

Abstract
Background
Alzheimer's disease (AD) is the second‐most frequent cause underlying corticobasal syndrome (CBS). However, a reliable diagnosis using clinical, neuropsychological, or neuroimaging approaches has not yet been achieved.
Methods
Clinical, neuropsychological, imaging, and neuropathology studies were undertaken in a large Spanish family with early‐onset familial AD (EOFAD) carrying a Met233Leu mutation linked to presenilin‐1 gene (PSEN‐1).
Results
Two of three examined members of this family presented with the usual amnestic pattern. At the age of 47 years, a third family member, in whom pathology was later confirmed, developed prominent CBS combined with severe neuropsychiatric and behavioral disturbances resembling those often found in EOFAD.
Conclusion
Although CBS in EOFAD appears to be rare, demonstration of a linkage to PSEN‐1 gene mutations may permit in vivo diagnosis.
Keywords: corticobasal syndrome, genetics, early‐onset familial Alzheimer's disease, presenilin‐1
Corticobasal degeneration (CBD) was first described around 50 years ago as a primary motor disorder that exhibits an asymmetric akinetic‐rigid syndrome and apraxia along with characteristic pathology findings.1, 2 Diverse clinical presentations have now been identified,3 and neuropathological criteria have hence been modified.4, 5 Because the underlying pathology in patients displaying symptoms similar to CBD is heterogeneous,6, 7, 8, 9 the term corticobasal syndrome (CBS) was introduced for patients awaiting pathological confirmation.10
In 23% to 25% of all patients, CBS is linked to Alzheimer‐type pathology (CBS‐AD),11, 12, 13, 14 and therefore represents the second‐most common type of clinically diagnosed CBS.15 To date, it has not been possible to establish specific features that might allow a definite diagnosis of CBS‐AD before pathological confirmation, despite proposals based on selected sets of clinical symptoms, neuropsychological profiles, or neuroimaging findings.16 In consequence, the diagnosis during the patient's lifetime remains uncertain.17 Tau/Abeta ratio in cerebrospinal fluid (CSF) may represent a useful way to detect CBS‐AD in vivo, although neuropathological confirmation is not yet forthcoming18 and the technique's sensitivity for detecting CBS‐AD at an early stage has yet to be determined.19
CBS‐AD series published to date correspond to genetically sporadic patients. We describe a Spanish family with pathologically confirmed cases of early‐onset, autosomal‐dominant familial Alzheimer's disease (EOFAD) linked to a Met233Leu mutation of the presenilin‐1 gene (PSEN‐1). Three family members carrying the mutation were examined by us, and 2 presented the amnestic form of the disease. In contrast, the third member studied presented with neuropsychiatric symptoms, a severe behavior disorder and a clear‐cut CBS, as documented in Video 1, and AD was pathologically confirmed. We believe this combination of clinical and genetic features, as well as its relevance in CBS‐AD diagnosis, has not been previously described.
Methods
Genetics and Neuropathology
We describe a large Spanish family in which 8 members representing four generations developed progressive dementia with onset at an early age (Fig. 1). The diagnosis of AD in case III5 has been confirmed by a neuropathology study at another institution. Genetic testing in 3 family members (III6, III7, and IV14) personally examined by us identified a M233L mutation in exon 7B of PSEN‐1 after analysis of exons 4, 5, 6, 7B, 10, 11, and 12 by DNA‐polymerase chain reaction using specific oligonucleotides. No mutations were found in tau protein associated with microtubule‐associated protein tau (MAPT) gene. Subjects III7 and IV14, a mother and son, had been studied in our neurology department's memory clinic some years before and has undergone long‐term follow‐up since being diagnosed with EOFAD. Patient III6, subsequently attended by the movement disorders unit in the same department, presented a prominent CBS combined with severe neuropsychiatric disturbances. Owing to lack of detailed information at time of admission, the initial studies of this patient were carried out without knowledge of his family history. Shortly thereafter, we were able to draw up a detailed family tree using multiple sources of information.
Figure 1.
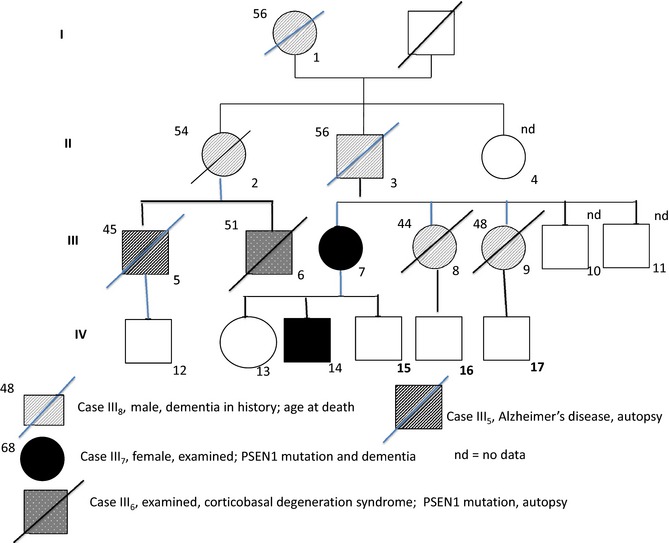
Family tree showing the 3 examined PSEN‐1 patients: III 6 (CBS‐AD, arrow); III 7 (AD); and IV 14 (AD).
Results
Patient Characteristics
Case 1
This 48‐year‐old right‐handed man (III6), a retired former clerk, lived alone. He had been observed oddly dressed and wandering near his apartment building. When questioned, he reported that foreigners were living in his flat and would attack him if he entered it. He spoke softly on the phone alleging he was being spied on. Workers in a restaurant he frequented noticed changes in his eating habits, including unusual rituals such as eating calamari almost exclusively or breaking bread into small pieces and mashing them into lumps before eating. The local parish priest who knew him well provided this information.
He expressed that his obvious difficulties moving his left arm were the result of violent assault. A close friend reported gradual onset of his behavior disorder beginning around 1 year before. He had been dismissed from his job because of numerous unjustified absences and abnormal behavior. Finally, Community Social Services brought him to the hospital after they noticed the inaccuracy of his complaints and his poor living conditions.
Results from his general physical examination were unremarkable. During the neurological examination, he was cooperative and oriented in time, place, and person, but he seemed apathetic and slow. His spontaneous speech was sparse, but he answered in well‐articulated short sentences. He was able to describe in detail the route from his home to the hospital. The patient felt that nothing was really wrong with him and explained that he would return to his flat once its illegal occupants were gone. He held his left arm immobile and markedly flexed, with no associated movements while walking, although he was able to perform elementary movements with this limb when prompted to do so.
Spontaneous left‐sided gaze was rare. He displayed prominent apraxic defects in sequencing complex symbolic gestures with his left arm, and in placing a sheet of paper in an envelope or putting on his jacket. Marked grasping was noticed in both upper limbs. Continuous, brief, irregular twitching of the fingers could be observed in his left hand, which increased in amplitude on action or when the hands were outstretched. Twitching could be provoked easily by applying light pinpricks to the dorsal aspects of the fingers. Tapping the forearm muscles or the sternum elicited a sudden jerk of the entire left upper limb. High‐amplitude, short‐duration (<50 ms) spontaneous potentials were recorded on electromyography simultaneously with the muscular jerks. No large somatosensitive cortical potentials were recorded on stimulation of the median nerve.
The patient was able to carry out simple sequential movements with his left upper limb with marked slowness. His left extremities displayed no cogwheel rigidity, but instead severe paratonic rigidity when doctors attempted to manipulate the limb. Deep tendon reflexes were normal in all four extremities and plantar responses were flexor. Sensory testing was unreliable. There were no cerebellar signs in the right upper limb or either lower extremity. In summary, abnormal neurological motor findings were confined to the left upper extremity and included abnormal flexor posturing, spontaneous and reflex myoclonus, severe paratonic rigidity, and bradykinesia. In addition, the patient displayed anosognosia, left limb kinetic apraxia, and left hemispatial neglect.
Routine hematological and urinary analyses, folic acid and vitamin B12 levels, thyroid antibodies, thyroglobulin and cardiolipin, antinuclear antibodies, and antiextractable nuclear antibodies, serum copper, ceruloplasmin, immunoglobulin measurements, and tests for acquired immune deficiency syndrome and syphilis yielded normal results. CSF examination showed no abnormalities, including absence of 14‐3‐3 protein and mutations in the PRNP gene encoding the PrP protein. MRI of the brain revealed marked asymmetric (right greater than left) cortical atrophy and mild‐to‐moderate subcortical and periventricular white matter signal changes. Ventricular enlargement was particularly marked in the right occipital horn (Fig. 2A,B). Marked right frontotemporal hypometabolism was identified on single‐photon emission CT (SPECT) imaging (Fig. 2C). Genetic testing revealed that the patient was a carrier of the mutation met233leu of the PSEN‐1 gene.
Figure 2.
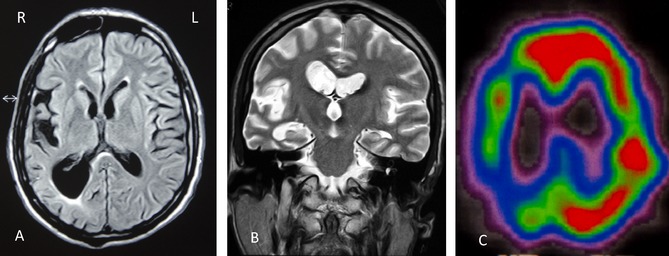
Axial T1‐weighted head MRI of CBS patient (III6) reveals severe asymmetric cortical atrophy (right more than left) and mild‐to‐moderate subcortical and periventricular white matter signal changes (A). Marked right occipital horn enlargement can be seen in coronal T‐weighted images (B). Brain imaging with SPECT shows pronounced right temporoparietal hypoperfusion (C).
Neuropsychology
Neuropsychological assessment at admission had some limitations owing to lack of proxy informants and patient apathy and slowness. He scored 2 points below the expected level for age and education on the Mini–Mental State Examination (MMSE)20 and obtained low scores on the Wechsler Adult Intelligence Scale‐Revised digit span subtest.21 No specific language abnormalities were found in a Spanish adaptation of the Goodglass and Kaplan battery.22 He scored 69/100 on the Addenbrooke Cognitive Examination (ACE)23 revealing impairment in multiple cognitive domains, particularly those items related to learning (0/21). The patient also showed very low scores on most Frontal Assessment Battery subtests,24 as well as perseverations on the Wisconsin Card Sorting Test (WCST),25 which is consistent with severe frontal executive dysfunction. Visuoperceptive and ‐spatial abnormalities were evident in the copy of pentagons and different geometric designs. The patient omitted the left half of designs, suggesting left visual neglect.26
Neuropathology
Legal permission was obtained to carry out a diagnostic cerebral biopsy. A sample from the right anterior frontal lobe, including cortex and adjacent white matter, was sent to the tissue bank for neurological research at Complutense University of Madrid. Findings included mild‐to‐moderate neuronal loss and superficial spongiosis and gliosis. Immunostains for ß‐amyloid showed numerous plaques with amyloid deposits with frequent density according to Consortium to Establish a Registry for Alzheimer's Disease (CERAD) criteria. Amyloid deposits were also found in meningeal and cortical vessels. Sections were also immunostained with a mouse monoclonal antibody against hyperphosphorylated tau (AT100 and AT8), and ubiquitin demonstrated the neuritic nature of the plaques. No astrocytes with tau deposits were observed, and no thick filaments were found in the white matter. Ubiquitin and tau immunostains did not show balloon neurons. Histological findings were consistent with AD and excluded frontotemporal degeneration and CBD.
Postmortem examination was limited to the cranial cavity (Brain Bank, Alcorcón Foundation, Madrid, Spain). There was marked cortical atrophy and global ventricular dilatation. The right hemisphere was frozen at −80°C for future molecular and biochemical studies. Histological examinations of the left hemisphere were performed on 4‐μm‐thick sections using several staining methods, including hematoxylin and eosin, Klüver‐Barrera, and Gallyas‐Braak. Sections were also immunostained with a mouse monoclonal antibody against hyperphosphorylated tau, ß‐amyloid, RD4, and RD3. There was marked neuronal loss, astrocytosis, and frequent neuritic plaques (CERAD score) in primary motor and sensory cortices. Intense astrocytosis, neuronal loss, and neuropil rarefaction were also evident in the amygdala and entorhinal cortex. Neuronal loss and astrocytosis were moderate in the basal ganglia, with a lower density of tau pathology and neuritic plaques. No astrocytic plaques or tufted astrocytes were observed. In addition, mild alpha‐synuclein pathology was found in the insula. The findings were consistent with a high level of AD neuropathological changes (A2 B3 C3) according to the Hyman et al. criteria27 and high probability of AD (criteria NIA/Reagan, Braak stage VI; Fig. 3).
Figure 3.
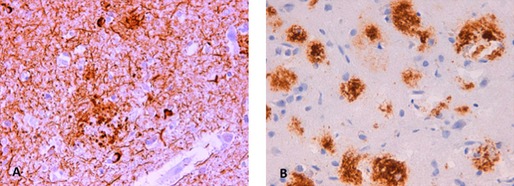
Left primary visual cortex, tau‐immunostain (40×), showing numerous neurofibrillary tangles (Braak VI) and neuropil threads (A). Frontal cortex (40×), immunostain for ß‐amyloid (B).
Case 2
This female patient (III7) was evaluated at the age of 48 years with a 6‐month history of memory loss, as reported by her husband. Her neurological examination, routine laboratory tests, and a CT scan were normal, but she scored 7 points below the cut‐off level on the MMSE. Four years later, she required help performing daily activities and her MMSE had decreased by 14 points. Examination showed nonfluent aphasia, frequent phonemic paraphasias, and impaired repetition. Her MRI showed symmetrical frontal and temporal cortical atrophy, and SPECT imaging revealed bilateral parietal and temporal hypoperfusion, with mild anterior frontal involvement. At the age of 54, she suffered a generalized seizure and has since developed spontaneous axial myoclonus. Two years later, she was confined to a wheelchair. The patient was a carrier of the met233leu mutation of the PSEN‐1 gene.
Case 3
This 38‐year‐old male patient (IV14), a professional nurse, was seen because of progressive loss of memory in the preceding 2 years. His memory loss had led to problems at work, such as administering the wrong medication or not administering it when requested. MMSE score was normal (29/30); ACE score was 96/100 with one error on the subtraction subtest, but 3 errors of 21 on the immediate memory subtest. He showed moderate‐to‐mild impairment for perseverative and nonperseverative items on the WCST,25 scoring 36 (percentile >90) on the Rey‐Osterrieth Complex Figure Test,28 but 19 (25th percentile) on the delayed visual memory subtest. A brain MRI was considered normal. SPECT images showed bilateral temporo‐parietal‐occipital perfusion defects that were more marked on the left, including left posterior frontal involvement. He asked to know his genetic status and was confirmed as a carrier of the met233leu mutation of the PSEN‐1 gene.
Discussion
Patients III7 and IV14 in the extended family presented here display typical findings of EOFAD linked to PSEN‐1 mutations, and the disease was confirmed pathologically in 1 family member (III5). Essential features of the family consist of autosomal‐dominant inheritance pattern, memory problems at presentation, and the occasional association of myoclonus, seizures, or aphasia in advances stages of the disease.29, 30
A Met‐Leu amino acid substitution at codon 233 had been identified in early‐onset forms of AD in patients from the same city as the family we describe.31 Although no relationship could be demonstrated between the two families, a remote common founder cannot be ruled out. The mutation was also found in a referral‐based series screening for PSEN‐1 mutations in early‐onset AD.32 Interestingly, severe frontal dysfunction at presentation, similar to that in patients with CBS‐AD, has been reported in 2 Met‐233‐Leu PSEN‐1 patients with sporadic early‐onset AD.33, 34
Some characteristics of our patient with CBS‐AD, such as family history, very early disease onset, and severe neurobehavioral disorders at presentation are more in keeping with features often found in families with EOFAD and PSEN‐1 mutations, whose genetics he shared, rather than the sporadic, comparatively late onset of CBS‐AD cases reported to date. Familial AD secondary to PSEN‐1 mutations may therefore exhibit initially psychiatric or behavioral traits,35, 36 sometimes mimicking the behavioral variant of frontotemporal dementia,37 whereas myoclonus and seizures are also well known.38, 39, 40 Some patients clinically diagnosed with CBS and showing Alzheimer‐type pathology lacked the usual clinical asymmetry for motor and sensory signs,15, 41 which has been suggested as a predictive feature of CBS‐AD. In this regard, we must emphasize the marked clinical and neuroimaging asymmetry displayed by our case.
Patients clinically resembling our case III6 were reported on in 1966 by Heston et al.42 Those researchers presented a family of 19 patients spanning four generations with progressive dementia combined with prominent parkinsonism and pyramidal tract signs. Focal motor findings represented the first sign of neurological abnormality in 2 of the patients personally examined by the authors, manifesting as progressive clumsiness and weakness of their right hands and legs. Symptoms at onset in another patient (IV2) consisted of stiffness and tremor in the left extremities. AD was confirmed pathologically in 4 of the 19 individuals affected. Some EOFAD variants linked to PSEN‐1 mutations may express early motor disturbances, such as spastic paraparesis43, 44 or parkinsonism.45, 46 Case III6 and those from the Heston et al. family42 might thus represent another motor disturbance, such as CBS related to PSEN‐1 mutations.
Thus far, genetic CBS has been reported in families with autosomal‐dominant frontotemporal lobar degeneration (FTLD) linked to progranulin (PGRN) gene mutations, which represent between 5% and 7.9% of all cases in large series.47 As in our patient III6, CBS in PGRN families may occur in young adults48 and neurobehavioral abnormalities may be associated at onset.49 FTLD may underlie a wide range of clinical presentations50; in contrast, 2 of the 3 family members described here presented with a pure amnestic disorder. Mutations in the MAPT gene have been demonstrated in a sporadic case of CBD51 and in 2 elderly first cousins, 1 of whom had additional pathological findings consistent with AD.52
Pathogenic C9orf72 repeat expansions are rarely found in patients with CBS and diverse forms of parkinsonism.53 A study screening 37 patients with CBS features for this gene found repeat expansions in only 3 patients. However, a positive family history of FTLD or of motor neuron disease in a CBS patient would justify screening for this mutation.54As a result, we propose testing for PSEN‐1, PGRN, MAPT, and C9of72 gene mutations, particularly in patients with CBS and a positive family history suggesting a neurodegenerative condition.55, 56
Predicting the underlying pathology in CBS‐AD has proved difficult,17 although it might be theoretically possible in cases linked to mutations in the presenilin genes.57 We conclude that CBS may represent an atypical clinical presentation in autosomal‐dominant EOFAD. In addition, evidence of mutations linked to the PSEN‐1 gene may represent a unique opportunity to predict Alzheimer‐type pathology in vivo.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
E.N.: 1A, 1C, 3B C.A.: 1C, 3B C.N.: 1C, 3B S.G.‐R.: 1A, 1B, 1C, 3A
Disclosures
Funding Sources and Conflicts of Interest: The authors report no sources of funding and no conflicts of interest.
Financial Disclosures for previous 12 months: The authors declare that there are no disclosures to report.
Supporting information
A video accompanying this article is available in the supporting information here.
Video 1. Segment 1: Patient shows marked postural flexion of left upper limb. Segment 2: Spontaneous jerking of fingers of left hand. Segment 3: Muscular jerks increase during activity (on the left), but are not seen on the right. Segment 4: Left‐sided jerks are also evident when the patient's hands are outstretched. Segment 5: Light pinpricks on the dorsal aspect of fingers of his left hand triggers a circumscribed jerking movement of that finger. A tap on left forearm and chest triggers a jerk of the entire left upper limb. Segment 6: Paratonic rigidity with attempts to mobilize the patient's left arm.
Acknowledgments
The authors thank Drs. García de Yébenes and Ampuero Sánchez for completing genetic studies. Neurophysiological studies of case 1 were carried out by Dr. A. Esteban. Dr. David Muñoz performed the neuropathology study of the brain biopsy for case 1.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Rebeiz JJ, Kolodny EH, Richardson EP Jr. Corticodentatonigral degeneration with neuronal achromasia: a progressive disorder of late adult life. Trans Am Neurol Assoc 1967;92:23–26. [PubMed] [Google Scholar]
- 2. Rebeiz JJ, Kolodny EH, Richardson EP Jr. Corticodentatonigral degeneration with neuronal achromasia. Arch Neurol 1968;18:20–33. [DOI] [PubMed] [Google Scholar]
- 3. Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013;80:496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dickson DW, Bergeron C, Chin SS, et al. Office of rare diseases of the National Institutes of Health. Neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 2002;61:935–946. [DOI] [PubMed] [Google Scholar]
- 5. Ouchi H, Toyoshima Y, Tada M, et al. Pathology and sensitivity of current clinical criteria in corticobasal syndrome. Mov Disord 2014;29:238–244. [DOI] [PubMed] [Google Scholar]
- 6. Boeve BF, Maraganore DM, Parisi JE, et al. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology 1999;53:795–800. [DOI] [PubMed] [Google Scholar]
- 7. Bhatia KP, Lee M, Rinne JO, et al. Corticobasal degeneration look‐alikes. Adv Neurol 2000;82:169–182. [PubMed] [Google Scholar]
- 8. Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol 2003;54(suppl 5):S15–S19. [DOI] [PubMed] [Google Scholar]
- 9. Lee SE, Rabinovici GD, Mayo CM, et al. Clinicopathological correlations in corticobasal degeneration. Ann Neurol 2011;70:327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Doran M, Du Plessis DG, Enevoldson TP, Fletcher NA, Ghadiali E, Larner AJ. Pathological heterogeneity of clinically diagnosed corticobasal degeneration. J Neurol Sci 2003;216:127–134. [DOI] [PubMed] [Google Scholar]
- 11. Lleó A, Rey MJ, Castellví M, Ferrer I, Blesa R. Asymmetric myoclonic parietal syndrome in a patient with Alzheimer's disease mimicking corticobasal degeneration. Neurología 2002;17:223–226. [PubMed] [Google Scholar]
- 12. Imamura A, Wszolek ZK, Lucas JA, Dickson DW. Corticobasal syndrome with Alzheimer's disease. Mov Disord 2009;24:152–153. [DOI] [PubMed] [Google Scholar]
- 13. Hu WT, Rippon GW, Boeve BF, et al. Alzheimer's disease and corticobasal degeneration presenting as corticobasal syndrome. Mov Disord 2009;24:1375–1379. [DOI] [PubMed] [Google Scholar]
- 14. Hassan A, Whitwell JL, Josephs KA. The corticobasal syndrome‐Alzheimer's disease conumdrum. Expert Rev Neurother 2011;11:1569–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ling H, O'Sullivan SS, Holton JL, et al. Does corticobasal degeneration exist? A clinicopathological re‐evaluation Brain 2010;133:2045–2057. [DOI] [PubMed] [Google Scholar]
- 16. Josephs KA, Whitwell JL, Boeve BF, et al. Anatomical differences between CBS‐corticobasal degeneration and CBS‐Alzheimer's disease. Mov Disord 2010;25:1246–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shelley BP, Hodges JR, Kipps CM, Xuereb JH, Bak TH. Is the pathology of corticobasal syndrome predictable in life? Mov Disord 2009;24:1593–1599. [DOI] [PubMed] [Google Scholar]
- 18. Borroni B, Premi E, Agosti C, et al. CSF Alzheimer's disease‐like pattern in corticobasal syndrome: evidence for a distinct disorder. J Neurol Neurosurg Psychiatry 2011;82:834–838. [DOI] [PubMed] [Google Scholar]
- 19. Shaw LM, Vanderstichele H, Knapik‐Czaijka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009;66:382–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Folstein MF, Folstein SE, McHugh PR. “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 21. Wechsler Adult Intelligence Scale , fourth ed, 2008. [Google Scholar]
- 22. Goodglass M. Kaplan E. Assessment of Aphasia and Related Disorders. Philadelphia: Lea & Febiger; 1962. [Google Scholar]
- 23. Mathuranath PS, Nestor PJ, Berrios GE, Rakowicz W, Hodges JR. A brief cognitive test battery to differentiate Alzheimer's disease and frontotemporal dementia. Neurology 2000;55:1613–1620. [DOI] [PubMed] [Google Scholar]
- 24. Dubois B, Slachevsky A, Litvan I, Pillon B. The FAB: a frontal assessment battery at bedside. Neurology 2000;55:1621–1626. [DOI] [PubMed] [Google Scholar]
- 25. Heaton RK, Chelune GJ, Talley JL, Kay GG, Curtis G. Wisconsin Card Sorting Test. Manual. Odessa: Psychological Assessment Resources Inc; 1993. [Google Scholar]
- 26. Marshall JC, Halligan PW. Visuo‐spatial neglect: a new copying test to assess perceptual parsing. J Neurol 1993;240:37–40. [DOI] [PubMed] [Google Scholar]
- 27. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Ageing‐Alzheimer's Association guidelines for the neuropathological assessment of Alzheimer's disease. Alzheimers Dement 2012;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Osterrieth PA. The Complex Figure. Arch Psychol 1944;28:1021–1034. [Google Scholar]
- 29. Sherrington R, Ragoaev EL, Llang Y, et al. Cloning a gene bearing missense in early‐onset familial Alzheimer's disease. Nature 1995;375:754–760. [DOI] [PubMed] [Google Scholar]
- 30. Larner AJ, du Plessis DG. Early‐onset Alzheimer's disease with presenilin‐1 M139V mutation: clinical, neuropsychological and neuropathological study. Eur J Neurol 2003;10:319–323. [DOI] [PubMed] [Google Scholar]
- 31. Aldudo J, Bullido MJ, Valdivieso F. DGGE method for the mutation analysis of the coding and proximal promoter region of Alzheimer's disease presenilin‐1 gene: two novel mutations. Hum Mutat 1999;14:433–439. [DOI] [PubMed] [Google Scholar]
- 32. Rogaeva EA, Fafel KC, Song YQ, et al. Screening for PS1 mutations in a referral‐based series of AD cases. Neurology 2001;57:621–625. [DOI] [PubMed] [Google Scholar]
- 33. Portet F, Dauvilliers Y, Campion D, et al. Very early onset AD with a de novo mutation in the presenilina 1 gene (Met233Leu). Neurology 2003;61:1136–1137. [DOI] [PubMed] [Google Scholar]
- 34. Mendez MF, McMurtray A. Frontotemporal dementia‐like phenotypes associated with presenilin‐1 mutations. Am J Alzheimer Dis Other Demen 2006;21:281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Raux G, Gantier R, Thomas‐Anterion C, et al. Dementia with prominent frontotemporal features associated with L113P presenilin 1 mutation. Neurology 2000;55:1577–1578. [DOI] [PubMed] [Google Scholar]
- 36. Tedde A, Forleo P, Nacmias B, et al. A presenilin mutation (Leu392Pro) in a familial AD kindred with psychiatric symptoms at onset. Neurology 2000;55:1590–1591. [DOI] [PubMed] [Google Scholar]
- 37. Queralt R, Ezquerra M, Lleó A, et al. A novel mutation (V89L) in the presenilin 1 gene in a family with early onset Alzheimer's disease and marked behavioural disturbances. J Neurol Neurosurg Psychiatry 2002;72:266–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Larner AJ, Doran M. Clinical phenotypic heterogeneity of Alzheimer's disease associated with mutations of the presenilin‐1 gene. J Neurol 2006;253:139–158. [DOI] [PubMed] [Google Scholar]
- 39. Ezquerra M, Carnero C, Blesa R, Gelpí JL, Ballesta F, Oliva R. A presenilin 1 mutation (Ser169Pro) associated with early‐onset AD and myoclonic seizures. Neurology 1999;52:566–570. [DOI] [PubMed] [Google Scholar]
- 40. Lampe TH, Bird TD, Nochlin D, et al. Phenotype of chromosome 14‐linked familial Alzheimer's disease in a large kindred. Ann Neurol 1994;36:368–378. [DOI] [PubMed] [Google Scholar]
- 41. Mathew R, Bak TH, Hodges JR. Diagnostic criteria for corticobasal syndrome: a comparative study. Neurology 2012;83:405–410. [DOI] [PubMed] [Google Scholar]
- 42. Heston LL, Lowther DLW, Leventhal CM. Alzheimer's disease. A family study. Arch Neurol 1966;15:225–233. [DOI] [PubMed] [Google Scholar]
- 43. Verkkoniemi A, Perez‐Tur J, Mehta N, et al. A variant of Alzheimer's disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat Med 1998;4:452–455. [DOI] [PubMed] [Google Scholar]
- 44. Houlden H, Baker M, McGowan E, et al. Variant Alzheimer's disease with spastic paraparesis and cotton wool plaques is caused by PS‐1 mutations that lead to exceptionally high amyloid‐beta concentrations. Ann Neurol 2000;48:806–808. [PubMed] [Google Scholar]
- 45. Takao M, Ghetti B, Hayakawa I, et al. A novel mutation (G217D) in the presenilin 1 gene (PSEN 1) in a Japanese family: presenile dementia and parkinsonism are associated with cotton wool plaques in the cortex and striatum. Acta Neuropathol 2002;104:155–170. [DOI] [PubMed] [Google Scholar]
- 46. Homma T, Takuboc H, Takahashib K, et al. Lateralized cortical involvement and contralateral parkinsonism without basal ganglia involvement in two autopsy cases of corticobasal syndrome‐Alzheimer's disease. J Alzheimers Dis 2014;40:51–55. [DOI] [PubMed] [Google Scholar]
- 47. Goldman JS, Farmer JM, Wood EM, et al. Comparison of family histories in FTLD subtypes and related tauopathies. Neurology 2005;65:1817–1819. [DOI] [PubMed] [Google Scholar]
- 48. Pickering‐Brown SM, Rollinson S, Du Plessis D, et al. Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain 2008;131:721–731. [DOI] [PubMed] [Google Scholar]
- 49. Spina S, Murrell JR, Huey E, et al. Corticobasal syndrome associated with the A9D progranulin mutation. J Neuropathol Exp Neurol 2007;66:892–900. [DOI] [PubMed] [Google Scholar]
- 50. Masellis M, Momeni P, Meschino W, et al. Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain 2006;129:3115–3312. [DOI] [PubMed] [Google Scholar]
- 51. Josephs KA, Petersen RC, Knopman DS, et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology 2006;66:41–48. [DOI] [PubMed] [Google Scholar]
- 52. Kouri N, Carlomagno Y, Baker M. Novel mutation in MAPT exon 13 (p.N410H) causes corticobasal degeneration. Acta Neuropathol 2014;127:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lesage S, Le Ber I, Condroyer C, Brousolle E, Gabelle A, Thobois S, et al. The analysis of C9orf72 repeat expansions are a rare genetic cause of parkinsonism. Brain 2013;136:385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schottlaender LV, Polke JM, Ling H, MacDonald ND, Tucci A, Nanji T, et al. The analysis of C9orf72 repeat expansions in a large series of clinically and pathologically diagnosed cases with atypical parkinsonisms. Neurobiol Aging 2015;36:1221.e1–1221.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fekete R, Bainbridge M, Baizabal‐Carvallo JF, Rivera A, Miller B, Du P, et al. Exome sequencing in familial corticobasal degeneration. Parkinsonism Relat Disord 2013;19:1049–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stamelou M, Quinn NP, Bhatia KP. “Atypical” atypical parkinsonism: new genetic conditions presenting with features of progressive supranuclear palsy, corticobasal degeneration, or multiple system atrophy–a diagnostic guide. Mov Disord 2013;28:1184–1199. [DOI] [PubMed] [Google Scholar]
- 57. Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG‐2 crieria. Lancet Neurol 2014;14:614–629. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A video accompanying this article is available in the supporting information here.
Video 1. Segment 1: Patient shows marked postural flexion of left upper limb. Segment 2: Spontaneous jerking of fingers of left hand. Segment 3: Muscular jerks increase during activity (on the left), but are not seen on the right. Segment 4: Left‐sided jerks are also evident when the patient's hands are outstretched. Segment 5: Light pinpricks on the dorsal aspect of fingers of his left hand triggers a circumscribed jerking movement of that finger. A tap on left forearm and chest triggers a jerk of the entire left upper limb. Segment 6: Paratonic rigidity with attempts to mobilize the patient's left arm.