

Acquired neuromyotonia (NMT) is a part of the spectrum of peripheral nerve hyperexcitability (PNH) syndrome, characterized by spontaneous and continuous muscle fiber activity, cramps and stiffness of peripheral nerve origin.1 We report the interesting case of a patient with myasthenia gravis (MG), who developed isolated facial and lingual NMT.
A 72‐year‐old man developed non‐progressive dysphagia and dysphonia over a period of 2 months. He described a feeling of tightening of his tongue and throat that interfered with swallowing and speech. He denied weakness or stiffness of facial and limb muscles apart from frequent calf cramps.
Past medical history included arterial hypertension and well‐controlled, acetylcholine receptor antibody seropositive MG since 2013 manifested by episodic diplopia and ptosis. There was no history of insomnia, excessive sweating, sensory symptoms or CNS disease.
Neurological examination demonstrated prominent undulating rippling of orbicularis oris and continuous, irregular twitching of the tongue (see accompanying Video 1). There was no evidence either of tongue atrophy or involuntary palate movement. Limb and facial muscle strength was normal and there were no abnormalities on ocular, deep tendon reflexes and sensory testing.
EMG sampling of the genioglossus and right orbicularis oris revealed sustained neuromyotonic discharges (see Fig. 1), whereas occasional fasciculations were recorded from the right tibialis anterior and upper portion of the trapezius muscle. Motor and sensory nerve conduction studies, as well as repetitive stimulation of the right facial nerve proved within normal limits.
Figure 1.
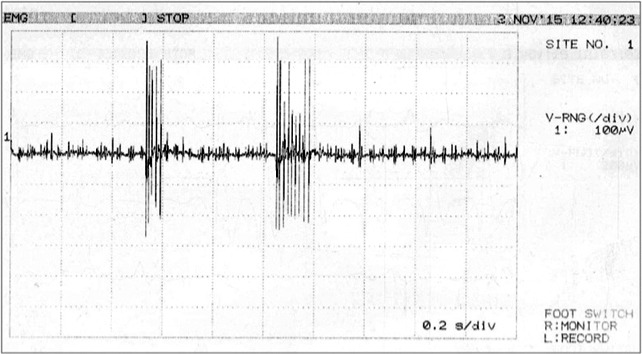
Spontaneous discharges of a single motor unit potential at high frequency (>120 Hz). Note the decrementing response.
Further investigations including MRI of the brain and base of the scull disclosed no abnormalities. Serological studies for antibodies against muscle‐specific kinase (MuSK), voltage‐gated potassium channel (VGKC), and contactin‐associated protein 2 (Caspr2) were similarly negative. Finally, CT scan of the thorax, abdomen and serum tumor markers did not demonstrate evidence of thymoma or other malignancy.
The patient was treated with prednisolone 1 mg/kg/alternate days without clinical response. Subsequently, symptomatic treatment with carbamazepine 600 mg/day was initiated leading to sustained clinical benefit during an 18‐month follow‐up period.
It is well established that MG, among other autoimmune, axonopathic and paraneoplastic disorders, may be associated with NMT.1, 2 In our patient, we detected neuromyotonic discharges defined by their extreme frequency (>120 Hz) and the decrementing pattern of the amplitude of the motor unit action potentials within the burst.1
In this context, our patient's symptoms might be attributed to oropharyngeal muscle stiffness. Alternatively, neuromuscular transmission failure resulting from continuous high rates of motor units firing could theoretically be implicated, although single‐fiber EMG recordings are normal in patients with NMT.1
Simon et al. described two MG patients positive for MuSK antibodies who developed PNH.3 Interestingly, their first patient developed facial NMT similar to our case. The authors hypothesized that these antibodies might have induced a relative deficiency of acetylcholinesterase (AChE) activity by disrupting MuSK‐AChE binding, resulting in increased synaptic ACh concentration. Accumulation of ACh could potentially initiate ectopic nerve discharges by modifying the balance of presynaptic nicotinic and muscarinic channel activation.3
In our patient, NMT was confined to cranial muscles. Of note, isolated bulbar myokymia or NMT is a well‐described complication following head and neck radiotherapy for nasopharyngeal carcinoma.4 Other acquired disorders that have been traditionally associated with faciobulbar NMT include multiple sclerosis, pontine tumors and Guillain‐Barre syndrome.5 The differential diagnosis of facial fasciculations and/or myokymia/NMT is even broader including genetic disorders, such as episodic ataxia type 1, Machado‐Joseph disease and bulbo‐spinal muscular atrophy.6, 7
Separately, acquired NMT may be clinically misdiagnosed as amyotrophic lateral sclerosis (ALS), particularly in the early stages of ALS, during which widespread fasciculations may be evident in the absence of other clinical and electrophysiological features of ALS.8 Interestingly, Vucic et al. have demonstrated that the threshold tracking transcranial magnetic stimulation technique may be of diagnostic use in differentiating NMT from ALS.8
We conclude that isolated faciobulbar NMT may exceptionally emerge in the context of MG related PNH.
Author Roles
1. Research project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript: A. Writing of the first draft, B. Review and Critique.
D.P.: 1A, 2A, 2B, 2C, 3B
P.I.: 3B
G.P.: 1B, 1C, 2A, 3A
D.K.: 2C, 3B
Disclosures
Ethical Compliance Statement: The authors confirm that the approval of an institutional review board was not required for this work. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: The authors report no sources of funding and no conflicts of interest.
Financial Disclosures for the previous 12 months: The authors report no additional financial disclosures.
Supporting information
A video accompanying this article is available in the supporting information here.
Video 1. The video segment demonstrates persistent and arrhythmic contractions of the patient's tongue musculature having a duration ranging from 0.6 to 3.7 seconds. Electrophysiologically, this spontaneous activity corresponded to typical neuromyotonic discharges.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Maddison P, Mills K, Newsom‐Davis J. Clinical electrophysiological characterization of the acquired NMT phenotype of autoimmune peripheral nerve hyperexcitability. Muscle Nerve 2006;33:801–808. [DOI] [PubMed] [Google Scholar]
- 2. Rubio‐Agusti I, Perez‐Miralles F, Sevilla T, et al. Peripheral nerve hyperexcitability: a clinical and immunologic study of 38 patients. Neurology 2011;76:172–178. [DOI] [PubMed] [Google Scholar]
- 3. Simon N, Reddel S, Kiernan M, Layzer R. Muscle‐specific kinase antibodies: a novel cause of peripheral nerve hyperexcitability? Muscle Nerve 2013;48:819–823. [DOI] [PubMed] [Google Scholar]
- 4. Rison RA, Beydoun SR. Teaching Video NeuroImages: tongue myokymia following head and neck radiotherapy for nasopharyngeal carcinoma. Neurology 2009;72:e65. [DOI] [PubMed] [Google Scholar]
- 5. Gutmann L. AAEM minimonograph #37: facial and limb myokymia. Muscle Nerve 1991;14:1043–1049. [DOI] [PubMed] [Google Scholar]
- 6. Tomlinson SE, Rajakulendran S, Tan SV, et al. Clinical, genetic, neurophysiological and functional study of new mutations in episodic ataxia type 1. J Neurol Neurosurg Psychiatry 2013;84:1107–1112. doi: 10.1136/jnnp-2012-304131. Epub 24 January 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. França MC Jr, D'Abreu A, Nucci A, et al. Muscle excitability abnormalities in Machado‐Joseph disease. Arch Neurol 2008;65:525–529. doi: 10.1001/archneur.65.4.525. [DOI] [PubMed] [Google Scholar]
- 8. Vucic S, Cheah B, Yiannikas C, Vincent A, Kiernan M. Corticomotoneuronal function and hyperexcitability in acquired neuromyotonia. Brain 2010;133:2727–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A video accompanying this article is available in the supporting information here.
Video 1. The video segment demonstrates persistent and arrhythmic contractions of the patient's tongue musculature having a duration ranging from 0.6 to 3.7 seconds. Electrophysiologically, this spontaneous activity corresponded to typical neuromyotonic discharges.