

Abstract
Xylans are the second most abundant form of hemicelluloses and are the second most abundant polysaccharide in nature after cellulose. To degrade xylan, microbes produce mainly xylanase enzyme. Wide range of microorganisms like fungi, bacteria, yeast, marine algae etc. are capable of producing xylanase. Main source of xylanase is fungi but industrial production of bacterial xylanase is low cost, easy downstream process and high production rate. To understand primary, secondary and tertiary structure of xylanase, in silico composition of amino acids, basic physiological characteristics; viz., pI, molecular weight, instability index, GRAVY, molar extinction coefficient, secondary structure, presence of functional domain and motifs, phylogenetic tree, salt bridge compositions are determined. In silico study of xylanase focused on 36 different bacterial sources are performed by retrieving FASTA and PDB sequences using RCSB PDB. FASTA and PDB files are proceed further in ExPASy-ProtParam, RAMPAGE, QMEAN, MEME, PSIPRED, InterProScan, MOTIF scan, ERRAT, Peptide cutter, ESBRI and MEGA 7. The instability index range (16.90–38.78) clearly indicates that the protein is highly stable. α-helix mean value (27.11%) infers the protein is dominated by α-helix region. The aliphatic index (39.80–90.68) gives information that the protein is highly thermostable, prevalence by alanine amino acid in aliphatic side chain. No transmembrane domain was found in the protein which confirms the enzyme is extracellular in nature. Ancestor chart analysis confirmed that it is a part of carbohydrate metabolic process and more specifically a member of glycoside hydrolase super family.
Keywords: Bacterial xylanase, Phylogenetic relationships, Physical parameters, Structural and functional analysis
1. Introduction
The enzyme xylanase catalyzes the hydrolysis of xylans. Xylans are 1,4-linked β-D xylose heteropolysaccharides, the principal constituent of plant cell present mainly in middle lamella. Xylan is present mainly in the secondary cell wall of softwood and participates in formation of interphase between lignin and other polysaccharides [1]. It is composed of various proportion of monosaccharide units such as d-mannose, d-glucose, d-xylose, d-arabinose, d-galactose, d-glucoronic acid and d-galactorunic acid. Till date, filamentous fungi are major source of xylanase synthesis. Mesophilic fungal strains like Aspergillus, Trichoderma have the ability to produce thermostable xylanase. Xylanase are also reported from thermophilic fungi including Chaetomium thermophile, Humicola lanuginose and Thermomyces lanuginosus [2]. However, bacteria have an advantage over fungi for xylanase production because bacteria produce xylanase at neutral pH or alkaline pH but in case of fungi it works in the acidic range [3]. Marine algae, protozoans, crustaceans, insects, snails derived xylanases have been reported too.
In recent years, the use of xylanase has increased remarkably. Xylanase are used from 1980 in the preparation of animal feed along with glucanase, pectinase, galactosidase and lipase [2]. These enzymes have property to break down arabinoxylans, thus reducing the viscosity of raw grains and make it better to poultry feed by improving easy digestion process [1]. In bread baking procedure, xylanase are introduced to increase bread volumes, greater absorption of water and improved resistance to fermentation [2]. Later it has been used in food and textile industries too. Recently, xylanase and cellulase, together with pectinase account for ∼20% of the world enzyme market. The synergistic action [2] improves the stabilization of fruit pulp during juice making, increased recovery of aroma, pigments and color. In textile industries, degumming of plant fiber such as flax, hemp, jute, ramie prebleaching of kraft pulps [4], [5] with xylanase is essential for minimum use of harsh chemicals. Wide application of xylanase is found in cellulose pulp and paper industries [3]. Thermostable xylanase has a plus point in industrial practices as it helps to increase the substrate and product standardization [6]. Thermotolerant xylanases have distinct catalytic and cellulose binding domains, with a different non-catalytic domain that gives thermostability [7]. Xylanase is able to remove the xylan derived chromophores such as hexonuronic acid during kraft cooking of wood [8], thereby increasing brightness of pulp. Xylanase have been used in traditional fields such as food, paper [9], textile, pharmaceutical uses since long time but more attention is needed to pay in producing other industry friendly materials. A recent example includes hydrolytic products of xylan such as β-D-xylopyrasonyl residues converted in bioethanol or artificial sweetener. Xylitol, a polyalcohol used as sugar free sweetener can prevent tooth decay and ear infections. To magnify commercial production of xylanase recombinant DNA techniques may offer new aspects. Several attempts have been made to express xylanase from bacteria such as Bacillus subtilis, reported by Bernier et al.[10], Acidobacterium capsulatum by Inagaki et al. [11] and Bacillus sp. Jeong et al.[12] into a non-cellulose producing strain of Escherichia coli. As cell wall maceration is a vital step in plant protoplast extraction, xylanase can be potentially used during the process [13] Heterologous xylanase proteins of diverse origins are produced using rhizosecration method, observed by Borisjuk et al. [14].
For a successful integration of xylanases in large scale industrial processes, a detailed understanding of the mechanism of enzyme action is required. In the present study, an in silico approach has been taken to understand the overall physical parameters, primary, secondary and tertiary structure of proteins, functional analysis, domains and motifs, protein model and Phylogenetic analysis of bacterial xylanases.
2. Materials and methods
2.1. Retrieval of sequences from RCSB.org
A total of 36 different xylanase sequences of bacterial origin have been retrieved from RCSB data bank (http://www.rcsb.org). These 36 sequences have been selected based on the overall quality parameters. The accession numbers of the respective proteins from different bacterial sources are provided in Supplementary Table 1.
2.2. Determination of physical parameters of the proteins
Analysis of physical parameters of bacterial xylanase is done by studying physiochemical parameters viz. number of amino acids and composition, molecular weight, theoretical pI, extinction coefficient, instability index, aliphatic index, grand average of hydrophobicity (GRAVY) by using ExPASy–ProtParam tool (http://web.expasy.org/protparam) [15], [16].
2.3. Primary structure analysis
For primary structure analysis; viz. the amino acids present in basic polypeptide chain of bacterial xylanases, ExPASy–ProtParam tool has been used. MEME Suite has been used to determine if any signature sequence of bacterial xylanase can be detected for in silico identification or not (meme-suite.org).
2.4. Secondary structure analysis
Secondary structure analysis of retrieved bacterial xylanase includes number of α-helices, β-turn, extended strand, β-sheet, coils which are performed by ExPASy SIB Bioinformatics SOPMA tool (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page = npsa_sopma.html). To utilize SOPMA, sequences have been submitted in FASTA format to run whereas Ramachandran plot is constructed using RAMPAGE tool (mordred.bioc.cam.ac.uk/∼rapper/rampage.php). Energetically allowed regions for backbone dihedral angles ψ against φ of amino acid residues of proteins are visualized through generated Ramachandran plot.
2.5. Tertiary structure analysis
SWISS-MODEL QMEAN tool is used to build and validate the 3D models of all the retrieved bacterial xylanase sequences (https://swissmodel.expasy.org/qmean/). Three different approaches under the subset of QMEAN models have been taken. The different geometric aspects of protein structure have been observed through QMEAN. QMEANDisCo computes by adding distance constraint score to QMEAN to improve local quality predictions. Statistical potentials targeted at local quality estimation of membrane protein in their naturally occurring oligomeric state have been performed by QMEANBrane. By analyzing the statistics of non-bonded interactions between different atom types, plotting the value of the error function versus position of a 9-residue sliding window has been done through ERRAT server. (services.mbi.ucla.edu/ERRAT). The result helps to verify protein crystallographic structure.
ESBRI is a web tool (http://bioinformatica.isa.cnr.it/ESBRI/introduction.html) which analyses the salt bridges in a protein structure, starting from the atomic coordinates.
2.6. Functional analysis
To find all the possible motifs that occur overall in bacterial xylanases, SIB myhits Motif scan has been used (myhits.isb-sib.ch/cgi-bin/motif_scan). Functional analysis of protein is further performed by InterProScan server (www.ebi.ac.uk/interpro/search/sequence-search) and the proteins are classified according to the protein families. GO (gene ontology) term predicts the ancestral details of the biological process and molecular function of protein. TMHMM tool is used to understand membrane protein topology, more specifically if the protein is membrane spanning or extracellular in nature (www.cbs.dtu.dk/services/TMHMM). Peptide Cutter tool reveals potential cleavage site of a protein by protease or chemicals (web.expasy.org/peptide_cutter/) [17].
The STRING database provides protein-protein association. The score indicate the functional network among the set of proteins of a given organism (https://string-db.org/)
2.7. Construction of phylogenetic tree and evolutionary analysis
Phylogenetic tree of all the total 36 bacterial xylanases protein sequences has been constructed through maximum likelihood method based on JTT matrix based model [18] Evolutionary distances among the proteins are also understood using MEGA7 software. Different groups or branches of bacterial xylanases are observed well to understand the evolutionary lineages.cDNA (reverse translated) is constructed by converting protein sequences into its complementary DNA sequences using Bioinformatics Reverse Translation Tool (http://www.gregthatcher.com/Bioinformatics/ReverseTranslate.aspx). Phylogenetic tree is also constructed based on cDNA (reverse translated) of xylanase protein by MEGA7 software.
3. Results and discussion
3.1. Retrieval of sequences from RCSB.org
A total of 36 different bacterial xylanase sequences have been retrieved. These 36 sequences have been selected based on the overall quality parameters in both FASTA and PDB formats.
3.2. Determination of physical parameters of the proteins
Physical parameters of proteins, viz, pI value, molecular weight, extinction coefficient, instability index, aliphatic index or average hydrophobicity are the preliminary characteristics to determine the uniqueness of any protein or enzyme molecule [19]. Isoelectric point (pI) is the pH at which the surface of protein is covered with charge but net charge of protein is zero. Theoretically pI value is more than 7 indicate the alkaline nature of the protein and below 7 depicts the acidic nature. In present study, theoretical pI of all the xylanases range 4.56–9.21 indicating diverse acidic or alkaline nature. Average molecular weight of the protein is 50870.73 Da. According to the previous study, instability index value below 40 confirms the protein structure to be stable. Above 40 values tends to instability in the protein structure. In our study, the range remains 16.2–38.78, indicating that all the bacterial origin xylanases are highly stable. Average extinction coefficient is 11238.61 refers the quantity of light that may be absorbed by protein in 280 nm. The aliphatic index is the relative volume of the protein occupied by the aliphatic amino acids like alanine in the side chain [20]. A high aliphatic index range 42.08–90.68 gives information that the protein has a wide range of temperature stability. Higher the aliphatic index greater the thermostability of the protein. Hydropathicity is the relative hydrophobicity or hydrophilicity of amino acids that are present in the protein sequence. Negative GRAVY value (Grand average of hydrophobicity) indicates that the protein is non polar and its better interaction with water that is hydrophilic in nature.
3.3. Primary structure analysis
Primary structure analysis reveals a set of characteristics of all the xylanase proteins. The amino acid composition of different proteins has been studied. The five predominant amino acids are found (Fig. 1A) to be threonine (9.5%), followed by Glycine (8.8%), alanine (8.2%), serine (7.9%) and aspartic acid (6.54%). Hydrophilic amino acids are those seeking contact with aqueous phase. Since the hydrophilic amino acids, viz. serine, threonine occur in high amount, depicts the protein might be extracellular in nature. Alanine amino acid being hydrophobic in nature has low propensity to be in contact with water and normally seldom buried inside the protein core. However, glycine does not have a side chain and often found at the surface of the protein, providing high flexibility to the polypeptide chain. Being charged and polar, Aspartate prefers to be on the surface of proteins and also involved in salt bridge when buried within the protein. Lysine and histidine are positively charged amino acids and prefers to be in the side chain of proteins and develop salt bridges during higher order tertiary structure formation.
Fig. 1.

(A) Column graph representing the primary structure contributing dominant amino acids present in all 36 different xylanse enzymes; while (B) Column graph showing contributing salt bridge building amino acids in all available 36 different bacterial xylanases.
MEME suite is the tool for motif elicitation for a particular group of proteins. Presence of any motif for a particular protein group has potential application as signature sequence may help in initial in silico identification of any protein. The obtained MEME result for xylanase group of enzyme is not found to be satisfactory and overall disparity of the resulted motifs cannot clearly indicate the uniqueness of the sequence.
3.4. Secondary structure analysis
α-helix mean value of 27.08% clearly indicates that the protein secondary structure of xylanase is dominated by α-helix region, followed by beta turn region of 26.66% and finally less amount of extended sheet region of 11.86% (Fig. 2A). Thus the result shows that the protein is highly stable in structure as it predominantly composed of α-helices. Ramachandran angles are the two torsion angles in the polypeptide chain, which describes the rotations of the polypeptide backbone around the bonds between N-Cα (called Phi, φ) and Cα-C (called Psi, ψ) [21]. The torsion angles ϕ and ψ provides the variable flexibility required for a polypeptide backbone to take up certain fold and steric effects from their side-chains. The Ramachandran plot generated by RAMPAGE tool provides the percentage of amino acid residues in various regions of the ϕ–ψ plot of a polypeptide chain; like favored region, allowed region and outlier region. The more number of residues in favored region indicates more stability of the protein. The plot shows (Fig. 2B) average 413.52% of the residues are falling in favored region whereas a less amount (11.02%) of residues belongs to glycine allowed region.
Fig. 2.

(A) Column graph predicting dominant secondary structures present in bacterial xylanases and (B) Ramachandran plot of xylanase (PDB ID 3GTN) generated from RAMPAGE.
3.5. Tertiary structure analysis
The spatial arrangement of secondary structures would results in development of tertiary structures of protein. SWISS-MODEL workspace provides experimental three-dimensional structures which guides in building protein homology models at different levels of complexity [22]. Most of the proteins in this study adopt well defined 3D structures, indicating soluble nature of protein. Some disordered regions inform that the region may possess biological function or could be involved in signaling and regulation processes, studied by others [15], [23]. QMEAN result reveals geometric aspects of the protein structure; i.e. the global arrangement of variable residues of xylanases. QMEAN PDB shows 3D structure of the protein which are represented in (Fig. 3A–C) and depicts the protein is properly folded into a compact three-dimensional field [24]. ERRAT program verify protein 3D structured determined through crystallography. It is also useful for studying protein structures from the numbers of non-bounded residues within a cut-off distance of 3.5 Å between different pairs of atom .The overall quality factor of ERRAT value is the highest (97%) and is within the accepted range, the value that resides below 95% are considered as rejected (Fig. 4). Salt bridging denotes combination of both non-bonded and hydrogen-bonded paired electrostatic interactions between acidic carboxyl groups and basic amino groups on different parts of a protein [25], [26]. In our study, we get five different pairs of positively charged amino acids: arg/asp, lys/asp, His/asp, arg/glu and lys/glu which form salt bridges (Fig. 1B). From software based experimental data almost all the force fields overestimate the strengths of the salt bridges. Average number of arg/asp is 16.36, lys/asp present in 9.6 and his/asp with 7.7 numbers. Average mean distance between lys and asp is 3.18, in case of arg and asp it is 3.12 and 3.08 in his-asp. Xylanase, being thermophilic have a higher propensity to form of salt bridges than of mesophilic one. Salt bridges between side chains of Arg(+) and Glu(-) are most favorable for formation of α helix, indicating the impact of salt bridges on the folding kinetics of a protein. [27]
Fig. 3.

(A) QMEAN PDB structure result of bacterial xylanase protein (PDB ID 3GTN) showing Z- score value, (B) QMEAN PDB 3D model of bacterial xylanase protein (PDB ID 3GTN) structure and (C) QMEAN DisCo chain results of PDB ID 3GTN; xylanase from Bacillus subtilis.
Fig. 4.

ERRAT value result showing satisfactory stability value in xylanase (PDB ID 3GTN) of bacterial origin.
3.6. Functional analysis
Most Xylanases belong to two structurally different glycoside hydrolases family. Sequence-based glycoside hydrolase classification has placed xylanase into two families GH10 and GH11 [28]. GO tool (gene ontology) compiles genes from different species and provides description of the biological process, molecular function and cellular component of gene products. The xylanase originate from carbohydrate metabolic pathway then diverged to form primary metabolic process and organic substance metabolic route. The two branches meet up at metabolic process ultimately going to biological process to produce xylanase enzyme. But in Bacillus subtilis (Supplementary Fig. 1) the pathway originated from sphingolipid metabolic process then diverged to membrane lipid metabolic process and organonitrogen compound metabolic pathway which is basically a biological process having multiple biological pathway in it. Carbohydrate metabolic pathway basically includes hydrolase activity more specifically a member of glycoside hydrolase super family acting on glycosyl group. Catalytic activity formed by metabolic processes.
Motif Scan data provides the functional motifs occurring in each xylanase sequence. From the reference xylanase protein produced by organism (PDB ID 3GTN) a total 23 unique motifs are found consisting two highly significant motifs and seven moderately significant motifs (Fig. 6).
Fig. 6.

Motif scan results showing the functional motifs present in xylanase enzyme (PDB ID 3GTN).
TMHMM tool indicates there is no transmembrane domain present in the protein, again confirming the extracellular production nature of bacterial xylanases.
To know proper metabolism, enzymatic digestion and simplification of high order protein structure, it has been hydrolyzed in silico with digestive enzymes like pepsin, thermolysin, chymotrypsin, proteinase K to yield into peptides and free amino acids. Peptide Cutter results indicates that average cutting site of proteinase K has high proficiency of average 218.8 cutting sites, followed by Pepsin (pH > 2) 115.8 sites. Thermolysin cut 110.2 sites and chymotrypsin at low specificity has cutting site of average of 94.1
STRING visualizes weighted and integrated and a confidence score of protein’s functional associations in a network of genome-wide connectivity [29]. The STRING server has detected eleven interacting proteins viz., yesX, yesW, gld, xynB, cah, gmuG, ganB, xynC, xynD, xynA, bglS which are involved in different hydrolyzing pathways (Fig. 5). Among them XynC protein cleaves beta-1,4 xylosidic bond of the xylose residue substituted with alpha-1,2 likned 4-O-methyl-D-glucuronate.
Fig. 5.
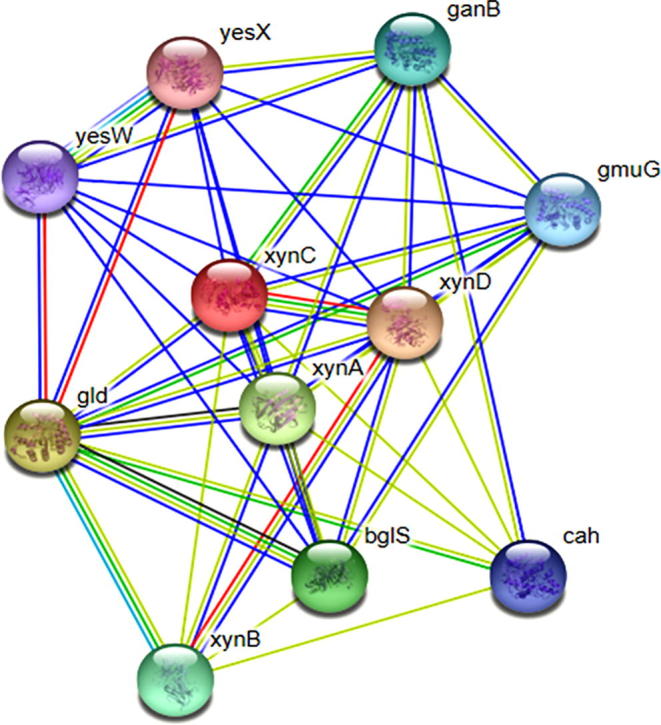
Protein-protein interaction network of visualized by STRING (PDB ID 3GTN).
3.7. Construction of phylogenetic tree
Phylogenetic tree has been constructed with MEGA 7, using maximum likelihood method based on JTT matrix based model.
Tree shows (Fig. 7A) overall similar bacterial xylanases are distributed into two main nodes and one outgroup. Dominant node consist of nineteen nodes and are marked as blue lines where as the second group consists of sixteen nodes and are represented in green line This can be broken down into branches. The branches are further diverged into small branches. The horizontal branches represent evolutionary lineages changing over time. The longer the branch in the horizontal dimension, the larger is the amount of change. The vertical lines which are connected with horizontal lines speak to how long they are irrelevant. The 36 bacterial strains diverged into two main daughter lineages and an out group named Vibrio sp., has maximal branch length 89.3, which arises from the ancestral lineage. From daughter branches many small branches have developed. The tip portion of the branch depicts the actual bacterial strain. Each lineage has a history that is unique of its alone and parts that are shared with other lineages. Length of branch represents the amount genetic change among the strains. The sequence similarities between different bacteria are a clear indication about the interconnected evolutionary lineages among them. The evolutionary tree implies that the xylanase of different bacterial origin appear to be related to each other which eventually form different groups. Thus the bacterial strains make up a family that diverged from a common evolutionary ancestor [6].
Fig. 7.

(A) Phylogenetic tree prepared with 36 different xylanases of bacterial origin by maximum likelihood method using MEGA 7 and (B) Phylogenetic tree cDNA retrieved from 36 different xylanase proteins.
In Fig. 7B phylogenetic tree is reconstructed to find out if there was any correlation among their protein sequences compared with respective cDNA. Tree shows three clusters of the 36 xylanase proteins each group consisting twenty, five and eleven proteins respectively. Evolutionary forces may affect the length of protein which ultimately alters the functional diversity of a particular protein of any organism.
4. Conclusion
From the present study of in silico bacterial xylanase that bacteria can be used as a potent alternative source of xylanase production other than using fungal source only. All the in silico tools are pre-requisite in predicting structural and functional characteristics of any proteins thereby building up overall clarity of desired enzyme sequences. Considering the high industrial demand of xylanase enzyme, it is necessary in the future to produce more enzymes from novel alternative sources. Our present in silico investigation clearly indicates the bacterial origin xylanases are highly stable, hydrophilic and extracellular in nature with molecular weight ranging between 0.438 kDa and 101.83342 kDa. Also microbial production is less labor oriented, cheaper cost and easy downstream process. From previous knowledge, it has been already shown that the bacteria can be grown from renewable sources or substrates too; therefore, to fulfill the demand of overall industrial xylanase needs, bacterial origin xylanase can be a potential novel alternative in future. This in silico study is significant in terms of further isolation and characterization of bacterial xylanase enzyme in wet lab to establish more helpful data, further crystal structure assessment using X-ray Crystallography may be suggested.
Conflict of interest
The authors declare that they have no conflict of interest in the publication.
Acknowledgments
Acknowledgement
Authors are thankful to UGC-Center of Advanced Study, Department of Botany, The University of Burdwan for pursuing research activities. The first author is also thankful to Department of Science and Technology, New Delhi for PURSE PHASE II project for the financial assistance as Research Fellow No RTI/PURSE Phase 2.
Footnotes
Peer review under responsibility of National Research Center, Egypt.
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.jgeb.2018.05.003.
Appendix A. Supplementary material
Supplementary Fig. 1.

PSIPRED analysis results representing the signature domains and repeats present in xylanase enzymes.
References
- 1.Kamble R.D., Jadhav A.R. Int J Microbiol. 2012;2012:128. doi: 10.1155/2012/683193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Polizeli M.L.T.M., Rizzatti A.C.S., Monti R. Appl Microbiol Biotechnol. 2005;67:577–591. doi: 10.1007/s00253-005-1904-7. [DOI] [PubMed] [Google Scholar]
- 3.Chakdar H., Kumar M., Pandiyan K. 3 Biotech. 2016;6:150. doi: 10.1007/s13205-016-0457-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Viikari L., Kantelinen A., Sundquist J. FEMS Microbiol Rev. 1994;13:335–350. [Google Scholar]
- 5.Beg Q.K., Kapoor M., Mahajan L., Hoondal G.S. Appl Microbiol Biotechnol. 2001;56:326–338. doi: 10.1007/s002530100704. [DOI] [PubMed] [Google Scholar]
- 6.Kumar V., Marín-Navarro J., Shukla P. World J Microbiol Biotechnol. 2016;32:34. doi: 10.1007/s11274-015-2005-0. [DOI] [PubMed] [Google Scholar]
- 7.Kulkarni N., Shendye A., Rao M. FEMS Microbiol Rev. 1999;23:411–456. doi: 10.1111/j.1574-6976.1999.tb00407.x. [DOI] [PubMed] [Google Scholar]
- 8.Yang V.W., Zhuang Z., Elegir G. J Ind Microbiol. 1995;15:434–441. [Google Scholar]
- 9.Padre R.A. Biotechnol Genet Eng Rev. 1996;13:101–131. doi: 10.1080/02648725.1996.10647925. [DOI] [PubMed] [Google Scholar]
- 10.Bernier R., Jr, Driguez H., Desrochers M. Gene. 1983;26:59–65. doi: 10.1016/0378-1119(83)90036-7. [DOI] [PubMed] [Google Scholar]
- 11.Inagaki K., Nakahira K., Mukai K. Biosci Biotechnol Biochem. 1998;62:1061–1067. doi: 10.1271/bbb.62.1061. [DOI] [PubMed] [Google Scholar]
- 12.Jeong K.J., Park I.Y., Kim M.S. Appl Microbiol Biotechnol. 1998;50:113–118. doi: 10.1007/s002530051264. [DOI] [PubMed] [Google Scholar]
- 13.Wong K.K., Tan L.U., Saddler J.N. Microbiol Rev. 1988;52:305–317. doi: 10.1128/mr.52.3.305-317.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borisjuk N.V., Borisjuk L.G., Logendra S. Nat Biotechnol. 1999;17:466–469. doi: 10.1038/8643. [DOI] [PubMed] [Google Scholar]
- 15.Dunker A.K., Obradovic Z. Nat Biotechnol. 2001;19:805–806. doi: 10.1038/nbt0901-805. [DOI] [PubMed] [Google Scholar]
- 16.Pramanik K., Ghosh P.K., Ray S. J Genet Eng Biotechnol. 2017;15:527–537. doi: 10.1016/j.jgeb.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Appaiah P., Vasu P. J Proteomics Bioinform. 2016;9:287–297. [Google Scholar]
- 18.Jones D.T., Taylor W.R., Thornton J.M. Bioinformatics. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- 19.Gasteiger E., Hoogland C., Gattiker A. In: The proteomics protocols handbook. Walker, editor. Humana Press; 2005. pp. 571–607. [Google Scholar]
- 20.Ikai A. Biochem J. 1980;88:1895–1898. [PubMed] [Google Scholar]
- 21.Ramachandran G.N., Ramakrishnan C., Sasisekharan V. J Mol Biol. 1963;7:95–99. doi: 10.1016/s0022-2836(63)80023-6. [DOI] [PubMed] [Google Scholar]
- 22.Arnold K., Bardoli L., Kopp J. Bioinformatics. 2006;15:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 23.Iakoucheva L.M., Brown C.J., Lawson J.D. J Mol Biol. 2002;323:573–584. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 24.Mahgoub E.O., Ahmed B. Open J Genet. 2013;3:183–194. [Google Scholar]
- 25.Musafia B., Buchner V., Arad D. J Mol Biol. 1995;254:761–770. doi: 10.1006/jmbi.1995.0653. [DOI] [PubMed] [Google Scholar]
- 26.Costantini S., Colonna G., Facchiano A.M. Bioinformation. 2008;3:137–138. doi: 10.6026/97320630003137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Panja A.S., Bandopadhyay B., Maiti S. PLoS ONE. 2015;10(7):e0131495. doi: 10.1371/journal.pone.0131495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collins T., Gerday C., Feller G. FEMS Microbiol Rev. 2005;29:3–23. doi: 10.1016/j.femsre.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Szklarczyk D., Morris J.H., Cook H. Nucl Acids Res. 2016;45:D362–D368. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.