

The V1-ATPase motor forms metastable asymmetrical structures that suggest the basis for the cooperative transition mechanism.
Abstract
V1-ATPase is an ATP-driven rotary motor that is composed of a ring-shaped A3B3 complex and a central DF shaft. The nucleotide-free A3B3 complex of Enterococcus hirae, composed of three identical A1B1 heterodimers, showed a unique asymmetrical structure, probably due to the strong binding of the N-terminal barrel domain, which forms a crown structure. Here, we mutated the barrel region to weaken the crown, and performed structural analyses using high-speed atomic force microscopy and x-ray crystallography of the mutant A3B3. The nucleotide-free mutant A3B3 complex had a more symmetrical open structure than the wild type. Binding of nucleotides produced a closely packed spiral-like structure with a disrupted crown. These findings suggest that wild-type A3B3 forms a metastable (stressed) asymmetric structure composed of unstable A1B1 conformers due to the strong constraint of the crown. The results further the understanding of the principle of the cooperative transition mechanism of rotary motors.
INTRODUCTION
The quaternary structure of a protein is thought to be the basis of the cooperativity of its subunits, as seen in hemoglobin and multimeric ring complexes including helicases and AAA-type adenosine triphosphatases (ATPases) (1). F-, A-, and V-ATPases are unique rotary motors that also contain these ring structures in their catalytic domains and are capable of pumping ions by using adenosine 5′-triphosphate (ATP) hydrolysis energy (2). F-ATPases work as ATP synthases in mitochondria, chloroplasts, and oxidative bacteria (3). In Archaea, A-ATPases work as ATP synthases like F-ATPase, although their structure and subunit composition resemble those of V-ATPases (4). V-ATPases work as proton-transporting ATPases in various organelles and plasma membranes of eukaryotic cells (2). These ATPases consist of hydrophilic globular catalytic domains (F1, A1, or V1) and hydrophobic membrane-embedded domains (Fo, Ao, or Vo), which facilitate ion translocation across membranes (2–4). The rotational catalysis mechanisms of these ATPases have been proposed on the basis of single-molecule observations (5, 6, 7) and structural studies (8–13). However, the principle of the cooperative transition mechanism of the rotary motors remains unclear.
V-ATPases are also found in bacteria (14, 15). We have been studying the structure and function of bacterial V-ATPase from Enterococcus hirae (15). This enzyme physiologically functions as a primary ion (Na+) pump (16) and is composed of nine subunits, with amino acid sequences that are homologous to those of the corresponding subunits of eukaryotic V-ATPases (15). We have established the in vitro expression, purification, and reconstitution systems of E. hirae V1-ATPase from A3B3 and DF complexes (17) and have elucidated several crystal structures (18, 19) and direct rotational dynamics of the V1 complex (20, 21) by x-ray crystallography and single-molecule observations, respectively. On the basis of these findings, we have proposed a rotation mechanism model of the V1-ATPase, in which the fundamental principle of the cooperative function is not yet defined (19).
We have also solved the crystal structures of A3B3 complex of E. hirae V1-ATPase (Fig. 1A), which is composed of three A1B1 pairs that form a hexamer ring. Each A1B1 pair as a unit consists of a catalytic subunit (A) and a noncatalytic subunit (B). To form the A3B3 ring, the N-terminal β-barrel domain is postulated to operate like as a headband (18) to form a crown structure (Fig. 1B). The A3B3 complex, consisting of three identical A1B1 units, has an asymmetrical hexamer ring structure even in the absence of bound nucleotides (eA3B3) (18). The central domains of the A and B subunits in the hexamer are in close contact and are densely packed, probably because of the tight packing of the crown structure, which may impart stress to the subunits that, in turn, produces a strain that forms the asymmetrical arrangement (Fig. 1C). As a result, the three A1B1 units are composed of three different conformations with different nucleotide binding affinities: “empty” (ATP-unbound form incapable of nucleotide binding), “bindable” (ATP-accessible form), and “bound” (ATP-bound form) forms (Fig. 1D) (18). The asymmetrical structure of nucleotide-free A3B3 complex (eA3B3) is changed upon the binding of the nonhydrolyzable ATP analog, adenosine 5′-(β, γ-imino) triphosphate (AMP-PNP). The binding of AMP-PNP to the bound and bindable forms triggers a conformational change of the bindable form to the bound form (bA3B3; Fig. 1E). These structures suggest that the A3B3 complex cooperatively changes its conformation from one asymmetrical structure to another by the binding and dissociation of nucleotides in one rotational direction, which determines the order of ATP hydrolysis (18). Furthermore, the structure of bA3B3 is changed by the binding of the DF complex inside the ring, which results in the formation of a more tightly packed “tight” structure compared to the bound structure formed in A3B3 complex with bound AMP-PNP (bV1; Fig. 1F). The tight form presumably corresponds to the “catalytic dwell” waiting for ATP hydrolysis to occur in the rotary cycles (18, 19).
Fig. 1. Crystal structures of the A3B3 and A3B3DF (V1) complexes.
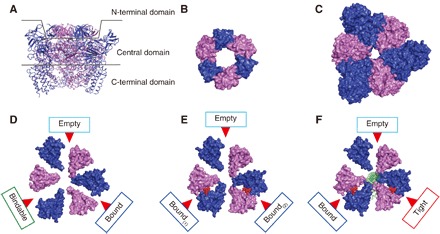
(A) Side view of the nucleotide-free A3B3 (eA3B3) structure divided into three domains as shown with two lines: N-terminal [residues A (1–71) and B (1–77)], central [residues A (72–449) and B (78–362)], and C-terminal [residues A (450–593) and B (363–458)] domains. A and B subunits are depicted in blue and purple, respectively. (B to F) Top views of N-terminal β-barrel domain (crown) (B), central domain (C), and C-terminal domains of eA3B3 (D), AMP-PNP bound A3B3 (bA3B3) (E), and AMP-PNP bound V1 (bV1) (F) as surface representations viewed from the N-terminal β-barrel side. Red arrows indicate the nucleotide binding sites. The bound AMP-PNP molecules are shown as red spheres. The DF shaft is shown in green.
To elucidate the mechanisms of the asymmetrical structure formation and the cooperative conformational change depending on nucleotide binding to the A3B3 complex, we designed a loosely packed A3B3 mutant of E. hirae by weakening the crown structure. The three-dimensional (3D) structure of the mutant was studied using high-speed atomic force microscopy (HS-AFM) and x-ray crystallography. By comparing the structures of the mutant and wild-type A3B3, we clarified the mechanism of the asymmetrical structure formation. Owing to the weak force exerted by the mutant crown, the mutant A3B3 in the absence of bound nucleotides had a nearly symmetrical structure, with more open A1B1 units than the wild type, indicating that the crown is the key determinant of the asymmetry of the A3B3 structure. In addition, a tight form having a closely packed A1B1 structure was observed in the mutant A3B3 in the presence of bound AMP-PNP, even without the DF shaft. On the basis of these findings, we propose that the origin of the cooperative activity of the subunits, as observed for V-ATPase, is from the stress-strain within the molecular rotary motors, which work by transitioning between metastable conformations.
RESULTS
Sample preparation and biochemical properties
To weaken the crown structure of E. hirae A3B3, we mutated an amino acid at the hydrophobic boundary of the β-barrel domain connecting the three A1B1 units by referring to the 3D structures of the complex (18). We replaced the leucine residue at amino acid 65 (Leu65) in the B subunit with tyrosine (Tyr), which is more hydrophilic and bulky. The L65Y mutant B has weaker hydrophobic interactions within the crown structure, which was expected to produce a less stable ring formation of the composite units in the mutant. We expressed the mutant B (L65Y) and unchanged A proteins in an Escherichia coli cell-free protein expression system. The mutant B (L65Y) and unchanged A subunits were purified similar to the wild-type A3B3 complex, even with a weakened crown (fig. S1). To evaluate the subunit stoichiometry of those complexes in solution, negative staining electron microscopy (EM) was done using a Tecnai F20 device. The examinations revealed a ring-shaped hexamer with an A and B subunit stoichiometry of 3:3 (fig. S2).
ATPase activities of the purified wild-type A3B3 and mutant A3B(L65Y)3 complexes were measured to compare their integrity (fig. S1). The initial velocity of the specific activity of the mutant was approximately 40% that of the wild-type activity. The activity of the mutant decreased rapidly, in contrast to the gradual decrease observed in the wild-type activity. Following incubation with the DF complex, the activity of wild-type enzyme increased by approximately 2.9 times, suggesting that the V1 complex was reconstituted with the DF shaft. The mutant A3B(L65Y)3 showed similar enhancement of activity following incubation with DF, suggesting that the mutation had not affected the interaction with the shaft, thus maintaining its integrity. However, the mutant activity was lower than that of the wild type, possibly because the weakened crown caused a gradual loss of complex stability. These results indicate that the mutant A3B(L65Y)3 retains the ability to interact with DF shaft but is less stable.
HS-AFM of wild-type and mutant A3B3
To compare the structural differences between the wild-type A3B3 and mutant A3B(L65Y)3 complexes in the absence of nucleotides, we used HS-AFM, which is a unique tool capable of visualizing the structure and dynamics of proteins, nucleic acids, cells, and organelles (22). HS-AFM can capture images of individual biomolecules in aqueous solution at a lateral resolution of approximately 2 nm, a vertical resolution of approximately 0.1 nm, and a time resolution of approximately 100 ms (22, 23). It can observe a single molecule for many frames in solution. Therefore, HS-AFM is a useful tool to understand the structure of individual molecules, thus providing a different perspective from other techniques such as crystallography, which averages structures from thousands of different molecules. In addition, HS-AFM is very sensitive to height (approximately 1 Å within the same AFM image), in some cases providing a higher resolution than a cryo-EM. For these reasons, evaluating the structural differences using HS-AFM is a reasonable choice. Previous HS-AFM observations of the α3β3 hexamer ring of F1-ATPase, which has a shape resembling the A3B3 hexamer ring of V1-ATPase, showed changes of the α3β3 complex from symmetrical to asymmetrical ring shape upon the addition of ATP (7) [unlike the A3B3 complex, the α3β3 complex has a symmetrical structure in the absence of nucleotides (24)]. Therefore, it seemed likely that HS-AFM would also detect the structural differences between the wild-type A3B3 and the mutant A3B(L65Y)3.
The A3B3 complex was deposited on the mica surface using the same condition as α3β3 hexamer ring of F1-ATPase, where the mica surface was chemically treated with 3-aminopropyl-triethoxysilane (APTES) followed by glutaraldehyde fixation to attach the N-terminal side of the A3B3 complex to the surface (7). This permitted the examination of the C-terminal side of the A3B3 complex, which is the region where the complex interacts with the central DF complex. Consistent with the crystal structure of the A3B3 complex and the known size difference between the A and B subunits (the A subunit is larger than the B subunit by approximately 140 amino acids) (Fig. 2A), HS-AFM examination of the A3B3 complex revealed the hexamer ring structure with three higher (bright) spots corresponding to the A subunits for both the wild-type and weak mutant (Fig. 2, B and C). The height difference between the A and B subunits was 0.1 to 0.5 nm for the wild-type and 0.55 nm for the mutant. To eliminate the noise derived from HS-AFM and positional fluctuations of the molecules, we averaged successive HS-AFM images for each molecule. In addition, by enhancing the image contrast, we were able to visualize each subunit of the A3B3 hexamer (Fig. 2, D and F, and movie S1). Enhancing the contrast further revealed that one of the three A subunits was lower (darker) in the wild-type A3B3 (Fig. 2E). All the images used for averaging were aligned with the same orientation of the A subunit height. The difference in the lowest and highest height of the A subunits was 0.3 ± 0.1 nm. Considering the vertical resolution of HS-AFM, this difference in size was significant and allowed us to conclude that the wild-type A3B3 complex took a similar asymmetric form in aqueous solution and in the crystals (Fig. 1D). By contrast, the mutant A3B(L65Y)3 did not show a clear difference in height among the three A subunits (Fig. 2G), suggesting that the L65Y mutation in the B subunits makes the A3B(L65Y)3 structure more symmetrical, as expected from the reduced interaction between the A and B subunits in the mutant complex.
Fig. 2. HS-AFM images of the wild-type and mutant A3B3 complexes.
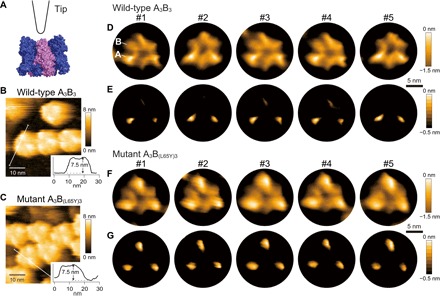
(A) Schematic image of HS-AFM imaging observation of A3B3 from the side with the N-terminal domain at the bottom. A and B subunits are shown in blue and purple, respectively, as a surface representation. The tip of the short cantilever is also drawn. (B and C) HS-AFM images of wild-type A3B3 (B) and mutant A3B(L65Y)3 (C) showing 50 nm × 50 nm area. The insets (30 nm × 8 nm) are the height line profiles from the path indicated by a white line in the images with the height of the highest peak marked. (D and E) Five representative HS-AFM images of wild-type A3B3 as averaged images, with the contrast of the height color table adjusted (see Materials and Methods). An A subunit (higher subunit) and a B subunit (lower subunit) are indicated at #1 in (D). The orientations of averaged images were adjusted to be the same for all molecules. Scale bar is indicated by a black bar and denotes 5 nm. (F and G) Five representative averaged images of the mutant A3B(L65Y)3. Zero nanometer of the color maps in (B) and (C) is the lowest point of the images, whereas in (D) to (G) it is the highest.
Crystal structure of nucleotide-free mutant A3B(L65Y)3
We crystallized the mutant A3B(L65Y)3 complex to elucidate detailed structural differences from wild-type eA3B3 and obtained a crystal structure in the absence of nucleotides at 3.4 Å resolution [Protein Data Bank (PDB) ID: 5ZEA; table S1, denoted as eA3B(L65Y)3]. In the crystal lattice, three of the A1B(L65Y)1 units formed a hexagonal arrangement similar to that of the wild-type eA3B3 (Fig. 3, A and B), which indicated the tendency of the A1B(L65Y)1 overall geometrical structure to form a ring, as in A3B(L65Y)3. Two hexamer A3B(L65Y)3 molecules, which were designated eA3B(L65Y)3-1 (chains A to F) and eA3B(L65Y)3-2 (chains G to L), were contained in the asymmetric unit of the crystal lattice. These two structures closely resembled each other [root mean square deviation (RMSD) = 1.30 Å]. Therefore, we used the data from the eA3B(L65Y)3-1 (chains A to F) in all of the figures and in the subsequent discussion.
Fig. 3. Crystal structure of nucleotide-free A3B(L65Y)3.
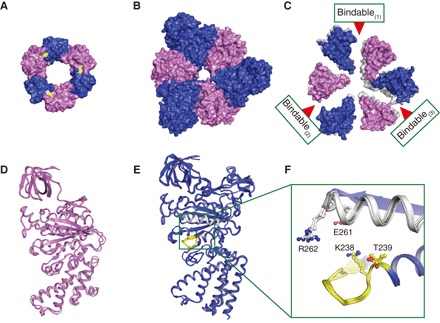
(A) Top view of N-terminal β-barrel (or crown) structure shown as in Fig. 1B. The mutated residues are shown in yellow. (B) Top view of the central domain of eA3B(L65Y)3 shown as in Fig. 1C. (C) Top view of the C-terminal domain of eA3B(L65Y)3 as in Fig. 1D, which is superimposed at the N-terminal β-barrel domain of the bindable form (chains B and E) of A1B(L65Y)1 units onto that of eA3B3 (gray). (D and E) Superimposed structures using Cα atoms of three A (blue) and B (purple) subunits in eA3B(L65Y)3. The P-loop and arm are shown in yellow and white, respectively. The green box indicates the nucleotide binding site. (F) Magnified views of the nucleotide binding sites with conserved residues, corresponding to the green box in (E).
The mutated amino acid Leu65 site had an electron density corresponding to a Tyr residue (fig. S3). Comparison of the crown structures of wild-type and mutant A3B3 complexes revealed several differences in the side-chain orientations around the mutated site, which suggested the loss of hydrophobic interaction (movie S2). This mutation seemed to loosen the crown structure, with its C-terminal halves appearing to be more open, resulting in a slightly looser shape with weaker associations (Fig. 3C and movie S3). The three individual A and B subunit structures in the mutant were very similar (Fig. 3, D to F). Consequently, the mutant A3B3 had a nearly symmetrical open structure as observed in the HS-AFM images (Fig. 2, C, F, and G), while the wild-type eA3B3 displayed the asymmetrical C-terminal halves (empty, bindable, and bound) in the absence of nucleotides (Fig. 1D) (18). All three A1B(L65Y)1 units in the mutant complex were found to be most similar to the bindable form among the possible A1B1 unit forms (empty, bindable, bound, and tight) by comparing the RMSD values (table S2), contact surface areas (table S3), and structures of the substrate binding sites (fig. S4). From the previous (18, 19) and present findings, we propose that the bindable form is a preferable default structure for the A1B1 unit when connected with the β-barrel domain in the absence of bound nucleotides. Thus, the weak crown of the mutant relaxed the dense packing stress of the central domain (Fig. 3B and table S4) by maintaining similarly close interunit contact surface area (approximately 2700 Å2), but with marginally smaller intraunit area (4547.3 Å2) than that of the wild-type (4837.0 Å2), and finally expanded the complex to allow more open symmetrical C-terminal halves in all bindable forms (Fig. 3C and movie S3).
Crystal structure of AMP-PNP bound mutant A3B(L65Y)3
The crystal structure of the wild-type bA3B3 in the presence of excess AMP-PNP revealed a hexameric ring structure similar to that of eA3B3, and two of the three nucleotide binding sites were occupied with AMP-PNP (Fig. 1E). We crystallized the mutant A3B(L65Y)3 complex [bA3B(L65Y)3] in the presence of excess AMP-PNP and determined the crystal structure at a resolution of 2.1 Å (table S1 and Fig. 4A; PDB ID: 5ZE9). The crystal structure revealed a hexamer similar to that of bA3B3; however, one of the three mutated sites in the crown was disrupted. As a consequence, the three A1B(L65Y)1 units formed a spiral-like hexameric structure (Fig. 4, A to D, and movie S4). The nucleotide binding sites of A1B(L65Y)1 units were all occupied with AMP-PNP and magnesium ion (Mg2+), as shown by the electron density maps (Fig. 4, E to G). The electron densities for the γ-phosphate of the AMP-PNP molecules in the two A1B(L65Y)1 units were rather weak (Fig. 4, F and G), probably because AMP-PNP was partially hydrolyzed. Thus, both the native AMP-PNP and the hydrolyzed product, AMP-PN, coexisted in the same crystal, because AMP-PNP tended to be hydrolyzed to AMP-PN and phosphate under the weakly acidic crystallization condition of pH 6.0 (25).
Fig. 4. Crystal structure of AMP-PNP bound A3B(L65Y)3.
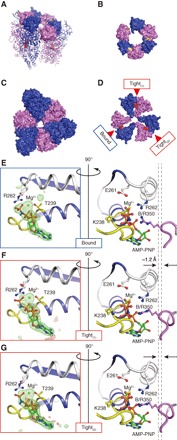
(A) Side-top view of AMP-PNP bound mutant A3B(L65Y)3 [bA3B(L65Y)3]. N-terminal domain is shown in surface representation, and the central and C-terminal domains are in cartoon representations. Colors are the same as in Fig. 1E. (B and C) Top views of the N-terminal (B) and central (C) domains as in Fig. 3, A and B, respectively. (D) Top view of the C-terminal domain of bA3B(L65Y)3 as in Fig. 1E. (E to G) Magnified views of the nucleotide binding sites. The sites corresponding to the green box in Fig. 3E are shown for bA3B(L65Y)3. The P-loop and arm are shown in yellow and white, respectively. The right panels show the A-B interface rotated 90° around a vertical axis from the left panels, with conserved residues of the “bound” (E), “tight(1)” (F), and “tight(2)” (G) forms in bA3B(L65Y)3. To compare structural differences, two dotted vertical lines were drawn from the Cα position of R350 in bound form (E) and tight forms (F and G) to show the closer location (approximately 1.2 Å) of the tight form to the bound nucleotides. The |Fo| − |Fc| map calculated without AMP-PNP: Mg2+ at the binding pockets at 4.0σ are shown in red (negative) and green (positive) meshes.
The bound nucleotide molecules interacted with Lys238 of the P-loop, Arg262 of the “arm” region in the A subunit, and Arg350 (Arg finger) in the B subunit of the three A1B(L65Y)1 units of the mutant (Fig. 4, E to G). One of the three A1B(L65Y)1 units displayed a slightly different interaction between its Arg350 and the nucleotide from the other two units and more closely resembled the bound form observed in bA3B3 (Fig. 4E and fig. S5). This difference in interaction was also supported by the comparison of the contact surface areas (table S3) and RMSD values (table S5). The other two units had Arg350 Cα located approximately 1.2 Å closer to the bound nucleotide than the bound form (Fig. 4, E to G). The A and B subunits of these units were closely packed (table S3), corresponding to that of a tight form detected only in the A1B1 unit interacting with the DF complex in the V1 complex (18). Comparisons of the RMSD values and nucleotide binding sites also supported the assignment of the tight form (table S5 and fig. S5). From these findings, we propose that the tight form is a stable default structure for the AMP-PNP bound A1B1 unit. The packing force of the tight form is so strong (table S3) that the weak crown of the mutant cannot withstand the stress of the constricting structure caused by a hexamer ring with three bound nucleotides. Thus, the crown breaks to release the stress, which results in the formation of the spiral-like structure (Fig. 4A). A bound form in the bA3B(L65Y)3 would have been produced by the additional stress of stacking on the opposite side of the broken pairs; the bound form is probably less stable than the tight form.
DISCUSSION
The assembly of identical units is generally expected to form a symmetrical structure. However, the A3B3 complex in the absence of bound nucleotides shows an asymmetrical hexagonal arrangement, which involves the formation of a crown of the β-barrel domain (18). We confirmed the asymmetrical hexagonal arrangement of the wild-type A3B3 by HS-AFM imaging, which contrasted with the more symmetrical arrangement of the mutant A3B(L65Y)3. X-ray crystallography of the mutant eA3B(L65Y)3 revealed a nearly open symmetrical structure with all bindable forms in the absence of bound nucleotides. We suggest that the bindable form is the preferred structure of the A1B1 unit in the nucleotide-free A3B3 complex. Furthermore, the A3B(L65Y)3 mutant with bound AMP-PNP displayed a closely packed spiral-like hexameric structure with a disrupted crown. We further suggest that the tight form is the stable structure of the A1B1 unit in the AMP-PNP bound A3B3 complex.
On the basis of these findings, we can propose how V-ATPase A3B3 forms an asymmetrical structure in the absence of bound nucleotides. The mutated weak crown is weak enough to relax the stress imposed by the close packing at the central domains of the A and B (L65Y) subunits. Therefore, all three A1B(L65aY)1 units take the preferable default open structure as the bindable form (Fig. 5A). The wild-type crown is strong enough to constrain the structure and form the stringent ring size and shape of the wild-type A3B3. Thus, the A3B3 complex becomes densely packed and closely contacts the central domains under strong stress (Fig. 5B). If the units can change their conformations flexibly and arbitrarily, the hexamer should remain symmetrical, even under the strong constraint of the crown. However, the A1B1 units might have discrete structures, as observed in eA3B3 and bA3B3. Therefore, the combination of the different structures when they assemble into the A3B3 complex results in the formation of an asymmetrical structure with various open or closed units. In this scenario, the wild-type asymmetrical structure of A3B3 may have been produced as a metastable structure with an open, preferable default, bindable form and with less preferable empty (ATP-unbound form with low affinity for nucleotide) and bound forms, which are forced to close by the strong constraint of the crown (Fig. 5B).
Fig. 5. Schematic structure models of the wild-type and mutant A3B3.
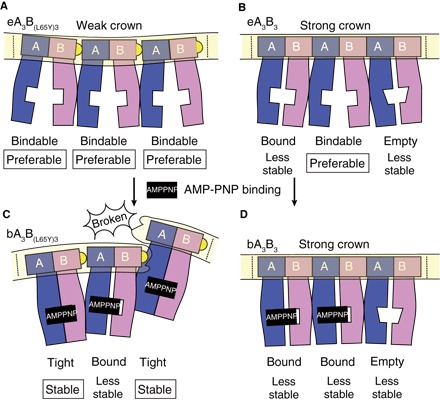
The A3B3 complexes are pictured as a side view from the inside, with the ring cut and laid flat so that all the subunits can be viewed simultaneously. A and B subunits are colored in blue and purple, respectively. The crown is highlighted by a light yellow box. The dotted lines signify where the ends would connect to form the ring. Mutant (A and C) and wild-type A3B3 (B and D) models are drawn showing the conformational changes induced by the nucleotide binding [from (A) to (C) and (B) to (D), respectively]. Yellow half-circles show the mutated amino acid, L65Y, in the crown. Corresponding to the weak crown, mutant models show a convex crown (A) and a concave broken crown (C). AMP-PNP molecules are shown as black squares. The nucleotide binding sites are depicted as white square boxes that are open, half-filled, or filled. The empty form has a specifically low affinity for nucleotides. Therefore, the square boxes indicating the binding sites are distorted to look like a trapezoid. The form name is presented under each A1B1 unit with a description of the preferable (or stable) form or the less stable form.
In the presence of AMP-PNP, A1B1 tends to be in a tight form. The mutated weak crown cannot withstand the stress of forming the ring shape with three tight forms, and the breakage at one site on the crown results in the formation of a spiral-like hexamer (Fig. 5C). The wild-type crown is strong enough to maintain the ring shape of the A3B3 complex, and resists the stress of the closely contacting central domains of the three tight forms. Instead, the crown forces one A1B1 unit to release its bound nucleotide to form a weakly packed empty form. The ring shape might be too constricted to allow the preferable open structure, bindable form, and instead might allow only a slightly closed empty form (Fig. 5D). The other two units can retain the bound nucleotides and adopt the bound form instead of the tight form for the sake of size restriction and symmetry. It is functionally advantageous to attain the empty form (18) in the presence of nucleotides. If the bindable or bound form is attainable, all the nucleotide binding sites in A3B3 might be occupied by nucleotides at high concentrations, which could disrupt the crown structure and the ring. The wild-type crown should be strong enough to keep the ring constricted, as well as to form the empty form instead of the bindable form, expelling bound nucleotides and stabilizing the A3B3 ring structure. Thus, the crown appears to work to control and restrict the behavior of the entire structure, as if it were Sun Wukong’s headband in the Chinese folk tale, Journey to the West: The headband keeps him to behave properly and considerately, preventing him from behaving freely and violently. The necessity of the quaternary structure of a cooperative protein like the A3B3 complex seems to produce a stress and/or strain response within the complex.
The crystal structures of the shaftless motors of V/A-ATP synthase [Thermus thermophilus A3B3 complex, (26)] and F-ATP synthase [Bacillus PS3 α3β3 complex, (24)], on the contrary, showed exact threefold symmetry related by a crystallographic threefold axis in those structures. There are two possibilities regarding this issue. The electron density map in which any difference in the conformation of the complex had been averaged was not suitable for detecting subtle differences between the AB or αβ pairs. Thus, further trials to determine the electron density maps of their whole structures are necessary to settle the symmetry or asymmetry issue. Another possibility is that the asymmetry of the shaftless motor of E. hirae V-ATPase could be just an exceptional observation among rotary motors, but the asymmetry of the E. hirae shaftless motor provides a comprehensive insight for one directional rotation mechanism of the rotary motors as described above. Therefore, we believe that the stress concept proposed for the V-ATPase asymmetry should be the underlying mechanism for any rotary motors. In this respect, the differences in the function of ATP-synthase/ATPase may have a relationship to the differences in those structures. These F- and A-ATPases function as ATP synthases physiologically with reversible ATPase activity, while V-ATPases function specifically as ATP-hydrolyzing enzyme.
In turn, the structures with their respective shafts, not only bacterial V1/A1-ATPase (27) and F1-ATPase (28) but also eukaryotic V1-ATPase (12) and F1-ATPase (8), showed asymmetry with three different forms, similar to those found in E. hirae V1-ATPase (18). E. hirae V1-ATPase showed empty, bound, and tight forms irrespective of nucleotide binding (18). The two empty and bound forms seem to be unstable and stressed conformations within the complex as described above. Thus, the cooperative function of V1-ATPase, and probably of other ATPases, must also be operative by transitioning among the metastable structures produced by the stress and/or strain mechanism as proposed in this study. The rotary motors might be able to continuously perform their hydrolyzing function by transitioning between several metastable asymmetrical structures without ever attaining the most stable structure. This stress and/or strain mechanism might be the fundamental principle for any cooperative proteins with quaternary structures to achieve their cooperative functions.
MATERIALS AND METHODS
Mutagenesis and preparation of proteins
To obtain weak crown mutants, we designed a mutation in the crown structure of the A3B3 ring. On the basis of the 3D structure of A3B3 and V1 in E. hirae (18), we selected the amino acid for mutation at the contact interfaces of three A1B1 units in the β-barrel region (B-L65Y, which is not conserved in F-, A-, and V-ATPases). Mutagenesis of the L65Y of B subunit was performed using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA) (20).
An E. coli cell-free protein expression system was used to synthesize the A, B [or B(L65Y)], and DF complexes [D(T60C/L67C)F mutant for single-molecule analysis (20)] using mixtures of respective plasmids harboring the corresponding genes with a modified natural poly-histidine (MKDHLIHNHHKHEHAHAEH) affinity tag, tobacco etch virus cleavage site (EHLYFQG), and linker (SSGSSG) sequences at the N terminus (17, 18). The cell-free lysate reaction mixture was loaded onto a HisTrap HP column (GE Healthcare, Little Chalfont, UK) equilibrated with buffer A [20 mM tris-HCl, 1 M NaCl, and 20 mM imidazole (pH 8.5)], and bound proteins were eluted with buffer B [20 mM tris-HCl, 500 mM NaCl, and 500 mM imidazole (pH 8.0)]. To obtain nontagged samples, the proteins were treated with tobacco etch virus protease at 4°C. The reaction solution was dialyzed overnight against buffer C [20 mM tris-HCl, 500 mM NaCl, 20 mM imidazole, and 2 mM dithiothreitol (DTT) (pH 8.5)]. The dialysate was loaded onto a second HisTrap HP column equilibrated with buffer A, and the flow-through fractions containing the nontagged proteins were pooled. The sample buffer was exchanged with buffer D [20 mM tris-HCl, 20 mM NaCl, and 2 mM DTT (pH 8.5)] using a HiPrep 26/10 desalting column (GE Healthcare), loaded onto a HiTrap Q HP column (GE Healthcare) equilibrated with buffer D, and eluted with a linear gradient of 20 to 1000 mM NaCl.
For wild-type A3B3 and mutant A3B(L65Y)3 complexes
Eluted samples were further purified using a HiLoad 16/60 Superdex 200 pg column (GE Healthcare) equilibrated with buffer E [20 mM tris-HCl, 10% glycerol, 100 mM NaCl, and 2 mM DTT (pH 7.5)]. Purified samples were concentrated with an Amicon Ultra-10 filter with a molecular weight cutoff of 10,000.
For the DF complex
The eluted sample was further purified using a HiLoad 16/60 Superdex 75 pg column (GE Healthcare) equilibrated with buffer F [20 mM tris-HCl, 150 mM NaCl, and 2 mM DTT (pH 8.0)]. Purified sample was concentrated with an Amicon Ultra-30 filter with a molecular weight cutoff of 3000.
EM and image processing
Purified samples were dispensed on glow-discharged carbon-coated copper grids. Each grid was negatively stained with 2.0% (w/v) uranyl acetate. Micrographs were collected on a direct electron detector (FEI Falcon II) using FEI Tecnai G2 F20 (FEI, Hillsboro, OR, USA) operating at 200 kV. For reference-free 2D class averaging, particle images were boxed out with e2boxer.py in the EMAN2 package (29) and then processed with RELION software (30).
Measurement of ATPase activity
A3B3 or A3B(L65Y)3 (10 mg/ml protein) was reconstituted with the DF complex (5 mg/ml protein) in buffer G [100 mM MES, 5 mM MgSO4, 100 mM NaCl, and 10% glycerol (pH 6.0)] by incubating the mixture on ice for 1 hour (18, 19). The ATPase activity of A3B3 or the reconstituted A3B3DF was measured using an ATP-regenerating system (18, 19). The reaction mixture contained 1 mM ATP/MgSO4, 2.5 mM phosphoenolpyruvate, pyruvate kinase (50 μg/ml), lactate dehydrogenase (50 μg/ml), 0.2 mM β-NADH (nicotinamide adenine dinucleotide) (dipotassium salt), and 2 to 20 μg of protein in 1 ml of reaction mixture (buffer G). The reaction was initiated by adding ATP/MgSO4. The rate of ATP hydrolysis was monitored at 23°C by measuring the rate of oxidation of NADH, as determined by a decrease in absorbance at 340 nm.
Fabrication of electron beam deposit tip on the cantilever for HS-AFM imaging
Electron beam deposit tip was grown on the original tip of a short cantilever (model BL-AC7DS, Olympus, Tokyo, Japan) by focusing the electron beam in an atmosphere of sublimated ferrocene gas using a field-emission scanning electron microscope (model SUPRA 40VP, Carl Zeiss, Jena, Germany) with the following conditions: working distance, 4 nm; beam aperture, 10 μm; deposition time, 20 to 30 s (31). The grown tip was sharpened by etching using a radiofrequency plasma etcher (model PE-2000, South Bay Technology, San Clemente, CA, USA) with the following conditions: gas composition, Ar and O2 in a 1:1 ratio; gas pressure, approximately 180 mtorr; plasma power, 15 W; etching time, approximately 2 min. The resulting tip had a length of 200 to 500 nm and a tip end diameter of 5 to 10 nm.
High-speed atomic force microscopy
HS-AFM observation was performed in the tapping mode using a laboratory-made HS-AFM apparatus (23) equipped with a dynamic feedback controller, in which the feedback gain was automatically tuned during imaging (32). The A3B3 proteins were immobilized on a mica surface that had been coated with APTES and then treated with glutaraldehyde because the protein tends to move on the mica, and hence, it is difficult to obtain enough good data for analysis. In the previous HS-AFM publication of α3β3 of F1-ATPase (7), the immobilization condition was the same as in this experiment. In that condition, the cooperative unidirectional rotation was observed in the presence of ATP. Therefore, we consider that the immobilization of α3β3 does not affect the protein structure so much (if the symmetry changes due to the immobilization, the protein cannot function properly). Because A3B3 of V1-ATPase is a similar protein, we make the same assumption. The mica substrate was freshly cleaved immediately before use. APTES was diluted 1:100,000 in Milli-Q water, deposited on the mica surface, and incubated for approximately 3 min. The APTES-coated substrate was thoroughly washed with Milli-Q water. Glutaraldehyde diluted to 0.025% was deposited on the APTES-treated mica and incubated for 3 min. The surface was rinsed with an observation buffer [20 mM tris-HCl (pH 7.5), 100 mM NaCl, 2 mM DTT, and 10% glycerol]. A drop (approximately 5 μl) of the A3B3 protein solution diluted to concentrations of 100 to 300 nM was loaded onto the substrate surface for 5 min. The sample was rinsed with the observation buffer and examined. The HS-AFM imaging conditions were as follows: cantilever free oscillation amplitude, 0.8 to 2 nmp-p; set-point amplitude, 80 to 90% of the free oscillation amplitude; frame rate, 3 to 5 frames/s; scan and pixel sizes, 50 nm × 50 nm with 150 × 150 pixels or 50 nm × 40 nm with 150 × 120 pixels.
Processing and analysis of HS-AFM images
HS-AFM image data were processed by a laboratory-made software program written in Igor Pro (WaveMetrics, Lake Oswego, OR, USA). The number of pixels of each HS-AFM image was increased twice by linear interpolation. The molecule of interest was tracked to compensate for drift of sample position. The tracking was conducted by calculating a coefficient of correlation between a template image and each frame image under various lateral shifts of the latter image. The lateral shift providing the maximum coefficient value was adopted. Successive HS-AFM images were stacked and averaged, where frames with unclear images were removed.
Crystallization conditions
All crystallization trials were performed using the sitting-drop vapor diffusion method at 296 K. The crystals were soaked in cryoprotectant by incrementally increasing the glycerol concentration up to 20%. The crystals were then mounted on cryo-loops (Hampton Research Corp., Aliso Viejo, CA, USA), flash-cooled, and stored in liquid nitrogen.
For A3B(L65Y)3 without nucleotides, crystals of nucleotide-free A3B3 [eA3B(L65Y)3] [10 mg/ml in 20 mM tris-HCl (pH 7.5), 100 mM NaCl, and 10% (v/v) glycerol] were grown at 23°C by vapor diffusion after an equal volume (0.3 μl) was mixed with 0.1 M NaCl, 0.1 M ammonium acetate, 0.1 M bis-tris propane (pH 6.5), and 28% (w/v) PEG-3350 (polyethylene glycol, molecular weight 3350).
For A3B(L65Y)3 with AMP-PNP, crystals of AMP-PNP bound A3B3 [bA3B(L65Y)3] [12.1 mg/ml in 20 mM tris-HCl (pH 7.5), 100 mM NaCl, 10%(v/v) glycerol incubated with addition of 120 mM MES (pH 6.0), 6 mM AMP-PNP, and 6 mM MgCl2 for 1 hour on ice] were grown at 23°C by vapor diffusion after an equal volume (0.5 μl) was mixed with 0.2 M NaCl, 0.1 M MES (pH 5.5), and 27% (w/v) PEG-3350.
Structure determination
All x-ray diffraction data were collected from a single crystal at a cryogenic temperature (100 K). For eA3B(L65Y)3, x-ray diffraction data were collected on beamline BL-17A (λ = 0.98 Å) at the Photon Factory (Tsukuba, Japan). The collected data were processed to 3.4 Å by x-ray detector software (XDS) (33). The structure was solved by molecular replacement with MOLREP (34) using the wild-type A3B3 without bound nucleotides (PDB ID: 3VR2) as a search model.
For bA3B(L65Y)3, x-ray diffraction data were collected on beamline BL-1A (λ = 1.1 Å) at the Photon Factory. The collected data were processed to 2.1 Å by XDS (33). The structure was solved by molecular replacement with MOLREP (34) using the E. hirae V1 tight form without bound nucleotide (PDB ID: 3VR4; chains C to F) as a search model.
The atomic models were manually built using Coot (35) and iteratively refined using REFMAC5 (36) and PHENIX (37). The refined structures were validated with RAMPAGE (38). The crystallographic and refinement statistics are summarized in table S1. All RMSD values were calculated using Cα atoms. The RMSD values for the superimpositions for each A1B1 unit in the crystal structures are listed in tables S2 and S5. PDBePISA was used for contact surface area calculation (39). Figures were prepared using PyMOL (The PyMOL Molecular Graphics System, version 1.0, Schrödinger LLC, New York, NY, USA). The video was generated by morphing the x-ray crystal structures of nucleotide-free wild-type (eA3B3) and mutant [eA3B(L65Y)3] A3B3 using Chimera [UCSF Chimera, an Extensible Molecular Modeling System, version 1.12.0, University of California, San Francisco (UCSF) Resource for Biocomputing, Visualization, and Informatics] (40) and PyMOL.
Other methods
Protein concentrations were determined with the BCA Protein Assay Kit (Bio-Rad Laboratories, Hercules, CA, USA and Thermo Fisher Scientific, Waltham, MA, USA), with bovine serum albumin used as the standard. SDS–polyacrylamide gel electrophoresis was carried out according to Laemmli, and gels were stained with Coomassie Brilliant Blue. All other chemicals were obtained from Sigma-Aldrich Japan KK (Tokyo, Japan) or Wako Pure Chemicals Industry Ltd. (Osaka, Japan). For crystallization screening, chemicals were obtained from Hampton Research Corp.
Supplementary Material
Acknowledgments
The synchrotron radiation experiments were performed at Photon Factory (proposals 2014G-171, 2014R-51, and 2016G048). We would like to thank the beamline staff at BL-1A and BL-17A of the Photon Factory (Tsukuba, Japan) for help during data collection. We would also like to thank A. Imamura who assisted with English editing, K. Hanada for technical support with plasmid preparations, and N. Kodera who gave us advice regarding the HS-AFM observation and analysis. Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311). Funding: This work was supported in part by Grants-in-aid for Scientific Research (26291009, 17H03638, and 18H05425 to T.M.; 16H00830, 16H00758, and 15H03540 to T.U.; and 26119003 and 24227005 to T.A.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT); by Platform Project for Supporting Drug Discovery and Life Science Research [Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)] from Japan Agency for Medical Research and Development (AMED) under grant nos. JP18am0101082 and JP18am0101083; and by CREST Program (JPMJCR13M1 to T.A.) from the Japan Science and Technology Agency (JST). Author contributions: T.M. designed the study. Y.S. designed and obtained the mutant. Y.S., Y.I.-K., T.K.-S., and M.S. prepared the proteins. S.M. and H.S. crystallized the proteins. S.M., H.S., and K.M. collected x-ray data. S.M., K.S., and H.S. processed and refined x-ray data. H.M. and M.S. performed EM analysis. M.I., T.U., and T.A. performed HS-AFM imaging. S.M., K.S., and F.L.I. performed functional analysis. S.M., M.I., H.S., I.Y., and T.M. analyzed the results. S.M., M.I., and H.M. prepared figures. S.M., M.I., I.Y., and T.M. wrote the paper. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Coordinates and structure factors are deposited in the PDB with ID: 5ZEA and 5ZE9.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaau8149/DC1
Fig. S1. Purification and ATPase activity of wild-type A3B3, mutant A3B(L65Y)3, and DF complexes.
Fig. S2. Electron micrographs of negatively stained wild-type and mutant A3B3 complexes.
Fig. S3. Comparison of the crown structures of wild-type and mutant A3B3 complexes.
Fig. S4. Magnified views of the nucleotide binding sites of the bindable forms in eA3B(L65Y)3.
Fig. S5. Comparison of the nucleotide binding sites of wild-type and mutant complexes.
Table S1. Data collection and refinement statistics.
Table S2. RMSD values in superimpositions for each A1B1 unit of eA3B(L65Y)3 in the crystal structures of A3B3 and V1 complexes.
Table S3. Contact surface areas between A and B subunits in various A1B1 forms (Å2).
Table S4. Sum of contact surface areas at the central domain in A3B3 (Å2).
Table S5. RMSD values in superimpositions for each A1B1 unit of bA3B(L65Y)3 in the crystal structures of A3B3 and V1 complexes.
Movie S1. AFM scan movie of the C-terminal side of the wild-type and mutant A3B3.
Movie S2. Conformational changes at the β-barrel domain induced by the mutation.
Movie S3. Conformational changes at the C-terminal domain of eA3B3 induced by the mutation.
Movie S4. Conformational changes of eA3B(L65Y)3 induced by binding AMP-PNP molecules.
REFERENCES AND NOTES
- 1.Snider J., Thibault G., Houry W. A., The AAA+ superfamily of functionally diverse proteins. Genome Biol. 9, 216 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Forgac M., Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 8, 917–929 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Walker J. E., The ATP synthase: The understood, the uncertain and the unknown. Biochem. Soc. Trans. 41, 1–16 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Grüber G., Manimekalai M. S. S., Mayer F., Müller V., ATP synthases from archaea: The beauty of a molecular motor. Biochim. Biophys. Acta Bioenerg. 1837, 940–952 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Noji H., Yasuda R., Yoshida M., Kinosita K., Direct observation of the rotation of F1-ATPase. Nature 386, 299–302 (1997). [DOI] [PubMed] [Google Scholar]
- 6.Suzuki T., Tanaka K., Wakabayashi C., Saita E.-i., Yoshida M., Chemomechanical coupling of human mitochondrial F1-ATPase motor. Nat. Chem. Biol. 10, 930–936 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Uchihashi T., Iino R., Ando T., Noji H., High-speed atomic force microscopy reveals rotary catalysis of rotorless F1-ATPase. Science 333, 755–758 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Abrahams J. P., Leslie A. G. W., Lutter R., Walker J. E., Structure at 2.8 Â resolution of F1-ATPase from bovine heart mitochondria. Nature 370, 621–628 (1994). [DOI] [PubMed] [Google Scholar]
- 9.Kabaleeswaran V., Puri N., Walker J. E., Leslie A. G. W., Mueller D. M., Novel features of the rotary catalytic mechanism revealed in the structure of yeast F1 ATPase. EMBO J. 25, 5433–5442 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cingolani G., Duncan T. M., Structure of the ATP synthase catalytic complex (F1) from Escherichia coli in an autoinhibited conformation. Nat. Struct. Mol. Biol. 18, 701–707 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morales-Rios E., Montgomery M. G., Leslie A. G. W., Walker J. E., Structure of ATP synthase from Paracoccus denitrificans determined by X-ray crystallography at 4.0 Å resolution. Proc. Natl. Acad. Sci. U.S.A. 112, 13231–13236 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao J., Benlekbir S., Rubinstein J. L., Electron cryomicroscopy observation of rotational states in a eukaryotic V-ATPase. Nature 521, 241–245 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Oot R. A., Kane P. M., Berry E. A., Wilkens S., Crystal structure of yeast V1-ATPase in the autoinhibited state. EMBO J. 35, 1694–1706 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imamura H., Takeda M., Funamoto S., Shimabukuro K., Yoshida M., Yokoyama K., Rotation scheme of V1-motor is different from that of F1-motor. Proc. Natl. Acad. Sci. U.S.A. 102, 17929–17933 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kakinuma Y., Yamato I., Murata T., Structure and function of vacuolar Na+-translocating ATPase in Enterococcus hirae. J. Bioenerg. Biomembr. 31, 7–14 (1999). [DOI] [PubMed] [Google Scholar]
- 16.Murata T., Yamato I., Kakinuma Y., Leslie A. G., Walker J. E., Structure of the rotor of the V-type Na+-ATPase from Enterococcus hirae. Science 308, 654–659 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Saijo S., Arai S., Hossain K. M. M., Yamato I., Suzuki K., Kakinuma Y., Ishizuka-Katsura Y., Ohsawa N., Terada T., Shirouzu M., Yokoyama S., Iwata S., Murata T., Crystal structure of the central axis DF complex of the prokaryotic V-ATPase. Proc. Natl. Acad. Sci. U.S.A. 108, 19955–19960 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arai S., Saijo S., Suzuki K., Mizutani K., Kakinuma Y., Ishizuka-Katsura Y., Ohsawa N., Terada T., Shirouzu M., Yokoyama S., Iwata S., Yamato I., Murata T., Rotation mechanism of Enterococcus hirae V1-ATPase based on asymmetric crystal structures. Nature 493, 703–707 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Suzuki K., Mizutani K., Maruyama S., Shimono K., Imai F. L., Muneyuki E., Kakinuma Y., Ishizuka-Katsura Y., Shirouzu M., Yokoyama S., Yamato I., Murata T., Crystal structures of the ATP-binding and ADP-release dwells of the V1 rotary motor. Nat. Commun. 7, 13235 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minagawa Y., Ueno H., Hara M., Ishizuka-Katsura Y., Ohsawa N., Terada T., Shirouzu M., Yokoyama S., Yamato I., Muneyuki E., Noji H., Murata T., Iino R., Basic properties of rotary dynamics of the molecular motor Enterococcus hirae V1-ATPase. J. Biol. Chem. 288, 32700–32707 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iino R., Ueno H., Minagawa Y., Suzuki K., Murata T., Rotational mechanism of Enterococcus hirae V1-ATPase by crystal-structure and single-molecule analyses. Curr. Opin. Struct. Biol. 31, 49–56 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Ando T., High-speed atomic force microscopy and its future prospects. Biophys. Rev. 10, 285–292 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ando T., Uchihashi T., Scheuring S., Filming biomolecular processes by high-speed atomic force microscopy. Chem. Rev. 114, 3120–3188 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shirakihara Y., Leslie A. G. W., Abrahams J. P., Walker J. E., Ueda T., Sekimoto Y., Kambara M., Saika K., Kagawa Y., Yoshida M., The crystal structure of the nucleotide-free α3β3 subcomplex of F1-ATPase from the thermophilic Bacillus PS3 is a symmetric trimer. Structure 5, 825–836 (1997). [DOI] [PubMed] [Google Scholar]
- 25.Ferguson A. D., Sheth P. R., Basso A. D., Paliwal S., Gray K., Fischmann T. O., Le H. V., Structural basis of CX-4945 binding to human protein kinase CK2. FEBS Lett. 585, 104–110 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Maher M. J., Akimoto S., Iwata M., Nagata K., Hori Y., Yoshida M., Yokoyama S., Iwata S., Yokoyama K., Crystal structure of A3B3 complex of V-ATPase from Thermus thermophilus. EMBO J. 28, 3771–3779 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Numoto N., Hasegawa Y., Takeda K., Miki K., Inter-subunit interaction and quaternary rearrangement defined by the central stalk of prokaryotic V1-ATPase. EMBO Rep. 10, 1228–1234 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shirakihara Y., Shiratori A., Tanikawa H., Nakasako M., Yoshida M., Suzuki T., Structure of a thermophilic F1-ATPase inhibited by an ε-subunit: Deeper insight into the ε-inhibition mechanism. FEBS J. 282, 2895–2913 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., Ludtke S. J., EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Scheres S. H. W., RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uchihashi T., Kodera N., Ando T., Guide to video recording of structure dynamics and dynamic processes of proteins by high-speed atomic force microscopy. Nat. Protoc. 7, 1193–1206 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Kodera N., Sakashita M., Ando T., Dynamic proportional-integral-differential controller for high-speed atomic force microscopy. Rev. Sci. Instrum. 77, 083704 (2006). [Google Scholar]
- 33.Kabsch W., XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vagin A., Teplyakov A., Molecular replacement with MOLREP. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 22–25 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Murshudov G. N., Vagin A. A., Dodson E. J., Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 53, 240–255 (1997). [DOI] [PubMed] [Google Scholar]
- 37.Adams P. D., Afonine P. V., Bunkoczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lovell S. C., Davis I. W., Arendall W. B. III, de Bakker P. I. W., Word J. M., Prisant M. G., Richardson J. S., Richardson D. C., Structure validation by Cα geometry: ϕ,ψ and Cβ deviation. Proteins Struct. Funct. Genet. 50, 437–450 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Krissinel E., Henrick K., Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaau8149/DC1
Fig. S1. Purification and ATPase activity of wild-type A3B3, mutant A3B(L65Y)3, and DF complexes.
Fig. S2. Electron micrographs of negatively stained wild-type and mutant A3B3 complexes.
Fig. S3. Comparison of the crown structures of wild-type and mutant A3B3 complexes.
Fig. S4. Magnified views of the nucleotide binding sites of the bindable forms in eA3B(L65Y)3.
Fig. S5. Comparison of the nucleotide binding sites of wild-type and mutant complexes.
Table S1. Data collection and refinement statistics.
Table S2. RMSD values in superimpositions for each A1B1 unit of eA3B(L65Y)3 in the crystal structures of A3B3 and V1 complexes.
Table S3. Contact surface areas between A and B subunits in various A1B1 forms (Å2).
Table S4. Sum of contact surface areas at the central domain in A3B3 (Å2).
Table S5. RMSD values in superimpositions for each A1B1 unit of bA3B(L65Y)3 in the crystal structures of A3B3 and V1 complexes.
Movie S1. AFM scan movie of the C-terminal side of the wild-type and mutant A3B3.
Movie S2. Conformational changes at the β-barrel domain induced by the mutation.
Movie S3. Conformational changes at the C-terminal domain of eA3B3 induced by the mutation.
Movie S4. Conformational changes of eA3B(L65Y)3 induced by binding AMP-PNP molecules.