

Abstract
Background
Spinal cord demyelination can cause several movement disorders. Although these abnormal movements could be the presenting symptom of the disease and, at times, the major source of disability, they are often overlooked, mislabeled, or undertreated. The aims of this study were to clearly define and establish common terminology for spinal movement disorders (SMDs) and characterize their full spectrum in patients with neuromyelitis optica (NMO).
Methods
We chart reviewed 37 patients with NMO or NMO spectrum disorder. We classified spinal movement disorders under five categories: tonic spasms; focal dystonia; spinal myoclonus; spontaneous clonus; and tremors of spinal origin. We examined clinical, MRI, and medication data of symptomatic patients.
Results
Of the 37 patients (86.4% female; mean age: 51 ± 17 years; mean disease duration: 9.4 ± 5.3 years), 16 (43.2%) had one or more form of SMDs. Compared to those without SMDs, patients with SMDs were generally older at presentation and were less likely to be African Americans. An abnormal movement was the main complaint in at least one posthospitalization visit in all symptomatic patients. Thirteen (35.1%) patients had paroxysmal tonic spasms, 2 (5.4%) had focal dystonia, 3 (8%) had postural/action tremors, and no patient had spinal myoclonus or spontaneous clonus. In 9 patients, spasms were painful. There was no signal abnormality in the basal ganglia or the brainstem/cerebellum in any of the symptomatic patients.
Conclusions
SMDs are common in NMO and are often a major source of disability. Using clear, unified terminology to describe SMDs is crucial for both clinical and research purposes.
Keywords: neuromyelitis optica, neuromyelitis optica spectrum disorders, movement disorders, spinal movement disorders, tonic spasms
The prototype pathological substrates for movement disorders are the basal ganglia, other deep subcortical nuclei, the cerebellum, and their connections in the brainstem.1
Comparatively, spinal movement disorders (SMDs) are under‐recognized despite their commonness in clinical practice, their significant contribution to patients' morbidity, and their potential diagnostic utility.2, 3 SMDs can result from any spinal cord pathology, but are most commonly described in patients with cord demyelination.4 SMDs have been described in neuromyelitis optica (NMO),2, 5 multiple sclerosis (MS),6 idiopathic transverse myelitis (ITM),7 and secondary transverse myelitis.8 Historically, studies describing movement disorders of spinal origin have used different terminologies to describe the same movement disorder. For example, spinal seizures,9 painful tonic spasms,2 paroxysmal dystonia,10 and paroxysmal kinesogenic dyskinesia11 have all been used to describe tonic spasms. Moreover, the same terminology had been used to describe two different entities, for example, using the term paroxysmal dystonia to describe both tonic spasm in simple flexion and a more complex dystonic posture of the involved limb.10, 12 One of the aims of the current study was to clearly define and establish common terminology for the categories of SMDs for clinical and academic purposes. The second aim was to characterize the full spectrum of SMDs in patients with NMO using our proposed definitions and classification algorithm.
Materials and Methods
Proposed definitions, classification, and unified terminology for SMDs in NMO
Based on our clinical experience as well as previous reports in the literature, we classified SMDs into five main types (see Table 1): (1) tonic spasms: subclassified to extensor, flexor, isometric, and unspecified spasms; (2) focal dystonia: subclassified to paroxysmal or nonparoxysmal dystonia; (3) spinal myoclonus: subclassified to focal/segmental myoclonus and propriospinal myoclonus; (4) tremors of spinal origin; and (5) spontaneous (noninducible) clonus.
Table 1.
Proposed definitions of SMDs in NMO
| SMD | Definition |
|---|---|
| Flexor tonic spasm | Paroxysmal sustained increase in muscle tone resulting in visible tonic posturing of the affected body part (often the whole limb or part of the limb) in flexion secondary to spinal cord pathology |
| Extensor tonic spasm | Paroxysmal sustained increase in muscle tone resulting in visible tonic posturing of the affected body part (often the whole limb or part of the limb) in extension secondary to spinal cord pathology |
| Isometric tonic spasm | Paroxysmal sustained increase in muscle tone that can be felt by the patient and palpated by the examiner, but does not result in visible change in posture (e.g., abdominal wall muscles) secondary to spinal cord pathology. |
| Unspecified tonic spasm | A tonic spasm not meeting criteria for other spinal movement disorders or not specified in documentation secondary to spinal cord pathology |
| Paroxysmal focal dystonia | Paroxysmal involuntary sustained muscle contraction of antagonistic muscle groups resulting in abnormal posture (other than simple flexion or extension) secondary to spinal cord pathology |
| Nonparoxysmal focal dystonia | Persistent (nonparoxysmal) sustained muscle contraction of antagonistic muscle groups resulting in a fixed abnormal posture secondary to spinal cord pathology |
| Focal/Segmental spinal myoclonus | Sudden, brief (nonsustained), shock‐like focal muscle contraction of one or more adjacent body parts secondary to spinal cord pathology |
| Propriospinal myoclonus | Sudden, brief, shock‐like, arrhythmic jerks of the trunk, hips, and knees (sparing the head) often in flexion secondary to spinal cord pathology |
| Tremors of spinal origin | Postural and/or action tremors occurring after spinal cord pathological event in absence of brain/brainstem/cerebellar lesions and personal or family history of essential tremors |
| Spontaneous clonus | Spontaneous (noninduced) involuntary, rhythmic muscle contractions and relaxations associated with spasticity secondary to spinal cord pathology. It commonly involves the ankle or the wrist and appears in certain positions or with certain movements. |
After determining the SMD type and subtype, we further classified each SMD according to the presence of pain (painful or painless), its relation to stimulation (stimulus sensitive or stimulus insensitive), and its relation to movement (kinesogenic or nonkinesogenic).
To characterize the spectrum of SMDs in NMO patients, we chart reviewed a sample of NMO/NMO spectrum disorder (NMOSD) patients. The patient cohort was identified from a data registry of NMO inpatient admissions from 2005 to 2013 that were treated with plasmapharesis for one or more acute relapses during that period and continued to follow at the NMO clinic in 2014 at the time of data collection. Only patients with spinal cord involvement were included in the analysis whereas patients with isolated optic nerve and/or brain/brainstem involvement were excluded. Demographic, clinical, radiological, and treatment data were collected and analyzed. Patients' charts were surveyed for documentation or description of SMDs, and each SMD type was subsequently categorized according to the classification algorithm. When clear documentation of the exact type of movement disorder was lacking, the description of the abnormal movement was reviewed by a movement disorder specialist and subsequently categorized.
Chi‐square and t tests were used to compare demographic data of patients with SMDs and those without SMDs. A level of significance <0.05 was utilized. This study was approved by the Johns Hopkins Institutional Review Board (Baltimore, MD).
Results
The patient cohort consisted of 37 patients (mean age: 51 ± 17 years; mean disease duration: 9.4 ± 5.3 years; 86% females). According to the Wingerchuck 2006 and 2007 diagnostic criteria,13, 14 73% met criteria for antiaquaporin 4/immunoglobulin G (AQP4‐IgG)‐seropositive NMO, 19% met criteria for AQP4‐IgG‐seropositive NMOSD, and 8% met criteria for seronegative NMO. Based on the new 2015 International Panel NMO Diagnostic criteria, 92% of the patients met criteria for NMOSD with AQP4‐IgG and 8% met criteria for NMOSD without AQP4‐IgG.15 .
Of 37, there were 16 patients (43.2%) with one or more types of SMDs (see Fig. 1). Patients with SMDs had a mean age of 55.9 ± 14.5 years, mean disease duration of 5.6 ± 4.4 years, and 93.7% of them were female. They were significantly older at presentation, compared to patients without SMDs (P = 0.020727), and were less likely to be African Americans compared to patients without SMDs (P = 0.000649). There was no significant difference in sex, disease duration, or AQP4‐IgG serostatus between patients with SMDs and those without SMDs (see Table 2). SMDs usually started an average of 3 months after a spinal cord attack. In 4 patients (25%), SMDs occurred in the setting of acute relapse (clinical and radiological), manifesting with tonic spasms in all 4. There were no lesions in the basal ganglia, brainstem (excluding the cervicomedullary junction [CMJ]), or cerebellum in any of the symptomatic patients before or at the time of the development of the SMD. The movement disorder was the main complaint, at least at one posthospitalization visit, in all 16 symptomatic patients.
Figure 1.
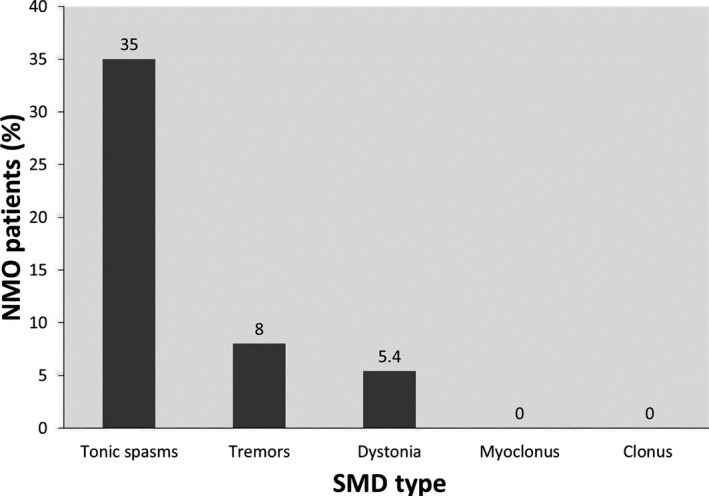
Frequency of SMDs in entire NMO cohort. Tonic spasms were the most frequent SMD in the NMO patient cohort, followed by action tremors, then focal dystonia. There were no documented cases of spinal myoclonus or spontaneous clonus. S. myoclonus, spinal myoclonus; Spont. Clonus, spontaneous clonus.
Table 2.
NMO with SMDs vs. NMO without SMDs
| Characteristics | NMO Study Cohort | NMO With SMD | NMO Without SMD | P Value |
|---|---|---|---|---|
| No. patients | 37 | 16 | 21 | |
| Mean age, years | 51 ± 17 | 55.9 ± 14.5 | 40.7 ± 16.4 | 0.020727 |
| Female (%) | 31 (86) | 15 (93.7) | 16 (76.2) | 0.151122 |
| Duration of disease, years | 9.4 ± 5.3 | 3.8 ± 3.6 | 5.6 ± 4.4 | 0.406825 |
| Race (%) | ||||
| African American | 25 (67) | 6 (37.5) | 19 (90.4) | 0.000649 |
| Caucasian | 9 (24) | 7 (43.7) | 2 (9.5) | 0.016218 |
| Hispanic | 3 (8) | 3 (18.7) | 0 (0) | 0.038452 |
| AQP4‐IgG (%) | ||||
| Seropositive | 34 (92) | 15 (93.75) | 19 (90.4) | 0.717778 |
| Seronegative | 3 (8) | 1 (6.25) | 2 (9.6) | |
Tonic spasms
Of the 16 NMO/NMOSD with SMDs, 13 patients had tonic type spasms: 8 unspecified, 3 flexor, 2 extensor, and 1 isometric in the chest who also had unspecified spams in the arms and legs (Fig. 2). In 9 patients (69%), the spasms were painful. In 5 patients (38%), the spasms were kinesogenic and stimulus sensitive. In 3 patients, the spams involved one side of the body (arm and leg); in 1 patient, both arms were involved sparing the legs; in 1 patient, only neck flexors were involved; and in 1 patient, the chest muscles were involved. In 8 patients, all four limbs were involved. When documented, the duration of tonic spasms lasted for an average of 30 to 60 seconds and occurred several times a day up to 100 times in 1 patient. All patients with tonic spasms had associated neuropathic pain, and 4 had an associated truncal band‐like feeling at the part corresponding to their sensory‐level “MS hug.” In 3 patients, urinary tract infection was identified at the time of worsening of tonic spasms.
Figure 2.
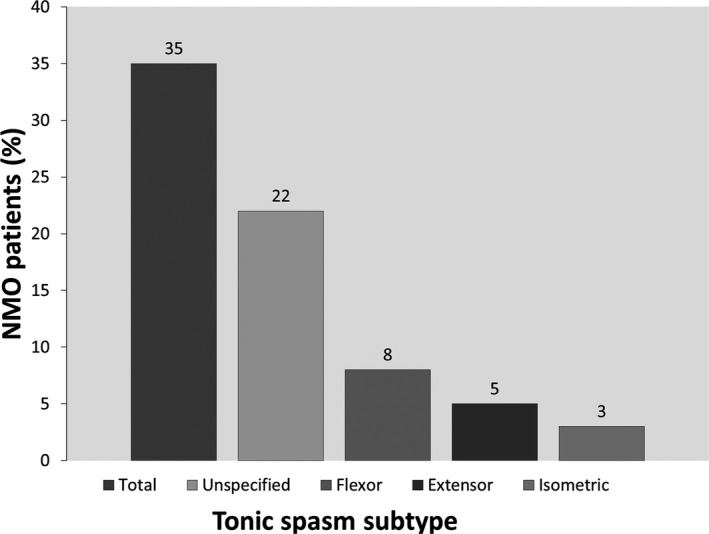
Frequency of tonic spasm subtypes in the entire NMO cohort. Unspecified spasms were the most common, followed by flexor spasms, extensor spasms, and isometric spasms.
Tremors and dystonia
Three patients had postural/action tremors of the hands with acute/subacute onset subsequent to a spinal relapse in the cervical spine. Two patients had focal nonparoxysmal dystonia (cervical and left arm). Tremors and nonparoxysmal dystonia were painless and stimulus insensitive.
Spinal myoclonus and spontaneous clonus
There were no documented cases of spinal myoclonus or spontaneous clonus in this cohort.
Radiological Findings
In all 16 patients with SMDs, spinal cord lesions where located anywhere from the level of the CMJ rostrally to the eighth thoracic vertebra caudally. The average lesion length was 4.75 segments (range, 1–11). Four patients with SMDs had lesion lengths equal or less than two segments. Nine patients had their lesions in the cervical region (including 3 with cervicothoracic lesions), 6 in the thoracic region only, and 1 in the CMJ. All patients with dystonia and hand tremor had lesions in the upper cervical region without lesions in the cerebellum, basal ganglia, or brainstem. Otherwise, there was no correlation between lesion location and type of SMD. Data on lesion locations on the axial cuts (anterior vs. posterior and gray vs. white matter) were not collected for this study.
Treatment Response
All patients were treated with either antiepileptics, muscle relaxants, or a combination of both classes (Fig. 3). Most patients were treated with gabapentin or pregabalin to address both neuropathic pain and spasms. Excellent treatment response (resolution of SMD) was documented in 3 patients, 1 of whom was treated with carbamazepine alone, 1 with baclofen pump after failing other oral agents, including carbamazepine, and 1 who received botulinium toxin injection for cervical dystonia (Fig. 4). Poor response (persistence of SMD) was documented in 2 patients with tonic spasms: 1 treated with gabapentin and muscle relaxants and 1 treated with medication combination, including oxcarbazepine. The rest of the patients reported fair response to treatment (some improvement in SMD frequency). In the 6 patients where documentation of SMDs at latest follow‐up was available, 3 patients reported resolution or significant improvement of their SMDs, including the patient who had excellent initial response to carbamazepine. Three patients reported persistent tonic spasms despite treatment, including the patient who had initial excellent response to baclofen pump. Median latest available follow‐up was 4 years.
Figure 3.
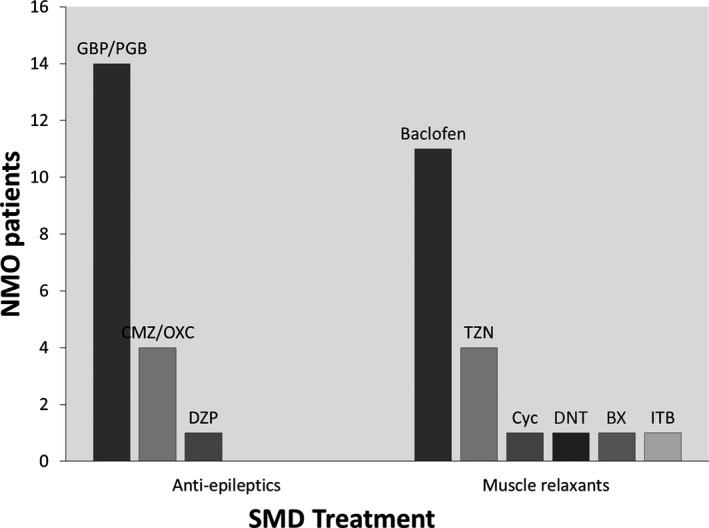
Symptomatic medications used for treatment of SMDs. GBP, gabapentin; PGB, pregabalin; CMZ, carbamezapine; DZP, diazepam; TZN, tizanadine; CYC, cyclobenzabrine; DNT, dantrolene; BX, botulinium toxin; ITB, intrathecal baclofen.
Figure 4.
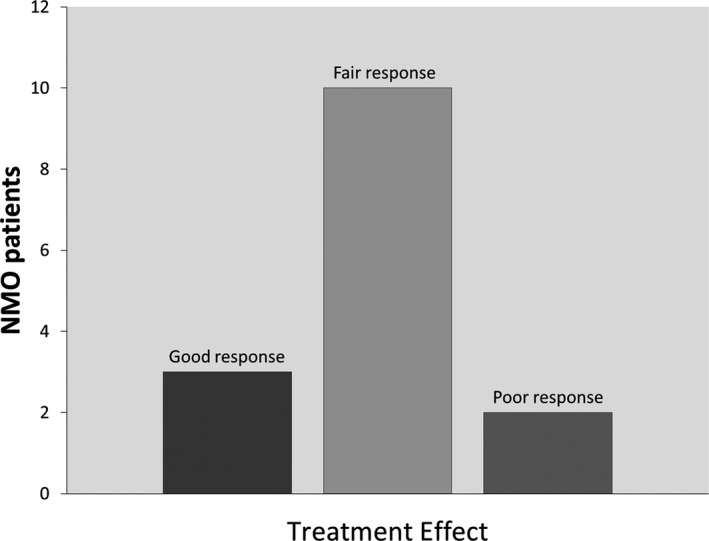
Response to symptomatic treatment. Good response = resolution of SMDs; fair response = improvement in SMDs frequency; poor response = persistence of SMDs at the same frequency.
Figure 5.
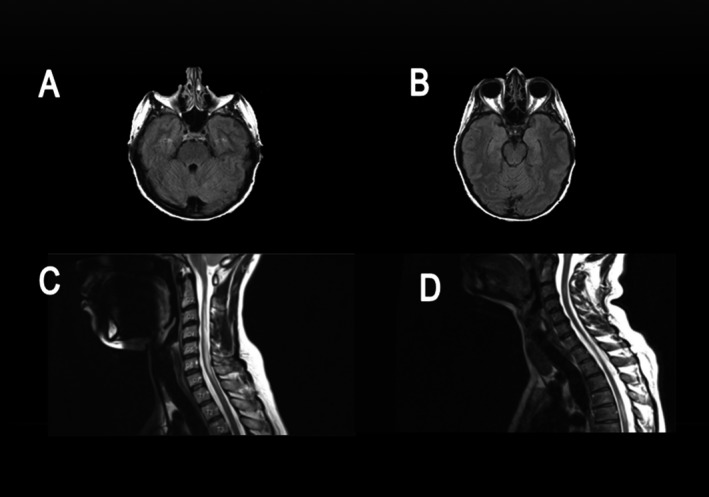
Sample MRI images from the NMO cohort. (A and B) Axial fluid‐attenuated inversion recovery images from patient 1 showing absence of demyelinating lesions in the brainstem and cerebellum. (C) Sagittal T2 Image from patient 2 showing ongitudinally extensive transverse myelitis lesion in the cervical spine. (D) Sagittal T2 image from patient 3 showing multifocal plaques in the cervical and thoracic spine.
Discussion
In this study, we attempted to clarify the ambiguity with which movement disorders of spinal origin have been reported in the literature. We proposed clear definitions and a classification algorithm, and we applied it to a sample of NMO patients in whom SMDs are expected to be prevalent. We classified SMDs in NMO into five major categories: tonic spasms; dystonia; myoclonus; tremors; and spontaneous clonus. We subsequently subclassified each category into subcategories based on the most commonly observed SMD subtypes. We further differentiated among the overlapping entities of tonic spasms, paroxysmal dystonia, and spinal myoclonus. To avoid artificial generalization (e.g., labeling all tonic spasms as painful tonic spasms), we evaluated each SMD subtype for three additional variables: presence of pain; stimulus sensitivity, and relation to motion.
Previous studies of movement disorders in NMO have focused mainly on tonic spasms where prevalence ranged from 14% to 95%.16, 17 Our data come in the midpoint of this wide range with a prevalence rate of 43.2% for any SMD and 35% for tonic spasms. We realize that our cohort of patients requiring admission and plasmapharesis may represent the more severe end of the NMO spectrum, where SMDs may be over‐represented compared to patients with milder disease course. However, NMO is known to be the most severe of all neuroinflammatory conditions, and NMO patients often require hospitalization for their acute relapses and are commonly treated with plasmapharesis for that purpose.18, 19 In that sense, we believe that our sample was representative of the majority of NMO patients.
Some studies used the term paroxysmal dystonia interchangeably with tonic spasms.12 We believe that the term paroxysmal dystonia should be reserved for cases where there is tonic contraction of antagonistic muscles resulting in an abnormal complex posture of the affected limb or body part. Simple flexion or extension of the limb is less complex and involves activation of one set of muscle group (either flexors or extensors), making the simple terms flexor spasm or extensor spasm more appropriate.
Repeated reports found a positive correlation between tonic spasms and anti AQP‐seropositivity.2, 20 In our study, all but 1 patient with SMDs were seropositive. However, there was no significant difference in serostatus between those with SMDs and those without, probably because of the generally limited number of seronegative cases in our cohort. Patients with SMDs were significantly older at the time of presentation, compared to those without SMDs, and were more likely to be Caucasians or Hispanics.
It has been suggested that tonic spasms occur more frequently in NMO compared to other demyelinating diseases.15, 21 Kim et al. suggested that painful tonic spasms are 98.7% specific for seropositive NMO compared to MS and ITM. They also found that transverse myelitis at disease onset was predictive of future occurrence of painful tonic spasm, compared to optic neuritis onset, and that painful tonic spasms were more likely to occur during recovery from relapse.2 Unlike the study by Kim et al., our study shows that tonic spasms can be painless (31% of all tonic spasms in our cohort were painless). Although the majority of SMDs in our cohort occurred after a relapse, a substantial portion (25%) occurred during an acute relapse as part of the presenting symptoms.
We identified 2 cases of nonparoxysmal or fixed dystonia after a myelitis relapse. Previous reports of dystonia in NMO described mainly paroxysmal dystonia with substantial overlap with tonic spasms.10, 12 To our knowledge, our study reports, for the first time, the development of new‐onset action tremors subsequent to spinal cord attacks in NMO in the absence of lesions in the cerebellum, brainstem, thalami, or basal ganglia. The anatomical origin of these tremors could be the spinocerebellar tracts within the spinal cord. Alternatively, the tremors could be dystonic in nature, but exploiting the exact nature and type of the tremor from medical records was not possible in the absence of video documentation. There was a single report of propriospinal myoclonus in NMO,5 and although we did not find any such cases in our cohort, we expect more reports of spinal myoclonus in the future as physicians become more familiar with the concept of movement disorders of spinal origin in NMO and other demyelinating diseases of the spinal cord. Although there were no cases of spontaneous clonus documented in our study cohort, this common SMD might have been under‐reported or overlooked as well.
Most previous reports described favorable response of paroxysmal symptoms in general and tonic spasms in particular to carbamazepine in patients with demyelinating diseases.2, 9, 10, 12 In our cohort, there was no evidence for greater efficacy among the many treatment options used; however, most patients showed fair to good response with a combination of an antiepileptic and a muscle relaxant or an antiepileptic alone. The use of muscle relaxants alone may not be sufficient to address paroxysmal SMDs in NMO and other demyelinating diseases. In addition to their proposed efficacy for paroxysmal dystonia in demyelinating diseases,22 botulinium toxin injections may also be an option for nonparoxysmal SMDs, as in the case of cervical dystonia in our cohort. In the 1 patient with tonic spasms who had a baclofen pump installed, the initial improvement was eventually followed by recurrence of tonic spasms upon follow‐up. No specific treatment was offered to address tremors in the 3 patients who developed this type of SMD in our cohort. Given the action nature of these tremors, primidone or propranolol may have been reasonable treatment options.23 .
Our study has several limitations. First, the retrospective nature of the study and our reliance on extracting data from medical records may have resulted in some degree of reporting bias. Given that there was no systematic method to inquire about and document SMDs, some SMDs may have been missed, making our prevalence numbers under‐representative. Also, documentation of the type of each movement disorder was made by the treating team, which did not include a movement disorder specialist in most cases, and no video recording was obtained. This might have resulted in mislabeling, in some cases. However, a movement disorder specialist reviewed all cases and compared the documented movement disorder type to the available description in the history and physical. Only cases where the terms flexor or flexion and extensor or extension appeared in documentation were labeled as such. All other spasms resulting in visible movement of the involved body part were labeled as unspecified. Only In 2 unlabeled cases, namely, the isometric tonic spasm of the chest wall and the fixed dystonia of the left arm, the movement disorder specialist made the diagnosis himself based on the available description. Otherwise, the terms documented by the inpatient team were felt to be appropriate for all other cases. Second, we did not compare SMDs between different etiologies of spinal cord demyelination, such as MS and ITM, nor did we study SMDs in noninflammatory cord pathology. This limits generalization of our SMD classification to conditions other than NMO. However, we are currently studying the utility of this classification in a prospective longitudinal study of movement disorders in different neuroinflammatory conditions. We also did not study movement disorders in NMO/NMOSD with subcortical brain lesions. Although these patients may indeed have nonspinal or prototypic movement disorders, this was simply beyond the scope of our study because we mainly aimed at studying and characterizing the entity of movement disorders of spinal origin. Last, although we tried to shed some light on the most effective medications to treat SMDs, the retrospective nature of this study does not allow for a meaningful comparison between the different classes of symptomatic medications.
Conclusions
Clinicians are advised to actively look for and address SMDs in patients with spinal cord pathology. Using clear, unified terminology to describe and document SMDs is crucial for both clinical and research purposes. SMDs are common in spinal cord demyelination, especially in NMO, and can be the presenting symptom of relapse, but more commonly occur within 3 months after a relapse. When present, SMDs are often a major source of disability. Tonic spasms are the most common among the different types of SMDs. In addition to tonic spasms, focal dystonia, spinal myoclonus, and action tremors may also occur as a result of spinal cord demyelination. Some SMDs in NMO/NMOSD can be painful, kinesogenic, and/or stimulus sensitive. Antiepileptics with or without muscle relaxants seem to have the best therapeutic benefit for paroxysmal SMDs although randomized, controlled trials are lacking. Carbamazepine may be particularly effective and should be tried for patients with paroxysmal forms of SMDs. Botulinium toxin can be used for focal dystonia.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
H.A.: 1A, 1B, 1C, 2A, 2B, 2C, 3A, 3B
H.H.F.: 3B
M.A.M.: 1A, 1B, 1C, 3B
M.L.: 1A, 1B, 2B, 2C, 3B
Disclosures
Funding Sources and Conflicts of Interest: The authors report no sources of funding and no conflicts of interest.
Financial disclosures for past 12 months: Dr. Abboud is supported by a grant from the National Multiple Sclerosis Society. Dr. Fernandez has received research support from Abbott, Acadia, Biotie Therapeutics, EMD‐Serono, Huntington Study Group, Merck, the Michael J. Fox Foundation, The International Parkinson and Movement Disorder Society (MDS), National Parkinson Foundation, National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke, Novartis, Parkinson Study Group, Synosia, and Teva, but has no owner interest in any pharmaceutical company. Dr. Fernandez has received honoraria from Advanced Health Media, Cleveland Clinic CME, Medical Communications Media, MDS, and Vindico Medical Education as a speaker in CME events. Dr. Fernandez has received honoraria from Ipsen, Merz Pharmaceuticals, Pfizer, Teva Neuroscience, and Zambon Pharmaceuticals as a speaker and/or consultant. Dr. Fernandez has received royalty payments from Demos Publishing for serving as a book author/editor. The Cleveland Clinic has contracts with EMD Serono, Abbott, and Merz Pharmaceuticals for Dr. Fernandez' role as a member of the Global Steering Committee for Safinamide and LCIG studies; and head principal investigator for the Xeomin Registry Study. Dr. Fernandez also serves as the chair of the publication committee for Xeomin Studies (Merz Pharmaceuticals); a member of the publication committee for Dysport studies (Ipsen Pharmaceuticals); and a consultant for Prostrakan/KyowaHakko, Britannia, Knopp, and US WorldMeds, but he does not receive any personal compensation for these roles. Dr. Fernandez has received a stipend from MDS for serving as medical editor of the MDS website. Dr. Levy receives research support from the NIH, Guthy Jackson Charitable Foundation, Viropharma, Acorda, Sanofi, NeuralStem, and Genentech and serves as a consultant for Chugai Pharmaceuticals, GlaxoSmithKline, and Medimmune.
Acknowledgments
This article represents valid work, and neither this article nor one with substantially similar content has been published elsewhere. The results of this study were presented as a platform presentation at the 67th Annual Meeting of the American Academy of Neurology and appeared in abstract form in the official journal of the American Academy of Neurology. Hesham Abboud had full access to all the data in the study and takes responsibility for the integrity and the accuracy of the data analysis.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Obeso JA, Rodríguez‐Oroz MC, Rodríguez M, Arbizu J, Giménez‐Amaya JM. The basal ganglia and disorders of movement: pathophysiological mechanisms. Physiology 2002;17:51–55. [DOI] [PubMed] [Google Scholar]
- 2. Kim SM, Go MJ, Sung JJ, Park KS, Lee KW. Painful tonic spasm in neuromyelitis optica: incidence, diagnostic utility, and clinical characteristics. Arch Neurol 2012;69:1026–1031. [DOI] [PubMed] [Google Scholar]
- 3. Rae‐Grant AD. Unusual symptoms and syndromes in multiple sclerosis. Continuum (Minneap Minn) 2013;19(4 Multiple Sclerosis):992–1006. [DOI] [PubMed] [Google Scholar]
- 4. Mathews WB. Paroxysmal symptoms in multiple sclerosis. J Neurol Neurosurg Psychiatry 1975;38:617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vetrugno R, D'Angelo R, Alessandria M, et al. Axial myoclonus in Devic neuromyelitis optica. Mov Disord 2009;24:1708–1709. [DOI] [PubMed] [Google Scholar]
- 6. Spissu A, Cannas A, Ferrigno P, et al. Anatomic correlates of painful tonic spasms in multiple sclerosis. Mov Disord 1999;14:331–335. [DOI] [PubMed] [Google Scholar]
- 7. Chung EJ, Kim SJ. Tonic spasms in acute transverse myelitis. J Clin Neurosci 2009;16:165–166. [DOI] [PubMed] [Google Scholar]
- 8. Taguchi Y, Takashima S, Dougu N, Tanaka K. Two cases of myelitis associated with Sjögren syndrome without xerosis: characteristic MRI findings. No To Shinkei 2006;58:701–707. [PubMed] [Google Scholar]
- 9. Cherrick AA, Ellenberg M. Spinal cord seizures in transverse myelopathy: report of two cases. Arch Phys Med Rehabil 1986;67:129–131. [DOI] [PubMed] [Google Scholar]
- 10. Schmidt FR, Costa FH, Silva FM, et al. Paroxysmal dystonia and neuromyelitis optica. Arq Neuropsiquiatr 2012;70:271–272. [DOI] [PubMed] [Google Scholar]
- 11. Mehanna R, Jankovic J. Movement disorders in multiple sclerosis and other demyelinating diseases. J Neurol Sci 2013;328:1–8. [DOI] [PubMed] [Google Scholar]
- 12. Waubant E1, Alizé P, Tourbah A, Agid Y. Paroxysmal dystonia (tonic spasm) in multiple sclerosis. Neurology 2001;57:2320–2321. [DOI] [PubMed] [Google Scholar]
- 13. Wingerchuk DM, Lennon VA, Pittock SJ, et al. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485–1489. [DOI] [PubMed] [Google Scholar]
- 14. Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol 2007;6:805–815. [DOI] [PubMed] [Google Scholar]
- 15. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015;85:177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Usmani N, Bedi G, Lam BL, Sheremata WA. Association between paroxysmal tonic spasms and neuromyelitis optica. Arch Neurol 2012;69:121–124. [DOI] [PubMed] [Google Scholar]
- 17. Abaroa L, Rodríguez‐Quiroga SA, Melamud L, et al. Tonic spasms are a common clinical manifestation in patients with neuromyelitis optica. Arq Neuropsiquiatr 2013;71:280–283. [DOI] [PubMed] [Google Scholar]
- 18. Bichuetti DB, Oliveira EM, Souza NA, et al. Patients with neuromyelitis optica have a more severe disease than patients with relapsing remitting multiple sclerosis, including higher risk of dying of a demyelinating disease. Arq Neuropsiquiatr 2013;71:275–279. [DOI] [PubMed] [Google Scholar]
- 19. Abboud H, Petrak A, Mealy M, Sasidharan S, Siddique L, Levy M. Treatment of acute relapses in neuromyelitis optica: steroids alone versus steroids plus plasma exchange. Mult Scler 2015. pii: 1352458515581438. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Viswanathan S, Arip M, Mustafa N, et al. The frequency of anti‐aquaporin‐4 Ig g antibody in neuromyelitis optica and its spectrum disorders at a single tertiary referral center in malaysia. Mult Scler Int 2014;2014:568254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology 1999;53:1107–1114. [DOI] [PubMed] [Google Scholar]
- 22. Restivo DA, Tinazzi M, Patti F, Palmeri A, Maimone D. Botulinium toxin treatment of painful tonic spasms in multiple sclerosis. Neurology 2002;61:719–720. [DOI] [PubMed] [Google Scholar]
- 23. Abboud H, Ahmed A, Fernandez H. Essential tremor: choosing the right management plan for your patient. Cleve Clin J Med 2011;78:821–828. [DOI] [PubMed] [Google Scholar]