

Abstract
Melanoma incidences are increasing rapidly, and ultraviolet (UV) radiation from the sun is believed to be its major contributing factor. UV exposure causes DNA damage in skin which may initiate cutaneous skin cancers including melanoma. Melanoma arises from melanocytes, the melanin-producing skin cells, following genetic dysregulations resulting into hyperproliferative phenotype and neoplastic transformation. Both UVA and UVB exposure to the skin are believed to trigger melanocytic hyperplasia and melanomagenesis. Melanocytes by themselves are deficient in repair of oxidative DNA damage and UV-induced photoproducts. Nicotinamide, an active form of Vitamin B3 and a critical component of the human body’s defense system has been shown to prevent certain cancers including nonmelanoma skin cancers. However, the mechanism of nicotinamide’s protective effects is not well understood. Here, we investigated potential protective effects and mechanism of nicotinamide against UVA- and/or UVB- induced damage in normal human epidermal melanocytes. Our data demonstrated an appreciable protective effect of nicotinamide against UVA- and/or UVB- induced DNA damage in melanocytes by decreasing both cyclobutane pyrimidine dimers and 8-hydroxy-2′-deoxyguanine levels. We found that the photoprotective response of nicotinamide was associated with activation of nucleotide excision repair genes and NRF2 signaling. Further studies are needed to validate our findings in in vivo models.
Keywords: Melanocyte, UV, Nicotinamide, DNA damage repair
Graphical abstract
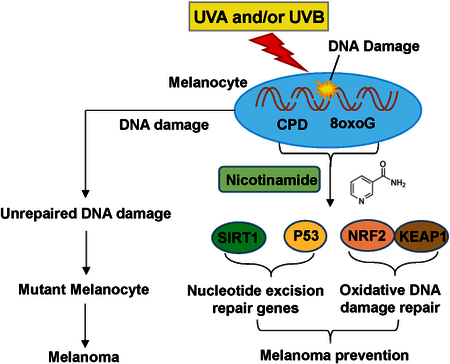
Schematic illustration of the mechanism of nicotinamide against UVA- and/or UVB- mediated DNA damages in normal melanocytes
INTRODUCTION
Ultraviolet (UV) radiation is a predominant environmental carcinogen and a major risk factor for skin cancers including melanoma (1). Solar UV radiation is the primary initiator of skin cancer by causing cutaneous DNA damage. Solar UV radiation that infiltrates the atmosphere and reaches the surface of the Earth is a mixture of UVA (320–400 nm) and UVB (280–320 nm) (2), and each UV component is capable of exerting a variety of effects on the skin cells, including DNA damage and oxidative stress (1). Although 95% of UVB radiation is absorbed in the protective ozone layer in our outer environment, the remaining 5% inbound UVB is a potent genotoxic agent to impart considerable response in skin cells, including melanocytes. UVB penetrates as deep as the epidermal basal cell layer of the skin and induces DNA damage (2), particularly in the form of cyclobutane pyrimidine dimers (CPDs), which are an important source of mutations in UV-induced melanoma (3). On the other hand, UVA is a potent driver of oxidative DNA damage in skin cells and can cause mutations by generating reactive oxygen species (ROS) (4) that oxidize guanine at the 8th position to produce 8-hydroxy-2′-deoxyguanine (8oxoG) (1). Interestingly, UVB also has been shown to produce 8oxoG in Chinese hamster cells (5) and primary human skin fibroblasts (6). Moreover, it has been shown that both UVA and UVB exposure to the skin can trigger melanocytic hyperplasia and melanomagenesis (7, 8). Melanocytes are melanin-producing skin cells that are primarily responsible for melanoma initiation. Wang et al. have shown that melanocytes by themselves are deficient in repair of oxidative DNA damage and UV-induced photoproducts (4). Thus, agents that can modulate DNA repair and prevent UV-induced DNA damage in melanocytes may reduce the risk of melanoma initiation and progression.
Nicotinamide is an amide form of Vitamin B3 or niacin and a critical component of the defense system of the human body which has been shown to enhance repair of genetic damage from various sources including UV radiation (9). Several studies have shown that nicotinamide protects against non-melanoma skin cancer (10, 9). However, there is limited literature available on the protective effects of nicotinamide against UV-induced damages in melanocytes. Additionally, the exact mechanism of DNA damage repair by nicotinamide is not fully understood.
In this study, we determined the potential protective effects of nicotinamide against UVA-, and/or UVB- mediated damages in human melanocytes. Further, the mechanisms of nicotinamide- mediated protection against UV-induced DNA damage was explored at both gene and protein levels.
MATERIALS AND METHODS
Melanocyte culture.
Primary melanocytes isolated from neonatal human skin were obtained from the University of Wisconsin Skin Disease Research Center (UWSDRC) core facility. Melanocytes were cultured in Medium 254 (Gibco, Invitrogen, Carlsbad, CA, USA) supplemented with 1% human melanocyte growth supplement (HMGS) (Gibco, Invitrogen, Carlsbad, CA, USA).
UV exposure and nicotinamide treatment.
Melanocytes were grown on either 60 mm dishes or chambered slides and at a confluency of 70–80% were washed twice with HBSS prior to UV exposure. Cells were pre-treated with 50 µM nicotinamide (3-Pyridinecarboxamide), obtained from Sigma-Aldrich (St Louis, MO, USA) for 24 h followed by exposure to a radiation dose of 3 J/cm2 of UVA or 30 mJ/cm2 of UVB light or a combination of both 3 J/cm2 UVA and 30 mJ/cm2 UVB (UVAB) with a custom designed 120V Spectra UV instrument (Daavlin, Bryan, OH). The UV doses used in our study were selected based on the published studies on UVA- and UVB- mediated cutaneous responses (11, 12). Immediately after UV exposure, HBSS was replaced with fresh growth medium. Following UV exposure, cells were treated with 50 µM nicotinamide for various time points and incubated at 37°C and 5% CO2 incubator. Cell pellets were stored at −80°C for protein or RNA isolation.
Immunostaining of 8oxoG and CPD.
Melanocytes (5×104/chamber) were seeded and grown in 8-well glass chambered slides and pre-treated with 50 µM nicotinamide in culture medium for 24 h followed by UV exposure as described earlier. Post-UV-exposure, cells were maintained with or without 50 µM nicotinamide for up to 3 h at 37°C and 5% CO2. Immunostaining of 8oxoG and CPD was performed as described by Thompson et al. (13) using anti-thymine dimer (CPD) antibody or anti-8oxoG antibody (See Supporting Information, Table S1). The staining was visualized by incubating the slides in the dark with Alexa Fluor 594 diluted 1:100 (red fluorescence) for 30 minutes at 37°C. Counterstaining was performed using 4’, 6-diamidino-2-phenylindole (DAPI) (blue fluorescence) containing mounting media (Vector laboratories, Burlingame, CA, ISA). The images were visualized and captured using EVOS FL Auto Imaging System (Life Technologies, CA, USA).
Western blot analysis.
For Western blot analysis, melanocytes (5×105 cells/2mL media) were seeded and grown for 24 h in 60 mm dishes. Cells were pre-treated with 50 μM nicotinamide for 24 h before exposing them to UVA and/or UVB. After UV exposure, cells were washed again and treated with 50 μm nicotinamide for 24 h. Cell lysates were prepared in ice-cold RIPA lysis buffer and subjected to SDS-PAGE. Proteins were transferred to a 0.2 μm nitrocellulose membrane (Bio-Rad, Hercules, CA), then blocked with 5% non-fat dry milk in TBS-T. Membranes were probed with appropriate primary antibody and secondary horseradish peroxidase (HRP) conjugated antibody (Cell Signaling Technology, Danvers, MA). Proteins were detected by chemiluminescence using Pierce ECL Western Blotting Substrate, or SuperSignal West Femto Chemiluminescent Substrates (Pierce, Rockford, IL). The quantification of the Western blot bands was performed using ImageJ (US National Institutes of Health, Bethesda, MD, USA). The list of primary antibodies used in this study is provided in Table S1.
Reverse transcription-quantitative real-time PCR (RT-qPCR) analysis.
RT-qPCR analysis was done as previously described (14). Briefly, RNA isolation was performed using Qiagen RNA isolation kit. RT-qPCR analysis was performed for genes associated with nucleotide excision repair (NER) and base excision repair (BER) pathways. RNA was transcribed using first strand cDNA synthesis with random primers, dNTPs and M-MLV reverse transcriptase (Promega, WI). RT-qPCR was run using QuantStudio 3 (Thermo Fisher Scientific) with SYBR Premix Ex Taq II (TaKaRa), first strand cDNA, forward and reverse primers. Primer pairs for human SIRT1, P53, DDB1, DDB2, OGG1, ERCC1, ERCC2, CDK7 and GAPDH retrieved from PrimerBank (15) and ACTB (Origene, #HP204660) are detailed in Table S2. RT-qPCR data were analyzed using QuantStudio™ design & analysis software (Thermo Fisher Scientific) and relative mRNA expression levels were calculated using the ΔΔCT comparative method. ACTB and GAPDH were used as endogenous controls.
Statistical analysis.
The experiments were performed in multiple replicates and data are expressed as the mean±SEM. The statistical test used in data analysis to calculate statistical significance are indicated in respective figure legends.
RESULTS
Effects of nicotinamide on UVA- and/or UVB- mediated modulations in 8oxoG in melanocytes
The level of 8oxoG in nucleus is widely used as a valuable biomarker for endogenous oxidative damage to DNA (16). The immunostaining of 8oxoG in primary human melanocytes showed that both UVA and/or UVB radiations significantly increased the levels of nuclear 8oxoG when compared to no UV, as non-irradiated cells incubated with or without nicotinamide showed limited 8oxoG staining. The UVA- and/or UVB- mediated increase in 8oxoG levels were markedly reduced by nicotinamide in primary human melanocytes as observed from reduced 8oxoG staining compared to without nicotinamide-supplemented cells (Fig. 1). Thus, the results suggest that nicotinamide increased repair of oxidative DNA damage in the form of 8oxoG resulting from the UVA and/or UVB (Fig. 1).
Figure 1. Effects of nicotinamide on UVA- and/or UVB- mediated modulations in 8oxoG in melanocytes.

Immunostaining of 8oxoG in melanocytes was performed 1 h post-UV-exposures with or without nicotinamide (50 µM) treatments. Representative images (20x magnification) of DAPI (blue color), 8oxoG staining (red color), and merged are shown for each group of cells. Scale bar = 200 µm. The data shown here represent the experiments done twice in duplicates.
Effects of nicotinamide on UVA- and/or UVB- mediated modulations in CPDs in melanocytes
CPDs are highly mutagenic and formed by exposure of skin cells to UVB radiation. CPDs are a measure of direct DNA damage responsible for mutations associated with skin cancer (17). The immunostaining of CPDs in the primary human melanocytes showed that the levels of nuclear CPD were noticeably increased following UVB exposure when compared to no UV or only UVA exposure. Further, the UVB-induced CPD levels were markedly reduced by nicotinamide in primary human melanocytes after UVB or a combination of UVA and UVB (Fig. 2). Thus, our data suggested that nicotinamide enhances repair of DNA damage in terms of CPD formation resulting from the UVB exposure.
Figure 2. Effects of nicotinamide on UVA- and/or UVB- mediated modulations in CPD in melanocytes.

Immunostaining of CPDs in melanocytes was performed 3 h post-UV-exposures with or without nicotinamide (50 µM) treatments. Representative merged images (20x magnification) of DAPI (blue color), CPD staining (red color) and merged are shown for each group of cells. Scale bar = 200 µm.
Effects of nicotinamide on NER and/or BER pathways
To further determine the mechanism of nicotinamide- mediated protection against DNA damage caused by UVA and/or UVB at the gene level, we performed RT-qPCR analysis of important genes involved in DNA damage repair pathways (NER and BER) (Fig. 3). Sirtuin1 (SIRT1), a founding member of a family of nicotinamide adenine dinucleotide (NAD)-dependent histone deacetylases (18), and known to facilitates the NER pathway in UV-induced DNA damage repair (19), was significantly upregulated in response to nicotinamide in both UVB and UVAB groups. Interestingly, UVAB itself increased SIRT1 level, possibly to act as the first responder against DNA damage. Tumor suppressor protein P53, which plays a major role as an architect of DNA damage repair by pausing the cell cycle to allow time for the repair (20), was upregulated in response to UVB and UVAB in melanocytes with relatively greater levels in nicotinamide treated cells after UVB- and UVAB-irradiation. Damage-specific DNA binding protein 1 (DDB1) recognizes UV-induced DNA lesions and initiates the NER process by binding to DDB2 to form the UV-damaged DNA-binding protein complex (21). Though DDB1 and DDB2 were upregulated in response to UVB and UVAB, nicotinamide treatment further enhanced their levels in these two groups. Additionally, 8-Oxoguanine glycosylase (OGG1), involved in both NER and BER pathways, was one of the DNA repair genes found to be markedly upregulated in all the treatment groups compared to no UV control. OGG1’s primary role is the excision of 8-oxo-2’-deoxyguanosine (8oxo-dG), which is a form of oxidative DNA damage related to cause mutations and cancer (22). Our study further identified the upregulation of excision repair cross-complementation group 1 and 2 (ERCC1 and ERCC2) genes in UVB and UVAB as well as in response to nicotinamide in UVB and UVAB groups. Interestingly, the upregulation of ERCC2 was significantly higher in UVB with nicotinamide treated group compared to UVB alone, suggesting that ERCC2 play significant roles in nicotinamide mediated NER pathway to effectively repair DNA damages. ERCC2 also work as DNA helicase subunit of transcription factor TFIIH that unwinds the DNA in the 5′ to 3′ direction at the damaged location (23). Cyclin-dependent kinase 7 (CDK7) is an important component of the TFIIH, which play a major role in transcription initiation and DNA repair (24). CDK7 was found to be significantly upregulated in UVAB alone and in the UVB and UVAB groups treated with nicotinamide. Importantly, CDK7 was markedly higher in UVB and UVAB groups treated with nicotinamide than UVB and UVAB alone groups, elucidating the role of nicotinamide in the protection of UVB-induced DNA damages. Interestingly, for all these genes, compared to no UV group, no significant changes were observed in nicotinamide alone, and UVA group with or without nicotinamide treatment.
Figure 3. Effects of nicotinamide on nucleotide excision repair (NER) and/or base excision repair (BER) pathways.

RT-qPCR analysis of genes involved in DNA damage repair. Nicotinamide enhances repair of UV-induced DNA damage in melanocytes by modulating genes associated with the NER process. Statistical analyses were performed using one-way ANOVA with Tukey’s multiple comparison test (GraphPad PRISM 5.0 software). The results are expressed as mean±SEM with statistical significance (* P<0.05, ** P<0.01, *** P<0.001, **** P<0.001) compared to no UV control. A star(s) above the lines (*) represents the statistical significance compared in respective treatment groups. The data shown here represent the experiments done thrice in duplicates.
Next, we uploaded these significantly altered genes of the DNA damage repair pathway into Ingenuity Pathway Analysis (IPA, Qiagen) and identified an interaction network of nicotinamide-modulated genes. The network indicated links to other genes with potential involvement in DNA damage repair function. In addition, the potential participation of other key targets such as Histone H1, H3 and H4, RNA polymerase II, transcription factor CREB and growth arrest and DNA damage (GADD45) gene was found in the nicotinamide modulated gene network (Fig. 4). Overall, we found that nicotinamide mediated protection against DNA damage was mostly associated with nucleotide excision repair, as all the modulated genes were of NER pathway.
Figure 4. Ingenuity Pathway Analysis (IPA, Qiagen) analysis of significantly modulated genes involved in DNA damage repair.

The predicted gene-gene interaction network was generated and analyzed using inputs of significantly modulated genes involved in DNA damage repair. Upregulated genes are displayed in red. The solid lines denote a robust correlation with partner genes, and dashed lines indicate statistically significant but less frequent correlations.
Effects of nicotinamide on NRF2-KEAP1 pathway in melanocytes after UVA- and/or UVB- treatment
NRF2–KEAP1 pathway has been shown to play a key role in the protection of the skin cells against UV induced oxidative DNA damage (25). To determine the potential mechanism of nicotinamide in protection from oxidative DNA damage caused by UVA and/or UVB, we used western blot analysis for NRF2 and KEAP1 proteins using cell lysates of UVA- and/or UVB- treated primary human melanocytes supplemented with or without nicotinamide. As shown in Figure 5, UVA- and/or UVB- treatment decreased NRF2 expression in melanocytes when compared to no UV treatment. Whereas, nicotinamide supplementation significantly increased NRF2 expression in UVA- and/or UVB- treated melanocytes (Fig. 5A). Further, KEAP1, that inhibits NRF2 signaling, was decreased by nicotinamide in no UV, UVB- and UVAB- and increased in UVA- treated melanocytes, however, the modulations in KEAP1 were not statistically significant (Fig. 5B).
Figure 5. Effects of nicotinamide on NRF2-KEAP1 pathway in melanocytes after UVA- and/or UVB- induced DNA damages.

Western blot analysis for NRF2 and KEAP1 proteins involved in oxidative DNA damage repair 24 h post-UVA- and/or UVB- exposures in cell lysates (40 µg) of melanocytes supplemented with or without nicotinamide (50 µM). β-Actin was used as a loading control. Representative blots are shown here for the experiments performed in triplicates. Bar diagram shows the quantitative analysis of the bands using ImageJ. Statistical analyses were performed using one-way ANOVA (GraphPad PRISM 5.0 software) with Fisher’s LSD test. The results are expressed as mean±SEM with statistical significance (* P<0.05, ** P<0.01, *** P<0.001). A star above the lines (*) represents the statistical significance compared in respective treatment groups.
DISCUSSION
This study was undertaken to determine the efficacy of nicotinamide against UVA- and/or UVB- mediated damaging responses in melanocytes as well as the mechanisms involved. Nicotinamide has been shown to prevent certain cancers including skin cancer (specifically non-melanoma skin cancers) possibly by facilitating DNA damage repair. In a recent phase III clinical trial, oral nicotinamide was found to be safe and effective in reducing the rates of new non-melanoma skin cancers and actinic keratoses in high-risk patients (10). However, there are limited studies on the efficacy of nicotinamide against melanoma. In this regard, a study by Thompson et al. showed that 50 μM nicotinamide protected melanocytes against UV-induced DNA damage in which melanocytes were irradiated with 2 J/cm2 solar‐simulated UV which was delivered from a 1000‐W xenon arc lamp (13). We were interested in determining the distinct effects of UVA and UVB radiations and their combination in melanocytes to determine the protective effects and mechanisms of nicotinamide.
For this purpose, we used a custom-designed 120V Spectra UV instrument (Daavlin, Bryan, OH) that allows exposing cells to each UV region of the terrestrial solar spectrum (UVA and UVB) and provides precise control of UV wavelength and dose delivery. Using this unit, we determined the protective effects of nicotinamide on primary human neonatal melanocytes against UVA- and/or UVB- mediated DNA damages. In accordance to the study by Thompson et al. (13), we also observed that nicotinamide reduced CPD and 8oxoG levels in melanocytes, which were exposed to UVA and/or UVB radiations. However, the reduction in CPD and 8oxoG levels were greater in the case of UVB treated melanocytes compared to UVA or combined UVAB treatments. This trend was also observed in our further mechanistic investigations of nicotinamide mediated protection of photodamage in melanocytes.
Further, our study explored the involvement of several key genes associated with DNA damage signaling pathways in the facilitation of nicotinamide-mediated protection from UV-induced DNA damage. NER is an important DNA damage repair process, which detects and repairs lesions that cause both chemical and structural alterations of the DNA helix such as CPDs. Whereas, the BER is the main process by which cells repair free radical damage in DNA to avoid oxidative mutagenesis (26). We found significant upregulation in SIRT1, P53, DDB1, DDB2, OGG1, ERCC1, ERCC2 and CDK7 in response to UVB and UVAB irradiation possibly to cope with DNA damages, which was further enhanced by nicotinamide treatment conceivably for better protection. Interestingly, as shown in Figure 4, SIRT1 and P53 appeared to be the main modulators of DNA damage repair pathway as they show direct and indirect interactions with most of DNA damage repair genes. Recent studies suggest that SIRT1 has important functions in DNA damage repair (reviewed in (18)). SIRT1 facilitates the NER pathway in UV-induced DNA damage repair and overexpression of SIRT1 protects from UV-induced damage (reviewed in (19)). Although nicotinamide was previously known as a SIRT1 inhibitor, recent studies suggest that it can also act as SIRT1 stimulator depending on context (reviewed in (27)). Whereas, p53 regulates the NER pathway by functioning as a transcription factor for various genes involved in NER or via direct response to DNA damage (28). In our study, we observed that nicotinamide enhanced expression of many NER pathway genes including SIRT1 and p53 after UVB and UVAB exposure, however, we did not observe any change in UVA-exposed groups. UVB radiation is the most mutagenic component of the UV spectrum causing DNA damage. Our data suggest that nicotinamide treatment significantly modulated DNA damage repair genes mostly related to NER pathway largely in response to UVB-induced DNA damages as well as in repair of DNA damages, which could be a major contributing factor in melanomagenesis, if left unrepaired.
Additionally, UVA and UVB radiation are also known to generate oxidative stress in human skin cells via increased cellular levels of reactive oxygen species (ROS) leading to DNA damages (25). The NRF2 signaling pathway is known to reduce oxidative DNA damage by transcriptional modulation of various antioxidant enzymes. Our data demonstrated that melanocytes exposed to UVA and/or UVB showed a reduction in NRF2 levels, which is in accordance with previously published studies (29, 30). Whereas, NRF2 upregulation has been shown to reduce UVB-induced damage in melanocytes (31). In our study, nicotinamide upregulated NRF2 expression after UVA- and/or UVB- treatments in primary human melanocytes. Thus, NRF2 signaling appears to be associated with nicotinamide’s repair capabilities against UV-induced damages. Further, KEAP1 is a negative regulator of NRF2 and NRF2–KEAP1 pathway is known to play an important role in the protection of the skin cells against UVB induced DNA damage (32, 25). Interestingly, we observed that there was a decreasing trend in KEAP1 levels after nicotinamide treatment in melanocytes in no UV, UVB- and UVAB- irradiated groups. Moreover, a non-significant increase in KEAP1 after nicotinamide treatment in UVA-irradiated cells suggest that the effects of nicotinamide may not be the same as in UVB and UVAB treated groups.
Overall, understanding of the molecular mechanisms by which nicotinamide aid in repair of DNA damages caused by UVA and/or UVB may provide new insights and novel means towards protection against cutaneous damages, including melanoma. Further studies are required to validate these findings in in vivo models.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by funding from the NIH (grant numbers R01AR059130 and R01CA176748 to NA), and the Department of Veterans Affairs (VA Merit Review Awards I01BX001008 and I01CX001441; and a Research Career Scientist Award IK6BX003780 to NA). We thank Samantha Fedorowicz for providing technical help in this study. We also acknowledge the core facilities supported by the Skin Diseases Research Center (SDRC) Core Grant P30AR066524 from NIH/NIAMS.
Footnotes
This article is part of a Special Issue celebrating Photochemistry and Photobiology’s 55th Anniversary.
SUPPORTING INFORMATION
Additional Supporting Information is available in the online version of this article:
REFERENCES
- 1.D’Orazio J, Jarrett S, Amaro-Ortiz A and Scott T (2013) UV radiation and the skin. Int J Mol Sci 14, 12222–12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chhabra G, Ndiaye MA, Garcia-Peterson LM and Ahmad N (2017) Melanoma Chemoprevention: Current Status and Future Prospects. Photochem Photobiol 93, 975–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brash DE (2016) UV-induced Melanin Chemiexcitation: A New Mode of Melanoma Pathogenesis. Toxicol Pathol 44, 552–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang HT, Choi B and Tang MS (2010) Melanocytes are deficient in repair of oxidative DNA damage and UV-induced photoproducts. Proc Natl Acad Sci U S A 107, 12180–12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kielbassa C, Roza L and Epe B (1997) Wavelength dependence of oxidative DNA damage induced by UV and visible light. Carcinogenesis 18, 811–816. [DOI] [PubMed] [Google Scholar]
- 6.Kappes UP, Luo D, Potter M, Schulmeister K and Runger TM (2006) Short- and long-wave UV light (UVB and UVA) induce similar mutations in human skin cells. J Invest Dermatol 126, 667–675. [DOI] [PubMed] [Google Scholar]
- 7.Noonan FP, Dudek J, Merlino G and De Fabo EC (2003) Animal models of melanoma: an HGF/SF transgenic mouse model may facilitate experimental access to UV initiating events. Pigment Cell Res 16, 16–25. [DOI] [PubMed] [Google Scholar]
- 8.von Thaler AK, Kamenisch Y and Berneburg M (2010) The role of ultraviolet radiation in melanomagenesis. Exp Dermatol 19, 81–88. [DOI] [PubMed] [Google Scholar]
- 9.Nazarali S and Kuzel P (2017) Vitamin B Derivative (Nicotinamide)Appears to Reduce Skin Cancer Risk. Skin Therapy Lett 22, 1–4. [PubMed] [Google Scholar]
- 10.Chen AC, Martin AJ, Choy B, Fernandez-Penas P, Dalziell RA, McKenzie CA, Scolyer RA, Dhillon HM, Vardy JL, Kricker A, St George G, Chinniah N, Halliday GM and Damian DL (2015) A Phase 3 Randomized Trial of Nicotinamide for Skin-Cancer Chemoprevention. N Engl J Med 373, 1618–1626. [DOI] [PubMed] [Google Scholar]
- 11.Syed DN, Afaq F and Mukhtar H (2012) Differential activation of signaling pathways by UVA and UVB radiation in normal human epidermal keratinocytes. Photochem Photobiol 88, 1184–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu QM, Yi WJ, Su MY, Jiang S, Xu SZ and Lei TC (2017) Induction of retinal-dependent calcium influx in human melanocytes by UVA or UVB radiation contributes to the stimulation of melanosome transfer. Cell Prolif 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thompson BC, Surjana D, Halliday GM and Damian DL (2014) Nicotinamide enhances repair of ultraviolet radiation-induced DNA damage in primary melanocytes. Exp Dermatol 23, 509–511. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Peterson LM, Ndiaye MA, Singh CK, Chhabra G, Huang W and Ahmad N (2017) SIRT6 histone deacetylase functions as a potential oncogene in human melanoma. Genes Cancer 8, 701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Spandidos A, Wang H and Seed B (2012) PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res 40, D1144–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fortini P, Pascucci B, Parlanti E, D’Errico M, Simonelli V and Dogliotti E (2003) 8-Oxoguanine DNA damage: at the crossroad of alternative repair pathways. Mutat Res 531, 127–139. [DOI] [PubMed] [Google Scholar]
- 17.Berube R, Drigeard Desgarnier MC, Douki T, Lechasseur A and Rochette PJ (2018) Persistence and Tolerance of DNA Damage Induced by Chronic UVB Irradiation of the Human Genome. J Invest Dermatol 138, 405–412. [DOI] [PubMed] [Google Scholar]
- 18.Singh CK, Chhabra G, Ndiaye MA, Garcia-Peterson LM, Mack NJ and Ahmad N (2018) The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid Redox Signal 28, 643–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan W and Luo J (2010) SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol Cell 39, 247–258. [DOI] [PubMed] [Google Scholar]
- 20.Williams AB and Schumacher B (2016) p53 in the DNA-Damage-Repair Process. Cold Spring Harb Perspect Med 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iovine B, Iannella ML and Bevilacqua MA (2011) Damage-specific DNA binding protein 1 (DDB1) is involved in ubiquitin-mediated proteolysis of p27Kip1 in response to UV irradiation. Biochimie 93, 867–875. [DOI] [PubMed] [Google Scholar]
- 22.Yasui M, Kanemaru Y, Kamoshita N, Suzuki T, Arakawa T and Honma M (2014) Tracing the fates of site-specifically introduced DNA adducts in the human genome. DNA Repair (Amst) 15, 11–20. [DOI] [PubMed] [Google Scholar]
- 23.Oksenych V and Coin F (2010) The long unwinding road: XPB and XPD helicases in damaged DNA opening. Cell Cycle 9, 90–96. [DOI] [PubMed] [Google Scholar]
- 24.Rimel JK and Taatjes DJ (2018) The essential and multifunctional TFIIH complex. Protein Sci 27, 1018–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kawachi Y, Xu X, Taguchi S, Sakurai H, Nakamura Y, Ishii Y, Fujisawa Y, Furuta J, Takahashi T, Itoh K, Yamamoto M, Yamazaki F and Otsuka F (2008) Attenuation of UVB-induced sunburn reaction and oxidative DNA damage with no alterations in UVB-induced skin carcinogenesis in Nrf2 gene-deficient mice. J Invest Dermatol 128, 1773–1779. [DOI] [PubMed] [Google Scholar]
- 26.Rastogi RP, Richa, Kumar A, Tyagi MB and Sinha RP (2010) Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J Nucleic Acids 2010, 592980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hwang ES and Song SB (2017) Nicotinamide is an inhibitor of SIRT1 in vitro, but can be a stimulator in cells. Cell Mol Life Sci 74, 3347–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fitch ME, Cross IV and Ford JM (2003) p53 responsive nucleotide excision repair gene products p48 and XPC, but not p53, localize to sites of UV-irradiation-induced DNA damage, in vivo. Carcinogenesis 24, 843–850. [DOI] [PubMed] [Google Scholar]
- 29.Marrot L, Jones C, Perez P and Meunier JR (2008) The significance of Nrf2 pathway in (photo)-oxidative stress response in melanocytes and keratinocytes of the human epidermis. Pigment Cell Melanoma Res 21, 79–88. [DOI] [PubMed] [Google Scholar]
- 30.Kokot A, Metze D, Mouchet N, Galibert MD, Schiller M, Luger TA and Bohm M (2009) Alpha-melanocyte-stimulating hormone counteracts the suppressive effect of UVB on Nrf2 and Nrf-dependent gene expression in human skin. Endocrinology 150, 3197–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janjetovic Z, Jarrett SG, Lee EF, Duprey C, Reiter RJ and Slominski AT (2017) Melatonin and its metabolites protect human melanocytes against UVB-induced damage: Involvement of NRF2-mediated pathways. Sci Rep 7, 1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMahon M, Itoh K, Yamamoto M and Hayes JD (2003) Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem 278, 21592–21600. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.