

Gerstmann‐Sträussler‐Scheinker (GSS) is a rare disease caused by a mutation in the prion protein (PRNP) gene. The incidence is around 5 cases per 100 million inhabitants per year, and its clinical onset is usually in the fifth decade of life.1 GSS is generally characterized by ataxia or spastic paraparesis followed by cognitive decline.1 We report on a patient with GSS who presented with atypical parkinsonism in which MRI findings led to the final diagnosis.
Case Report
A 74‐yr‐old woman was referred to our hospital for hypomimia, generalized slowness of movements, and shuffling gait, which had developed over the past 3 yr. Subsequent to diagnosis of Parkinson's disease (PD) at another center, she had been prescribed levodopa and motor function had improved slightly. On examination, blepharospasm, freezing of gait, a hint of apraxia on the right side, cognitive impairment, and frontal disinhibition were detected. She had neither ocular movements abnormalities nor backward falls (see Video 1, Segment 1).
Workup for treatable causes of dementia and parkinsonism was negative. Neuropsychological examination showed a Mini–Mental State Examination score of 24/30, temporal and spatial disorientation, mild impairment in immediate and delayed recall verbal memory, and marked visuospatial and ‐constructive dysfunction.
Taking into account the presence of atypical clinical signs for PD, together with the non‐l‐dopa responsive parkinsonism and visuospatial abnormalities in the neuropsychological examination, a syndromic diagnosis of atypical parkinsonism with overlapping features of progressive supranuclear palsy and corticobasal syndrome was established.
MRI (Fig. 1) showed marked loss of volume of the cerebellum, brainstem, and frontal and temporal lobes; and MRI diffusion‐weighted imaging (DWI) showed hyperintense signals and decreased apparent diffusion coefficients of the cortical ribbon, left caudate and putamen, bilateral thalami, and cerebellar vermis and follia. Although the pattern resembled that described in frontotemporal lobar degeneration,2 the atypical features of volume loss of the cerebellum and midbrain, and the presence of DWI hyperintensities, raised the possibility of a prion disease.
Figure 1.
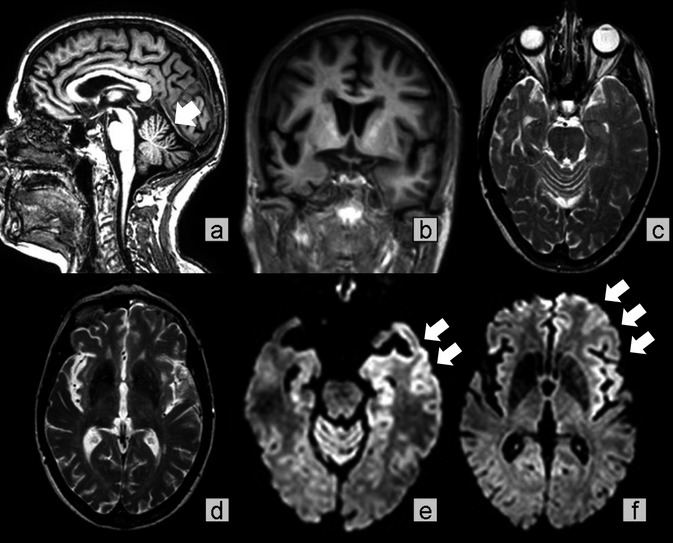
MRI findings. (A and B) T1‐weighted images show volume loss of the cerebellum, brainstem, and brain, particularly in the right frontal and temporal lobes. (C and D) T2‐weighted images show ill‐defined white matter hyperintensities. (E and F) Diffusion‐weighted imaging reveals hyperintensities within the cortical ribbon, involving the left temporal and frontal cortex, left caudate and putamen, medial thalami (mainly left), and cerebellar vermis.
After consulting the family about possible familial disorders, an autosomal‐dominant, hereditary, neurological disease was disclosed. Fifteen relatives in 3 generations had clinical symptoms resembling frontotemporal dementia. Three of them had already been diagnosed with GSS and were carriers of a novel PRNP mutation, Y218N in heterozygosis with 129V in homozygosis.3 This mutation was confirmed in our patient, but was associated with methionine/valine heterozygosis at codon 129. A final diagnosis of GSS was made.
At follow‐up assessment of the patient 10 mo later, motor function, cognition, and behavior had worsened (see Video 1, Segment 2). The patient showed postural instability and presented frequent backward falls when turning; left‐limb dystonia had appeared; she had developed repetitive behaviors and stereotypies; well‐structured visual hallucinations and delusional ideas had arisen; and anxiety, irritability, and apathy had increased.
During the last year, the patient has become unable to walk owing to unsteadiness and the progression of dystonia on her left foot. Moreover, she has developed seizures that have been controlled with antiepileptic drugs and aphasia.
Discussion
GSS is one of the presentations of the 10% to 15% of prion diseases caused by mutations in the PRNP gene, usually presenting with an autosomal‐dominant pattern of inheritance. However, family history can be absent in up to one third of cases because of a de novo mutation.4
Besides the classical clinical picture in GSS, less‐common clinical presentations, such as dementia of Alzheimer's type, frontotemporal‐like dementia, parkinsonism, and atypical psychiatric disorders, have been reported.1, 3, 5 This clinical heterogeneity has also been found in families with the same mutation and has been attributed to the distinct abnormal isoforms of prion protein and polymorphisms at codons 129 and 219.1
Our case highlights the clinical heterogeneity of GSS and is one of the few patients with GSS mimicking PSP reported in the literature.6, 7, 8, 9, 10 Despite carrying the same Y218N mutation in the PRNP gene as the other affected members of her family, our patient showed different clinical features. Whereas symptoms in her relatives developed at a younger age (median age: 60 yr) and presented with a frontotemporal dementia syndrome, in our patient, parkinsonism was the only neurological manifestation during the first 2 yr of the disease. These phenotypical differences could have been influenced by our patient having 129V in heterozygosis whereas her relatives had valine in homozygosis.
Our case illustrates the importance of MRI when an atypical parkinsonian syndrome is suspected. Volume loss of the cerebellum and brainstem and cortical and subcortical DWI findings on MRI raised the suspicion of prion disease. However, the sensitivity of DWI‐MRI in genetic prionopathies is low, compared to that in sporadic Creutzfeldt Jakob disease.4 This point is reflected by the fact that no DWI abnormalities were described in any of the previously reported GSS cases mimicking PSP.6, 7, 8, 9, 10
In conclusion, we report on a patient who presented with an atypical parkinsonism, but was diagnosed with GSS after findings on MRI. This case reinforces the diverse clinical spectrum of genetic prion diseases, and shows that suspicion of GSS should be raised in patients with familial frontotemporal or atypical parkinsonian symptoms and suggestive MRI.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
R.R.‐N.: 3A, 3B
J.P.: 3B
B.G.‐A.: 3B
E.G.‐M.: 3B
R.S.‐V.: 3B
J.K.: 3B
Disclosures
Funding Sources and Conflict of Interest: The authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosures for previous 12 months: J.P. has received honoraria for lecturing or consultation from Boehringer, UCB, Allergan, IPSEN, and Lundbeck. J.K. receives public grants from CIBERNED and the Fundació La Marató de TV3.
Supporting information
A video accompanying this article is available in the supporting information here.
Video 1. Segment 1: First visit. Slight hypomimia and dysarthria, blepharospasm with mild apraxia of eyelid opening, mild left‐sided parkinsonian motor features, and a hint of apraxia on the right side can be seen. No supranuclear gaze palsy is detected, and postural reflexes are normal. Luria premotor series are mildly impaired, but the “applause sign” is absent. Segment 2: Follow‐up (10 mo later). Motor stereotypies (fidgeting with clothes) and their consequences (forehead bruise) are present. Blepharospasm and parkinsonism have progressed in severity and a left‐limb dystonia, more marked in the arm, has appeared. No supranuclear gaze palsy is detected, but postural instability is evident.
Acknowledgments
The authors thank the patient and her family for their permission to publish clinical data and video footage. The authors are also grateful to Carolyn Newey for help with English.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Ghetti B, Bugiani O, Tagliavini F, Piccardo P. Gerstmann‐Sträussler‐Scheinker disease In: Dickson D, ed. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. Basel, Switzerland: ISN Neuropath; 2003:318–325. [Google Scholar]
- 2. Boccardi M, Laakso MP, Bresciani L, et al. The MRI pattern of frontal and temporal brain atrophy in fronto‐temporal dementia. Neurobiol Aging 2003;24:95–103. [DOI] [PubMed] [Google Scholar]
- 3. Alzualde A, Indakoetxea B, Ferrer I, et al. A novel PRNP Y218N mutation in Gerstmann‐Sträussler‐Scheinker disease with neurofibrillary degeneration. J Neuropathol Exp Neurol 2010;69:789–800. [DOI] [PubMed] [Google Scholar]
- 4. Kovács GG, Puopolo M, Ladogana A, et al. Genetic prion disease: the EUROCJD experience. Hum Genet 2005;118:166–174. [DOI] [PubMed] [Google Scholar]
- 5. Webb TE, Poulter M, Beck J, et al. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain 2008;131:2632–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamazaki M, Oyanagi K, Mori O, et al. Variant Gerstmann‐Sträussler syndrome with the P105L prion gene mutation: an unusual case with nigral degeneration and widespread neurofibrillary tangles. Acta Neuropathol 1999;98:506–511. [DOI] [PubMed] [Google Scholar]
- 7. Oba N, Fujimoto Y, Hirata K, Ando N, Saida K. A case of Gerstmann‐Sträussler‐Scheinker disease with severe muscular atrophy and vertical gaze palsy. Rinsho Shinkeigaku 2000;40:726–731. [PubMed] [Google Scholar]
- 8. Rowe DB, Lewis V, Needham M, et al. Novel prion protein gene mutation presenting with subacute PSP‐like syndrome. Neurology 2007;68:868–870. [DOI] [PubMed] [Google Scholar]
- 9. Fleming AB, Ghetti B, Murrell JR, et al. Gerstmann‐Sträussler‐Scheinker disease with progressive supranuclear palsy presentation. Dement Geriatr Cogn Disord 2010;30(suppl 1):43–44.20689282 [Google Scholar]
- 10. Plate A, Benninghoff J, Jansen GH, et al. Atypical Parkinsonism due to a D202N Gerstmann‐Sträussler‐Scheinker prion protein mutation: first in vivo diagnosed case. Mov Disord 2013;28:241–244. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A video accompanying this article is available in the supporting information here.
Video 1. Segment 1: First visit. Slight hypomimia and dysarthria, blepharospasm with mild apraxia of eyelid opening, mild left‐sided parkinsonian motor features, and a hint of apraxia on the right side can be seen. No supranuclear gaze palsy is detected, and postural reflexes are normal. Luria premotor series are mildly impaired, but the “applause sign” is absent. Segment 2: Follow‐up (10 mo later). Motor stereotypies (fidgeting with clothes) and their consequences (forehead bruise) are present. Blepharospasm and parkinsonism have progressed in severity and a left‐limb dystonia, more marked in the arm, has appeared. No supranuclear gaze palsy is detected, but postural instability is evident.