

Abstract
The sympathetic nervous system (SNS) serves to maintain homeostasis of vital organ systems throughout the body, and its dysfunction plays a major role in human disease. The SNS also links the central nervous system to the immune system during different types of stress via innervation of the lymph nodes, spleen, thymus and bone marrow. Previous studies have shown that pituitary adenylate cyclase-activating polypeptide (PACAP, gene name adcyap1) exhibits anti-inflammatory properties in the experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis. Because PACAP is known to regulate SNS function, we hypothesized that part of the immunoprotective action of PACAP is due to its neuromodulatory effects on sympathetic neurons. To examine this, we used an inducible, targeted approach to conditionally disrupt the PACAP-preferring PAC1 receptor gene (adcyap1r1) in dopamine β-hydroxylase-expressing cells, which includes postganglionic sympathetic neurons, but also catecholaminergic neurons in the brain and adrenomedullary chromaffin cells. In contrast to our previous EAE studies using PACAP global knockout mice which developed severe and prolonged EAE, we found that mice with conditional loss of PAC1 receptors in catecholaminergic cells developed a delayed time course of EAE with reduced helper T-cell type 1 (Th1) and Th17 and enhanced Th2 cell polarization. At later time points, similar to mice with global PACAP loss, mice with conditional loss of PAC1 exhibited more severe clinical disease than controls. The latter was associated with a reduction in the abundance of thymic regulatory T cells (Tregs). These studies indicate that PAC1 receptor signaling acts in catecholaminergic cells in a time-dependent manner. At early stages of disease development, it enhances the ability of the SNS to polarize the Th response towards a more inflammatory state. Then, after disease is established, it enhances the ability of the SNS to dampen the inflammatory response via Tregs. The lack of concordance in results between global PACAP KO mice and mice with the PAC1 deletion targeted to catecholaminergic cells during early EAE may be explained by the fact that PACAP acts to regulate inflammation via multiple receptor subtypes and multiple targets, including inflammatory cells.
Keywords: Multiple sclerosis, experimental autoimmune encephalomyelitis, PACAP, PAC1, sympathetic nervous system, inflammation, regulatory T cells, Th cells, tamoxifen
INTRODUCTION
Multiple sclerosis (MS) affects about 2.5 million people worldwide and is a leading cause of disability in young adults. In this disease, auto-reactive immune cells target oligodendrocytes and neurons in the central nervous system (CNS). Chronic inflammation leads to neurodegeneration and causes symptoms including vision loss, paralysis, and cognitive decline, which can severely interfere with quality of life. Most commonly prescribed drugs target inflammation, and thereby reduce flare-ups and slow progression of the disease, but do not lead to repair or restoration of neuronal function. Additionally, these drugs can cause mild side effects such as aches, fever, fatigue, diarrhea, hair loss, and nausea, as well as more severe effects such as liver damage, infertility, birth defects, depression, and a suppressed immune system. Thus, there is a need to investigate alternative treatment options to modulate inflammation in MS.
The sympathetic nervous system (SNS) links the central nervous system and the immune system (Bellinger et al., 2008; Kenney and Ganta, 2014), and has tremendous importance in human disease. The SNS regulates processes which underlie cardiovascular and metabolic disorders, obesity, psychiatric disorders associated with stress, autoimmune and other inflammatory diseases. Drugs that modify sympathetic function currently include those that target catecholamine synthesis, metabolism, uptake, or receptor activity. Strategies to modify symptoms of MS and progression of disease by regulating sympathetic activity have undergone testing in preclinical models (Sternberg, 2016).
It is well documented that peripheral lymphoid organs, including the spleen, lymph nodes, thymus, and probably bone marrow, receive abundant innervation from sympathetic ganglia, with nerve terminals projecting to immune and stromal cells. The role of this innervation has been examined primarily by peripheral administration of 6-hydoxydopamine (6-OHDA), which does not appreciably penetrate the blood-brain barrier, and thus selectively ablates sympathetic neurons in the periphery. Most of these studies, as well as studies in which immune cells were treated with norepinephrine (NE) receptor analogs in vitro, suggested that NE acts primarily via β−2 adrenergic receptors (β−2AR), and mainly inhibits innate inflammatory responses, and either promotes or inhibits adaptive immunity (Bellinger et al., 2008; Nance and Sanders, 2007). The advent of gene targeting has provided a new approach to examine the role of the SNS on various functions at a molecular level. Unfortunately, limited illuminating data are available so far which address how the SNS regulates inflammation using this approach. For example, a comprehensive analysis of the immune phenotype of β−2AR-deficient mice reported essentially normal immune responses (Nance and Sanders, 2007). On the other hand, dopamine β-hydroxylase deficient mice were found to exhibit diminished Th1-responses to pathogen challenge (Sanders et al., 2003), providing evidence that SNS actions on adaptive immune cells might support inflammation. Overall, the studies using sympathectomy, and genetically-engineered mice have generally not examined the immunomodulatory of other signaling molecules, such as pituitary adenylyl cyclase activating peptide (PACAP), vasoactive intestinal peptide (VIP), galanin, and neuropeptide Y, that are released by sympathetic neurons or their presynaptic innervation in naïve or stressed rodents (Beaudet et al., 1998; Shadiack et al., 2001).
Our laboratory has been investigating immunomodulatory actions of PACAP, VIP, and their receptors in the EAE model using mice globally deficient in these proteins (Abad et al., 2010, 2016; Tan et al., 2009, 2013, 2015). PACAP was originally isolated and shown to induce adenylyl cyclase to produce cyclic AMP in rat pituitary cell cultures (Miyata et al., 1989). Its receptors include VPAC1, VPAC2, and PAC1. While VPAC1 and VPAC2 also bind the homologous protein VIP with equal high affinity, the PAC1 receptor binds PACAP with 100-fold greater affinity than it binds VIP. PACAP is highly conserved through evolution, with the human PACAP sequence sharing 90% homolog with cnidarians, the earliest phyla to develop tissue layers. PACAP is upregulated in response to injury and inflammation and has been shown to protect neurons and reduce inflammation in the CNS in animal models of neurodegenerative diseases such as multiple sclerosis, Alzheimer’s Disease, Parkinson’s Disease, and stroke (Brifault et al., 2014; Chen and Tzeng, 2005; Dejda et al., 2008; Lee and Seo, 2014; Seaborn et al., 2011; Waschek, 2013). Using the EAE model, our lab previously demonstrated that PACAP-deficient mice have more severe clinical disease, greater inflammation, and higher mortality compared to wild type mice (Tan et al., 2009, 2013). Moreover, PACAP deficient mice had impaired Treg responses during EAE, along with a skewing of Teffectors towards more inflammatory phenotypes (Th1 and Th17), and a reduction of anti-inflammatory Th2 cells. The results imply that PACAP primarily plays an anti-inflammatory role during EAE. However, the relevant anatomical sites of PACAP action cannot be ascertained in these global KO mice studies.
One potential site of PACAP action during EAE is the SNS. PACAP is expressed in neurons in the intermediolateral column of the thoracic spinal cord that provide the presynaptic innervation of sympathetic ganglia (Beaudet et al., 1998; Pettersson et al., 2004). PACAP in these projections appear to modulate SNS function via action on PAC1 receptors expressed on sympathetic neurons (Braas and May, 1999), Diane et al., 2014). We hypothesized that immunoprotective actions of PACAP in EAE are in part mediated through modulation of the SNS (Fig. 1a). More specifically, we hypothesized that mice with conditional elimination of PAC1 receptors from postganglionic sympathetic neurons would mimic PACAP KO mice with respect to EAE, and thus result in more severe clinical disease and increased inflammation.
Fig. 1.
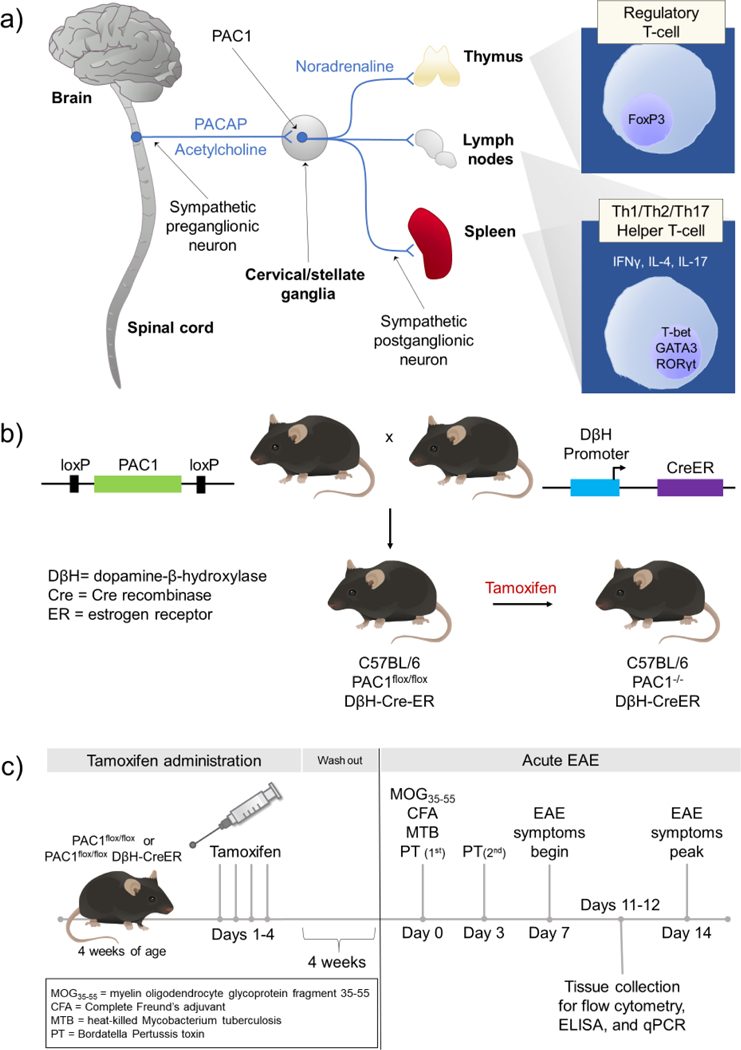
(a) Potential neural circuitry by which PACAP modulates peripheral immune cell activity during inflammatory stress. During inflammatory stress, neurons in the brain stem and hypothalamus activate preganglionic sympathetic neurons in the spinal cord. PACAP is expressed along with acetylcholine in the preganglionic neurons in thoracic spinal cord. When released during stress or inflammation, PACAP acts via PAC1 receptors expressed on sympathetic neurons in cervical/stellate ganglia to alter the immune response in the thymus, lymph nodes, and spleen. (b) Approach to conditionally knockout PAC1 receptors in postganglionic neurons in the SNS. C57BL/6 mice were genetically engineered to introduce loxP sites that flank critical sequences in the PAC1 receptor gene. Such mice (PAC1flox/flox mice) are bred to C57BL/6 mice which express Cre enzyme physically linked to the ligand-binding portion of the estrogen receptor driven by the dopamine β-hydroxylase (DβH) promoter (DβH-CreER). Administration of tamoxifen to PAC1flox/flox DβH-CreER mice allows Cre recombinase to translocate into the nucleus specifically in DβH-expressing cells, and subsequently disrupt the endogenous PAC1 receptor gene. (c) Experimental timeline to conditionally knockout PAC1 and induce EAE. PAC1flox/flox and PAC1flox/flox DβH Cre-ER mice are administered tamoxifen (Sigma Aldrich, St. Louis, MO, USA) (100 μL of 20 mg/mL dissolved in peanut oil) by daily oral gavage for four consecutive days at 4 weeks of age, the age at which the pups are weaned. Because tamoxifen is a known estrogen receptor agonist, we allow four weeks for tamoxifen washout to minimize any confounding effects of tamoxifen before proceeding with immunization to MOG35–55.
MATERIALS AND METHODS
Animals.
Mice were housed under environmentally controlled conditions in a 12-hour light/dark cycle with access to food and water ad libitum. All animal studies were approved by the UCLA institutional animal care and use committee (IACUC) and Animal Research Committee (ARC).
Conditional knockout of PAC1 on catecholaminergic neurons.
We used tamoxifen-inducible CreER-Lox recombination to delete PAC1 receptors from dopamine-β-hydroxylase-expressing neurons, including postganglionic SNS neurons (Fig. 1b). The tamoxifen-regulated approach was selected so that we could induce gene deletion in adult mice, thereby avoiding disrupting known effects of PACAP on developing sympathetic neurons (DiCicco-Bloom et al., 2000; Lu et al., 1998). We commissioned the National Institutes of Health Knockout Mouse Project (KOMP) to generate mice with loxP sites flanking the PAC1 gene. Such mice (PAC1flox/flox mice), appeared healthy and showed no obvious behavioral abnormalities. PAC1flox/flox mice were bred with mice obtained from Dr. Hermann Rohrer (Max Planck Institute for Brain Research, Frankfurt, Germany) which express Cre fused to the ligand binding domain of the estrogen receptor (ER) driven by the dopamine β-hydroxylase promoter (Stubbusch et al., 2011), resulting in PAC1flox/flox DβH-CreER double transgenic mice (Fig. 1b). For all experiments, PAC1flox/flox DβH-CreER mice were bred with PAC1flox/flox mice, so that litters would include both PAC1flox/flox DβH-CreER and PAC1flox/flox control mice. Tamoxifen treatment of PAC1flox/flox DβH-CreER mice results in translocation of Cre recombinase into the nucleus where it targets loxP sites and mediates excision of the PAC1 gene in catecholaminergic neurons. From here onwards, the tamoxifen-treated PAC1flox/flox DβH-CreER mice will be referred to as PAC1 receptor conditional knockdown (cKD) mice, whereas PAC1flox/flox littermates, which do not express Cre, will be referred to as control mice. In some cases, PAC1flox/flox DβH-CreER mice were bred to an Ai9 reporter mouse line obtained from Dr. Anton Maximov (Scripps Research Institute, La Jolla, CA, USA) which expresses the red fluorescent reporter TdTomato under a CAG promoter downstream of a loxP flanked STOP cassette. Resulting mice (PAC1flox/flox DβH-CreER Ai9) only express the TdTomato reporter when active Cre targets the loxP sites and excises the STOP cassette.
We used a conditional, rather than a global knockout approach to focus relatively specifically on the effects of PAC1 signaling in the SNS, i.e., to avoid confounding effects from lack of PAC1 on other PAC1 receptor-expressing cell types, which include multiple populations of CNS neurons, astrocytes, and macrophages. Additionally, it is known that C57BL/6 mice deficient in PAC1 receptor exhibit high neonatal mortality (Jamen et al., 2000; Otto, 2004). Although PAC1 deficient mice appear normal at birth, within the first two postnatal weeks, pups develop pulmonary hypertension and right ventricle heart failure (Otto, 2004). It is not feasible to induce EAE in these mice.
Experimental autoimmune encephalomyelitis (EAE).
To induce the acute form of EAE, mice are subjected to a well-characterized protocol in which MOG35–55, a fragment of myelin oligodendrocyte glycoprotein, is administered peripherally in adjuvant (Constantinescu et al., 2011). 100 μg of MOG35–55 in phosphate-buffered saline (PBS) is emulsified 1:1 with Difco Complete Freund’s Adjuvant (BD, Franklin Lakes, NJ, USA) supplemented with an additional 100 mg of Difco Mycobacterium tuberculosis H37RA (BD, Franklin Lakes, NJ, USA). The emulsion is split and subcutaneously injected in the left and right flanks posterior to the forelimbs. This was specified as day 0 of EAE (Fig. 1c). This results in the expansion of T-cells autoreactive to myelin (Constantinescu et al., 2011). Additionally, the mice receive an intraperitoneal injection of 200 ng of pertussis toxin (List Biological Laboratories, Campbell, CA, USA) dissolved in PBS. Pertussis toxin causes permeabilization of the blood brain barrier, thus allowing the T-cells targeting MOG to enter the CNS. After two days, the mice are given an additional 200 ng booster injection of pertussis toxin. Mice reproducibly begin to develop demyelination and paralysis within 7–10 days. Clinical scores of EAE severity were given based on a 5-point scale where 0=asymptomatic, 1=limp tail, 2=partial paralysis or failure to resist inversion, 3=complete paralysis of one hind limb, 4=complete paralysis of both hind limbs, and 5=moribund or death. EAE was monitored for 21 days. In cases where mice reached the moribund state, they were euthanized as dictated by federal and university policy.
Lymph node, spleen, and thymus cell suspension preparation.
On days 11 and 12 post-MOG35–55 immunization lymph nodes (auxiliary, brachial, and inguinal), spleens, and thymi were collected into Complete Medium (2% fetal bovine serum, 10,000 I.U./mL Penicillin, 10,000 μg/mL Streptomycin in Roswell Park Memorial Institute (RPMI) 1640 medium) on ice. The tissues were mechanically dissociated into a single cell suspension using the plunger from a 5 mL syringe to gently grind the tissues through a 40 μm cell strainer basket. The cell suspension was centrifuged at 400 rpm at 4˚C for 7 min using a Sorvall RT7 (Thermo Scientific, Canoga Park, CA, USA) refrigerated centrifuge. The supernatant was aspirated, and the cell pellet was resuspended in 2% fetal bovine serum, 10,000 I.U./mL Penicillin, 10,000 μg/mL Streptomycin in RPMI 1640.
Flow cytometry.
Cells were incubated in 2% fetal bovine serum, 10,000 I.U./mL Penicillin, 10,000 μg/mL Streptomycin in RPMI 1640 containing 50 ng/ml phorbol 12-myristate 13-acetate (PMA) (Sigma Aldrich, St. Louis, MO, USA), 1 ug/ml ionomycin (Sigma Aldrich, St. Louis, MO, USA), 1X brefeldin A (BioLegend, San Diego, CA, USA), 1x monesin (BioLegend, San Diego, CA, USA) for 4 hours at 37°C. The cell cultures were centrifuged at 400 rpm at 4°C for 7 min, and then washed with phosphate-buffered saline (PBS). To distinguish live/dead cells, cell were stained with Zombie UV™ dye (BioLegend, San Diego, CA, USA) diluted in 1xPBS, and incubated at room temperature (RT) in the dark for 30 min. The cells were washed with 1% bovine serum albumin (BSA) in 1xPBS to quench the reaction and then centrifuged at 400 rpm at 4°C for 7 min. The supernatant was aspirated, and the cells were washed with 1xPBS. The cells were centrifuged again at 400 rpm at 4°C for 7 min, and the supernatant aspirated. For extracellular staining, the cell pellet was resuspended in 1% BSA 1XPBS containing an extracellular antibody cocktail (anti-CD4 VioBlue, anti-CD8a FITC, Miltenyi Biotec, Bergisch Gladbach, Germany). The cells were incubated at 4°C for 15 min shielded from light. The cells were washed with 1xPBS, then centrifuged at 400 rpm at 4°C for 7 min. The supernatant was aspirated and the cells were fixed with 2% paraformaldehyde (PFA) in 1xPBS at 37°C for 10min. Then, the cells were washed with 0.2% Tween-20 in 1xPBS. Following centrifugation at 400 rpm at 4°C for 7 min, the supernatant was aspirated. For intracellular staining, the cell pellets were resuspended with 0.2% Tween-20 in 1XPBS containing an intracellular antibodies cocktail (anti-IFNy PE, anti-IL-4 Vio515, anti-IL-17A APC, Miltenyi Biotec, Bergisch Gladbach, Germany). For detection of nuclear proteins FoxP3 and Ki67, the antibodies (anti-FoxP3 PE, anti-Ki67 APC, Miltenyi Biotec, Bergisch Gladbach, Germany) were diluted in FoxP3 Staining Kit buffers (Miltenyi Biotec, Bergisch Gladbach, Germany). The samples were incubated at 4°C for 15 min shielded from light. The cells were washed twice with 1XPBS and resuspended in 1XPBS for analysis by flow cytometry using a BD LSRII flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Antigen recall assay.
Lymph node and spleen cells are plated at 1 × 106 cells/mL in 2% fetal bovine serum, 10,000 I.U./mL Penicillin, 10,000 μg/mL Streptomycin in RPMI 1640 containing 10 μg/mL myelin oligodendrocyte glycoprotein fragment 35–55 (MOG35–55) (GL Biochem, Shanghai, China) or ovalbumin (Sigma Aldrich, St. Louis, MO, USA) antigen control and incubated in 5% CO2 at 37°C. After 48 hours, the culture media was collected, and flash frozen in liquid nitrogen. Cytokine levels in the culture media were analyzed by sandwich enzyme-linked immunosorbent assay (ELISA) using Ready-Set-Go! kits for IFNγ, IL-5, and IL-17 (eBioscience, San Diego, CA, USA).
RNA Extraction.
TRIzol (Invitrogen, Carlsbad, CA, USA) was added to dissociated lymph node or spleen cell pellets (see cell suspension preparation) at approximately a ratio of 1 mL TRIzol buffer per 50–100 mg of tissue. The samples were passed through a 21-gauge needle 10x, and the homogenate was incubated at RT for 5 min. 200 μL of RNAse-free chloroform was added and the samples were vortexed vigorously for 15 secs. The samples were incubated at RT for 3 min, then centrifuged at 13,000 g for 10 min at 4°C. The aqueous phase was transferred to new 1.5 mL microcentrifuge tube. One volume of isopropanol was added to tube to precipitate the RNA. The tube was incubated at −20°C for 1 hour to facilitate precipitation. Then, the samples were centrifuged at 20,000 g for 15 min at 4°C to pellet the precipitated RNA. The resulting RNA pellet was washed twice with 70% ethanol before air-drying the pellet at RT for about 20 mins. The RNA pellet was resuspended with nuclease-free water. All centrifugation steps were performed with an Eppendorf 5417R (Eppendorf, Hamburg, Germany) refrigerated centrifuge.
RNA concentration and purity (determined by A260/280nm and A260/230nm ratios) were measured using a Thermo Scientific NanoDrop One Microvolume UV-Vis Spectrophotometer. 250 ng of each sample of RNA was denatured with 2x RNA Loading Dye (Thermo Scientific, Canoga Park, CA, USA) for 70°C at 10 min using a MJ Mini Thermal Cycler (Bio-Rad, Hercules, CA, USA). Denatured RNA samples were run alongside 1 μg of RiboRuler Low Range RNA Ladder (Thermo Scientific, Canoga Park, CA, USA) in a 1.2% agarose gel in Tris-Buffered EDTA buffer pre-cast with 5% GelRed (Biotium, Fremont, CA, USA) at 90V for 20 mins to check for intact 28S and 18S RNA bands. Gels were imaged using a Universal Hood Gel Doc System (Bio-Rad, Hercules, CA, USA).
Real-time qRT-PCR.
Complementary DNA (cDNA) was synthesized from 500 ng of purified RNA in a 20 μL reaction using SuperScript IV VILO Master Mix (Invitrogen, Carlsbad, CA, USA). Quantitative polymerase chain reaction (qPCR) was performed using PowerUp SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA). cDNA was diluted 10x and 7.5 ng of cDNA was used per 20 μL qPCR reaction. The target genes and the primers used are listed in Table 1. PCR primers were used at a final concentration of 500 nM. Amplifications were performed using the following cycling protocol: UDG activation at 50°C for 2 min, Dual-Lock™ DNA polymerase hot-start at 95°C for 2 min, then 40 cycles of denaturing at 95°C for 15 min and annealing/extending at 60°C for 1 min. Reactions were performed and read using an StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). A melt curve was performed at the end of each assay to check for single product amplification. A standard curve made by sequential 10-fold dilution from the most concentrated standard (containing 5 μL from all cDNA samples and diluted to a total volume of 200 μL with nuclease-free water) was used to determine amplification efficiency. Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) was used as the housekeeping gene for all qPCR assays.
Table 1.
List of primers used in qPCR assays.
| Gene | Forward primer | Reverse primer |
|---|---|---|
| FoxP3 | CACACCTCTTCTTCCTTGAACC | GATCATGGCTGGGTTGTCCA |
| GAPDH | GGCCTTCCGTGTTCCTAC | TGTCATCATACTTGGCAGGTT |
| GATA3 | CAGATAGCATGAAGCTGGAG | CCTTCTGTGCTGGATCGTG |
| IFNy | CAATGAACGCTACACACTGC | GCTTTCAATGACTGTGCCG |
| IL-4 | GTCATCCTGCTCTTCTTTCTCG | CTTCTCCTGTGACCTCGTTC |
| IL-6 | ACAACCACGGCCTTCCCTACTT | CACGATTTCCCAGAGAACATGTG |
| IL-7 | CCTCCACTGATCCTTGTTCTGC | GCAGCTTCCTTTGTATCATCAC |
| IL-10 | CAGCCGGGAAGACAATAAC | CATTTCCGATAAGGCTTGGC |
| IL-17 | GTGTCTCTGATGCTGTTGCTG | CATTCTGGAGGAAGTCCTTGG |
| IL-33 | CTACTGCATGAGACTCCGTTC | GTGTCAACAGACGCAGCAAATG |
| RORγt | CAGTGTAATGTGGCCTACTCC | CTTGACAGCATCTCGGGAC |
| SIRT1 | CCTTGGAGACTGCGATGTTA | GTGTTGGTGGCAACTCTGAT |
| T-bet | GCAGTGTGGAAAGGCAGAAG | CTGGGTCACATTGTTGGAAGC |
Tissue collection and sectioning for Immunofluorescence and RNA in situ hybridization assays.
Mice were euthanized by isoflurane overdose. For immunofluorescence assays, super cervical ganglia were collected and fixed overnight at 4˚C. The next day the tissues were cryoprotected in 15% sucrose 0.05% sodium azide in 1xPBS. The following day, the tissues were incubated in 30% sucrose 0.05% sodium azide in 1xPBS until they no longer floated in the solution. Cryoprotected tissues were embedded in Tissue-Tek Optimum Cutting Temperature compound (Sakura Finetek, Torrance, CA, USA) and cryosectioned using a Leica CM1950 (Wetzlar, Germany) cryostat at 10 μm onto Fisherbrand Super Frost Plus slides (Fisher Scientific, Hampton, NH, USA). Slides were stored at −80˚C until use.
For RNA in situ hybridization, brain and superior cervical ganglion were harvested and fresh frozen on dry ice. The tissues were cryosectioned using a Leica CM1950 (Wetzlar, Germany) cryostat at 10 μm onto Fisherbrand Super Frost Plus slides (Fisher Scientific, Hampton, NH, USA). Slides were stored at −80˚C until use.
Immunofluorescence assays.
For the immunofluorescence detection assay, slides were blocked and permeabilized with 10% normal goat serum 0.5% TritonX-100 1% BSA in 1xPBS for 1 hour at RT. Following a rinse slide with 1xPBS, slides were incubated with an appropriate dilution ((1:1000) Rabbit anti-tyrosine hydroxylase (Abcam, Cambridge, UK)) of primary antibody in 5% goat serum 1% BSA in 1xPBS overnight at 4°C. The next day, slides were washed 3×10min with 1xPBS then incubated in an appropriate dilution of fluorophore-conjugated antibody ((1:500) Goat anti-rabbit IgG (H+L) polyclonal cross-adsorbed secondary Alexa Fluor 488 (Invitrogen, Carlsbad, CA, USA)) in 5% goat serum 1% BSA 1xPBS for 1 hour at RT. The slides are then stained with Hoechst 33342 (20mM at 1:1000) (Pierce, WI, USA) in 1XPBS for 5 min at RT, then washed 3×10 min with 1xPBS. Finally, Prolong Gold Antifade mounting media (Invitrogen, Carlsbad, CA, USA) and glass coverslips were applied and sealed with nail polish.
RNA in situ hybridization.
RNA in situ hybridization was performed following standard instructions accompanying the Advanced Cell Diagnostics RNAScope Fluorescent Multiplex kit (Biotechne, Minneapolis, MN, USA). Following, RNA in situ hybridization, an immunofluorescent assay was performed starting from the blocking step as described above, except the blocking buffer contained no TritonX-100.
Statistical Analysis.
R (R Foundation for Statistical Computing, Vienna, Austria) and Microsoft Excel (Redmond, WA, USA) were used for statistical analysis and for generating graphs. Analysis of variance (ANOVA) and Student’s t-test were used to determine statistical significance.
Microscopy.
Fluorescent microscope images were captured using an Axio Imager 2 (Carl Zeiss, Oberkochen, Germany), and analyzed using ZEN lite (Carl Zeiss, Oberkochen, Germany).
RESULTS
Conditionally knockout of PAC1 receptors.
To determine the efficiency of tamoxifen-induced recombination in catecholaminergic neurons, we crossed the PAC1flox/flox DβH-CreER line to the Ai9 reporter line. These Ai9 mice were designed to express the red TdTomato fluorescent reporter in cells following Cre recombination. The DβH-CreER line has previously been shown to induce recombination with greater than 90% recombination efficiency in the locus coeruleus and adrenal medulla, and partial recombination in the SCG (Stubbusch et al., 2011). Using the tdTomato reporter and a similar tamoxifen-treatment protocol, we detected about 50% recombination in tyrosine hydroxylase (TOH)-positive neurons SCG (Fig. 2a). To determine if the PAC1 receptor gene underwent similar recombination, we combined RNA in situ hybridization for PAC1 receptor with immunofluorescence assay of TOH. Results demonstrated a partial loss of PAC1 gene transcripts in the SCG (Fig. 2b) and a nearly complete loss in the locus coeruleus (Fig. 2c) (Stubbusch et al., 2011). The estimated percentage of TOH- expressing SCG neurons in which PAC1 receptor gene transcripts were eliminated was 20–30%, which is comparable to that observed with a lacZ reporter (Stubbusch et al., 2011), but less than that observed when DβH-CreER mice were crossed with mice containing conditional alleles of choline acetyl transferase (ChAT) (Olivas et al., 2016) and GATA3 (Tsarovina et al., 2010). In these cases, gene expression in SCG remained at about 20% of control levels after targeted deletion. Nonetheless, strong phenotypic alterations were reported in those mice, corroborating that incomplete elimination of a targeted gene can be sufficient to significantly impair gene function.
Fig. 2.
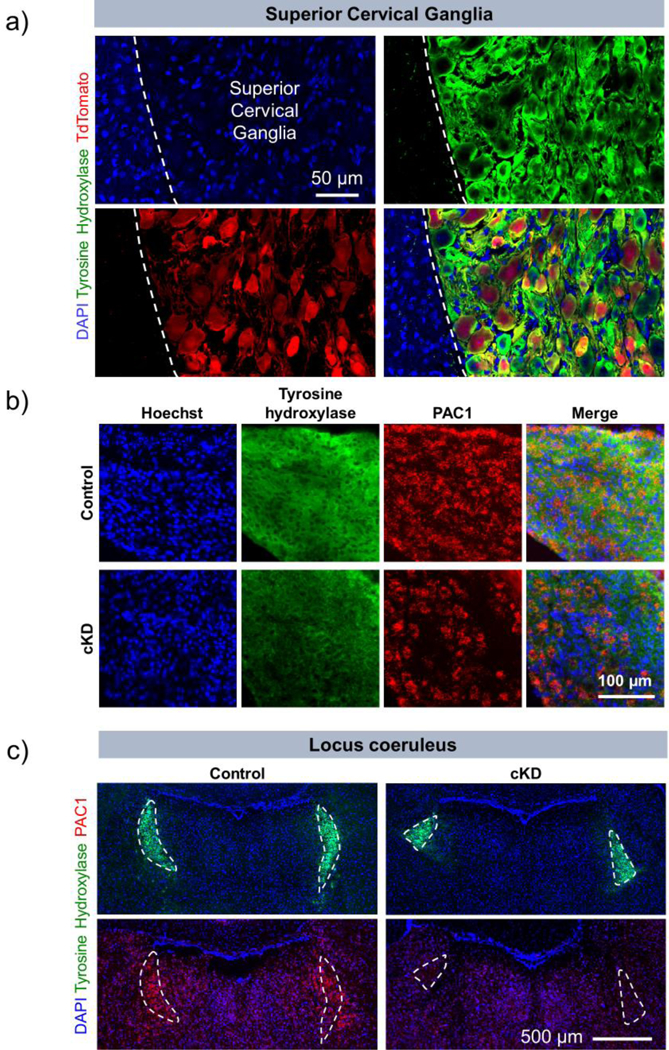
PAC1 receptor gene expression is specifically knocked down in catecholaminergic cells. (a) Use of PAC1flox/flox DβH-CreER Ai9 reporter triple transgenic mouse confirms cell-specific targeting of Cre in the superior cervical ganglion. We generated a triple transgenic mouse which only expresses the red reporter protein, TdTomato, when an upstream floxed stop sequence is excised by tamoxifen-induced Cre. Within the superior cervical ganglion, TdTomato is expressed only in cells which express tyrosine hydroxylase, an enzyme which is highly expressed in DβH+ cells. This demonstrates specific targeting of Cre activity. The mice were euthanized, and brains and superior cervical ganglia were collected 4 weeks after the last dose of tamoxifen. (b) Superior cervical ganglion sections and (c) coronal brain sections from control and cKD mice at the level of the locus coeruleus were labeled by RNA in situ hybridization using with a DNA probe against PAC1 mRNA and co-labeled by immunofluorescence assay using antibody against tyrosine hydroxylase to label catecholaminergic neurons
Conditional knockdown (cKD) of PAC1 led to early inhibition, then enhanced, severity of EAE clinical scores.
Because tamoxifen is known to affect the clinical severity of EAE, tamoxifen-treated PAC1flox/flox mice were used as controls rather than vehicle-treated PAC1flox/flox DβH-CreER mice. Tamoxifen was thus administered to PAC1 cKD and PAC1flox/flox control mice at two months of age. After a four-week tamoxifen washout, acute EAE was induced with MOG35–55 as described in our prior studies (Abad et al., 2016). Conditional deficiency of PAC1 receptors resulted in a statistically significant inhibition of EAE during both the induction and peak periods of EAE (Fig. 3). Thereafter, beginning on day 17, disease progression curves crossed, with clinical disease being more severe in PAC1 cKD mice compared to controls. PAC1 receptor signaling cells thus appears to facilitate disease in the early inductive phase of EAE and to promote recovery once disease is fully established.
Fig. 3.
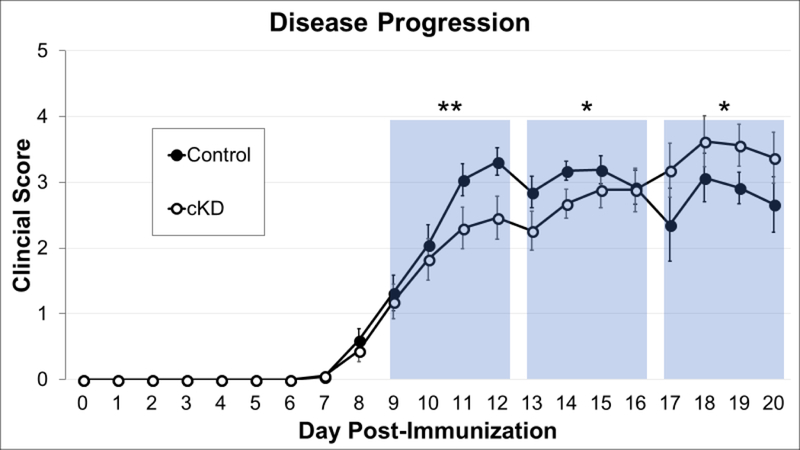
Conditional knockdown of PAC1 receptors in catecholaminergic cell led to early inhibition of, then enhanced, EAE severity. PAC1flox/flox DβH-CreER and PAC1flox/flox control mice were treated with tamoxifen at 4 weeks of age to induce CreER-dependent excision of PAC1 gene. At 8 weeks of age, the mice were immunized against MOG35–55 to induce acute EAE. Data shown combines data from 4 independent experiments, including a total of 36 WT and 32 cKD mice. EAE was established by day 9 post MOG35–55-immunization and allowed to progress until day 20, a time point in which clear differences in Th1/Th2/Th17 CD4+ phenotype shifts were observed in PACAP deficient mice (Tan et al., 2013). The twelve days of EAE disease were divided into three bins aggregating scores from four consecutive days. This binning strategy divides the twelve days of EAE evenly for analysis and avoids combing data from EAE scores before day 16 with scores after day 16, the time point at which the EAE clinical scores curves for the two groups cross. *p<0.05, **<0.01. Statistical significance was determined by 2-tailed t-tests. Standard error bars are shown
Analysis of cytokine expression in lymph nodes and spleen.
Because the SNS provides major innervation of lymphoid organs and regulates Th differentiation, we determined the effect of sympathetic PAC1 receptor diminution on helper T-cells (Th) subtype abundance in the spleen and lymph nodes. Th cells are characterized based on the cytokines they produce. The most common phenotypes are Th1, Th2, and Th17. In autoimmune diseases, it is thought that Th1 and Th17 helper T-cells are generally pathogenic while Th2 cells are protective (Damsker et al., 2010; Lafaille, 1998; Simmons et al., 2013). Previous research has shown that PACAP modulates Th phenotype in a way that protects against EAE. PACAP promotes the recruitment, expansion, and survival of antigen specific Th2 T-cells by modulating chemotactic molecules released by dendritic cells (Delgado, 2004). Moreover, we previously showed that PACAP deficient mice exhibited a skewing of CD4+ Th phenotypes away from a Th2 phenotype towards the more pro-inflammatory Th1 and Th17 phenotypes (Tan et al., 2009, 2013).
To determine if PACAP regulated Th polarity through PAC1 receptors on postganglionic SNS neurons that target lymphoid tissues, we characterized by flow cytometry CD4+ Th phenotypes (Th1, Th2, and Th17) in spleen and lymph nodes of tamoxifen-treated mice 11–12 days after EAE induction. In PACAP cKD mice, in contrast to that observed in global PACAP KO mice, we detected a significant reduction in the relative abundance of Th17 cells in the spleen and a significant increase in Th2 cells (Fig. 4a upper panels). This indicates a shift to a less inflammatory state. No change was observed in the abundance of Th1 cells. Analyses of gene expression similarly showed a shift to less inflammatory state, in this case showing a reduction in T-bet and RORγt, transcription factors that are relatively specific for Th1 and Th17 cells, respectively (Fig. 4a upper panels). No significant differences between genotypes were observed in splenic gene expression of IFNγ (a Th1 cytokine), IL-4 (a Th2 cytokine), GATA-3 (a Th2 transcription factor), or IL-17 (a Th17 cytokine). In the lymph nodes, no significant differences between genotypes were observed in the flow cytometric (Fig. 4b) or by gene expression assays (data not shown).
Fig. 4.
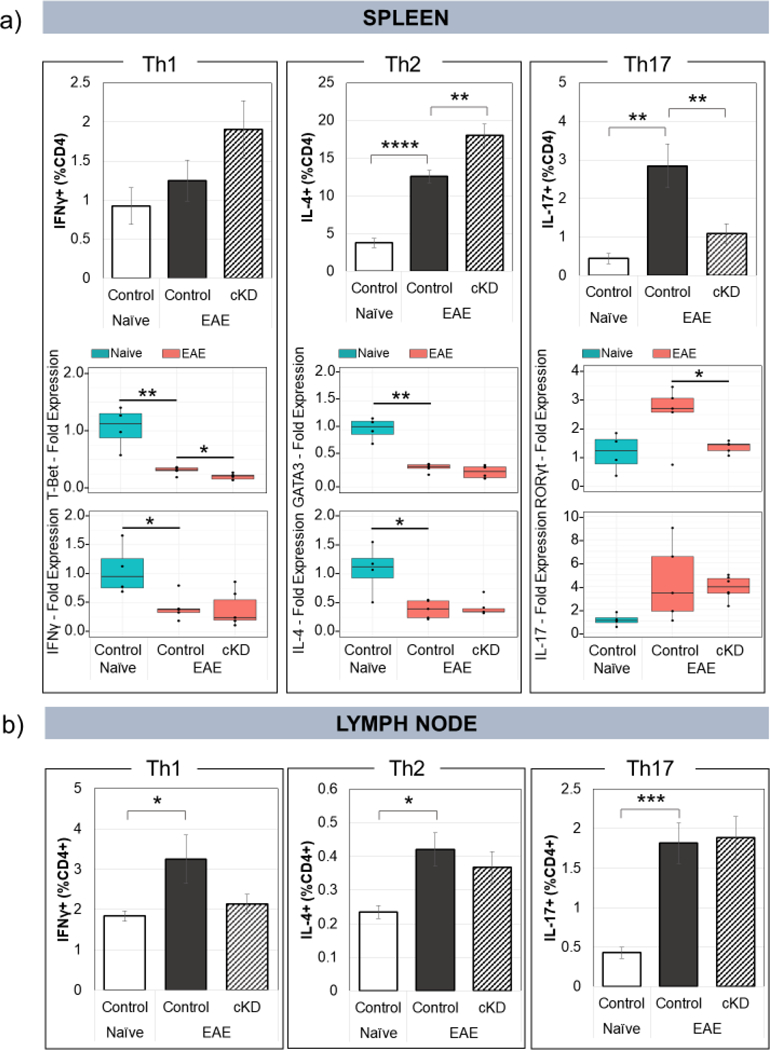
Conditional knockdown of PAC1 in sympathetic ganglia alters CD4+ helper T-cell polarization during EAE. Cell suspensions from (a, top portion) spleen and (b) lymph nodes were stained with antibodies against CD4, IFNγ, IL-4, and IL-17 and analyzed by flow cytometry to characterize the populations of Th1, Th2, and Th17 CD4+ helper T-cells. Naïve floxed=5, EAE floxed=14, EAE cKD=9. RNA was extracted from whole lymph nodes (data not shown) and spleen (a, bottom portion), and analyzed by qPCR for signature transcription factors and cytokines associated with Th1, Th2, and Th17 CD4+ T-cells. Naïve floxed=4, EAE floxed=5, EAE cKD=6. *p<0.05, **<0.01. Statistical significance was determined by 2-tailed t-tests. Standard error bars are shown
To obtain antigen-specific information on Th differentiation, we performed ex vivo antigen recall assays on spleen and lymph node single cell suspensions cultured in the presence of purified MOG35–55 or control stimulus ovalbumin. We observed robust MOG35–55-induced inductions of IFNγ production in lymph node and spleen cultures of cKD and control mice (Fig. 5). In cultures from cKD mice, mean IFNγ levels were decreased compared to controls, but only in the case of lymph nodes was the reduction significant. We also observed robust MOG-induced inductions of IL-17 production in spleen and lymph node cultures of cKD and control mice, but no differences were observed between genotypes (data not shown). IL-4 in spleen and lymph node cultures was undectable by ELISA. Overall, the data obtained by FACS, qPCR, and antigen recall assay indicate that loss of sympathetic PAC1 receptor signaling results in a shift in the immune response to a less inflammatory state, with lower Th1 and Th17, and higher Th2 activities.
Fig. 5.
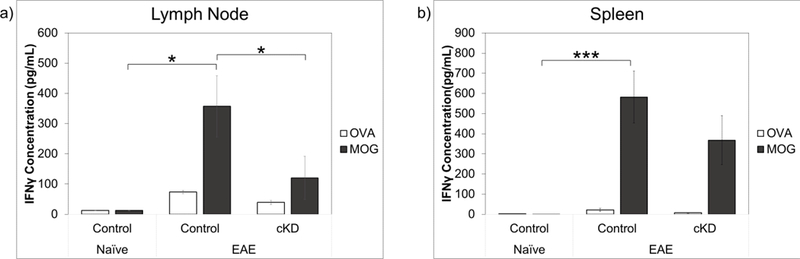
Conditional knockdown of PAC1 in catecholaminergic neurons led to decreased production of IFNγ from spleen and lymph node cells cultured with MOG35–55. Lymphocytes (a) and splenocytes (b) were harvested from naïve and EAE animals and cultured in media containing the control antigen ovalbumin control stimulus or specific antigen MOG35–55 stimulus. Media was collected and analyzed for IFNγ secretion by ELISA. Standard error bars shown. *<0.05. Statistical significance was determined by 1-tailed t-tests. Standard error bars are shown. Naïve floxed=4, EAE floxed=6, EAE cKD=5
PAC1 loss in the catecholiminergic cells led to a defect in Treg production during EAE.
We previously found that PACAP deficient mice exhibited a deficit in thymic Treg proliferation (Tan et al., 2009, 2013). We hypothesized that the effects of PACAP in Th proliferation observed in those studies were in part mediated by PACAP/PAC1 receptor modulation of the SNS. We thus analyzed Treg abundance in the thymus, the site where Tregs are produced de novo. Thymi from PAC1flox/flox DβH CreER mice induced with EAE and were indeed found to have a reduction in Tregs during EAE (Fig. 6).
Fig. 6.
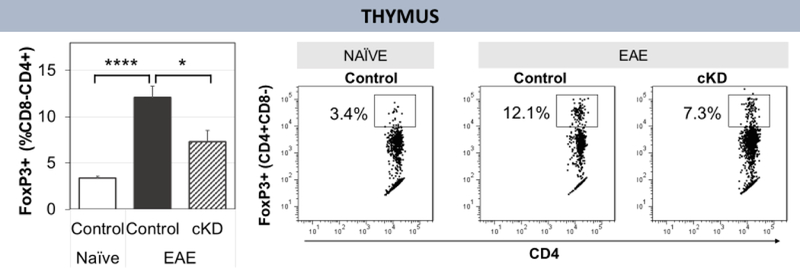
Flow cytometry analysis of Tregs in thymus. On day 11 post-EAE induction, thymi were harvested for analysis by flow cytometry. There are fewer Tregs in the thymus of tamoxifen-treated PAC1 cKD mice during EAE as compared to floxed mice. Naïve floxed=5, EAE floxed=14, EAE cKD=9. Statistical significance determined by two-tailed T-test. *p<0.05 **p<0.01 ***p<0.001 ****p<0.00001. Standard error bars shown
Other genes potentially affected in the inflammatory response.
To address potential downstream targets of PACAP/PAC1 in the control of the inflammatory response, severalcandidate genes were analyzed. IL-6, IL-10, and IL-33. IL-6 plays a key role in inhibiting production of Tregs during EAE (Korn et al., 2008), IL-6 deficient mice are resistant to developing EAE (Okuda et al., 1999; Samoilova et al., 1998), and administering neutralizing antibodies to IL-6 reduces EAE disease severity (Gijbels et al., 1995). We thus hypothesized that an increase in IL-6 may explain the reduction in Tregs. However no significant difference was observed between genotypes (data not shown),Tregs, It was previously demonstrated that Tregs from IL-10 deficient mice are unable to effectively suppress EAE, indicating that IL-10 is important for Treg function (Zhang et al., 2004). We found a significant decrease in IL-10 gene expression in the lymph node—but not spleen—of cKD EAE mice as compared to control mice (data not shown)
We also examined expression of NAD-dependent deacetylase sirtuin-1 (SIRT1), a histone deacetylase, which in EAE, is known to protect against oxidative stress and reduce retinal ganglion cell loss (Khan et al., 2012). Additionally, in EAE, SIRT1 overexpression leads to lower peak disease severity, and reduces immune cell infiltration, demyelination, and apoptosis in the spinal cord (Nimmagadda et al., 2013). SIRT1 was also previously found to deacetylate RORγt, thus promoting differentiation of Th17 T-cells (Lim et al., 2015), and to deacetylate and destabilize FoxP3, thus inhibiting Treg expansion during EAE (Kwon et al., 2012; van Loosdregt et al., 2010). In our study, SIRT1 significantly decreased (p<0.01) with EAE but does not differ between control and cKO mice in the spleen (data not shown).
Others have shown that mice with T-cells that cannot respond to TGF-β do not develop EAE and thus TGF-β is essential in EAE disease initiation (Veldhoen et al., 2006). On the other hand, TGF-β is also known to suppress Th1 and promote Th17 immune response (Lin et al., 2005; McGeachy et al., 2007). We found that levels of TGF-β decreased with EAE in the lymph nodes and spleen but was not significant different between control and cKO animals (data not shown).
DISCUSSION
We sought to determine if the well-known anti-inflammatory actions of PACAP are mediated in part by PAC1 receptor-expressing sympathetic neurons that innervate lymphoid tissue. To accomplish this objective, we used a gene targeting approach to interfere with this pathway by conditionally inducing PAC1 receptor gene deletion in postganglionic sympathetic neurons. This approach resulted in a partial deletion of PAC1 in postganglionic sympathetic neurons. The results reported here support a model in which PACAP/PAC1 receptor signaling in sympathetic neurons serves to promote the ability of the SNS to build an inflammatory response early in the phase of EAE, and then facilitate recovery by shifting the immune response to a less inflammatory, pro-repair state. Clinically, PAC1 receptor cKD mice showed less disease severity in the early inductive phase of EAE. This was associated with decreased Th1 and Th17, and increased Th2 responses in cKD mice. Later, as disease progressed, clinical severity scores of cKD mice surpassed those of control mice. Our analyses suggest that this reversal was due to impaired Treg production in PAC1 cKO mice. Tregs are known to increase in abundance during EAE and promote recovery (Kohm et al., 2003), but they do not prevent the onset of disease (Korn et al., 2007).
A time-dependent action of the SNS to promote the induction of inflammatory disease and then restrict inflammation has to our knowledge not yet been demonstrated in an acute monophasic EAE model such as employed here. However, this type of regulation is known to occur in other autoimmune disease models. For example, a time-dependent SNS enhancement, then inhibition of disease severity and Th polarization was observed in a murine collagen-induced arthritis model (Härle et al., 2005). Moreover, in an acute MOG35–55 C57BL/6 model such as ours, 6-OHDA-induced peripheral sympathetic innervation ablation prior to MOG35–55 immunization resulted in significantly diminished and delayed disease (Ebbinghaus et al., 2012), suggesting that sympathetic signaling facilitates inflammatory responses in the course of disease induction. In agreement, diminished Th1-responses to pathogen challenge were observed in dopamine β-hydroxylase (DβH) deficient mice (Sanders et al., 2003).
We used a tamoxifen-inducible Cre-Lox system to restrict PAC1 receptor loss to catecholaminergic cells. Such cells are located in various areas of the brain, in sympathetic neurons, and in the adrenal medulla. Thus, sympathetic neurons are not the sole cell type in which PAC1 receptors were eliminated on PAC1 cKD mice. Because of this, we cannot rule out that the phenotype we observed is due to loss of PAC1 receptors in these other sites rather than sympathetic neurons. On the other hand, our analyses of Th cells and Tregs were performed on lymphoid tissues that are directly innervated by postganglionic sympathetic neurons. Of course, the possibility exists that the effects on immune cells were mediated indirectly by catecholaminergic cells in the brain or adrenal medulla.
The results obtained in the present study were quite different from that we reported in our previous studies in which EAE was examined in mice globally deficient in PACAP (Tan et al., 2009, 2013). For example, the reduction of disease severity we observed at early time points in cKD mice was not observed in PACAP KO mice. Moreover, the Th balance was skewed toward greater inflammation in PACAP KO mice, whereas in PAC1 receptor cKD mice, the balance was tipped towards reduced inflammation. The differing results in PACAP KO vs. PAC1 receptor cKD mice can potentially be explained by the fact that PACAP acts in many different sites and on many cell types, including directly on inflammatory cells. PACAP is also known to regulate the hypothalamic-pituitary-adrenal axis via induction of corticotropic releasing factor gene expression in the hypothalamus. In addition, PACAP also acts on two other known PACAP receptors, VPAC1 and VPAC2, which are also expressed on many cell types including most types of immune cells. Use of mice in which a specific PACAP receptor (PAC1) is eliminated in a specific cell type (catecholaminergic), represents a step towards dissecting the complex actions of PACAP in autoimmune and other diseases modified by sympathetic function. The delineation of the mechanisms by which PACAP regulates sympathetic function could have therapeutic implications in other diseases impacted by the SNS, including diabetes and other metabolic diseases, obesity, and cardiovascular disorders.
ACKNOWLEDGEMENTS
We would like to thank Dr. Hermann Rohrer (Max Planck Institute for Brain Research, Frankfurt, Germany) for the DβH-CreER mouse line, and Dr. Anton Maximov (Scripps Research Institute, La Jolla, CA, USA) for the Ai9 reporter mouse line.
This project received support from the National Multiple Sclerosis Society (RG 1501–02646), the Cousins Center for Psychoneuroimmunology, UCLA Semel Institute, and the NIH/NCATS UCLA CTSI Grant Number UL1TR000124). The latter award helped fund the generation of the PAC1flox/flox DβH-CreER mice.
Microscopy and cryosectioning was performed using instruments made available through the UCLA Intellectual and Developmental Disabilities Research Center (IDDRC) Core. The IDDRC is supported by a grant from the Eunice Kennedy Shriver National Institute of Child Health (5U54HD087101–03) and is an Organized Research Unit supported by the Jane and Terry Semel Institute for Neuroscience and Human Behavior.
Flow cytometry was performed in the UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research Flow Cytometry Core Facility that is supported by National Institutes of Health awards P30 CA016042 and 5P30 AI028697, and by the JCCC, the UCLA AIDS Institute, the David Geffen School of Medicine at UCLA, the UCLA Chancellor’s Office, and the UCLA Vice Chancellor’s Office of Research.
REFERENCES
- Abad C, Tan Y-V, Lopez R, Nobuta H, Dong H, Phan P, Feng J-M, Campagnoni AT, and Waschek JA (2010). Vasoactive intestinal peptide loss leads to impaired CNS parenchymal T-cell infiltration and resistance to experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. 107, 19555–19560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abad C, Jayaram B, Becquet L, Wang Y, O’Dorisio MS, Waschek JA, and Tan Y-V (2016). VPAC1 receptor (Vipr1)-deficient mice exhibit ameliorated experimental autoimmune encephalomyelitis, with specific deficits in the effector stage. J. Neuroinflammation 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudet MM, Braas KM, and May V (1998). Pituitary adenylate cyclase activating polypeptide (PACAP) expression in sympathetic preganglionic projection neurons to the superior cervical ganglion. J. Neurobiol. 36, 325–336. [PubMed] [Google Scholar]
- Bellinger DL, Millar BA, Perez S, Carter J, Wood C, ThyagaRajan S, Molinaro C, Lubahn C, and Lorton D (2008). Sympathetic modulation of immunity: Relevance to disease. Cell. Immunol. 252, 27–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brifault C, Gras M, Liot D, May V, Vaudry D, and Wurtz O (2014). Delayed Pituitary Adenylate Cyclase–Activating Polypeptide Delivery After Brain Stroke Improves Functional Recovery by Inducing M2 Microglia/Macrophage Polarization. Stroke STROKEAHA.114006864. [DOI] [PubMed] [Google Scholar]
- Chen W-H, and Tzeng S-F (2005). Pituitary adenylate cyclase-activating polypeptide prevents cell death in the spinal cord with traumatic injury. Neurosci. Lett. 384, 117–121. [DOI] [PubMed] [Google Scholar]
- Constantinescu CS, Farooqi N, O’Brien K, and Gran B (2011). Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 164, 1079–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damsker JM, Hansen AM, and Caspi RR (2010). Th1 and Th17 cells. Ann. N. Y. Acad. Sci. 1183, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejda A, Jolivel V, Bourgault S, Seaborn T, Fournier A, Vaudry H, and Vaudry D (2008). Inhibitory effect of PACAP on caspase activity in neuronal apoptosis: a better understanding towards therapeutic applications in neurodegenerative diseases. J. Mol. Neurosci. MN 36, 26–37. [DOI] [PubMed] [Google Scholar]
- Delgado M (2004). VIP/PACAP preferentially attract Th2 effectors through differential regulation of chemokine production by dendritic cells. FASEB J. [DOI] [PubMed] [Google Scholar]
- DiCicco-Bloom E, Deutsch PJ, Maltzman J, Zhang J, Pintar JE, Zheng J, Friedman WF, Zhou X, and Zaremba T (2000). Autocrine Expression and Ontogenetic Functions of the PACAP Ligand/Receptor System during Sympathetic Development. Dev. Biol. 219, 197–213. [DOI] [PubMed] [Google Scholar]
- Ebbinghaus M, Gajda M, Boettger MK, Schaible H-G, and Bräuer R (2012). The anti-inflammatory effects of sympathectomy in murine antigen-induced arthritis are associated with a reduction of Th1 and Th17 responses. Ann. Rheum. Dis. 71, 253–261. [DOI] [PubMed] [Google Scholar]
- Gijbels K, Brocke S, Abrams JS, and Steinman L (1995). Administration of neutralizing antibodies to interleukin-6 (IL-6) reduces experimental autoimmune encephalomyelitis and is associated with elevated levels of IL-6 bioactivity in central nervous system and circulation. Mol. Med. Camb. Mass 1, 795–805. [PMC free article] [PubMed] [Google Scholar]
- Härle P, Möbius D, Carr DJJ, Schölmerich J, and Straub RH (2005). An opposing time-dependent immune-modulating effect of the sympathetic nervous system conferred by altering the cytokine profile in the local lymph nodes and spleen of mice with type II collagen-induced arthritis. Arthritis Rheum. 52, 1305–1313. [DOI] [PubMed] [Google Scholar]
- Jamen F, Persson K, Bertrand G, Rodriguez-Henche N, Puech R, Bockaert J, Ahrén B, and Brabet P (2000). PAC1 receptor–deficient mice display impaired insulinotropic response to glucose and reduced glucose tolerance. J. Clin. Invest. 105, 1307–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney M, and Ganta C (2014). Autonomic Nervous System and Immune System Interactions. Compr. Physiol. 4, 1177–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan RS, Fonseca-Kelly Z, Callinan C, Zuo L, Sachdeva MM, and Shindler KS (2012). SIRT1 activating compounds reduce oxidative stress and prevent cell death in neuronal cells. Front. Cell. Neurosci. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohm AP, Carpentier PA, and Miller SD (2003). Regulation of experimental autoimmune encephalomyelitis (EAE) by CD4+CD25+ regulatory T cells. Novartis Found. Symp. 252, 45–52; discussion 52–54, 106–114. [PubMed] [Google Scholar]
- Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Bäckström BT, Sobel RA, Wucherpfennig KW, Strom TB, et al. (2007). Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat. Med. 13, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, Vollmar P, Stritesky GL, Kaplan MH, Waisman A, et al. (2008). IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. 105, 18460–18465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H-S, Lim HW, Wu J, Schnolzer M, Verdin E, and Ott M (2012). Three Novel Acetylation Sites in the Foxp3 Transcription Factor Regulate the Suppressive Activity of Regulatory T Cells. J. Immunol. 188, 2712–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafaille JJ (1998). The role of helper T cell subsets in autoimmune diseases. Cytokine Growth Factor Rev. 9, 139–151. [DOI] [PubMed] [Google Scholar]
- Lee EH, and Seo SR (2014). Neuroprotective roles of pituitary adenylate cyclase-activating polypeptide in neurodegenerative diseases. BMB Rep. 47, 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HW, Kang SG, Ryu JK, Schilling B, Fei M, Lee IS, Kehasse A, Shirakawa K, Yokoyama M, Schnölzer M, et al. (2015). SIRT1 deacetylates RORγt and enhances Th17 cell generation. J. Exp. Med. 212, 607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JT, Martin SL, Xia L, and Gorham JD (2005). TGF-beta 1 uses distinct mechanisms to inhibit IFN-gamma expression in CD4+ T cells at priming and at recall: differential involvement of Stat4 and T-bet. J. Immunol. Baltim. Md 1950 174, 5950–5958. [DOI] [PubMed] [Google Scholar]
- van Loosdregt J, Vercoulen Y, Guichelaar T, Gent YYJ, Beekman JM, van Beekum O, Brenkman AB, Hijnen D-J, Mutis T, Kalkhoven E, et al. (2010). Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood 115, 965–974. [DOI] [PubMed] [Google Scholar]
- Lu N, Zhou R, and DiCicco-Bloom E (1998). Opposing mitogenic regulation by PACAP in sympathetic and cerebral cortical precursors correlates with differential expression of PACAP receptor (PAC1-R) isoforms. J. Neurosci. Res. 53, 651–662. [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, and Cua DJ (2007). TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain TH-17 cell–mediated pathology. Nat. Immunol. 8, 1390–1397. [DOI] [PubMed] [Google Scholar]
- Miyata A, Arimura A, Dahl RR, Minamino N, Uehara A, Jiang L, Culler MD, and Coy DH (1989). Isolation of a novel 38 residue-hypothalamic polypeptide which stimulates adenylate cyclase in pituitary cells. Biochem. Biophys. Res. Commun. 164, 567–574. [DOI] [PubMed] [Google Scholar]
- Nance DM, and Sanders VM (2007). Autonomic innervation and regulation of the immune system (1987–2007). Brain. Behav. Immun. 21, 736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmagadda VK, Bever CT, Vattikunta NR, Talat S, Ahmad V, Nagalla NK, Trisler D, Judge SIV, Royal W, Chandrasekaran K, et al. (2013). Overexpression of SIRT1 Protein in Neurons Protects against Experimental Autoimmune Encephalomyelitis through Activation of Multiple SIRT1 Targets. J. Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda Y, Sakoda S, Fujimura H, Saeki Y, Kishimoto T, and Yanagihara T (1999). IL-6 plays a crucial role in the induction phase of myelin oligodendrocyte glucoprotein 35–55 induced experimental autoimmune encephalomyelitis. J. Neuroimmunol. 101, 188–196. [DOI] [PubMed] [Google Scholar]
- Olivas A, Gardner RT, Wang L, Ripplinger CM, Woodward WR, and Habecker BA (2016). Myocardial Infarction Causes Transient Cholinergic Transdifferentiation of Cardiac Sympathetic Nerves via gp130. J. Neurosci. Off. J. Soc. Neurosci. 36, 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto C (2004). Pulmonary Hypertension and Right Heart Failure in Pituitary Adenylate Cyclase-Activating Polypeptide Type I Receptor-Deficient Mice. Circulation 110, 3245–3251. [DOI] [PubMed] [Google Scholar]
- Pettersson LME, Heine T, Verge VMK, Sundler F, and Danielsen N (2004). PACAP mRNA is expressed in rat spinal cord neurons. J. Comp. Neurol. 471, 85–96. [DOI] [PubMed] [Google Scholar]
- Samoilova EB, Horton JL, Hilliard B, Liu TS, and Chen Y (1998). IL-6-deficient mice are resistant to experimental autoimmune encephalomyelitis: roles of IL-6 in the activation and differentiation of autoreactive T cells. J. Immunol. Baltim. Md 1950 161, 6480–6486. [PubMed] [Google Scholar]
- Sanders VM, Kasprowicz DJ, Swanson-Mungerson MA, Podojil JR, and Kohm AP (2003). Adaptive immunity in mice lacking the beta(2)-adrenergic receptor. Brain. Behav. Immun. 17, 55–67. [DOI] [PubMed] [Google Scholar]
- Seaborn T, Masmoudi-Kouli O, Fournier A, Vaudry H, and Vaudry D (2011). Protective effects of pituitary adenylate cyclase-activating polypeptide (PACAP) against apoptosis. Curr. Pharm. Des. 17, 204–214. [DOI] [PubMed] [Google Scholar]
- Shadiack AM, Sun Y, and Zigmond RE (2001). Nerve growth factor antiserum induces axotomy-like changes in neuropeptide expression in intact sympathetic and sensory neurons. J. Neurosci. Off. J. Soc. Neurosci. 21, 363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons SB, Pierson ER, Lee SY, and Goverman JM (2013). Modeling the heterogeneity of multiple sclerosis in animals. Trends Immunol. 34, 410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg Z (2016). Promoting sympathovagal balance in multiple sclerosis; pharmacological, non-pharmacological, and surgical strategies. Autoimmun. Rev. 15, 113–123. [DOI] [PubMed] [Google Scholar]
- Stubbusch J, Majdazari A, Schmidt M, Schütz G, Deller T, and Rohrer H (2011). Generation of the tamoxifen-inducible DBH-Cre transgenic mouse line DBH-CT. Genes. N. Y. N 2000 49, 935–941. [DOI] [PubMed] [Google Scholar]
- Tan Y-V, Abad C, Lopez R, Dong H, Liu S, Lee A, Gomariz RP, Leceta J, and Waschek JA (2009). Pituitary adenylyl cyclase-activating polypeptide is an intrinsic regulator of Treg abundance and protects against experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U. S. A. 106, 2012–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y-V, Abad C, Wang Y, Lopez R, and Waschek JA (2013). Pituitary Adenylate Cyclase Activating Peptide Deficient Mice Exhibit Impaired Thymic and Extrathymic Regulatory T Cell Proliferation during EAE. PLoS ONE 8, e61200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y-V, Abad C, Wang Y, Lopez R, and Waschek JA (2015). VPAC2 (vasoactive intestinal peptide receptor type 2) receptor deficient mice develop exacerbated experimental autoimmune encephalomyelitis with increased Th1/Th17 and reduced Th2/Treg responses. Brain. Behav. Immun. 44, 167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsarovina K, Reiff T, Stubbusch J, Kurek D, Grosveld FG, Parlato R, Schütz G, and Rohrer H (2010). The Gata3 transcription factor is required for the survival of embryonic and adult sympathetic neurons. J. Neurosci. Off. J. Soc. Neurosci. 30, 10833–10843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, Flavell RA, and Stockinger B (2006). Signals mediated by transforming growth factor-β initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat. Immunol. 7, 1151–1156. [DOI] [PubMed] [Google Scholar]
- Waschek J (2013). VIP and PACAP: neuropeptide modulators of CNS inflammation, injury, and repair: VIP and PACAP in neural injury. Br. J. Pharmacol. 169, 512–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Koldzic DN, Izikson L, Reddy J, Nazareno RF, Sakaguchi S, Kuchroo VK, and Weiner HL (2004). IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int. Immunol. 16, 249–256. [DOI] [PubMed] [Google Scholar]