30 Approximately 50% of EA 2 subjects complain of frequent headaches, which fulfill the International Headache Society (IHS) criteria of migraine.30 Unusual features of EA 2 include cognitive deficits, abdominal pain, and possibly strabismus and hyperactivity disorders.31 Focal and segmental dystonia was reported in carriers of truncating CACNA1A mutations.32 Furthermore, there are several reports on paroxysmal torticollis and blepharospasm.33, 34, 35, 36 Animal models using CACNA1A‐mutant mice suggest that paroxysmal dystonia may result from disruption of a motor network involving both the basal ganglia and cerebellum.37
EA type 3 (EA 3), for which no specific mutations have been identified thus far, is characterized by attacks of vertigo, tinnitus, and ataxia.38 EA type 4 (EA 4) does not link to EA 1 and 2 loci and is accompanied by episodes of generalized ataxia, diplopia, imbalance, and vertigo.39 EA type 5 (EA 5) results from a mutation of a gene (CACNB4) encoding for voltage‐activated Ca2+ channels, which results in ACT‐responsive recurrent episodes of vertigo and ataxia.40 Mutations in the SLC1A3 gene impair glutamate transporter function and lead to EA type 6 (EA 6), which is associated with episodic and progressive ataxia.41 EA type 7 (EA 7) is associated with long‐lasting attacks of weakness and dysarthria; no specific mutations have been identified as yet.42 EA 2, EA 3, EA 5, and, to some extent, EA 1 are considered to be ACT‐responsive disorders.43 An overview of clinical features of EA 1 to 7 is given in Table 1 and Supporting Table 1.
Table 1.
Main features of episodic ataxia types 1 to 7
| EA1 | EA2 | EA3 | EA4 | EA5 | EA6 | EA7 | |
|---|---|---|---|---|---|---|---|
| Gene | KCNA1 | CACNA1a | Unknown | Unknown | CACNB4 | SLC1A3 | Unknown |
| Gene product | Kv1.1 | Cav2.1 | — | — | Cav2.1 | EAAT1 | — |
| Attack duration | <15 minutes | >10 minutes to hours | Minutes to hours | Brief | Hours | Hours to days | Hours to days |
| Frequent ictal symptomsa | Vertigo | Nystagmus, dysarthria, vertigo | Tinnitus, vertigo | Vertigo, diplopia, imbalance | Vertigo | Seizures, migraine, hemiplegia | Weakness, dysarthria |
| Interictal symptoms | Myokymia, epilepsy | Nystagmus, migraine epilepsy | Myokymia, nystagmus, epilepsy | Nystagmus | Nystagmus, epilepsy, ataxia | Ataxia, epilepsy | None |
| Acetazolamide response | Occasionally | Usually | Usually | No | Occasionally | No | Occasionally |
Excluding ataxia.
Furthermore, several reports on unclassified familial EAs have been published recently; Conroy et al. describe a three‐generation Irish family with an EA phenotype associated with ictal slurred speech and an unreported locus (candidate gene UBR4) on chromosome 1.44 Proline‐rich transmembrane protein gene (PRRT2) mutations have also rarely been associated with an EA phenotype.45, 46 Cha et al. reported late‐onset episodic vertical oscillopsia with interictal downbeat nystagmus and slowly progressive gait ataxia.47 Genetic testing for EA 1 and 2 and for SCA 1, 2, 3, and 6 were negative. A region of suggestive linkage was identified on chromosome 13.
Genetic and Molecular Aspects of EAs
The Online Mendelian Inheritance of Man (http://www.omim.org) currently lists seven clinical EA phenotypes (Table 1). Thus far, four causative genes have been identified.
EA 1 represents the first discovered human brain‐specific channelopathy.14 Heterozygous, usually missense mutations in the KCNA1 gene on chromosome 12p13 cause this autosomal‐dominant disorder.14 The KCNA1 gene consists of one single exon and encodes the alpha subunit of Kv1.1, a voltage‐gated potassium channel, which plays a crucial role in controlling neuronal excitability.14 Thus far, at least 28 (mostly missense) mutations have been described.4, 5, 11, 16, 18, 19 In vitro studies suggest that Kv1.1 mutations impair channel function and increase neuronal excitability.17, 48 The degree and severity of these dysfunctions may explain the various EA 1 phenotypes.4 Kv1.1 channels are preferentially localized on gamma‐aminobutyric acid (GABA)‐ergic cerebellar interneurons and are highly expressed in the hippocampus and in myelinated peripheral nerves.49 Electrophysiological recordings from cerebellar Purkinje cells (PCs) in a KCNA1 knock‐in mouse model suggest that KCNA1 mutations alter the excitability of the cerebellum. In myelinated nerves, Kv1.1 channels are densely expressed in the juxtaparanodal region of the axon.50 Activation of these K+ channels reduces resistance of the intermodal membrane and limits axonal hyperexcitability after an action potential.51 Mutant channels thus are expected to lead to excessive excitability of neurons.52
EA 2 is caused by a wide range of mutations in the CACNA1A gene, which contains 47 exons and is located on chromosome 19p13. CACNA1A encodes a voltage‐gated Cav2.1 P/Q‐type calcium channel (Cav2.1 channel).20, 53 In 1996, Ophoff et al. found four missense CACNA1A mutations in familial hemiplegic migraine type 1 (FHM1) and two CACNA1A mutations disrupting the reading frame in unrelated patients with EA 2.53 Thus, EA2 and FHM1 are allelic disorders. Subsequently, expansion of a polymorphic CAG repeat segment in CACNA1A was reported in patients with SCA6.54 The great majority of CACNA1A mutations associated with an EA 2 phenotype are nonsense mutations or defects in splice sites predicting a premature stop, resulting in a truncated protein product.55, 56 Cav2.1 channels are abundantly expressed in the cerebellum and at the neuromuscular junction. Because EA 2 is most often caused by loss‐of‐function mutations, it is associated with reduced calcium currents.57 Mutant channels probably lead to an altered or reduced calcium‐dependent neurotransmitter release, which, in turn, leads to altered intrinsic firing properties of the inhibitory effect of GABA‐ergic PCs.57, 58 However, markedly decreased calcium currents resulting from reductions of calcium conductance, as found in EA 2–causing mutations, have not been observed in mutations causing SCA6 or FHM1.21 Dysfunction of Cav2.1 channels has also been associated with altered time‐varying responses to transient increases in corticomotor excitability.59 Fluctuating muscle weakness of EA 2 patients is most probably the result of a presynaptic defect in neuromuscular transmission. In vitro microelectrode studies showed an increased jitter and blocking on single‐fiber electromyography (EMG).60 Most EA 2 cases are familial, but sporadic mutations were also described.24, 30 To date, at least 77 different mutations in CACNA1A have been associated with an EA 2 phenotype, but specific CACNA1A mutations do not strictly predict the EA 2 phenotype.25, 61 In line with this, a novel CACNA1A missense mutation has been recently associated with nonepisodic SCA with slow progression.62 To date, approximately only one third of suspected EA 2 cases could be associated with a detectable CACNA1A mutation.11, 35
Candidate genes have yet not been identified for EA 3, 4, and 7 (Supporting Table 1).
EA 5 is caused by a mutation in the CACNB4 gene on chromosome 2q22‐13.40 This gene encodes an isoform of the beta subunit of voltage‐activated Ca2+ channels. Mutations in CACNB4 are also found in generalized epilepsy and juvenile myoclonic epilepsy.63
EA 6 is caused by mutations in the SLC1A3 gene, encoding glial excitatory amino acid transporter 1 (EAAT1). EAAT1 is primarily expressed in the cerebellum, caudal brainstem, and diencephalon, mediates secondary active glutamate transport, and functions as an ion channel. A disease‐associated point mutation (P290R) has been shown to result in a reduced number of this transporter in the surface cell membrane and to impair its associated glutamate uptake.64 Nevertheless, the mutant channel shows larger anion currents than wild‐type cells, which argues for a gain of function. EA 6 represents the first human disease found to be associated with altered function of excitatory amino acid transporter anion channels.64
Recently reported EA phenotypes, which are associated with PRRT2 ‐ (and possibly UBR4 ‐) mutations demonstrate that ion channel dysfunction is not the unique pathophysiological mechanism of EA.45
Diagnostic Approach and Differential Diagnosis in EAs
Commercially genetic testing is currently available only for EA 1 and 2. In genetically negative EA 2 cases, mutations in CACNA1A promotor regions and introns, as other EA types (especially EA 6), have to be considered.
In EA 1, progressive ataxia occurs in a small proportion of individuals, but cerebellar atrophy seems to be rare.12, 16 EMG at rest of EA 1 subjects may show continuous spontaneous activity or afterpotentials and myokymic discharges.13, 52 In addition, the diagnostic utility of nerve excitability studies has been demonstrated in EA 1 recently.52 In EA 2, brain MRI may display an atrophy of the midline cerebellum, which is more accentuated in patients with long‐standing disease with persistent interictal ataxia.20 A small proportion of EA 2 patients complain of weakness during attacks, which is probably the result of presynaptic failure of neuromuscular transmission and synaptic remodeling. However, routine EMG and nerve conduction studies in EA 2 usually are unremarkable, but single‐fiber EMG and morphological studies of the neuromuscular junction may demonstrate signs of impaired neuromuscular transmission.65 Abnormal epileptiform EEG findings are frequent in EA 2 patients.29
The differential diagnoses of the EA spectrum diseases mainly include migraine, in particular, vestibular migraine, glucose transporter type 1 (GLUT1) deficiency syndrome, and PRRT2 mutations (Fig. 1). Preliminary studies show no higher frequencies of CACNA1A mutations in migraine with or without aura.66
Figure 1.
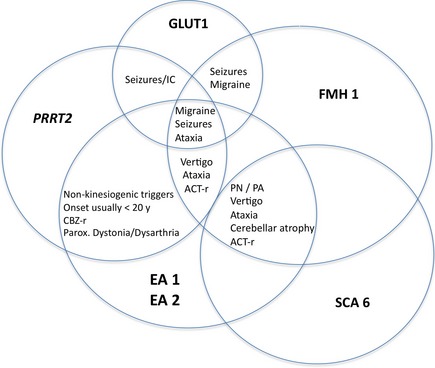
Clinical overlap of EA 1 and 2 and its differential diagnoses. ACT‐r, (possibly) acetazolamide responsive; CBZ‐r, (possibly) carbamazepine responsive; IC, infantile convulsions; PA, permanent ataxia; parox., paroxysmal; PN, permanent nystagmus; PRRT2, proline‐rich transmembrane protein gene (PRRT2) mutations.
Interictal clinical key features are of important diagnostic value. Because EA 1 and 2 represent the majority of diagnosed EA cases, one should systematically look for interictal myokymia, nystagmus, and ataxia. Onset of EA 1 and 2 is very rare after the age of 20. A negative family history does not rule out EA because sporadic cases have been described both in EA 1 and 2.24
CACNA1A allelic disorders such as FHM1 and SCA6 are common differential diagnoses of EA 2 and are sometimes (FHM1) or usually (SCA6) associated with progressive ataxia. Especially in FHM1 patients, a progressive cerebellar dysfunction may lead to a phenotype that is clinically indistinguishable from EA 2 (Fig. 1).67 Moreover, fluctuating disease course with treatment response to acetazolamide in SCA6 is possible.
Therapy
ACT, carbamazepine (CBZ), and valproic acid have been shown to be effective in some EA 1 subjects.68 Phenytoin is contraindicated in EA 2, but one article reports good control of symptoms in EA 1.69 Approximately two thirds of EA 2 patients respond to ACT treatment (250–1,000 mg/day).3, 30 However, placebo‐controlled trials about the efficacy of ACT are lacking, and side effects (e.g., kidney stones) limit its therapeutic use.
Efficacy of the potassium channel blocker, 4‐aminopyridine (4‐AP), in EA 2 was shown in an observational study in 2004.70 4‐AP treatment was safe and associated with a reduction of attacks. Furthermore, positive effects of 4‐AP on ataxia attacks have been confirmed in studies using an animal model of EA 2, the tottering mouse.58, 71 In 2011, a randomized, placebo‐controlled, double‐blind, crossover trial demonstrated a significant effect of this agent on the number of attacks and quality of life.72 Therefore, we recommend 4‐AP in a dosage of 5 to 10 mg 3 times a day in EA 2 patients with no, or a declining, ACT treatment response. Recent results from an observational study suggest that the sustained release form of 4‐AP is also effective and well tolerated.73 Furthermore, segmental dystonia in EA 2 has recently been successfully treated by DBS.36
Perspectives
There are several promising ongoing and proposed clinical trials in EA research. Performing a next‐generation sequencing study, E. Mantuano from Rome tries to identify new genes involved in EA syndromes. To achieve this, whole‐genome sequencing in a series of extended multigenerational EA pedigrees (in which known EA‐causative genes were excluded) will be used.
Furthermore, there are two ongoing placebo‐controlled trials with the sustained release form of 4‐AP (University of California, NCT01543750) and a randomized, controlled three‐arm phase III trial comparing the sustained release form of 4‐AP with ACT and placebo (EAT2TREAT; EudraCT‐Nr.: 2013‐000107‐17; University of Munich, EAT‐2‐TREAT, EudraCT‐Nr.: 2013‐000107‐17).
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Exe‐cution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the First Draft, B. Review and Critique.
S.K.: 1A, 1B, 1C, 3A
M.S.: 1A, 1B, 1C, 3B
Disclosures
Funding Sources and Conflicts of Interest: This work was supported by a grant to M.S. from the German Federal Ministry of Education and Research (BMBF) to the Integrierte Forschungs‐ und Behandlungszentren (IFB [integrated research and treatment centers]; grant code: 01 EO 0901). The authors report no conflicts of interest.
Financial Disclosures for previous 12 months: M.S. is joint chief editor of the Journal of Neurology, editor in chief of Frontiers of Neuro‐otology, and section editor of F1000. He has received speaker's honoraria from Abbott, Actelion, UCB, GlaxoSmithKline, Teva, Biogen Idec, Pierre‐Fabre, Eisai, and Hennig Pharma.
Supporting information
A video accompanying this article is available in the supporting information here.
SUPPORTING TABLE 1. Overview of genetic aspects and clinical features of episodic ataxia types 3 to 7.
Video 1. EA 2 patient history.
Video 2. Downbeat nystagmus syndrome: typical finding in patients with episodic ataxia type 2. A downbeat nystagmus occurs in the primary position; it intensifies during convergence or when looking to the side. It is typically associated with a gaze‐evoked nystagmus and saccadic smooth pursuit, in particular, in the vertical plane. These are typical ocular motor findings for a dysfunction of the cerebellum, namely, the flocculus and paraflocculus. Nystagmus is superimposed over gaze pursuit. When testing the vertical optokinetic nystagmus, the downbeat nystagmus appears to be lessened when the stimulus pattern is turned downward.
Stefan Kipfer and Michael Strupp contributed equally to the manuscript.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Gancher ST, Nutt JG. Autosomal dominant episodic ataxia: a heterogeneous syndrome. Mov Disord 1986;1:239–253. [DOI] [PubMed] [Google Scholar]
- 2. Parker HL. Periodic ataxia. Collect Papers Mayo Clinic Mayo Found 1946. –1947;38:642–645. [PubMed] [Google Scholar]
- 3. Griggs RC, Moxley RT III, Lafrance RA, McQuillen J. Hereditary paroxysmal ataxia: response to acetazolamide. Neurology 1978;28:1259–1264. [DOI] [PubMed] [Google Scholar]
- 4. Jen JC, Graves TD, Hess EJ, Hanna MG, Griggs RC, Baloh RW; CINCH3s investigators . Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain 2007;130:2484–2493. [DOI] [PubMed] [Google Scholar]
- 5. Graves TD, Rajakulendran S, Zuberi SM, et al. Nongenetic factors influence severity of episodic ataxia type 1 in monozygotic twins. Neurology 2010;75:367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. D'Adamo MC, Hanna MG, Di Giovanni G, Pessia M. Episodic ataxia type 1 In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 2010:1993–2013. [Google Scholar]
- 7. Brunt ER, van Weerden TW. Familial paroxysmal kinesigenic ataxia and continuous myokymia. Brain 1990;113:1361–1382. [DOI] [PubMed] [Google Scholar]
- 8. Klein A, Boltshauser E, Jen J, Baloh RW. Episodic ataxia type 1 with distal weakness: a novel manifestation of a potassium channelopathy. Neuropediatrics 2004;35:147–149. [DOI] [PubMed] [Google Scholar]
- 9. Lubbers WJ, Brunt ER, Scheffer H, et al. Hereditary myokymia and paroxysmal ataxia linked to chromosome 12 is responsive to acetazolamide. J Neurol Neurosurg Psychiatry 1995;59:400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee H, Wang H, Jen JC, Sabatti C, Baloh RW, Nelson SF. A novel mutation in KCNA1 causes episodic ataxia without myokymia. Hum Mutat 2004;24:536. [DOI] [PubMed] [Google Scholar]
- 11. Tomlinson SE, Rajakulendran S, Tan SV, et al. Clinical, genetic, neurophysiological and functional study of new mutations in episodic ataxia type 1. J Neurol Neurosurg Psychiatry 2013;84:1107–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Graves TD, Cha YH, Hahn AF, et al. Episodic ataxia type 1: clinical characterization, quality of life and genotype‐phenotype correlation. Brain 2014;137:1009–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. VanDyke DH, Griggs RC, Murphy MJ, Goldstein MN. Hereditary myokymia and periodic ataxia. J Neurol Sci 1975;25:109–118. [DOI] [PubMed] [Google Scholar]
- 14. Browne DL, Gancher ST, Nutt JG, et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet 1994;8:136–140. [DOI] [PubMed] [Google Scholar]
- 15. Zuberi SM, Eunson LH, Spauschus A, et al. A novel mutation in the human voltage‐gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain 1999;122:817–825. [DOI] [PubMed] [Google Scholar]
- 16. Demos MK, Macri V, Farrell K, et al. Novel KCNA1 mutation associated with global delay and persistent cerebellar dysfunction. Mov Disord 2009;24:778–782. [DOI] [PubMed] [Google Scholar]
- 17. Eunson LH, Rea R, Zuberi SM, et al. Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann Neurol 2000;48:647–656. [PubMed] [Google Scholar]
- 18. Imbrici P, Gualandi F, D'Adamo MC, et al. A novel KCNA1 mutation identified in an Italian family affected by episodic ataxia type 1. Neuroscience 2008;157:577–587. [DOI] [PubMed] [Google Scholar]
- 19. Shook SJ, Mamsa H, Jen JC, Baloh RW, Zhou L. Novel mutation in KCNA1 causes episodic ataxia with paroxysmal dyspnea. Muscle Nerve 2008;37:399–402. [DOI] [PubMed] [Google Scholar]
- 20. Spacey S. Episodic ataxia type 2 In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 2003:1993–2013. [Google Scholar]
- 21. Baloh RW. Episodic ataxias 1 and 2. Handb Clin Neurol 2012;103:595–602. [DOI] [PubMed] [Google Scholar]
- 22. Subramony SH, Schott K, Raike RS, et al. Novel CACNA1A mutation causes febrile episodic ataxia with interictal cerebellar deficits. Ann Neurol 2003;54:725–731. [DOI] [PubMed] [Google Scholar]
- 23. Imbrici P, Eunson LH, Graves TD, et al. Late‐onset episodic ataxia type 2 due to an in‐frame insertion in CACNA1A. Neurology 2005;65:944–946. [DOI] [PubMed] [Google Scholar]
- 24. Kipfer S, Jung S, Lemke JR, et al. Novel CACNA1A mutation(s) associated with slow saccade velocities. J Neurol 2013;260:3010–3014. [DOI] [PubMed] [Google Scholar]
- 25. Hu Y, Jiang H, Wang Q, Xie Z, Pan S. Identification of a novel nonsense mutation p.Tyr1957Ter of CACNA1A in a Chinese family with episodic ataxia 2. PLoS One 2013;8:e56362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rucker JC, Jen J, Stahl JS, Natesan N, Baloh RW, Leigh RJ. Internuclear ophthalmoparesis in episodic ataxia type 2. Ann N Y Acad Sci 2005;1039:571–574. [DOI] [PubMed] [Google Scholar]
- 27. Riant F, Vahedi K, Tournier‐Lasserve E. Hereditary episodic ataxia. Rev Neurol (Paris) 2011;167:401–407. [DOI] [PubMed] [Google Scholar]
- 28. Baloh RW, Yue Q, Furman JM, Nelson SF. Familial episodic ataxia: clinical heterogeneity in four families linked to chromosome 19p. Ann Neurol 1997;41:8–16. [DOI] [PubMed] [Google Scholar]
- 29. Rajakulendran S, Graves TD, Labrum RW, et al. Genetic and functional characterisation of the P/Q calcium channel in episodic ataxia with epilepsy. J Physiol 2010;588:1905–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jen J, Kim GW, Baloh RW. Clinical spectrum of episodic ataxia type 2. Neurology 2004;62:17–22. [DOI] [PubMed] [Google Scholar]
- 31. Bertholon P, Chabrier S, Riant F, Tournier‐Lasserve E, Peyron R. Episodic ataxia type 2: unusual aspects in clinical and genetic presentation. Special emphasis in childhood. J Neurol Neurosurg Psychiatry 2009;80:1289–1292. [DOI] [PubMed] [Google Scholar]
- 32. Spacey SD, Materek LA, Szczygielski BI, Bird TD. Two novel CACNA1A gene mutations associated with episodic ataxia type 2 and interictal dystonia. Arch Neurol 2005;62:314–316. [DOI] [PubMed] [Google Scholar]
- 33. Giffin NJ, Benton S, Goadsby PJ. Benign paroxysmal torticollis of infancy: four new cases and linkage to CACNA1A mutation. Dev Med Child Neurol 2002;44:490–493. [DOI] [PubMed] [Google Scholar]
- 34. Roubertie A, Echenne B, Leydet J, et al. Benign paroxysmal tonic upgaze, benign paroxysmal torticollis, episodic ataxia and CACNA1A mutation in a family. J Neurol 2008;255:1600–1602. [DOI] [PubMed] [Google Scholar]
- 35. Mantuano E, Romano S, Veneziano L, et al. Identification of novel and recurrent CACNA1A gene mutations in fifteen patients with episodic ataxia type 2. J Neurol Sci 2010;291:30–36. [DOI] [PubMed] [Google Scholar]
- 36. Harries AM, Sandhu M, Spacey SD, Aly MM, Honey CR. Unilateral pallidal deep brain stimulation in a patient with dystonia secondary to episodic ataxia type 2. Stereotact Funct Neurosurg 2013;91:233–235. [DOI] [PubMed] [Google Scholar]
- 37. Neychev VK, Fan X, Mitev VI, Hess EJ, Jinnah HA. The basal ganglia and cerebellum interact in the expression of dystonic movement. Brain 2008;131:2499–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Steckley JL, Ebers GC, Cader MZ, McLachlan RS. An autosomal dominant disorder with episodic ataxia, vertigo, and tinnitus. Neurology 2001;57:1499–1502. [DOI] [PubMed] [Google Scholar]
- 39. Damji KF, Allingham RR, Pollock SC, et al. Periodic vestibulocerebellar ataxia, an autosomal dominant ataxia with defective smooth pursuit, is genetically distinct from other autosomal dominant ataxias. Arch Neurol 1996;53:338–344. [DOI] [PubMed] [Google Scholar]
- 40. Escayg A, De Waard M, Lee DD, et al. Coding and noncoding variation of the human calcium‐channel beta4‐subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet 2000;66:1531–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jen JC, Wan J, Palos TP, Howard BD, Baloh RW. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology 2005;65:529–534. [DOI] [PubMed] [Google Scholar]
- 42. Kerber KA, Jen JC, Lee H, Nelson SF, Baloh RW. A new episodic ataxia syndrome with linkage to chromosome 19q13. Arch Neurol 2007;64:749–752. [DOI] [PubMed] [Google Scholar]
- 43. Kotagal V. Acetazolamide‐responsive ataxia. Semin Neurol 2012;32:533–537. [DOI] [PubMed] [Google Scholar]
- 44. Conroy J, McGettigan P, Murphy R, et al. A novel locus for episodic ataxia: UBR4 the likely candidate. Eur J Hum Genet 2014;22:505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gardiner AR, Bhatia KP, Stamelou M, et al. PRRT2 gene mutations: from paroxysmal dyskinesia to episodic ataxia and hemiplegic migraine. Neurology 2012;79:2115–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Labate A, Tarantino P, Viri M, et al. Homozygous c.649dupC mutation in PRRT2 worsens the BFIS/PKD phenotype with mental retardation, episodic ataxia, and absences. Epilepsia 2012;53:e196–e199. [DOI] [PubMed] [Google Scholar]
- 47. Cha YH, Lee H, Jen JC, Kattah JC, Nelson SF, Baloh RW. Episodic vertical oscillopsia with progressive gait ataxia: clinical description of a new episodic syndrome and evidence of linkage to chromosome 13q. J Neurol Neurosurg Psychiatry 2007;78:1273–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bretschneider F, Wrisch A, Lehmann‐Horn F, Grissmer S. Expression in mammalian cells and electrophysiological characterization of two mutant Kv1.1 channels causing episodic ataxia type 1 (EA‐1). Eur J Neurosci 1999;11:2403–2412. [DOI] [PubMed] [Google Scholar]
- 49. Herson PS, Virk M, Rustay NR, et al. Mouse model of episodic ataxia type‐1. Nat Neurosci 2003;6:378–383. [DOI] [PubMed] [Google Scholar]
- 50. Scherer SS, Arroyo EJ. Recent progress on the molecular organization of myelinated axons. J Peripher Nerv Syst 2002;7:1–12. [DOI] [PubMed] [Google Scholar]
- 51. Bostock H, Baker M. Evidence for two types of potassium channel in human motor axons in vivo. Brain Res 1988;462:354–358. [DOI] [PubMed] [Google Scholar]
- 52. Tan SV, Wraige E, Lascelles K, Bostock H. Episodic ataxia type 1 without episodic ataxia: the diagnostic utility of nerve excitability studies in individuals with KCNA1 mutations. Dev Med Child Neurol 2013;55:959–962. [DOI] [PubMed] [Google Scholar]
- 53. Ophoff RA, Terwindt GM, Vergouwe MN, et al. Familial hemiplegic migraine and episodic ataxia type‐2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 1996;87:543–552. [DOI] [PubMed] [Google Scholar]
- 54. Zhuchenko O, Bailey J, Bonnen P, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A‐voltage‐dependent calcium channel. Nat Genet 1997;15:62–69. [DOI] [PubMed] [Google Scholar]
- 55. Eunson LH, Graves TD, Hanna MG. New calcium channel mutations predict aberrant RNA splicing in episodic ataxia. Neurology 2005;65:308–310. [DOI] [PubMed] [Google Scholar]
- 56. Denier C, Ducros A, Vahedi K, et al. High prevalence of CACNA1A truncations and broader clinical spectrum in episodic ataxia type 2. Neurology 1999;52:1816–1821. [DOI] [PubMed] [Google Scholar]
- 57. Kullmann DM. The neuronal channelopathies. Brain 2002;125:1177–1195. [DOI] [PubMed] [Google Scholar]
- 58. Alvina K, Khodakhah K. KCa channels as therapeutic targets in episodic ataxia type‐2. J Neurosci 2010;30:7249–7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Helmich RC, Siebner HR, Giffin N, Bestmann S, Rothwell JC, Bloem BR. The dynamic regulation of cortical excitability is altered in episodic ataxia type 2. Brain 2010;133:3519–3529. [DOI] [PubMed] [Google Scholar]
- 60. Jen J, Wan J, Graves M, et al. Loss‐of‐function EA2 mutations are associated with impaired neuromuscular transmission. Neurology 2001;57:1843–1848. [DOI] [PubMed] [Google Scholar]
- 61. Barros J, Damásio J, Tuna A, et al. Cerebellar ataxia, hemiplegic migraine, and related phenotypes due to a CACNA1A missense mutation: 12‐year follow‐up of a large Portuguese family. JAMA Neurol 2013;70:235–240. [DOI] [PubMed] [Google Scholar]
- 62. Bürk K, Kaiser FJ, Tennstedt S, et al. A novel missense mutation in CACNA1A evaluated by in silico protein modeling is associated with non‐episodic spinocerebellar ataxia with slow progression. Eur J Med Genet 2014;57:207–211. [DOI] [PubMed] [Google Scholar]
- 63. Herrmann A, Braathen GJ, Russell MB. Episodic ataxias. Tidsskr Nor Laegeforen 2005;125:2005–2007. [PubMed] [Google Scholar]
- 64. Winter N, Kovermann P, Fahlke C. A point mutation associated with episodic ataxia 6 increases glutamate transporter anion currents. Brain 2012;135:3416–3425. [DOI] [PubMed] [Google Scholar]
- 65. Maselli RA, Wan J, Dunne V, et al. Presynaptic failure of neuromuscular transmission and synaptic remodeling in EA2. Neurology 2003;61:1743–1748. [DOI] [PubMed] [Google Scholar]
- 66. Meamar R, Ostadsharif M, Saadatnia M, et al. Mutation analysis of CACNA1A gene in Iranian migrainous and review literatures. J Res Med Sci 2013;18:S6–S10. [PMC free article] [PubMed] [Google Scholar]
- 67. Ducros A, Denier C, Joutel A, et al. The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N Engl J Med 2001;345:17–24. [DOI] [PubMed] [Google Scholar]
- 68. Scoggan KA, Friedman JH, Bulman DE. CACNA1A mutation in a EA‐2 patient responsive to acetazolamide and valproic acid. Can J Neurol Sci 2006;33:68–72. [DOI] [PubMed] [Google Scholar]
- 69. Vaamonde J, Artieda J, Obeso JA. Hereditary paroxysmal ataxia with neuromyotonia. Mov Disord 1991;6:180–182. [DOI] [PubMed] [Google Scholar]
- 70. Strupp M, Kalla R, Dichgans M, Freilinger T, Glasauer S, Brandt T. Treatment of episodic ataxia type 2 with the potassium channel blocker 4‐aminopyridine. Neurology 2004;62:1623–1625. [DOI] [PubMed] [Google Scholar]
- 71. Weisz CJ, Raike RS, Soria‐Jasso LE, Hess EJ. Potassium channel blockers inhibit the triggers of attacks in the calcium channel mouse mutant tottering. J Neurosci 2005;25:4141–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Strupp M, Kalla R, Claassen J, et al. A randomized trial of 4‐aminopyridine in EA2 and related familial episodic ataxias. Neurology 2011;77:269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Claassen J, Teufel J, Kalla R, Spiegel R, Strupp M. Effects of dalfampridine on attacks in patients with episodic ataxia type 2: an observational study. J Neurol 2013;260:668–669. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A video accompanying this article is available in the supporting information here.
SUPPORTING TABLE 1. Overview of genetic aspects and clinical features of episodic ataxia types 3 to 7.
Video 1. EA 2 patient history.
Video 2. Downbeat nystagmus syndrome: typical finding in patients with episodic ataxia type 2. A downbeat nystagmus occurs in the primary position; it intensifies during convergence or when looking to the side. It is typically associated with a gaze‐evoked nystagmus and saccadic smooth pursuit, in particular, in the vertical plane. These are typical ocular motor findings for a dysfunction of the cerebellum, namely, the flocculus and paraflocculus. Nystagmus is superimposed over gaze pursuit. When testing the vertical optokinetic nystagmus, the downbeat nystagmus appears to be lessened when the stimulus pattern is turned downward.