

Abstract
Antifreeze proteins (AFPs) are known to polypeptide components formed by certain plants, animals, fungi and bacteria which support to survive in sub-zero temperature. Current study highlighted the seven different antifreeze proteins of fish Ocean pout (Zoarces americanus), in which protein (amino acids sequence) were collected from National Centre for Biotechnology Information and finely characterized using several in silico tools. Such biocomputational techniques applied to figure out the physicochemical, functional and conformational characteristics of targeted AFPs. Multiple physicochemical properties such as Isoelectric Point, Extinction Coefficient and Instability Index, Aliphatic Index, Grand Average Hydropathy were calculated and analysed by ExPASy-ProtParam prediction web server. EMBOSS: pepwheel online tool was used to represent the protein sequences in a helical form. The primary structure analysis shows that most of the AFPs are hydrophobic in nature due to the high content of non-polar residues. The secondary structure of these proteins was calculated using SOPMA tool. SOSUI server and CYS_REC program also run for ideal prediction of transmembrane helices and disulfide bridges of experimental proteins respectively. The modelling of 3D structures of seven desired AFPs were executed by the homology modelling programmes; SWISS MODEL and ProSA web server. UCSF Chimera, Antheprot 3D, PyMOL and RAMPAGE were used to visualize and analysis of the structural variation of the predicted protein model. MEGA7.0.9 software used to know the phylogenetic relationship among these AFPs. These models offered excellent and reliable baseline information for functional characterization of the experimentally derived protein domain composition by using the advanced tools and techniques of Computational Biology.
Keywords: In-sillico, Antifreeze proteins, Ocean pout, Computational biology
1. Introduction
Antifreeze proteins (AFPs) are a special class of polypeptides and distinguished by their capacity to bind ice, prevent growth and recrystallization of newly from ice nucleus [1]. These proteins are found in certain animals, plants, fungi and bacteria species [2], [3], [4], which help these organisms to live in sub-zero environment of Polar Regions without freezing. The AFP was first discovered by DeVries in 1969 in the blood of fishes living in the frozen sea [5]. Depending on the biochemical features, AFPs in fishes are of five different types, viz. Antifreeze glycoprotein (AFGP), Antifreeze protein type I, type II, type III and type IV [6]; additionally there is subtype of AFP type I, known as type I hyperactive AFP. Antifreeze proteins have wide commercial applications including biotic components preservation [7], transgenic production [8], cryosurgery [9], ice cream production [10]. AFPs inhibit the freezing of living organisms by lowering the freezing temperature where the melting temperature remains constant and also through an adsorption-inhibition mechanism [11]. Last few years numerous Computational tools and online servers are frequently used to analysed and characterised protein sequences [12]. With the help of in silico tools, the physicochemical characteristics such as Extinction Coefficient (EC), Aliphatic Index (AI), Instability Index (II), Grand Average Hydropathy (GRAVY), Isoelectric Point (pI), secondary structures, homology modelling outputs and 3D structures of these AFPs can be easily characterised now a days. In this study, the in silico analysis of the physicochemical, functional and structural properties of seven numbers of type III Antifreeze protein has been workout which is found in the blood of Ocean pout (Zoarces americanus, Bloch & Schneider, 1801) also focused [13]. Ocean pout is most commonly found in the Northwest Atlantic Ocean, off the coast of New England and Eastern Canada. As Ocean pout is a groundfish and the average water temperature in these regions is −0.5 °C to 1.7 °C [14], AFP type III is too much importance in their survival through preventing hypothermic damage of body tissues in close to freezing temperature.
2. Materials and methods
2.1. Retrieval of antifreeze protein sequences
The sequences of seven numbers of Type III Antifreeze proteins were retrieved from the National Center for Biotechnology Information (NCBI) in random mode [15]. All these antifreeze protein sequences were stored in FASTA format and used for further dry lab analysis [16]. Table 1 shows the Protein Data Bank (PDB) IDs, sequence descriptions, sequence lengths and amino acid sequences of all these experimental proteins [17].
Table 1.
Antifreeze protein sequences retrieved from National Center for Biotechnology Information (NCBI).
| Sl No. | PDB ID of proteins | Sequence description | Length of proteins | Sequences of amino acids |
|---|---|---|---|---|
| 1 | 2MSJ | Chain A, Type III Antifreeze Protein Isoform HPLC12 N46s | 66 aa | ANQASVVAN QLIPINTALTL VMMRSEVVTPV GIPAEDIPRLVSMQVSRAVPLGTTLMPDMVKGYAA |
| 2 | 1KDF | Chain A, North-Atlantic Ocean Pout Antifreeze Protein Type III Isoform HPLC12 Mutant, NMR, Minimized Average Structure | 70 aa | MNQASVVANQLIPINTALTLVMMRSEVVTPVGIPAEDIPRLVSMQVNRAVPLGTTLMPDMVKGYAAKDEL |
| 3 | 1OPS | Chain A, Ice-Binding Surface On A Type III Antifreeze Protein From Ocean Pout | 64 aa | SQSVVATQLIPMNTALTPAMMEGKVTNPIGIPFAEMSQLVGKQVNTPVAKGQTLMPNMVKTYAA |
| 4 | 1AME | Chain A, Crystal Structure Of Type III Antifreeze Protein At 4 C | 67 aa | AANQASVVANQLIPINTALTLVMMRSEVVTPVGIPAEDIPRLVSMQVNRAVPLGTTLMPDMVKGYAA |
| 5 | 3QF6 | Chain A, Neutron Structure Of Type-III Antifreeze Protein | 66 aa | NQLIPINTALTLVMMRSEVVTPVGIPADDIPRLVSM QVNRAVPLGTTLMPDMVKGYPPA |
| 6 | 1GZI | Chain A, Crystal Structure Of Type III Antifreeze Protein From Ocean Pout | 65 aa | NQASVVANQLIPINTALTLVMMRSEVVTPVGIPAEDIPRLVSMQVNRAVPLGTTLMPDMVKGYAA |
| 7 | 7MSI | Chain A, Type III Antifreeze Protein Isoform HPLC12 | 66 aa | MAQASVVANQLIPINTYLTLVMMRSEVVTPVGIPAEDIPRLVSMQVNRAVPLGTTLMPDMVKG YAA |
2.2. Physicochemical properties prediction
The amino acid sequences are much more important to know the physicochemical, structural and functional properties of a protein [24]. The amino acid compositions of these proteins were computed by ExPASy-ProtParam prediction webserver (https://web.expasy.org/protparam/). The amino acid sequences of these proteins were graphically represented in wheel structure with the help of EMBOSS: pepwheel tool [18]. The physico-chemical properties such as Extinction Coefficient (EC) [19], Aliphatic Index (AI) [20], Instability Index (II) [21], Grand Average Hydropathy (GRAVY) [22], Isoelectric Point (pI) [23], molecular weight, total atoms, no. of positively charged and no. of negatively charged residues were calculated using ExPASy-ProtParam prediction server [24].
2.3. Structural and functional analysis
The secondary structures such as alpha helix, extended strand, beta turn and the random coil of antifreeze proteins were obtained by using SOPMA (self-optimized prediction method with alignment) tool [25], [26]. SOSUI server used to determine a part of secondary structure of these proteins whether these are soluble or transmembrane in nature (Sahay and Shakya 2010). The disulfide bridges in a protein can be calculated by CYS_REC tool [27].
2.4. Homology modelling
The three dimensional structure of antifreeze proteins also predicted by using homology modelling programmes: SWISS MODEL [28], [29] and ProSA web server [30]. These three dimensional structures were evaluated using UCSF Chimera ver. 1.12 [31], Antheprot 3D ver. 1.4.2 [32], PyMOL ver. 2.0.7 [33] and RAMPAGE [29].
2.5. Phylogenetic relationship analysis
The phylogenetic relationship based on amino acid sequences among these seven Antifreeze proteins was constructed by Unweighted Pair Group Method with Arithmetic Mean (UPGMA) method with the help of Molecular Evolutionary Genetics Analysis (MEGA) version 7.0.9 in global alignment.
3. Results and discussion
3.1. Physicochemical characterization of AFPs
A total no. of seven antifreeze protein type III sequences were collected from NCBI and analysed by using web based computational tools and techniques. The primary structure and different parameters were characterised by ExPASy-ProtParam tool prediction server. Table 2 represents the various amino acid compositions of every retrieved antifreeze protein sequences from which it can be concluded that all these proteins are hydrophobic in nature as these proteins contain higher amount of hydrophobic, non-polar amino acid residues than that of polar residues; table 3 also supports this statement. Fig. 1 represents the composition of amino acid sequences of AFP namely 2MSJ in wheel structure.
Table 2.
Amino acid compositions of Antifreeze proteins (in percentage) computed using ExPASy-ProtParam tool.
| Amino Acids | 2MSJ | 1KDF | 1AME | 3QF6 | 1OPS | 1GZI | 7MSI |
|---|---|---|---|---|---|---|---|
| Ala (A) | 12.1% | 10.0% | 13.4% | 9.1% | 10.9% | 10.8% | 10.6% |
| Arg (R) | 4.5% | 4.3% | 4.5% | 4.5% | 0.0% | 4.6% | 4.5% |
| Asn (N) | 4.5% | 5.7% | 6.0% | 4.5% | 6.2% | 6.2% | 4.5% |
| Asp (D) | 3.0% | 4.3% | 3.0% | 4.5% | 0.0% | 3.1% | 3.0% |
| Cys (C) | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| Gln (Q) | 4.5% | 4.3% | 4.5% | 4.5% | 7.8% | 4.6% | 4.5% |
| Glu (E) | 3.0% | 4.3% | 3.0% | 1.5% | 3.1% | 3.1% | 3.0% |
| Gly (G) | 4.5% | 4.3% | 4.5% | 4.5% | 6.2% | 4.6% | 4.5% |
| His (H) | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| Ile (I) | 6.1% | 5.7% | 6.0% | 6.1% | 4.7% | 6.2% | 6.1% |
| Leu (L) | 9.1% | 10.0% | 9.0% | 9.1% | 6.2% | 9.2% | 9.1% |
| Lys (K) | 1.5% | 2.9% | 1.5% | 1.5% | 6.2% | 1.5% | 1.5% |
| Met (M) | 7.6% | 8.6% | 7.5% | 7.6% | 9.4% | 7.7% | 9.1% |
| Phe (F) | 0.0% | 0.0% | 0.0% | 0.0% | 1.6% | 0.0% | 0.0% |
| Pro (P) | 9.1% | 8.6% | 9.0% | 12.1% | 9.4% | 9.2% | 9.1% |
| Ser (S) | 6.1% | 4.3% | 4.5% | 4.5% | 4.7% | 4.6% | 4.5% |
| Thr (T) | 7.6% | 7.1% | 7.5% | 7.6% | 10.9% | 7.7% | 7.6% |
| Trp (W) | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
| Tyr (Y) | 1.5% | 1.4% | 1.5% | 1.5% | 1.6% | 1.5% | 3.0% |
| Val (V) | 15.2% | 14.3% | 14.9% | 15.2% | 10.9% | 15.4% | 15.2% |
Table 3.
Hydrophobic and hydrophilic residues content of eight AFPs.
| Sl No. | PDB Id | Percentage of hydrophobic residues | Percentage of hydrophilic residues | Net hydrophobic residues content |
|---|---|---|---|---|
| 1 | 2MSJ | 63.70% | 21.20% | Very high |
| 2 | 1KDF | 61.50% | 20.00% | Very high |
| 3 | 1OPS | 64.30% | 19.50% | Very high |
| 4 | 1AME | 63.70% | 21.10% | Very high |
| 5 | 3QF6 | 57.70% | 23.40% | Very high |
| 6 | 1GZI | 63.10% | 20.00% | Very high |
| 7 | 7MSI | 63.70% | 19.60% | Very high |
Fig. 1.
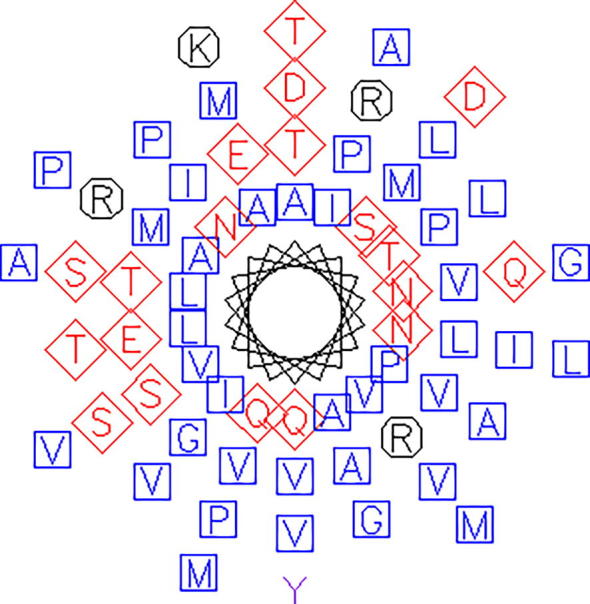
Wheel structure of 2MSJ showing the amino acid composition (blue squares indicate non-polar aliphatic residues, red diamonds indicate polar, uncharged and negatively charged residues, black octagons indicates positively charged and rest are aromatic residues.
Where table 4 represents the physicochemical parameters such as sequence length, atom content, molecular weight, theoretical Isoelectric Point (pI), no. of positively charged residues (+R), no. of negatively charged residues (−R), total no. of atoms, Extinction Coefficient (EC), Instability Index (II), Aliphatic Index (AI), Grand Average Hydropathy (GRAVY) of these targeted seven AFPs. Among these studied proteins, 1KDF has maximum sequence length (70), maximum atom content (1078); and thereby it has maximum molecular weight (7520.92 Da); while 1OPS protein has minimum sequence length (64), least number of atom content (9 6 1) and has minimum molecular weight (6746.99 Da). As ice binding mechanism depends on both sequence length and molecular weight of an AFP, therefore AFP 1KDF can bind to a newly formed ice crystal more efficiently than other experimental proteins and this protein is most suitable for cryopreservation than others [34]. Isoelectric Point (pI) indicates that pH value at which a protein or a particular molecule carries no net charge [35]. In this study, the lowest value of pI is 4.99 of antifreeze protein 1KDF and the highest value is 9.40 of 1OPS. The pI value of a protein below 7 (<7) indicates that the protein is acidic in nature and above 7 (>7) is basic in nature [36]. Here all studied proteins have pI less than 7 which are acidic in nature except 1OPS (pI = 9.40) that is basic. The total number of positively charged and negatively charged residues refers to the total no. of Lysine (K), Arginine (R) and Aspartate (D), Glutamate (E) respectively [37]. Extinction Coefficient (EC) value indicates at a certain wavelength how much light is absorbed by a protein. Extinction Coefficient can be determined by the amount of Tyrosine (W), Tryptophan (Y) and Cysteine (C) in that protein at 280 nm wavelength measured in water. The Extinction Coefficient values of these proteins are 1490 M−1 cm−1, except 7MSI protein whose EC value is 2980 M−1 cm−1. Instability index of a protein provides a crucial estimation whether it is stable or unstable. The Aliphatic Index (AI) is the relative volume occupied by Alanine (Ala), Leucine (Leu), Isoleucine (Ile) and Valine (Val) amino acids having aliphatic side chains. The highest and lowest AI values are 85.31 and 115.38, obtained from 1OPS and 1GZI proteins respectively. Aliphatic Index acts as a positive factor for molecular thermostability; therefore it can be stated that 1GZI is maximum thermostable than the other proteins. The Grand Average Hydropathy (GRAVY) value of a protein refers to the hydropathy of a protein whether it is hydrophilic or hydrophobic in nature. The positive value of GRAVY indicates the hydrophobicity and negative value indicates hydrophilicity of a protein. The protein 1OPS has lowest GRAVY value (0.172) and highest value (0.538) containing protein is 2MSJ. As all the studied proteins have GRAVY value greater than 0, then it can be stated that all these proteins are hydrophobic in nature.
Table 4.
Physico-chemical parameters computed by ExPASy-ProtParam tool.
| PDB ID | No. of aa | Mol. Wt. | Theoretical pI | No. of Negatively Charged Residues | No. of Positively Charged Residues | Total No. of Atoms | EC (M−1 cm−1) | II | AI | GRAVY |
|---|---|---|---|---|---|---|---|---|---|---|
| 2MSJ | 66 | 6948.25 | 6.26 | 4 | 4 | 999 | 1490 | 25.08 | 115.15 | 0.538 |
| 1KDF | 70 | 7520.92 | 4.99 | 6 | 5 | 1078 | 1490 | 21.46 | 112.71 | 0.369 |
| 1OPS | 64 | 6746.99 | 9.40 | 2 | 4 | 961 | 1490 | 32.82 | 85.31 | 0.172 |
| 1AME | 67 | 7046.35 | 6.26 | 4 | 4 | 1012 | 1490 | 21.98 | 114.93 | 0.516 |
| 3QF6 | 66 | 7013.32 | 6.16 | 4 | 4 | 1007 | 1490 | 22.07 | 112.12 | 0.394 |
| 1GZI | 65 | 6904.19 | 6.21 | 4 | 4 | 992 | 1490 | 22.35 | 115.38 | 0.477 |
| 7MSI | 66 | 7084.46 | 5.97 | 4 | 4 | 1016 | 2980 | 25.17 | 113.64 | 0.532 |
3.2. Structural and functional characterization of AFPs
The secondary structures of protein chains were analysed by SOPMA tool which is showed in different form likewise the alpha helix, extended strand, beta turn and random coil (Table 5). Alpha helical structure is composed of Methionine (M), Alanine (A), Leucine (L), Glutamate (E), Lysine (K) amino acids; whereas the beta strand composed of Tryptophan (W), Tyrosine (Y), Phenylalanine (F), Valine (V), Isoleucine (I), Threonine (T) furthermore, Glycine (G), Proline (P) amino acids help to build the relevant turns [38]. Such findings suggested that the amounts of amino acids are solely responsible for constructing respective secondary structure of protein sequences. In Table 5 percentage score of the amino acid distribution infers that alpha helix is dominated over other secondary structures followed by the random coil, extended strand and beta turn, except for two proteins having PDB IDs 3QF6 and 1OPSwhere the amount of random coil is maximum. Fig. 2 represents the secondary structure of 2MSJ protein where alpha helix is maximum than other structures and Fig. 3 represents AFP 3QF6, shows the dominancy of random coils over others. Such unstructured regions within the cellular protein molecules are essential for intracellular signalling pathways [39].
Table 5.
Calculated secondary structures (in percentage) by SOPMA.
| PDB ID | Alpha helix | Extended strand | Beta turn | Random coil |
|---|---|---|---|---|
| 2MSJ | 54.55% | 6.06% | 4.55% | 34.85% |
| 1KDF | 50.00% | 5.71% | 4.29% | 40.00% |
| 1AME | 50.75% | 8.96% | 2.99% | 37.31% |
| 3QF6 | 36.36% | 9.09% | 9.09% | 45.45% |
| 1OPS | 37.50% | 9.38% | 7.81% | 45.31% |
| 1GZI | 46.15% | 9.23% | 4.62% | 40.00% |
| 7MSI | 43.94% | 13.64% | 7.58% | 34.85% |
Fig. 2.

Secondary structure of 2MSJ (SOPMA) indicates alpha helix (blue), beta strands (red), turns (green) and coils (pink). From this it can be stated that in case of 2MSJ protein, alpha helices are formed in the residues ranges from 1–9, 15–22, 38–44, 57–66; random coils are constructed by the residues ranges from 10–14, 26–37, 45–53 and on 56th residue; extended strands and beta turns are minimum. Extended strands only present from the residues 26–28 and beta turns are present in the residues 54 and 55.
Fig. 3.

Secondary structure of 3QF6 indicates alpha helix (blue), beta strands (red), turns (green) and coils (pink). In this AFP, the following residues 1–7, 14–22, 37–44, 56–64 help to fabricate alpha helices; 10–13, 24–27, 30–36, 45–55 and 62–64 produce random coils; residues 23, 28 and 29 compose the extended strand structure and finally the beta structure is only composed of residues 8 and 9.
To identify the proteins either transmembrane or membrane soluble of the cellular component SOSUI server was run and outputs are analysed. No such score was generated as none of the transmembrane helices are found in these proteins which mean all the antifreeze proteins are soluble in nature.
Apart that, CYS_REC tool was used to determined the disulfide bridges within a protein and the result showing blank score. Where the disulfide bridges or bonds are built in between two adjacent Cysteine (C) amino acid residues. In current analysis, CYS_REC tool fails to find out any disulfide bridges of experimental proteins because these AFPs have no Cysteine (C) residues. Absence of disulfide bridges along with higher amount of random coil may facilitated the AFPs by not structuring solid structures and providing more actions to them for overcome from freezing [40].
3.3. Homology modelling and model validation
The three dimensional (3D) structures of experimented antifreeze proteins were predicted by Swiss model and ProSA web server. The SWISS MODEL is a homology modelling method applied to build models of an experimental protein structure (target) with an evolutionary related protein (template). SWISS MODEL shows the local model quality (Fig. 4) of protein 2MSJ and comparison with non-redundant set of PDB structures (Fig. 5). Here 1AME protein acts as the template for our experimental protein 2MSJ which have similarity of 98.48%. The local model quality (Fig. 4) indicates the similarity of the target protein (2MSJ) to its template or native protein (1AME) and the residues showing the score above 0.6 that in turn specifies the model quality is high. Fig. 5 is the graphical representation of comparison between 2MSJ and non redundant set of PDB structures where normalized QMEAN Z-scores are plotted against protein sizes. The normalized QMEAN Z-score of 2MSJ protein is 0.77 which is also supported by Fig. 5. ProSA web server reveals the Z-score and overall model quality of a protein. The Z-score confirms the quality of the homology model and is used to check whether the input protein structure is within the range of similar size. The Z-scores of all seven amino acids are listed in Table 6. The overall models for these seven proteins are prepared by ProSA web server, but here only one plot with the PDB id 2MSJ(Fig. 6) is mentioned, which is describing the best model as the Z-score lies within the range of scores typically found for similar sized proteins, that indicates a highly reliable structure. These three dimensional structures were evaluated using UCSF Chimera, Antheprot 3D, PyMOL and RAMPAGE software. RAMPAGE checks the stereochemical quality of a protein structure by analyzing residues by residues geometry and overall structure geometry. This tool has been used to determine the Ramachandran plot to give surety the excellence of the model. Ramachandran plot is a four quadrant plot, shows the distribution of two most important dihedral angles psi (plotted on Y-axis) and phi (plotted on X-axis) in a protein. The psi (ψ) angle is formed among Ni-Cαi-Ci-Ni+1, i.e., around the α-carbon-carbonyl carbon atom and phi (φ) angle is formed among Ci-1-Ni-Cαi-Ci, i.e., around the α-carbon-amide nitrogen bond in a polypeptide chain [41]. With the help of RAMPAGE, the percent value of the amino acid residues in proteins in favourable region, allowed region and outlier region calculated (Table 7). The ‘favourable region’ is that region where the amino acid residues mostly reside. The amino acid residues are sometimes found in the outside of the ‘favourable region’, that are also permitted for the stable protein structure, known as ‘allowed region’; and rest regions in the Ramachandran plot is known as ‘outlier region’. There is no residue in the outlier region and very few residues lie in the allowed region for all eight AFPs. Table 7 shows that numbers of amino acid residues in favourable region are more than 95%. A model having more than 90% residues in favourable region is considered as good quality model [42]. From this, it can be stated that all these protein models are reliable and good quality model as residues in favourable regions are more than 90% and among them two proteins having the PDB ids 1OPS and 7MSI are the best quality model as 100% residues lie in the favourable region for both the proteins. Fig. 7 and 8 signify the Ramachandran plots for 1OPS and 7MSI respectively. From Fig. 9, Fig. 10, Fig. 11 show the three dimensional structures of AFP 2MSJ constructed by using UCSF Chimera, Antheprot 3D and PyMOL respectively.
Fig. 4.
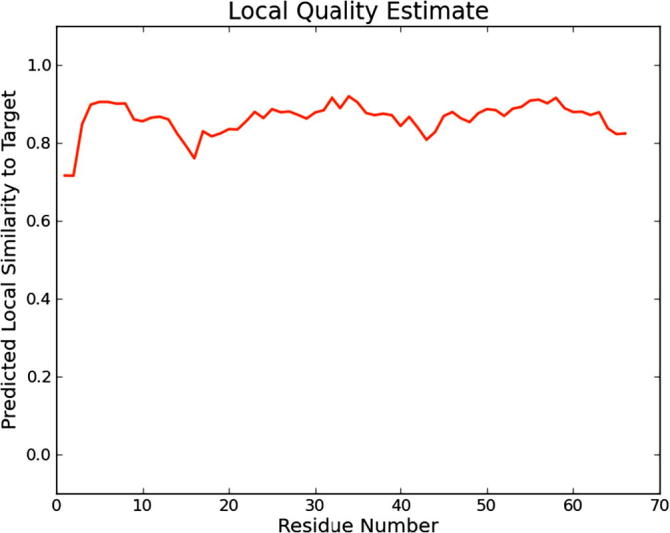
Graphical presentation of Homology modeling with special reference to the local model quality of antifreeze protein 2MSJ (red line indicates the local similarity of the target protein to its template).
Fig. 5.
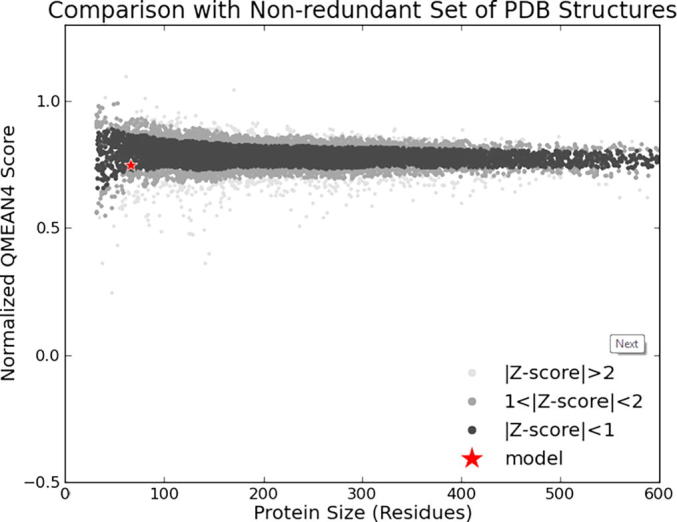
Graphical representation of the comparison of a target protein (red dot) with non-redundant set of PDB structures of AFP 2MSJ.
Table 6.
Calculated Z-scores of AFPs by ProSA web server.
| PDB ID | Z-Score |
|---|---|
| 2MSJ | −4.47 |
| 1KDF | −4.81 |
| 1AME | −4.69 |
| 3QF6 | −4.76 |
| 1OPS | −5.72 |
| 1GZI | −4.72 |
| 7MSI | −4.38 |
Fig. 6.
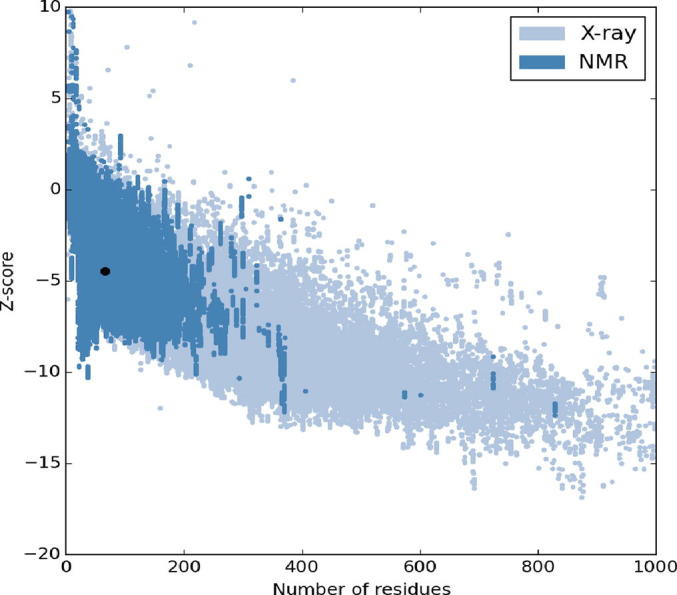
Graphical representation of comparative Z-score of antifreeze protein 2MSJ.
Table 7.
Ramachandran plot calculation with RAMPAGE Ramachandran plot tool.
| PDB ID | Residues in favoured region (%) | Residues in allowed region (%) | Residues in outlier region (%) |
|---|---|---|---|
| 2MSJ | 96.9 | 3.1 | 0.0 |
| 1KDF | 95.2 | 4.8 | 0.0 |
| 1OPS | 100 | 0.0 | 0.0 |
| 1AME | 98.4 | 1.6 | 0.0 |
| 3QF6 | 96.8 | 3.2 | 0.0 |
| 1GZI | 98.4 | 1.6 | 0.0 |
| 7MSI | 100 | 0.0 | 0.0 |
Fig. 7.

Ramachandran plot of 1OPS; dark blue and dark yellow coloured regions ‘favourable regions’ for amino acids, slightly blue and slightly yellow coloured regions are ‘allowed regions’ and rest are ‘outlier region’.
Fig. 8.

Ramachandran plot of 7MSI; dark blue and dark yellow colored regions ‘favorable regions’ for amino acids, slightly blue and slightly yellow colored regions are ‘allowed regions’ and rest are ‘outlier region’.
Fig. 9.
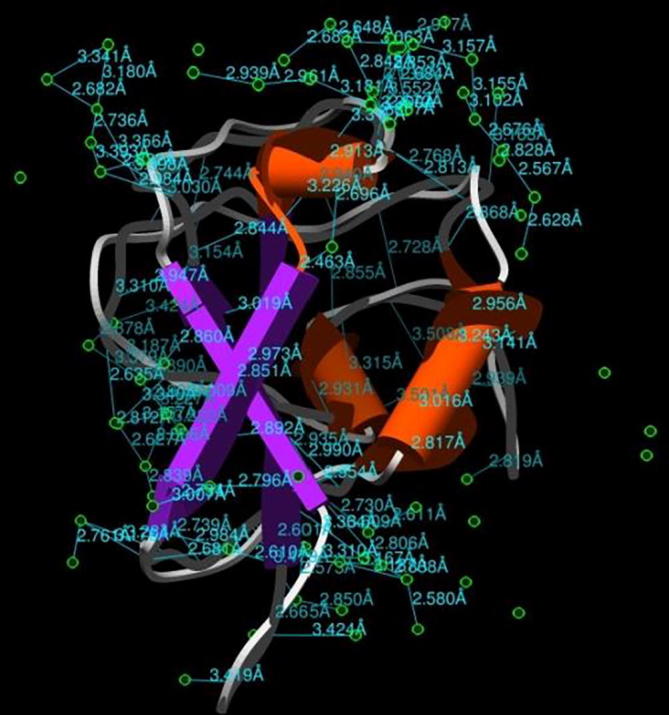
UCSF Chimera representation of 3D structure of fish AFP (2MSJ), pipes and planks model shows secondary structures (alpha helices orange, beta strands violet and coils are grey in colour), H-bonds (sky blue) with distances and selected (green) atoms are water molecules.
Fig. 10.
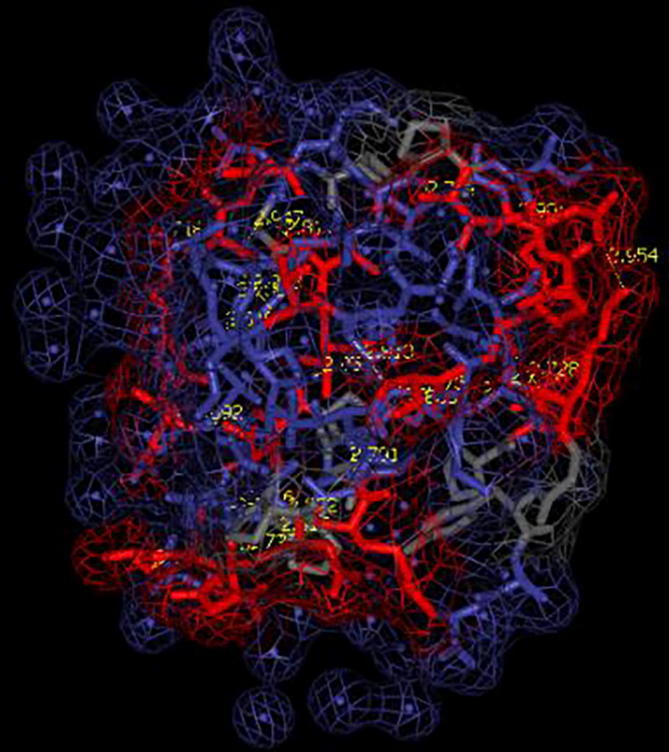
3D structure of fish AFP 2MSJ represented by Antheprot 3D, shows overall surface structure, hydrophobic (violet), hydrophilic (red), neutral (grey) parts and H-bonds (yellow).
Fig. 11.
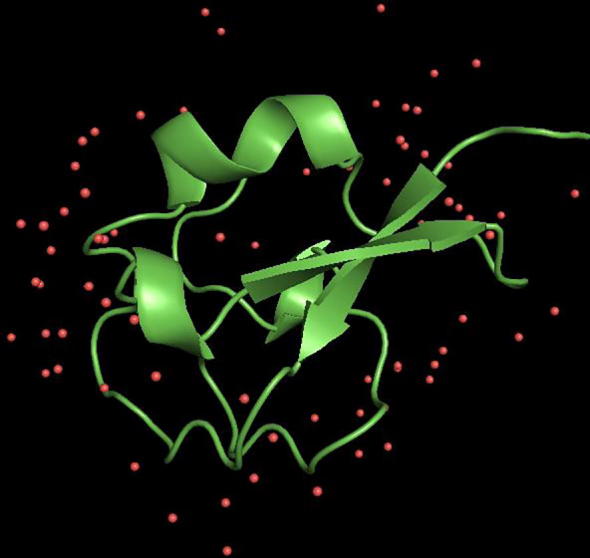
Three dimensional representation of 2MSJ; here green structure represents the main chain and warm pink colour represents the atoms, attached to the main chain.
3.4. Phylogenetic analysis
The phylogenetic tree of seven antifreeze protein sequences of Zoarces americanus was created using MEGA 7.0.9 [43] through UPGMA method, to elucidate the phylogenetic relationship among all the proteins and infer their evolutionary history (Fig. 12). The evolutions of these proteins are represented in this tree. The analysis reveals that all these AFPs are originated from a common ancestor and then divided into two major clusters, in which both the clusters divided into two sub-clusters in the course of evolution. Antifreeze protein 2MSJ, 1KDF, 7MSI, 1AME belongs to one group while 1OPS, 3QF6 and 1GZI belong to another group. There is a strong support for sister species relationship between (2MSJ + 1KDF), as well as in between (3QF6 + 1GZI). Antifreeze proteins 7MSI and 1AME are out branches of the first sister group 2MSJ + 1KDF; on the other hand, 1OPS is the out branch of the second sister group 3QF6 + 1GZI. From this tree it can be concluded that all these proteins are closely related to each other in the course of evolution.
Fig. 12.

Phylogenetic tree of seven AFPs of Zoarces americanus using MEGA 7.0.9.
4. Conclusion
The present investigation focuses total seven numbers of randomly selected antifreeze proteins (AFP type III) of Ocean pout which was finely studied by using in silico tools and servers. Findings also revealed their physicochemical properties, structural and functional features based on computational platforms. Several online programs, software packages, tools were utilised for prediction of structural characteristics as well as functional properties viz. the ExPASy-ProtParam server, SOPMA, SOSUI, CYS_REC tools along with the SWISS MODEL, ProSA, UCSF Chimera, Antheprot 3D, PyMOL and RAMPAGE. Based on experimental outcomes it has been clearly stated that all these antifreeze proteins are hydrophobic in nature, these proteins bind to the ice surface by hydrophobic interactions which are much more important in ice binding within tissue components. Apart that, the Instability Index (II) refers to the stability of these AFPs and as these proteins have such index value less than 40, it surely stated that these proteins are stable in any rapidly changing physical condition. Whereas, the Aliphatic Index (AI) acts as a positive factor for thermostability of protein molecules and shaping the recent observations it support that 1GZI protein has maximum stability in increasing or decreasing the water temperature range. From these experimental consequences it also found that all these proteins contain more alpha helices structure than other secondary structures except in case of 3QF6 and 1OPS proteins where random coils are dominant types. The AFPs (2MSJ, 1KDF, 1AME, 1GZI, 7MSI) containing alpha helices dominancy have greater interactions with the ice nucleus as the AFPs bind to the ice surface with the close interaction between the helix macro dipole along with the dipoles of water molecule in the lattice [6]. Moreover, these AFPs do not act as transporter molecules for any substances because all these are lipid soluble in nature. Consequently, the predicted 3D structures help to understand the overall functional configuration of these antifreeze proteins. A special drive also performed to generate an idea about the evolutionary distance of multiple AFPs in the light of the hierarchical clustering methods of Ocean pout. This may enhance the best knowhow practices of structural protein chemistry of such AFPs in further implications for superior research and development to the Ocean pout in near future.
Acknowledgments
Acknowledgement
The authors are thankful to Science and Engineering Research Board, Department of Science and Technology, Govt. of India for financial assistance to carry out the research work (Project file No Ref. No. PDF/2016/001776).
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Peer review under responsibility of National Research Center, Egypt.
References
- 1.Deswal R., Sharma B. Antifreeze proteins in plants: an overview with an insight into the detection techniques including nanobiotechnology. J Prot Proteom. 2014;5(2):89–107. [Google Scholar]
- 2.Li J., Ma J., Zhang F. Recent advances in research of antifreeze proteins. Chin J Biochem Mol Biol. 2005;21(6):717–722. [Google Scholar]
- 3.Kontogiorgos V. Isolation and characterization of ice structuring proteins from cold-acclimated winter wheat grass extract for recrystallization inhibition in frozen foods. J Food Biochem. 2007;31(2):139–160. [Google Scholar]
- 4.Petzold G., Aguilera J.M. Ice morphology: fundamentals and technological applications in foods. Food Biophys. 2009;4(4):378–396. [Google Scholar]
- 5.DeVries A.L., Wohlschlag D.E. Freezing resistance in some Antarctic fishes. Science. 1969;163(3871):1073–1075. doi: 10.1126/science.163.3871.1073. [DOI] [PubMed] [Google Scholar]
- 6.Davies P.L., Hew C.L. Biochemistry of fish antifreeze proteins. FASEB J. 1990;4(8):2460–2468. doi: 10.1096/fasebj.4.8.2185972. [DOI] [PubMed] [Google Scholar]
- 7.Wang J.-H. A comprehensive evaluation of the effects and mechanisms of antifreeze proteins during low-temperature preservation. Cryobiology. 2000;41(1):1–9. doi: 10.1006/cryo.2000.2265. [DOI] [PubMed] [Google Scholar]
- 8.Wang R. Expression of the antifreeze protein gene in transgenic goldfish (Carassius auratus) and its implication in cold adaptation. Mol Mar Biol Biotech. 1995;4(1):20–26. [PubMed] [Google Scholar]
- 9.Koushafar H. Chemical adjuvant cryosurgery with antifreeze proteins. J Surg Oncol. 1997;66(2):114–121. doi: 10.1002/(sici)1096-9098(199710)66:2<114::aid-jso8>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 10.B. Tharp Ice cream II. Proceedings of the Second IDF International Symposium on Ice Cream, Thessaloniki, Greece, 14-16 May 2003. in Ice cream II. Proceedings of the Second IDF International Symposium on Ice Cream, Thessaloniki, Greece, 14-16 May 2003. 2004. International Dairy Federation.
- 11.Raymond J.A., DeVries A.L. Adsorption inhibition as a mechanism of freezing resistance in polar fishes. Proc Natl Acad Sci. 1977;74(6):2589–2593. doi: 10.1073/pnas.74.6.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sivakumar K. Protein sequence analysis and characterization using Insilico methods. In: Trivedi P.C., editor. Medical Biotechnology. Aavishkar Publisher; Jaipur: 2007. pp. 211–235. [Google Scholar]
- 13.Keats D., Steele D., South G. Ocean pout (Macrozoarces americanus (Bloch and Schneider) (Pisces: Zoarcidae)) predation on green sea urchins (Strongylocentrotus droebachiensis (O.F Mull.)(Echinodermata: Echinoidea)) in eastern Newfoundland. Canad J Zool. 1987;65(6):1515–1521. [Google Scholar]
- 14.Emery W., Meincke J. Global water masses-summary and review. Oceanol Acta. 1986;9(4):383–391. [Google Scholar]
- 15.Pruitt K.D., Tatusova T., Maglott D.R. NCBI reference sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005;33(suppl_1):D501–D504. doi: 10.1093/nar/gki025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pearson W.R. Using the FASTA program to search protein and DNA sequence databases. Comput Anal Seq Data: Part. 1994;I:307–331. doi: 10.1385/0-89603-246-9:307. [DOI] [PubMed] [Google Scholar]
- 17.Kirchmair J. The protein data bank (PDB), its related services and software tools as key components for in silico guided drug discovery. J Med Chem. 2008;51(22):7021–7040. doi: 10.1021/jm8005977. [DOI] [PubMed] [Google Scholar]
- 18.Di Scala C. Mechanism of cholesterol-assisted oligomeric channel formation by a short Alzheimer β-amyloid peptide. J Neurochem. 2014;128(1):186–195. doi: 10.1111/jnc.12390. [DOI] [PubMed] [Google Scholar]
- 19.Gill S.C., Von Hippel P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182(2):319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 20.Ikai A. Thermostability and aliphatic index of globular proteins. J Biochem. 1980;88(6):1895–1898. [PubMed] [Google Scholar]
- 21.Guruprasad K., Reddy B.B., Pandit M.W. Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng Des Sel. 1990;4(2):155–161. doi: 10.1093/protein/4.2.155. [DOI] [PubMed] [Google Scholar]
- 22.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157(1):105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 23.Hossain M.M. Fish antifreeze proteins: Computational analysis and physicochemical characterization. Int Current Pharmaceut J. 2012;1(2):18–26. [Google Scholar]
- 24.Gasteiger E. Protein identification and analysis tools on the ExPASy server. In: Walker J.M., editor. The proteomics protocols handbook. Humana Press Inc.; Totowa, NJ: 2005. pp. 571–607. [Google Scholar]
- 25.Geourjon C., Deleage G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics. 1995;11(6):681–684. doi: 10.1093/bioinformatics/11.6.681. [DOI] [PubMed] [Google Scholar]
- 26.Geourjon C., Deleage G. SOPM: a self-optimized method for protein secondary structure prediction. Protein Eng Des Sel. 1994;7(2):157–164. doi: 10.1093/protein/7.2.157. [DOI] [PubMed] [Google Scholar]
- 27.Guz N. Identification of a putative antifreeze protein gene that is highly expressed during preparation for winter in the sunn pest, Eurygaster maura. J Insect Physiol. 2014;68:30–35. doi: 10.1016/j.jinsphys.2014.06.021. [DOI] [PubMed] [Google Scholar]
- 28.Schwede T. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31(13):3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arnold K. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22(2):195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 30.Wiederstein M., Sippl M.J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35(suppl_2):W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goddard T.D., Huang C.C., Ferrin T.E. Visualizing density maps with UCSF Chimera. J Struct Biol. 2007;157(1):281–287. doi: 10.1016/j.jsb.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 32.Deléage G. An interactive 3D viewer of molecules compatible with the suite of ANTHEPROT programs. J Biophys Chem. 2012;3(01):35. [Google Scholar]
- 33.Becerril J., Hamilton A.D. Helix mimetics as inhibitors of the interaction of the estrogen receptor with coactivator peptides. Angew Chem. 2007;119(24):4555–4557. doi: 10.1002/anie.200700657. [DOI] [PubMed] [Google Scholar]
- 34.Kim H.J. Marine antifreeze proteins: structure, function, and application to cryopreservation as a potential cryoprotectant. Mar Drugs. 2017;15(2):27. doi: 10.3390/md15020027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sivakumar K., Balaji S. In silico characterization of antifreeze proteins using computational tools and servers. J Chem Sci. 2007;119(5):571–579. [Google Scholar]
- 36.Hossain M.M. In silico characterization of plant and microbial antifreeze proteins. Int J Bioautomat. 2012;16(2):225–238. [Google Scholar]
- 37.Filiz E., Koç İ. In silico sequence analysis and homology modeling of predicted beta-amylase 7-like protein in Brachypodium distachyon. L. J BioSci Biotechnol. 2014;3(1):61–67. [Google Scholar]
- 38.Wikipedia, Protein secondary structure, https://en.wikipedia.org/wiki/Protein_secondary_structure, 2018.
- 39.Muthukumaran J. A framework for classification of antifreeze proteins in over wintering plants based on their sequence and structural features. J Bioinform Seq Analy. 2011;3(4):70–88. [Google Scholar]
- 40.Bansal H., Narang D., Jabalia N. Computational characterization of antifreeze proteins of Typhula ishikariensis–gray snow mould. Prot Proteom. 2014;5:169–179. [Google Scholar]
- 41.Kleywegt G.J., Jones T.A. Phi/psi-chology: Ramachandran revisited. Structure. 1996;4(12):1395–1400. doi: 10.1016/s0969-2126(96)00147-5. [DOI] [PubMed] [Google Scholar]
- 42.Idrees S. In silico sequence analysis, homology modeling and function annotation of Ocimum basilicum hypothetical protein G1CT28_OCIBA. Int J Bioautomat. 2012;16(2):111–118. [Google Scholar]
- 43.Kumar S., Stecher G., Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]