

Abstract
Soybean lipoxygenase (SLO) has served as a prototype for understanding the molecular origin of enzymatic rate accelerations. The double mutant (DM) L546A/L754A is considered a dramatic outlier, due to the unprecedented size and near temperature-independence of its primary kinetic isotope effect, low catalytic efficiency, and elevated enthalpy of activation. To uncover the physical basis of these features, we herein apply three structural probes: hydrogen-deuterium exchange mass spectrometry, room-temperature X-ray crystallography and EPR spectroscopy on four SLO variants (wild-type (WT) enzyme, DM, and the two parental single mutants, L546A and L754A). DM is found to incorporate features of each parent, with the perturbation at position 546 predominantly influencing thermally activated motions that connect the active site to a protein-solvent interface, while mutation at position 754 disrupts the ligand field and solvation near the cofactor iron. However, the expanded active site in DM leads to more active site water molecules and their associated hydrogen bond network, and the individual features from L546A and L754A alone cannot explain the aggregate kinetic properties for DM. Using recently published QM/MM-derived ground-state SLO-substrate complexes for WT and DM, together with the thorough structural analyses presented herein, we propose that the impairment of DM is the combined result of a repositioning of the reactive carbon of linoleic acid substrate with regard to both the iron cofactor and a catalytically-linked dynamic region of protein.
Graphical Abstract
INTRODUCTION
Enzymes catalyze chemical reactions with enormous rate enhancements nearing 1026-fold that are accompanied by high regio- and stereo-specificities relative to their uncatalyzed counterparts.1–2 The physical bases for such properties are of considerable ongoing interest and investigation.3–9 De novo designed protein catalysts, based on structural information of active sites and the principles of transition state theory, have generally achieved only moderate rate accelerations in the range of 103-109 fold, even after further functional optimization via directed evolution.10–15 The large gap between naturally occurring catalysts and designed systems underscores the need for increased understanding that can be used to inform greater success in designed catalysts.3–9 Soybean lipoxygenase (SLO) is one of several paradigmatic systems for studying the properties of enzymatic room-temperature hydrogen tunneling and their relevance to general, multidimensional models for the origins of enzymatic rate acceleration.5, 9, 16 SLO catalyzes the stereospecific oxidation of linoleic acid through a rate-limiting abstraction of a pro-S hydrogen atom from the C11 of the substrate linoleic acid (LA); this occurs via a proton coupled electron transfer (PCET) reaction in which a proton is transferred to the active site ferric-bound hydroxide and an electron reduces the ferric center to ferrous (Scheme 1).17–18 The initially measured and, at the time, unprecedented kinetic features of wild type (WT) enzyme,19–21 included a large (ca. 80) and nearly temperature-independent (ΔEa = 0.9 kcal/mol) hydrogen/deuterium kinetic isotope effect and a small enthalpy of activation (Ea(H)= 2 kcal/mol) on kcat. These features have stimulated more than two decades of experimental and theoretical efforts to establish an appropriate room temperature catalytic model.17, 22–29 The use of site specific mutagenesis has played an important role in building and developing a “deep-tunneling” model. Early studies were primarily focused on six single site mutants that reduce the size of native, bulky hydrophobic side chain, at positions either adjacent to the active carbon C-11 of the substrate (residues L546 and L754), or one helix away (residue I553).21, 27 While these mutations produce a range of catalytic impairment on kcat, five of the six mutants studied retain a primary kinetic isotope effect (KIE, = kcat-H/kcat-D) at 30° C similar to WT (with only a single variant, I553G, showing an increase in the KIE to ca. 180). A compelling feature of this series of mutations is an increase in the temperature-dependence of the KIE, as assessed as ΔEa = Ea(D)-Ea(H). (Table S1).
Scheme 1.

Reaction mechanism for the first and rate limiting step of the stereospecific oxidation of linoleic acid catalyzed by SLO, where the hydrogen atom transfer from substrate LA to ferric hydroxide cofactor occurs by a PCET process.
Significantly, the magnitude and temperature-dependence of the experimental KIEs for both WT SLO and each of the single mutants can be well reproduced and interpreted within the framework of vibronically nonadiabatic PCET models.17, 21–23 Within these deep tunneling models, the observed energy barrier (Ea(H)) of the C-H bond activation originates entirely from the heavy atom motions of the protein and substrate. When conformational sampling is optimal, as occurs in the WT enzyme, a compact and precisely positioned active site leads to a small energy difference for the H- vs. D-transfer (i.e. ΔEa 0).30 By contrast, the introduction of packing defects via the reduction in size of native hydrophobic side chains leads to an elongated donor-acceptor distance (DAD) for hydrogen transfer and, concomitantly, a more flexible active site structure that enables the restoration of the DAD to one that resembles native enzyme. Deuterium, with its less diffusive wave function, requires more sampling energy to recover WT-like function, generating a enthalpy of activation for D-transfer that can greatly exceed that for H-transfer (increased ΔEa).30 This “deep-tunneling” feature of SLO has allowed robust correlations to be made between active site motions and the kinetic parameter ΔEa.
More recent investigations of SLOs have assisted in forming important correlations between protein motions and the energy of activation for catalysis. Differences between experimental KIEs during the biomolecular reaction of enzyme with substrate (kcat/Km) and the unimolecular turnover rate (kcat) led to the proposal of perturbed conformational landscapes in selected mutants.31 The further application of various biophysical approaches that include studies of increased hydrostatic pressure,32 hydrogen-deuterium exchange mass spectrometry (HDXMS),33 and 13C ENDOR spectroscopy,34 has both enhanced our understanding of the structural origin of perturbed conformational landscapes and expanded the “deep-tunneling” model to include both local DAD sampling and more global conformational sampling coordinates.9, 35 For the majority of enzymes that have been studied (e.g.,32, 36–40), perturbations produce an elevated energy of activation for C-H cleavage that can be ascribed to an altered global conformational landscape with an increase in inactive (or poorly active) enzyme-substrate (E-S) sub-states. For native enzyme, one of the primary functions of conformational sampling is the transient achievement of hydrogen donor and acceptor distances that are sufficiently reduced from those of initial van der Waals complexes to enable productive wave function overlap.
Studies of a series of double mutants of SLO have further “pushed the envelope” in the ability of theoretical models to rationalize behavior.25, 28–29, 31 While the majority of double mutants show behavior similar to single mutants, i.e., analogous KIE values at RT that are accompanied by an increase in ΔEa (Table S1), a single dramatic outlier, L546A/L754A (DM), emerged with a kH/kD of 537±55 under pre-steady single turnover anaerobic measurements29 and an average kH/kD of 661±27 via steady-state measurement across six temperatures.28 Additionally, in contrast to the KIEs of other active site single and double mutants that show temperature sensitivity, the KIEs in DM change little if at all with temperature (Table S1). These properties of DM-SLO can be well-reproduced using the developed vibronically nonadiabatic PCET model, where donor-acceptor distance sampling within the active site appears to have become rigidified compared to WT and the other SLO variants,22, 28–29 preventing DM from recovering the tunneling-ready distance characteristic of native enzyme and other well characterized mutants. A related temperature-pressure perturbation study of DM-SLO also highlights the distinctive features of this variant.28, 32 Unlike the pressure-induced shifted conformational landscape in WT and three single mutants (L546A, L754A, and I553V), the conformational landscape of the enzyme-substrate complex of DM is more resistant to perturbations from elevated pressure, showing a consistently high energy barrier (Ea = 8 – 9.9 kcal/mol) and slow catalytic efficiency (104-fold decrease) relative to WT. At this juncture, a major unanswered question is the structural origin underlying the aggregated features of DM-SLO.
In this study we compare a suite of biophysical probes applied to DM, the two components of DM (L546A and L754A) and WT. These probes include HDXMS as a function of temperature, EPR spectroscopy and room temperature X-ray crystallography. The primary results show that DM adopts features from both parent single mutants, exhibiting the same HDXMS behavior as L546A and showing structural changes similar to L754A. Significantly, in no instance has evidence been found for enhanced structural rigidification in the resting form of enzyme, in contrast to the earlier inference of rigidification within the donor-acceptor distance sampling modes. As we discuss, the aggregate data indicate a disruption in the positioning of the reactive carbon of substrate with regard to the catalytic iron center that is further incapacitated via an impairment of its communication with a dynamical network that has been identified to extend from the active site to a remote protein/solvent interface.33
RESULTS
Hydrogen-Deuterium Exchange.
The time and temperature dependent analysis of HDXMS was carried out with DM for comparison to the previously characterized WT SLO.33 Using the same set of non-overlapping peptides selected for WT SLO, 78% of the protein sequence for the DM catalytic domain (C-terminal domain) was covered (compared to 89% for WT). The apparent averaged rate constants for exchange, kHDX(avg), from exponential fits to the HDXMS traces for WT and DM are presented in Table S2 and represent the protein in the absence of substrate and exchanged in buffer pL (L = H, D) of 8, conditions where EX-2 conditions have been demonstrated. EX-2 conditions enable us to extract thermodynamic information regarding regional peptide flexibility. The majority of DM-derived peptides display nearly identical exchange behavior to WT across the 5 temperatures (10, 20, 25, 30 and 40˚C) analyzed (cf. Figure S1). In a previous study, peptide 317–334, a solvent-exposed loop (Figure 1A), was shown to produce significant alterations in Ea for kHDX(avg) following mutation at positions 553 (I553G) and 546 (L546A).33 This peptide exhibited trends in the Ea for kHDX(avg) (EaHDX(avg)) that correlate with trends in the Ea for catalytic PCET (solid line in Figure 1C).33 In the current HDX analysis of peptide 317–334 in DM, we observe a value for EaHDX(avg), again distinct from WT (Figure 1B, Table S3); however, while DM-SLO displays the highest enthalpic barrier of catalysis among all SLO variants (Table S1), the value of EaHDX(avg) associated with DM is 14 kcal/mol, almost identical to the behavior of L546A (Figure 1C, top red arrow). This initially surprising result, of comparable EaHDX(avg) values in DM and L546A, implies that the added active site packing defect that arises from L754A in DM does not impact the thermally activated surface motions that correlate with catalysis.
Figure 1:

Peptide 317–334 (in orange) remote from the active site exhibits mutation-induced changes in the enthapic barrier for HDX rates (EaHDX(avg)) in L546A, I553G and DM but not in L754A. (A) The SLO structure with highlighted active site residues L546 (in green), L754 (in blue) and I553 (in red). In panel B, Arrhenius-like plots of the weighted average exchange rate, (ln(kHDX(avg)) for WT, I553G, L546A, L754A and DM are compared. In panel C, the relationship between EaHDX(avg) and the enthalpic barrier for hydrogen tunneling (Ea(kcat)) is represented. The solid line is the trend among I553G, WT and L546A previously reported.33 The bottom arrow indicates that L754A shows no difference in EaHDX(avg) relative to WT. The top arrow shows that there is no further difference in EaHDX(avg) when L754A is further introduced into L546A.
To further substantiate the lack of an impact of L754 on the long-range protein motional network, the single variant L754A was examined with regard to the time and temperature dependence of its HDXMS. The L754A demonstrates an elevated enthalpic barrier of catalysis (Ea(H) = 4.1 kcal/mol) and an even more impaired catalytic rate constant than L546A (Table S1),21 however the kHDX(avg) and EaHDX(avg) obtained from the HDXMS profiles of all L754A derived peptide across the full temperature range are within experimental error of WT (Table S2, S3), (Figure 1C, bottom red arrow). The absence of a linkage between the Ea(H) and the EaHDX(avg) in L754A supports an uncoupling between the packing defect at position 754 and motions at the protein-solvent interface, such that the observed rate impairment with this mutant must arise from other features (see below). We note that there were no noticeable changes in the extent of exchange as a function of temperature for L546A, L754A and DM (cf. full data sets in Figure S1), in marked contrast to the previously studied I553X series where the primary impact of mutation was seen as an increase in the extent of exchange in the 541–565 region of protein.33
These new comparisons between HDXMS profiles and catalytic parameters highlight the combinative yet distinct contributions from the residues L546 and L754 in defining the behavior of DM. As earlier noted, L546 is located within the identified network of structural communication between the surface loop (317–334) and active site residues (Figure S2),33 with a packing defect at position 546 influencing motions at the protein-solvent interface that are reflected both in Ea(H) and EaHDX(avg). This impact from L546A appears well preserved in DM. In contrast, L754 resides near the iron cofactor and spatially away from the identified network (Figure S2), suggesting that changes in the microenvironment around the metal, such as the degree of hydration, ligand field, and/or redox potential may be the major cause of the observed change in properties for L754A. These possibilities are explored below.
Room-temperature X-ray Crystallography of the SLOs.
To systematically reveal structural changes associated with the size-reduction at position 546 and/or position 754, room temperature X-ray crystallography was adopted for WT, L546A, L754A, and DM. Such room temperature structures can reveal a multiplicity of protein conformers that are undetectable under cryo-conditions.41–42 Additionally, in the context of visualizing solvent water, difference maps calculated from two room temperature structures are more reliable than cryogenic maps.43 High resolution (1.70–1.85 Å) data were obtained for L546A, L754A and DM, revealing practically superimposable protein backbones and largely unaltered side-chain conformations in comparison to the room temperature X-ray structure of WT.33 The most dramatic change in these three mutants is the expanded active site that is associated with changes in solvation. The size reduction of the side chain on position 546 and/or position 754 leads to a gradually enlarged active site. Defining a more specific active site pocket than in the earlier study33, the cavity in WT is calculated to be ≤ 21 Å3. Using this new value as a frame of reference, the variants described in this study indicate increases of 170 % in L546A, 76 % in L754A, and over 270 % in DM. The isomorphous difference map was further applied to individual mutants using phases from the room temperature WT model. The difference map between WT and DM indicates an increased electron density in the active site of DM (Figure S3A), resembling most closely the difference map between WT and L754A (Figure S3B). These density changes imply increased bound (or diffuse) water molecules near the iron cofactor. The other single mutant L546A demonstrates a slightly positive density, which also implicates the presence of extra water molecules (Figure S3C). However, the number of water molecules visualized is clearly less than for DM and L754A, and restricted to a position near 546 and H499. It is important to note that changes in solvation have not always been associated with mutation at hydrophobic side chains in SLOs: the I553X series failed to indicate X-ray evidence for the presence of any additional water molecules at low temperature and room temperature, albeit with a significantly enlarged volume near the mutated residue.26, 33
To further substantiate possible microenvironment changes around the iron cofactor, we turned to a detailed inspection of the impact of mutation on the active-site geometries for WT, L546A, L754A and DM. Figure 2A-D indicates no significant changes in the coordination geometry between the iron and first/second sphere residues in all SLOs studied. The major difference within the active site is the newly entered water molecules in each variant that may interact with other coordinating residues through hydrogen bond networks. In both L546A and L754A, one additional water molecule is seen to occupy the packing defect created near position 546 and 754, respectively. However, the newly entered water molecule in L754A forms hydrogen bonds with two first sphere coordinating residues (iron-bound water molecule and H499) as well as a second sphere coordinating residue Q495, while the extra water in L546A is far from the second sphere coordinating residue Q495, interacting dominantly with H499 (Figure 2B, 2C).The water molecules occupying these single site packing defects and their associated hydrogen bond network are retained in DM. Beyond these two water molecules, we further observe two additional water molecules in DM that are adjacent to position 754 and form hydrogen bond network to the first sphere coordinating residues I839 as well as Q697.
Figure 2:

Coordination geometry of the iron center of (A) WT SLO (PDB: 5T5V); (B) L546A (PDB:5TQN); (C) L754A (PDB:5TR0); and (D) DM (PDB: 5TQO). Oxygen atoms are colored in red, nitrogens in blue, iron center in orange and water in cyan. Carbons in first sphere ligands are colored in yellow, second-sphere ligands in pink, side chains on position 546 and 754 in grey. Bonds between iron and first-sphere ligands (yellow) are drawn as dashed grey lines, and hydrogen-bonding interactions between newly added water in the active site with the first sphere and second sphere ligands are shown as solid grey lines. There are no significant differences in side chain conformers for SLO variants based on the electron densities analysis.
EPR spectra of Fe(II) and Fe(III) SLOs.
In order to further explore changes in the microenvironment around the non-heme iron center of SLO that result from L546A, L754A and DM we employed EPR spectroscopy. The WT, L546A, L754A and DM isolated from E.coli all contain EPR-silent Fe(II), as shown in Figure S4, consistent with previous studies on SLOs and other lipoxygenases.44–47 The oxidized (“yellow”) Fe(III) SLOs were prepared through the addition of 2 equiv. of substrate, linoleic acid (LA), to the Fe(II) SLOs as described in Materials and Methods. The Fe(III) WT and L546A both demonstrate a complex multicomponent EPR signals with a pronounced feature around g = 7.3 and 6.2 and an additional weak feature at g =4.3, Figure 3. The previous EPR analysis of WT attributes the two groups of peaks to different geometric structures around the high-spin iron with the g =6.2 signal representing axial asymmetry, g ≈ 4.3 arising from a rhombic species, and g=7.3 as an intermediate between the above two species.47 These features indicate that the ferric form of WT and L546A both contain the above mentioned three species with axial asymmetry of the zero field splitting as the dominant feature.
Figure 3:

EPR spectra (X-band, 6.5K) of oxidized WT, L546A, L754A and DM SLO (140 μM) in 0.1 M potassium phosphate pH = 7.0 buffer. The peak intensities of L546A, L754A and DM have been corrected for the iron content of each mutant: WT (0.9), L546A (0.50), L754A (0.70), DM (0.70). A 10-x amplified EPR spectrum of Fe(III) DM SLO is presented for comparison. Expanded EPR spectra are shown in Figure S5.
Remarkably, once again the L754A shows distinct patterns from WT and L546A. The EPR spectrum of L754A shows dramatically increased intensity at the g ≈ 4.3 peak and a weak feature at g = 7.3 (Figure 3), indicating that the ferric active site in L754A contains a primarily rhombic species. The interconversion between axial to rhombic species zero field splitting in lipoxygenases indicates the alteration in the active site geometry, which is usually seen to arise as the result of either product coordination to the iron center or substitution of the iron ligands that impacts the ligand field strengths.44, 47 We propose that the unique EPR signals in L754A are directly related to the changes in microenvironment around the iron center, including the additional water molecule near the position 754 and its interaction with other iron ligands as shown in Figure 2C.
Unlike any previously reported high-spin ferric iron, the EPR studies of oxidized DM demonstrate a dramatically decreased EPR signal intensity (Figure 3). However, the increase in UV/Vis Fe(III) absorption between 330–350 nm for DM, representing the formation of Fe(III) following incubation of enzyme with linoleic acid, appears almost identical to WT after correction for their respective iron levels (Figure S6); this indicates that the active site in DM-SLO does contain an oxidized, Fe(III) species. The presence of Fe (III) in DM-SLO is further validated by the strong peak at g = 4.3 after addition of 4-nitrocatechol to free enzyme (Figure 4D, top two EPR spectra). As shown previously in spectroscopic, kinetic and structural analyses, 48–50 4-nitrocatechol can bind to the active site of SLO and form complexes with Fe(III) SLO in the absence of a redox reaction at pH=7.0. The dramatically increased intensity in the 4-nitrocatechol complex is, thus, attributed to a zero field splitting change caused by displacement of first sphere side chain ligands with catechol oxygens within the DM-4-nitrocatechol complex.48 As has been reported for SLO and homologs of lipoxygenase, the Fe(III) spectra can have a range of zero field splitting parameters (D = −0.3 to +1.7 MHz, E/D = 0.02 to 0.33) which is influenced by the microenvironment of the Fe and will greatly affect the intensity and shape of the EPR spectrum.46–47 We note that the 10-x EPR spectra of oxidized DM SLO displays features of both WT/L546A and L754A (Figure 3).
Figure 4:

EPR spectra after Fe(III)-SLOs are anaerobically incubated with catechol ligands in 0.1 M potassium phosphate pH = 7.0 buffer at 4°C for 12 hrs: (A) WT; (B) L546A; (C) L754A; (D) DM. The EPR spectra of oxidized Fe(III) SLO vs Fe(II) SLO are shown at the top and bottom of each panel for comparison.
Estimation of redox potential in SLOs.
Room temperature X-ray structures, together with the above EPR analyses, indicate significant disruption in both the microenvironment and electronic structures of the iron sites in L754A and DM. Such changes could impact the redox potential of the active site iron, that would further impact both the reaction free energy ΔG° and the corresponding rate constant kcat. A previously developed method by Nelson demonstrated that formation of complexes between oxidized WT Fe(III) SLO and a series of substituted catechols enables an estimate of the redox potentials of the active site irons.50 Specifically, conditions were established for the SLO-catechol complexes, in which less reducing catechols do not transfer an electron to the active site Fe(III), while the more reducing catechols reduce the ferric iron to EPR silent Fe (II), establishing a redox ruler for the active site iron of SLO.
Figure 4A shows a series of EPR spectra of the WT SLO-catechol complexes after an incubation time of 12 h in phosphate buffer (pH=7.0). Samples of complexes of WT with 4-nitrocatechol or 3,4-dihydroxybenzonitirile both show sharp EPR signals at g ≈ 4.3, that suggest a conversion from the dominant axial component of Fe(III) to a rhombic component of Fe(III) in the complexes. The EPR signals at g ≈ 4.3 are also seen in the complex of ferric WT SLO with 3,4-dihydroxybenzaldehyde albeit with largely decreased intensity, as evidence for a partially reduction of Fe(III) to Fe(II). In the case of the other five SLO-catechol complexes, the EPR spectra all demonstrate very minor peaks in the same region, indicating almost fully reduced Fe(II) in those complexes. The remaining very small peak have been suggested to arise from some portion of denatured protein that releases the free Fe(III).51 The trends of the reaction between active site iron and catechols at pH=7.0 are consistent with Nelson’s previous observations, and an assignment of a redox potential for iron in WT at pH =7.0 between the range of the redox potentials for the test ligands (0.57 V to 0.68 V).50
We proceeded to measure the redox potential of iron in L546A, L754A, and DM in a similar fashion, Figure 4B–4D. In the cases of L546A and DM (Figure 4B and 4D), the trends in the EPR with 4-nitrocatechol and the more reducing catechol are similar to WT, implicating redox potentials for iron in these two mutants within the 0.57V to 0.68V. The slight differences in the reducing ability of catechols in the middle range (Figure 4A, 4B and 4D, from 3,4-dihydroxybenzonitrile to caffeic acid) further imply a trend of redox potential that is DM > WT > L546A. Finally, in the case of L754A, the observation of a reduction of the ferric center by the least reducing reagent in this series, 4-nitrocatechol, indicates a potential that is somewhat above 0.68V, for a full trend in redox potential of L754A>DM>WT>L546A.
Even though the adopted method can only give an estimated range and relative trend of redox potentials for SLO variants, we can conclude that the redox potential at the iron center does not directly control C-H activation rate constants. This is supported first, by only minor alternations in the range of redox potential (less than 0.1V) for changes in rate constants of 102-104 fold. Second, the trend of the redox potential change (L754A > DM > WT > L546A) does not align with the trend of observed rate constants (WT > L546A > L754A > DM). Clearly, other factors dominate the enormous impact of the DM (and other variants) on the rate impairment of SLO.
DISCUSSION
Hydrogen tunneling offers a unique window into the role of protein conformational motions in facilitating optimal active site alignment that accounts for the rate enhancements in enzyme catalyzed reactions. SLO has served as an ideal model for understanding the role of protein structure and dynamics in generating highly efficient tunneling rates and properties relative to small molecules in condensed phase. Kinetic analyses of this system over decades have led to a predictive multi-dimensional kinetic model that incorporates a hierarchy of protein motions.5, 9, 16, 35 Further, recent biophysical studies of WT versus single site SLO mutants, 33,34 have begun to reveal the molecular basis of catalysis-linked protein motions. The unique kinetic features emerging from a nearly fatal double mutant, DM, earlier led to the proposal of an elongation of the hydrogen transfer distance within its E-S complex that is accompanied by an apparent rigidification of the enzyme active site that prevents recovery to a tunneling DAD that approximates WT. These parameters include a greatly reduced kcat together with an enlarged temperature dependence on kcat (Ea ~ 10 kcal/mol) and an enormous KIE (Dkcat ~ 500–700) that is nearly independent of temperature (ΔEa ~ 0 kcal/mol). 28–29 The present work addresses the structural origin of the enormous KIE and other aberrant properties of DM SLO via the interrogation of a range of biophysical and structural probes that include HDXMS,33 EPR34 and room temperature X-ray crystallography.28–29, 31, 33–34
Among the kinetic parameters characteristic of DM, kcat is found to be reduced 104-fold relative to WT. Previously, it had been shown that altering the ligands to the metal in SLO both reduced rates and decreased reduction potentials of the metal, implicating an altered chemical driving force, ΔG°, in catalytic rate impairment.52 To test this possibility for the present series of SLO mutants, we examined relative reduction potentials using an established EPR method involving analysis of Fe(III) SLO complexes with various catechol derivatives. As shown, the DM undergoes only a very modest variation in its redox potential, with no systematic trend in the magnitude of its kcat and estimated E° relative to WT (Figure 4). These experiments serve as an important frame of reference in ruling out significant alterations in the chemical driving force as the source of the impaired activity of DM (see below).
Origins of the elevated enthalpic barriers in SLO mutants.
Because wave function overlap from the rate-limiting C-H bond cleavage step in SLO catalysis is, by definition, barrier-less, the observed enthalpic barriers are derived from the conformational sampling of the enzyme-substrate complexes. We first examined the extent to which the aggregate behavior of DM could be rationalized from the two parent mutants, L546A and L754A. Though these single mutants have the same enthalpic barriers (EaH = 4 kcal/mol), the origins of the increased Ea values relative to the native enzyme (EaH = 2kcal/mol) are now concluded to be different, arising either from impairment of a catalytically-linked dynamic network in SLO (L546A33) or an alteration in the active site micro-environment (L754A, this study).
HDXMS previously showed that reduction in side chain volume at L546 or I553 produces changes in the enthalpic barrier for HDXMS rate constants within an identified region of solvated protein (317–334) that correlate with trends in the enthalpic barrier for the catalytic PCET process.33 The elevated catalytic EaH for L546A was, thus, assigned to an impact of this mutation on a catalytically-linked protein dynamical network that traverses a distance of ≥ 20 Å from the active site to the protein-solvent interface. By contrast, the current comparison of HDXMS profiles for L754A to WT shows these to be practically identical for every peptide including 317–334, ruling out perturbations to the dynamical network as the primary origin of the altered properties of L754A.
The present studies point instead toward a perturbation of the micro-environment surrounding the iron cofactor as the source of changes in catalysis for L754A. The estimated relative redox potential for L754A (Figure 5) is seen to exhibit a modest increase relative to the E° of WT (ΔE° < 100 mV), predicting an increase in rate, thereby making a change in redox potential an improbable source of its 103 lower kcat and elevated EaH. The position of the additional, structured water that infiltrates the expanded active site in L754A (Figure 2) may be a particularly significant feature that influences both the kinetic parameters and EPR spectrum of apo-L754A; the latter is clearly divergent from both WT and the L546A variant (Figure 3).
Figure 5.

Rate constants for enzyme catalyzed nonadiabatic hydrogen transfer can be formalized as kobs = Keq • kPCET.9, 28, 33 (A) Keq (<< 1) represents a stochastic ground state search through inactive conformations (b) to reach catalytically active E-S complexes (a conformer). Subsequent thermal sampling further reduces the distance between the H-donor and acceptor as seen in the pre-tunneling a* conformer. (B-D) illustrate contributions from kPCET: (B) Heavy atom protein motions produce transiently degenerate energies for the reactant and product wells, a prerequisite for wave function overlap at the tunneling ready state (TRS) (C). (D) The effective potential along the DAD sampling coordinate varies, starting with ΔEa ≅ 0 for native enzyme (top), becoming ΔEa >>0 when the DAD is elongated following single mutations (middle) and finally arriving at the catastrophic DM scenario in which the DAD is elongated, but coupled with a rigid DAD sampling potential (bottom). In Frame D, light purple refers to the shorter wave function distribution for deuterium, and dark purple represents the more distributive protium wave function.
Buried water molecules have been assigned to important roles in biological function, including ligand binding, protein stability and flexibility.53–57 In the case of buried waters that become displaced upon ligand binding,58 these can provide an important thermodynamic driving force for enhanced substrate affinity (see for examples, 55–57,59). WT SLO has at least five X-ray-resolved water molecules that line the putative ligand binding pocket29 and most, if not all, are likely to be expelled upon substrate acquisition. Because of the unique positioning of the new water introduced upon mutation at position 754, it is possible that this will remain bound in the E-S complex, leading to changes in the active site dielectric that impact both kcat and the accompanying Ea. While X-ray crystallography of complexes of LA with SLO could, in principle, resolve any ambiguity regarding the degree to which active site waters are retained in the E-S complex (WT or otherwise), decades of efforts to obtain such data have thus far been unsuccessful. We note that X-ray structures of the apo- form of I553G provide a reasonable control for the impact of L754A. While the 553G variant shows an enlarged active site cavity volume similar to 754A, it lacks any detectable added water molecules by either cryo- or room temperature X-ray studies26, 33 and exhibits a turnover number much closer to WT.26
The combination of impaired dynamics and altered active site hydration that are distinctive of L546A and L754A, respectively, begin to rationalize the greatly increased activation energy (~10 kcal/mol) and decreased rate of DM relative to WT. However, DM also shows two highly deviant experimental properties, absent in either L546A or L754A: these are an enormous primary deuterium room temperature isotope effect of 661±27 and its very low sensitivity to temperature (ΔEa ~ 0). 29,30
DM as an outlier: Enormous temperature independent KIEs can be rationalized by modifications in both the hydrogenic donor-acceptor distance (DAD) and the frequency of DAD sampling.
Based on the nonadiabatic PCET model that fits the temperature independent KIEs in many native enzymes,54, 60–62 the observed rate constants of SLO are determined by a conformational sampling coordinate and PCET reaction coordinate, that can be visualized in Figure 5 and formalized as Eq 1:9, 28, 33
| (Eq1) |
where Keq<1 represents the small fraction of active enzyme-substrate protein substates that arise from stochastic sampling processes (Fig 5A), multipled by kPCET, the nonadiabatic rate constant of the H-tunneling process (Fig. 5B-D). The kPCET is derived from a formulation that resembles the Marcus analysis for electron tunneling (including reorganization energy, λ, and driving force ΔG° parameters), in which the barrier-less hydrogenic wave function overlap coordinate depends on a temperature-dependent coordinate for achieving transient degenerate energy levels between reactant and product (Fig. 5B&C)). The addition of a thermally averaged DAD sampling mode completes the analysis (Fig 5D):63 The latter has emerged as an important ruler for the degree of active site optimization and can be quantitated by the magnitude of ΔEa.
In naturally evolved (i.e. WT) enzymes, active sites for enzymatic C-H activation reactions are finely tuned by way of conformational sampling, to achieve highly compacted DADs in pre-tunneling configurations, labelled conformer (a*) (Fig. 5A). When this occurs, the coordinate in Fig. 5B produces the tunneling ready state (TRS) (Fig. 5C) via thermal excitation that is largely independent of the labelling of substrate with deuterium; in this instance further optimization of the reaction coordinate via DAD sampling (Fig.5D) is either absent or minor. The DAD sampling coordinate becomes increasingly important for impaired enzymes systems, leading to an incremental increase for Ea(D) relative to Ea(H), and is easily detected from an increase in ΔEa away from a WT value that is generally small, close to zero. The isotope dependence of the DAD sampling coordinate, Fig. 5D, is often perceived as puzzling, but can be readily understood in the context of the smaller, less diffuse wave length for deuterium that introduces a requirement for shorter DADs; this can become an important source of the increased energetic barrier for achieving deuterium wave function overlap when the active site environment has been perturbed away from optimal alignment. The fact that mutation most generally also leads to softer active site potential energy surfaces enhances access to the DAD sampling process.
Historically, in the case of all catalytically-impairing single mutants of SLO that include L546A and L754A, the ΔEa increases relative to WT. However, the DM kinetic isotope effect is almost completely temperature independent (ΔEa ~ 0). While this would at first glance suggest a finely tuned active site, the significantly decreased kcat and enormous primary Dkcat of ~700 of DM can only be ascribed to an elongated, non-optimal DAD in the pre-tunneling conformer that prevents subsequent DAD sampling. When seen in this context, the collective kinetic data indicate that DM is simply unable to recover from a nearly catastrophically elongated DAD. Though the multiple biophysical probes in the present study were initially pursued with the goal of detecting and defining which aspects of protein structure/dynamics would be unique to DM and account for an implied rigidification, we have found no evidence for impairment to the dynamical network in L754A, which looks identical to WT in this regard. While a dynamical impairment does arise in the other component of DM, L546A, it alone is not serious enough to prevent DAD sampling and allows recovery to an active site configuration and KIE similar to WT. In the context of the behavior of these two parent mutants the most direct interpretation of the origin of the impairment in DM is seen as a combination of a serious mispositioning of substrate (due to L754A) that has simultaneously made it much more difficult to rearrange itself into a configuration that permits tunneling (due to the additional impairment of the protein dynamical network from L546A).
DM as an outlier: Mispositioning of the reactive carbon of substrate into a region of SLO that lies outside of the native protein dynamical network.
In light of the current findings, results from other methods were sought as a source of structural insight into the DM’s unique behavior. ENDOR studies have shown themselves to be a powerful probe for E-S complexes of SLO, enabling a direct interrogation of the dominant ground state H-donor and H-acceptor distances.34 Completed studies of WT show two such ground state structures, with one configuration (a) showing van der Waals distance for the hydrogen donor and acceptor and a second, inactive configuration (b) with a DAD almost 1 Å longer. Proficient wave function overlap in WT enzyme is proposed to arise via thermal sampling within the potential energy well of the “a” conformer to form DADs reduced from van der Waals (a*) (cf. Figure 5).
Importantly, ENDOR of the DM reveals only a single ground state ‘b’ configuration that corresponds to the more extended, catalytically inactive form of E-S, consistent with the sluggish rate of this variant. In the same experiment, a single ‘b’ configuration was also obtained with I553G, a mutation that like DM is predicted to exhibit an elongated DAD, but unlike DM, has a catalytic activity only 5-fold reduced from WT; the latter is accompanied by a highly temperature dependent KIE (ΔEa ~ 5 kcal/mol) that reflects active dynamical sampling back to a WT-like configuration, indicating that ENDOR alone cannot provide the elusive source of impairment for DM-SLO. MD simulations were initiated for the ground state E-S structures of WT, DM, and I553G using the metal to C11 and metal to C10 distances restrained by ENDOR measurements; significantly these studies suggested differences in the positioning of substrate within the DM form of SLO relative to WT and I553G.
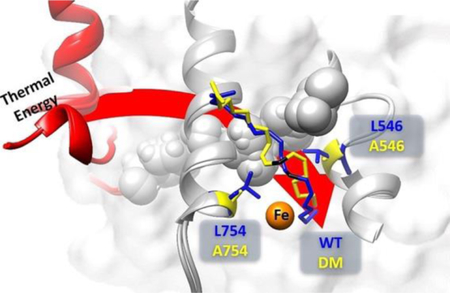
Pursuing this possibility, a recent QM/MM study of the E-S complexes for WT and DM SLO provides a potential glimpse into the molecular origins for the collective impairment of mutations at both L546 and L754.64 QM/MM models have the advantage over the restrained MD simulations, because these calculations do not require any input from experimental findings to generate active site conformations. Corresponding ground state models of the active site structures for WT and DM derived from these computations show an almost superimposable C1-C7 region of LA that subsequently diverges in the region of substrate that encompasses the reactive carbon and its flanking double bonds (C9-C13). Importantly, the computed ground state DADs between C11 of LA substrate and metal-bound oxygen, of 3.2 Å for WT and 3.6 Å for DM, are remarkably consistent with the distances estimated from ENDOR experiments of 3.1 Å and 3.8 Å, respectively.65 It has been noted that the two altered side chains in DM, L546 and L754, are highly conserved and act to clamp the C11 of LA in position for reaction with the adjacent Fe(III)-OH.66 It is, therefore, of considerable interest that simultaneous replacement of both leucine side chains for alanine in SLO leads to a new QM/MM-based configuration for C11 of substrate, which has moved into the space normally filled by L754 (Figure 6).
Figure 6:

A comparison between the ground state substrate binding mode in WT (Blue) and DM (yellow). The reactive carbon C11 is colored as black, iron colored as orange.
A second observation in support of substrate mispositioning in DM comes from the accompanying QM/MM computation of ΔG° for conversion of bound substrate to product. For DM, this ΔG° is calculated to be more uphill by 10 kcal/mol relative to WT, in the same direction as the experimentally observed increase in the energy of activation of ~10 kcal/mol for DM. Given an improbable ability of the SLO active site to alter the innate bond dissociation energy of bound substrate, together with our failure to detect significant changes in redox potential of the Fe cofactor in DM, a change in chemical driving force is ruled out as the source of the increased ΔG° for DM. The most reasonable interpretation is a stabilization of substrate within a binding mode that is highly compromised with regard to its distance and ability to promote wave function overlap from the C11 of substrate to the Fe(III)-OH acceptor. One caveat regarding the referenced QM/MM study is its inability to capture the experimental evidence for irreversible formation of pentadienyl radical under single turnover, anaerobic conditions.20, 29 Scheme 2 suggests a way to rationalize the discrepancy between computation (ΔG° > 0) and experimental findings (ΔG° < 0). As shown, completion of the DM reaction will entail a return of the initially formed radical intermediate (illustrated in Scheme 1) to a less active but thermodynamically more stable binding position (E-I). This leads to the net exergonic process observed experimentally for the conversion of E-S to E-I. In this context, the trajectory captured by QM/MM for DM represents only a portion of the reaction in which E-S has been converted to E-I’ via an uphill process.
Scheme 2.

The free energy profile for WT (red) is represented by a two state thermodynamic model involving E-S and the enzyme-bound pentadienyl intermediate, E-I. In the case of DM, the most stable E-S complex (Figure 6) must first isomerize to a pre-tunneling configuration (E-S’) that converts via PCET to the mispositioned pentadienyl intermediate (E-I’) with a driving force similar to WT. The return of the bound intermediate E-I’ to a more stable binding position completes the reaction. The dashed blue line has been added as a possible model for the QM/MM computation.
CONCLUSION
The paper presents the biophysical characterization of a kinetically impaired double mutant of soybean lipoxygenase L546A/L754A and its parent single mutants L546A and L754A. Beyond adopting features from both parent single mutants, DM demonstrates a distinctive substrate binding mode that mispositions the substrate into configurations that are unfavorable for hydrogen tunneling. In the WT SLO, L546 resides within a protein motional network communicating between the surface loop and active site (Figure S2).33 It is of considerable interest that while L546A alone disrupts the protein motional network connecting the catalytically-relevant surface loop and the active site,33 the enzyme is still able to undergo DAD sampling to achieve a KIE similar to WT.21 The movement of C-H of substrate into the “hole” created by the second mutation of L756 in DM has the additional effect of removing the reactive carbon of substrate from access to such thermal activation. This combination of a new substrate binding mode that is removed from access to thermally activated protein modes provides an explanation for the divergent features of DM that include its greatly impaired kcat, excessively elevated energy of activation and its enormous and close to temperature independent KIE. The current study, thus, provides a critical examination and insightful structural example of how relatively minor alterations in the placement of the reactive bond(s) of a bound substrate can cause dramatic alterations in the efficiency of enzymatic rate acceleration. In this context, the findings are highly relevant to the generic origins of substrate specificity in enzymes, whereby changes in the volume of active site side chains may either activate or impair an intended reaction course.
MATERIALS AND METHODS
Materials.
All reagents were purchased from commercial sources and used without further purification unless otherwise indicated. Wild-type and mutant SLOs were prepared, expressed and purified according to the previous report.28–29, 31
EPR Sample Preparation.
The EPR samples were prepared according to the previous protocols50 with several modifications. Each sample contained 140 μM SLO and 1 equiv of respective catechols in 0.1 M potassium phosphate (pH =7.0). The ferric (yellow) lipoxygenase was first prepared from the mixing purified ferrous lipoxygenase and linoleic acid. The concentrated, purified ferrous lipoxygenase (300 μM) was titrated with linoleic acid (10 mM) at 4°C, 0.1 M borate buffer (pH = 9.0) while monitoring the optical spectrum between 300 and 800 nm for development of the 330-nm peak characteristic of the ferric lipoxygenase. For WT and mutants, 2 equiv. of linoleic acid was required to fully oxidize the protein. The ferric lipoxygenase (ca. 280 μM) was removed from the cuvette and dialyzed overnight in 0.1 M potassium phosphate buffer (pH = 7.0) at 4°C. The sample was then stored at −80°C until use. A known amount of catechols was transferred to an anaerobic glovebox, then dissolved in deoxygenated 0.1 M potassium phosphate with a final concentration of 280 μM. The catechols solution (100 μL) was mixed with deoxygenated ferric lipoxygenase (100 μL) and transferred into a suitable EPR tube. The tube was sealed with rubber stopper and kept at 4°C. After 12 hrs, the tube was removed from the glovebox, and frozen in liquid nitrogen within 1 min of its removal. The samples were stored at liquid nitrogen temperature until data acquisition. For each SLO variant, two control EPR samples were prepared as described above but with minor variations: One only contains 140 μM ferric lipoxygenase; another only contains 140 μM ferrous lipoxygenase.
EPR Spectroscopy.
Samples for X-band (~9.4 GHz) EPR spectroscopy were measured at the CalEPR center at the University of California, Davis. Continuous wave (CW) spectra were collected using a Bruker Instruments EleXsys-II E500 CW EPR spectrometer (Bruker Corporation, Billerica, MA) equipped with an Oxford Instruments ESR900 liquid helium cryostat and an Oxford Instruments ITC503 temperature and gas-flow controller. Samples were measured under non-saturating slow-passage conditions using a Super-High Q resonator (ER 4122SHQE). Typical acquisition conditions were: T = 6.5 K; 9.38 GHz microwave frequency; 10G modulation amplitude; 0.6325 mW microwave power.
HDX Sample Preparation.
Purified aliquots of wild-type and variant SLO were thawed and diluted to 100 µM. Protein aliquots from this stock sample were diluted 10-fold in D2O buffer 10 mM HEPES pD 7.4; corrected pD = pHread + 0.4(65) to a pL of 8.0 (confirmed with pH electrode). Samples were incubated randomly at a specified temperature (temperatures studied: 10, 20, 30, and 40˚C; bath stability ± 0.1˚C), using a NESLAB RTE-111 water bath, for 14 time points (0, 10, 30, 45, 60, 180, 600, 1200, 1800, 2700, 3600, 7200, 10800, 14400 s) at each temperature. The same conditions were used in characterizing each mutant. Upon completion of the designated incubation time, samples were rapidly cooled (−20°C bath) and acid quenched (to pH 2.4, confirmed with pH electrode, with 0.32 M citric acid stock solution at 0°C). SLO derived peptides were generated from 2.5 minute incubations with pepsin after addition of guanidine HCl (ca. 0.5 M final concentration). The peptide fragments were removed from the immobilized pepsin with spin cups and flash frozen immediately in liquid N2. Samples were stored at −80°C until data collection.
Liquid chromatography-mass spectrometry for hydrogen/deuterium exchange measurements.
Peptide assignments (from the digestion in water) were previously determined for WT SLO.33 Deuterated, pepsin-digested samples of SLO from hydrogen/deuterium exchange experiments were analyzed using an Agilent 1200 LC (Santa Clara, CA) that was connected in-line with the LTQ Orbitrap XL mass spectrometer (Thermo). The LC was equipped with a reversed-phase analytical column (Viva C8, 30 mm length × 1.0 mm inner diameter, 5 µm particles, Restek, Bellefonte, PA) and guard pre-column (C8, Restek). Solvent A was 99.9% water/0.1% formic acid and solvent B was 99.9% acetonitrile/0.1% formic acid (v/v). Each sample was thawed immediately prior to injection onto the column. The elution program consisted of a linear gradient from 5% to 10% B over 1 min, a linear gradient to 40% B over 5 min, a linear gradient to 100% B over 4 min, isocratic conditions at 100% B for 3 min, a linear gradient to 5% B over 0.5 min, and isocratic conditions at 5% B for 5.5 min, at a flow rate of 300 µL/min. The column compartment was maintained at 4 °C and lined with towels to absorb atmospheric moisture condensation. The column exit was connected to the ESI source of the mass spectrometer using PEEK tubing (0.005” inner diameter × 1/16” outer diameter, Agilent). Mass spectra were acquired in the positive ion mode over the range m/z = 350 to 1800 using the Orbitrap mass analyzer, in profile format, with a mass resolution setting of 100,000 (at m/z = 400). Data acquisition was controlled using Xcalibur software (version 2.0.7, Thermo).
HDX data analysis.
Mass spectral data acquired for HDX measurements were analyzed using the software, HDX WorkBench. The percent deuterium incorporation was calculated for each of these peptides, taking into account the number of amide linkages (excluding proline residues) and the calculated number of deuterons incorporated. The values were normalized for 100% D2O and corrected for peptide-specific back-exchange, as determined previously.33 The data were plotted as deuterium exchange versus time using Igor Pro software. The rates and extents of exchange (at 4 h) were determined from one- or two-exponential fits to the analyzed time-resolved HDX data. The weighted average rates of HDX were determined from the individual rate constants of these two exponentials. Apparent enthalpies of activation from HDXMS rates (EaHDX(avg)) were determined for each peptide and mutation from the linear fits to the Arrhenius plots of the HDXMS weighted average exchange rates versus inverse temperature. These EaHDX(avg) values represent the mean ± s.d. from the five temperatures studied here.
Room-Temperature Crystallography.
All additional protein purification and crystallization steps were performed as previously described.33, 67 Diffraction data were collected at room temperature at Beamline 8.3.1 of the Advanced Light Source at Lawrence Berkeley National Laboratory. Data were processed by the ELVES program,68 with integration performed in MOSFLM69 and scaling and merging in SCALA, POINTLESS and TRUNCATE,70–71 or using XDS72 with scaling and merging using AIMLESS,73 POINTLESS and TRUNCATE in the CCP4 suite74 provided through SBGRID.75 An initial solution was found by molecular replacement using Phaser76 with PDB 3PZW as the search model. Manual refinement was performed in Coot77 and automated refinement using the PHENIX suite.78 Alternate conformers were modeled using qFit.79–80 We manually deleted waters that did not fit into the Fo-Fc map density and edited alternate side chain conformations as indicated in the positive density of the Fo-Fc map. Active site volumes were calculated using CASTp.81 The relative size of the pockets in each mutant was based on analysis of the most populated conformer of the residues lining the pocket and included residues Gln495, Leu496 His499, Trp500, His504, Ile538, Ala542, Leu546, Ile547, Ile553, Phe557, Val564, Ser567, Val750, Ile751, Leu754, and Ile839. See Table S4 for data processing and refinement statistics.
Supplementary Material
ACKNOWLEDGMENTS
We thank Prof. John Kuriyan, UC Berkeley for his provision of personnel and facilities in the X-ray crystallographic portion of this investigation. We also thank Prof. Sharon Hammes-Schiffer and Dr. Pengfei Li for assistance in the preparation of Figure 6, which was derived from their QM/MM analysis of the WT and DM forms of SLO (cf., Ref 64).
Funding Sources
This study was supported in part by funding from the following sources: National Institutes of Health grants to JPK (GM118117), ARO (GM113432 (F32)), RDB (1R35GM126961-01) and JSF (GM123159 and GM124149), National Science Foundation grant to JSF (STC-1231306), University of California Office of the President Laboratory Fees Research Program to JSF (LFR-17-476732), the David and Lucile Packard Foundation Packard Fellowship to JSF, and institutional support from the Howard Hughes Medical Institute to CLG. Data collection at BL831 at the Advanced Light Sources is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231, UC Office of the President, Multicampus Research Programs and Initiatives grant MR-15-32859, the Program Breakthrough Biomedical Research, which is partially funded by the Sandler Foundation, and the NIH P30GM124169.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. Tables S1-S4, Figures S1-S6, and supporting references.
REFERENCES
- (1).Radzicka A; Wolfenden R, A Proficient Enzyme. Science 1995, 267, 90–93. [DOI] [PubMed] [Google Scholar]
- (2).Edwards DR; Lohman DC; Wolfenden R, Catalytic Proficiency: The Extreme Case of S-O Cleaving Sulfatases. J. Am. Chem. Soc 2012, 134, 525–531. [DOI] [PubMed] [Google Scholar]
- (3).Bruice TC, A View at the Millennium: The Efficiency of Enzymatic Catalysis. Acc. Chem. Res 2002, 35, 139–148. [DOI] [PubMed] [Google Scholar]
- (4).Benkovic SJ; Hammes-Schiffer S, A Perspective on Enzyme Catalysis. Science 2003, 301, 1196–1202. [DOI] [PubMed] [Google Scholar]
- (5).Nagel ZD; Klinman JP, A 21st Century Revisionist’s View at a Turning Point in Enzymology. Nat. Chem. Biol 2009, 5, 543–550. [DOI] [PubMed] [Google Scholar]
- (6).Schwartz SD; Schramm VL, Enzymatic Transition States and Dynamic Motion in Barrier Crossing. Nat. Chem. Biol 2009, 5, 552–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Glowacki DR; Harvey JN; Mulholland AJ, Taking Ockham’s Razor to Enzyme Dynamics and Catalysis. Nat. Chem 2012, 4, 169–176. [DOI] [PubMed] [Google Scholar]
- (8).Hay S; Scrutton NS, Good Vibrations in Enzyme-Catalysed Reactions. Nat. Chem 2012, 4, 161–168. [DOI] [PubMed] [Google Scholar]
- (9).Klinman JP; Offenbacher AR; Hu S, Origins of Enzyme Catalysis: Experimental Findings for C–H Activation, New Models, and Their Relevance to Prevailing Theoretical Constructs. J. Am. Chem. Soc 2017, 139, 18409–18427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Burton AJ; Thomson AR; Dawson WM; Brady RL; Woolfson DN, Installing Hydrolytic Activity into a Completely De Novo Protein Framework. Nat. Chem 2016, 8, 837–844. [DOI] [PubMed] [Google Scholar]
- (11).Hilvert D, Design of Protein Catalysts. Annu. Rev. Biochem 2013, 82, 447–470. [DOI] [PubMed] [Google Scholar]
- (12).Obexer R; Godina A; Garrabou X; Mittl PRE; Baker D; Griffiths AD; Hilvert D, Emergence of a Catalytic Tetrad During Evolution of a Highly Active Artificial Aldolase. Nat. Chem 2017, 9, 50–56. [DOI] [PubMed] [Google Scholar]
- (13).Schultz PG; Lerner RA, From Molecular Diversity to Catalysis - Lessons from the Immune-System. Science 1995, 269, 1835–1842. [DOI] [PubMed] [Google Scholar]
- (14).Schultz PG; Yin J; Lerner RA, The Chemistry of the Antibody Molecule. Angew. Chem. Int. Ed 2002, 41, 4427–4437. [DOI] [PubMed] [Google Scholar]
- (15).Kries H; Blomberg R; Hilvert D, De Novo Enzymes by Computational Design. Curr. Opin. Chem. Biol 2013, 17, 221–228. [DOI] [PubMed] [Google Scholar]
- (16).Klinman JP, Importance of Protein Dynamics During Enzymatic C-H Bond Cleavage Catalysis. Biochemistry 2013, 52, 2068–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hatcher E; Soudackov AV; Hammes-Schiffer S, Proton-Coupled Electron Transfer in Soybean Lipoxygenase. J. Am. Chem. Soc 2004, 126, 5763–5775. [DOI] [PubMed] [Google Scholar]
- (18).Glickman MH; Klinman JP, Nature of Rate-Limiting Steps in the Soybean Lipoxygenase-1 Reaction. Biochemistry 1995, 34, 14077–14092. [DOI] [PubMed] [Google Scholar]
- (19).Glickman MH; Wiseman JS; Klinman JP, Extremely Large Isotope Effects in the Soybean Lipoxygenase-Linoleic Acid Reaction. J. Am. Chem. Soc 1994, 116, 793–794. [Google Scholar]
- (20).Jonsson T; Glickman MH; Sun SJ; Klinman JP, Experimental Evidence for Extensive Tunneling of Hydrogen in the Lipoxygenase Reaction: Implications for Enzyme Catalysis. J. Am. Chem. Soc 1996, 118, 10319–10320. [Google Scholar]
- (21).Knapp MJ; Rickert K; Klinman JP, Temperature-Dependent Isotope Effects in Soybean Lipoxygenase-1: Correlating Hydrogen Tunneling with Protein Dynamics. J. Am. Chem. Soc 2002, 124, 3865–3874. [DOI] [PubMed] [Google Scholar]
- (22).Soudackov AV; Hammes-Schiffer S, Proton-Coupled Electron Transfer Reactions: Analytical Rate Constants and Case Study of Kinetic Isotope Effects in Lipoxygenase. Faraday Discuss 2016, 195, 171–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hatcher E; Soudackov AV; Hammes-Schiffer S, Proton-Coupled Electron Transfer in Soybean Lipoxygenase: Dynamical Behavior and Temperature Dependence of Kinetic Isotope Effects. J. Am. Chem. Soc 2007, 129, 187–196. [DOI] [PubMed] [Google Scholar]
- (24).Edwards SJ; Soudackov AV; Hammes-Schiffer S, Impact of Distal Mutation on Hydrogen Transfer Interface and Substrate Conformation in Soybean Lipoxygenase. J. Phys. Chem. B 2010, 114, 6653–6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Sharma SC; Klinman JP, Experimental Evidence for Hydrogen Tunneling When the Isotopic Arrhenius Prefactor (a(H)/a(D)) Is Unity. J. Am. Chem. Soc 2008, 130, 17632–17633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Meyer MP; Tomchick DR; Klinman JP, Enzyme Structure and Dynamics Affect Hydrogen Tunneling: The Impact of a Remote Side Chain (1553) in Soybean Lipoxygenase-1. Proc. Natl. Acad. Sci. U.S.A 2008, 105, 1146–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Meyer MP; Klinman JP, Modeling Temperature Dependent Kinetic Isotope Effects for Hydrogen Transfer in a Series of Soybean Lipoxygenase Mutants: The Effect of Anharmonicity Upon Transfer Distance. Chem. Phys 2005, 319, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hu S; Soudackov AV; Hammes-Schiffer S; Klinman JP, Enhanced Rigidification within a Double Mutant of Soybean Lipoxygenase Provides Experimental Support for Vibronically Nonadiabatic Proton-Coupled Electron Transfer Models. ACS Catal 2017, 7, 3569–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hu S; Sharma SC; Scouras AD; Soudackov AV; Carr CAM; Hammes-Schiffer S; Alber T; Klinman JP, Extremely Elevated Room-Temperature Kinetic Isotope Effects Quantify the Critical Role of Barrier Width in Enzymatic C-H Activation. J. Am. Chem. Soc 2014, 136, 8157–8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Klinman JK; Offenbacher AR, Understanding Biological Hydrogen Transfer through the Lens of Temperature Dependent Kinetic Isotope Effects. Acc. Chem. Res 2018. 51, 1966–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sharma SC; Klinman JP, Kinetic Detection of Orthogonal Protein and Chemical Coordinates in Enzyme Catalysis: Double Mutants of Soybean Lipoxygenase. Biochemistry 2015, 54, 5447–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hu S; Cattin-Ortola J; Munos JW; Klinman JP, Hydrostatic Pressure Studies Distinguish Global from Local Protein Motions in C-H Activation by Soybean Lipoxygenase-1. Angew. Chem. Int. Ed 2016, 55, 9361–9364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Offenbacher AR; Hu S; Poss EM; Carr CAM; Scouras AD; Prigozhin DM; Iavarone AT; Palla A; Alber T; Fraser JS; Klinman JP, Hydrogen–Deuterium Exchange of Lipoxygenase Uncovers a Relationship between Distal, Solvent Exposed Protein Motions and the Thermal Activation Barrier for Catalytic Proton-Coupled Electron Tunneling. ACS Cent. Sci 2017, 3, 570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Horitani M; Offenbacher AR; Carr CAM; Yu T; Hoeke V; Cutsail GE; Hammes-Schiffer S; Klinman JP; Hoffman BM, C-13 Endor Spectroscopy of Lipoxygenase-Substrate Complexes Reveals the Structural Basis for C-H Activation by Tunneling. J. Am. Chem. Soc 2017, 139, 1984–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Klinman JP; Kohen A, Hydrogen Tunneling Links Protein Dynamics to Enzyme Catalysis. Annu. Rev. Biochem 2013, 82, 471–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kohen A; Cannio R; Bartolucci S; Klinman JP, Enzyme Dynamics and Hydrogen Tunnelling in a Thermophilic Alcohol Dehydrogenase. Nature 1999, 399, 496–499. [DOI] [PubMed] [Google Scholar]
- (37).Nagel ZD; Dong M; Bahnson BJ; Klinman JP, Impaired Protein Conformational Landscapes as Revealed in Anomalous Arrhenius Prefactors. Proc. Natl. Acad. Sci. U.S.A 2011, 108, 10520–10525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Singh P; Sen A; Francis K; Kohen A, Extension and Limits of the Network of Coupled Motions Correlated to Hydride Transfer in Dihydrofolate Reductase. J. Am. Chem. Soc 2014, 136, 2575–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Francis K; Kohen A, Protein Motions and the Activation of the Ch Bond Catalyzed by Dihydrofolate Reductase. Curr. Opin. Chem. Biol 2014, 21, 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Loveridge EJ; Tey LH; Behiry EM; Dawson WM; Evans RM; Whittaker SB; Gunther UL; Williams C; Crump MP; Allemann RK, The Role of Large-Scale Motions in Catalysis by Dihydrofolate Reductase. J. Am. Chem. Soc 2011, 133, 20561–20570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Fraser JS; van den Bedem H; Samelson AJ; Lang PT; Holton JM; Echols N; Alber T, Accessing Protein Conformational Ensembles Using Room-Temperature X-Ray Crystallography. Proc. Natl. Acad. Sci. U.S.A 2011, 108, 16247–16252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Keedy DA; Kenner LR; Warkentin M; Woldeyes RA; Hopkins JB; Thompson MC; Brewster AS; Van Benschoten AH; Baxter EL; Uervirojnangkoorn M; McPhillips SE; Song J; Alonso-Mori R; Holton JM; Weis WI; Brunger AT; Soltis SM; Lemke H; Gonzalez A; Sauter NK; Cohen AE; van den Bedem H; Thorne RE; Fraser JS, Mapping the Conformational Landscape of a Dynamic Enzyme by Multitemperature and XFEL Crystallography. Elife 2015, 4. [DOI] [PMC free article] [PubMed]
- (43).Keedy DA; van den Bedem H; Sivak DA; Petsko GA; Ringe D; Wilson MA; Fraser JS, Crystal Cryocooling Distorts Conformational Heterogeneity in a Model Michaelis Complex of Dhfr. Structure 2014, 22, 899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Slappendel S; Veldink GA; Vliegenthart JF; Aasa R; Malmstrom BG, Epr Spectroscopy of Soybean Lipoxygenase-1. Description and Quantification of the High-Spin Fe(Iii) Signals. Biochim. Biophys. Acta 1981, 667, 77–86. [DOI] [PubMed] [Google Scholar]
- (45).Chasteen ND; Grady JK; Skorey KI; Neden KJ; Riendeau D; Percival MD, Characterization of the Non-Heme Iron Center of Human 5-Lipoxygenase by Electron Paramagnetic Resonance, Fluorescence, and Ultraviolet-Visible Spectroscopy: Redox Cycling between Ferrous and Ferric States. Biochemistry 1993, 32, 9763–9771. [DOI] [PubMed] [Google Scholar]
- (46).Gaffney BJ; Mavrophilipos DV; Doctor KS, Access of Ligands to the Ferric Center in Lipoxygenase-1. Biophys. J 1993, 64, 773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Zhang Y; Gan Q-F; Pavel EG; Sigal E; Solomon EI, Epr Definition of the Non-Heme Ferric Active Sites of Mammalian 15-Lipoxygenases: Major Spectral Differences Relative to Human 5-Lipoxygenases and Plant Lipoxygenases and Their Ligand Field Origin. J. Am. Chem. Soc 1995, 117, 7422–7427. [Google Scholar]
- (48).Nelson MJ; Brennan BA; Chase DB; Cowling RA; Grove GN; Scarrow RC, Structure and Kinetics of Formation of Catechol Complexes of Ferric Soybean Lipoxygenase-1. Biochemistry 1995, 34, 15219–15229. [DOI] [PubMed] [Google Scholar]
- (49).Pham C; Jankun J; Skrzypczak-Jankun E; Flowers RA; Funk MO, Structural and Thermochemical Characterization of Lipoxygenase-Catechol Complexes. Biochemistry 1998, 37, 17952–17957. [DOI] [PubMed] [Google Scholar]
- (50).Nelson MJ, Catecholate Complexes of Ferric Soybean Lipoxygenase-1. Biochemistry 1988, 27, 4273–4278. [Google Scholar]
- (51).Kemal C; Louis-Flamberg P; Krupinski-Olsen R; Shorter AL, Reductive Inactivation of Soybean Lipoxygenase 1 by Catechols: A Possible Mechanism for Regulation of Lipoxygenase Activity. Biochemistry 1987, 26, 7064–7072. [DOI] [PubMed] [Google Scholar]
- (52).Holman TR; Zhou J; Solomon EI, Spectroscopic and Functional Characterization of a Ligand Coordination Mutant of Soybean Lipoxygenase-1: First Coordination Sphere Analogue of Human 15-Lipoxygenase. J. Am. Chem. Soc 1998, 120, 12564–12572. [Google Scholar]
- (53).Bellissent-Funel MC; Hassanali A; Havenith M; Henchman R; Pohl P; Sterpone F; van der Spoel D; Xu Y; Garcia AE, Water Determines the Structure and Dynamics of Proteins. Chem. Rev 2016, 116, 7673–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Abad JL; Camps F; Fabrias G, Is Hydrogen Tunneling Involved in Acylcoa Desaturase Reactions? The Case of a Delta(9) Desaturase That Transforms (E)-11-Tetradecenoic Acid into (Z,E)-9,11-Tetradecadienoic Acid. Angew. Chem. Int. Ed 2000, 39, 3279–3281. [DOI] [PubMed] [Google Scholar]
- (55).Biela A; Nasief NN; Betz M; Heine A; Hangauer D; Klebe G, Dissecting the Hydrophobic Effect on the Molecular Level: The Role of Water, Enthalpy, and Entropy in Ligand Binding to Thermolysin. Angew. Chem. Int. Ed. Engl 2013, 52, 1822–1828. [DOI] [PubMed] [Google Scholar]
- (56).Snyder PW; Mecinovic J; Moustakas DT; Thomas SW 3rd; Harder M; Mack ET; Lockett MR; Heroux A; Sherman W; Whitesides GM, Mechanism of the Hydrophobic Effect in the Biomolecular Recognition of Arylsulfonamides by Carbonic Anhydrase. Proc. Natl. Acad. Sci. U.S.A 2011, 108, 17889–17894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Chervenak MC; Toone EJ, A Direct Measure of the Contribution of Solvent Reorganization to the Enthalpy of Binding. J. Am. Chem. Soc 1994, 116, 10533–10539. [Google Scholar]
- (58).Barillari C; Taylor J; Viner R; Essex JW, Classification of Water Molecules in Protein Binding Sites. J. Am. Chem. Soc 2007, 129, 2577–2587. [DOI] [PubMed] [Google Scholar]
- (59).Abel R; Young T; Farid R; Berne BJ; Friesner RA, Role of the Active-Site Solvent in the Thermodynamics of Factor Xa Ligand Binding. J. Am. Chem. Soc 2008, 130, 2817–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Basran J; Sutcliffe MJ; Scrutton NS, Enzymatic H-Transfer Requires Vibration-Driven Extreme Tunneling. Biochemistry 1999, 38, 3218–3222. [DOI] [PubMed] [Google Scholar]
- (61).Francisco WA; Knapp MJ; Blackburn NJ; Klinman JP, Hydrogen Tunneling in Peptidylglycine Alpha-Hydroxylating Monooxygenase. J. Am. Chem. Soc 2002, 124, 8194–8195. [DOI] [PubMed] [Google Scholar]
- (62).Sikorski RS; Wang L; Markham KA; Rajagopalan PTR; Benkovic SJ; Kohen A, Tunneling and Coupled Motion in the Escherichia Coli Dihydrofolate Reductase Catalysis. J. Am. Chem. Soc 2004, 126, 4778–4779. [DOI] [PubMed] [Google Scholar]
- (63).Hammes-Schiffer S, Hydrogen Tunneling and Protein Motion in Enzyme Reactions. Acc. Chem. Res 2006, 39, 93–100. [DOI] [PubMed] [Google Scholar]
- (64).Li P; Soudackov AV; Hammes-Schiffer S, Fundamental Insights into Proton-Coupled Electron Transfer in Soybean Lipoxygenase from Quantum Mechanical/Molecular Mechanical Free Energy Simulations. J. Am. Chem. Soc 2018, 140, 3068–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Li P; Soudackov AV; Hammes-Schiffer S, Fundamental Insights into Proton-Coupled Electron Transfer in Soybean Lipoxygenase from Quantum Mechanical/Molecular Mechanical Free Energy Simulations. J. Am. Chem. Soc 2018, 140, 3068–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Newcomer ME; Brash AR, The Structural Basis for Specificity in Lipoxygenase Catalysis. Protein Sci 2015, 24, 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Carr CAM On the Role of Enzyme Structure and Structural Dynamics in Lipoxygenase Catalysis Doctoral Dissertation, University of California, Berkeley, 2013. [Google Scholar]
- (68).Holton J; Alber T, Automated Protein Crystal Structure Determination Using ELVES. Proc. Natl. Acad. Sci. U.S.A 2004, 101, 1537–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Leslie AGW; Powell HR, Processing Diffraction Data with Mosflm. In Evolving Methods for Macromolecular Crystallography; Read RJ, Sussman JL, Eds.; NATO Science Series; Springer: Dordrecht, 2007; Vol. 245, pp 41–51. [Google Scholar]
- (70).Kabsch W, Evaluation of Single-Crystal X-Ray Diffraction Data from a Position-Sensitive Detector. J. Appl. Crystallogr 1988, 21, 916–924. [Google Scholar]
- (71).Evans P, Scaling and Assessment of Data Quality. Acta Crystallogr. D 2006, 62, 72–82. [DOI] [PubMed] [Google Scholar]
- (72).Kabsch W, Xds. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Evans PR; Murshudov GN, How Good Are My Data and What Is the Resolution? Acta Crystallogr. D Biol. Crystallogr 2013, 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AG; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS, Overview of the Ccp4 Suite and Current Developments. Acta Crystallogr. D Biol. Crystallogr 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Morin A; Eisenbraun B; Key J; Sanschagrin PC; Timony MA; Ottaviano M; Sliz P, Cutting Edge: Collaboration Gets the Most out of Software. Elife 2013, 2, e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ, Phaser Crystallographic Software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Emsley P; Lohkamp B; Scott WG; Cowtan K, Features and Development of Coot. Acta Crystallogr. D 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung L-W; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH, Phenix: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).van den Bedem H; Dhanik A; Latombe J-C; Deacon AM, Modeling Discrete Heterogeneity in X-Ray Diffraction Data by Fitting Multi-Conformers. Acta Crystallogr. D 2009, 65, 1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Keedy DA; Fraser JS; van den Bedem H, Exposing Hidden Alternative Backbone Conformations in X-Ray Crystallography Using Qfit. PLOS Comput. Biol 2015, 11, e1004507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Dundas J; Ouyang Z; Tseng J; Binkowski A; Turpaz Y; Liang J, Castp: Computed Atlas of Surface Topography of Proteins with Structural and Topographical Mapping of Functionally Annotated Residues. Nucleic Acids Res 2006, 34, W116–W118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.