

Abstract
Leptospirosis is a widespread zoonotic disease caused by Leptospira interrogans. Symptoms of disease range from mild symptoms to serious complications including, jaundice, pulmonary hemorrhage, renal and hepatic failure, which may prove fatal. Clinical presentations of this disease are similar with other febrile illness. Therefore, rapid and appropriated laboratory diagnostic tests are needed to aid clinical case identification. As these reasons, objective of this study is to develop and evaluate a simple latex agglutination test coating with recombinant leptospiral antigens, LipL32 for serodiagnosis of human leptospirosis. Firstly, lipl32 gene was amplified from genomic DNA of Leptospira interogans serovar Pyrogenes. Then PCR product of lipl32 gene was ligated with pGEX-2T plasmid, generating pGRK32 recombinant plasmid. Recombinant GST-LipL32 protein was overexpressed and subsequently purified by using Glutathione-Agarose Resin. Recombinant GST-Lipl32 protein was coated on latex beads for development latex agglutination test (LAT). The relative sensitivity, specificity and accuracy of the developed LAT were compared with indirect immunofluorescences assay (IFA) for detection of anti-leptospiral antibodies in 30 human leptospirosis samples, 30 healthy blood donor samples, 10 dengue fever positive samples, 10 scrub typhus positive samples, and 10 melioidosis samples. Results showed that the developed LAT showed sensitivity, specificity and accuracy: 66.66%, 86.66%, and 80.00%, respectively, comparing with IFA method. Moreover, Kappa analysis showed agreement rate of the two methods were 0.421. It concluded that our developed gave compatible result with IFA. Additionally, Our LAT are simple, rapid and suitable for detection in the field. However, for better sensitivity, diagnostic specificity, positive predictive value, negative predictive value, accuracy and Cohen’s kappa comparison should be done in larger amounts of sera samples.
Keywords: Leptospirosis, LipL32, Latex agglutination test
1. Introduction
Leptospirosis is a widespread zoonotic disease caused by Leptospira interrogans (L. interrogans). An international survey of human leptospirosis conducted by World Health Organization (WHO) reported that there are annually approximately 100,000 severe cases. Leptospirosis was firstly reported in Thailand in 1942. It is an emerging health problem in Thailand. Number of cases have been dramatic increasing in reported incidence since 1996 [1]. This disease is transmitted either by direct contact to water or soil contaminated by urine of infected animals. Leptospires are highly motile bacteria that can penetrate abraded skin and mucous membranes and rapidly disseminate to other tissues shortly after infection [2].
Symptoms of this disease vary from mild symptoms to more serious complications including, jaundice, pulmonary hemorrhage, renal and hepatic failure, which may prove fatal [3]. Although leptospirosis can be effectively treated with antibiotics in the early stages of disease, clinical presentations in this stage are similar with other febrile illness including dengue fever, scrub typhus, and murine typhus. Therefore, rapid and appropriated laboratory diagnostic tests are needed to aid clinical case identification for optimal treatment and patient management and to facilitate the implementation of rapid outbreak investigations [4], [5].
Generally, clinical laboratory diagnoses leptospirosis using bacterial culture and serological method. The reference method for serodiagnosis of leptospirosis is the microscopic agglutination (MAT). Although, MAT is specific for leptospirosis detection, it is laborious and requires maintenance of live organism that is potentially dangerous for laboratory workers. Therefore, MAT is only performed in only reference labs and requires paired sera for confirmation [6]. Polymerase chain reaction (PCR) has been tested for early diagnosis of leptospriral infection [7]. However, this molecular technique requires trained technicians and sophisticated laboratory equipment. Therefore, serological assay are still cheaper and easier to perform, especially in tropical developing countries. Several serological diagnostic tests have been developed based on whole-cell leptospiral antigen preparations such as lepto Dipstick [7], Lepto Lateral flow, Lepto Dri Dot, ELISA, IFA, or Latex Agglutination Test [8]. In the present time, recombinant antigen-based serological test may achieve higher sensitivity and specificity than other tests because of the purity of antigen and lack of non-specific moieties presenting in whole-cell preparations [9].
Previously, several research groups studied on leptospiral lipopolysaccharide (LPS) molecular. LPS is the major component of the leptospiral cell surface [10]. However, LPS-mediated immunity is restricted to serovars which are antigenically related. Consequently, the research in protective antigens has shifted toward conserved outer membrane proteins (OMPs), which may be able to stimulate heterologous immunity [11]. Leptospiral OMPs have been identified into three classes such as transmembrane OMPs, peripheral membrane proteins, and envelope lipoproteins. Among the OMPs, lipoproteins are the most abundant proteins on the surface of leptospiral bacteria. Eight of the lipoproteins have been identified such as LipL21, LipL31, LipL32, LipL36, LipL41, LipL45, LipL46 and LipL48 [10]. The three major lipoproteins in order of abundance on the cell surface are LipL32, LipL21, and LipL41 [10]. All three lipoproteins are found in only pathogenic Leptospira species [12]. In addition, LipL32 is the major OMPs, of pathogenic leptospires, accounting for up to 75% of total OMPs [13]. Recombinant proteins of immunodominant OMPs LipL32 and LipL41 have been combined and evaluated in latex agglutination test for serodiagnosis of canine leptospirosis. This test is simple and rapid, and can be performed in situations where facilities or resources to perform more complicated tests are unavailable [14]. Accordingly, major objective of this study was to develop a latex agglutination test with leptospiral recombinant antigens, LipL32 for serodiagnosis of human leptospirosis and evaluate the developed test with IFA method.
2. Materials and methods
2.1. Leptospiral DNA, bacterial strains and plasmids
Leptospiral genomic DNA was extracted from killed L. interrogans serovar Pyrogenes using NucleoSpin® Tissue Kit (MACHEREY-NAGEL GmbH & Co.KG, Germany). This bacterial strains was kindly given from Institute of Animal Health, Thailand. Bacterial stains and plasmids are listed in Table 1.
Table 1.
Bacterial strains and plasmids used in this study.
| Strain or plasmid | Properties | Source |
|---|---|---|
| E. coli strains | ||
| BL21 (DE3) | F–ompT gal dcm lon hsdSB(rB–mB–) λ(DE3 [lacI lacUV5-T7p07 ind1 sam7 nin5]) [malB+]K-12(λS) | Novagen |
| Plasmids | ||
| pGEX-2 T | Expression vector with GST fusion, AmpR | GE Healthcare |
| pGRK32 | pGEX-2T derivative encoding secretion signal less LipL32 | This study |
2.2. Plasmid constructions
All recombinant DNA techniques used were based on standard protocols [15]. The PCR primers were synthesized by Biobasic, Canada. The PCRs were carried out using PhusionTM DNA Polymerase (Finnzymes, USA.), and ligations were performed using a T4 DNA ligase (Fermentas, USA.).
The amplification of DNA fragments encoding secretion-signal less LipL32 (LipL32Δ1-20) was conducted by PCR using forward primer, l32F: ggatccGCGAATTCGTTGTGGTGCTTTCGGTG and reward primer, l32R: gaattc TTACTTAGTCGCGTCGGAAGC. Lower-case type indicates an engineered restriction enzyme site. The PCR products were digested with BamHI and EcoRI and cloned into pGEX-2T by utilizing the same restriction enzymes to produce pGRK32 recombinant vector. The recombinant plasmids were verified by restriction analysis and DNA sequencing (Macrogen, South Korea). After that, the recombinant plasmid was transformed into E. coli BL21 (DE3) cells.
2.3. Recombinant protein expression and optimization of protein expression conditions
A single colony of E. coli BL21(DE3) cells containing pGRK32 plasmids was used to inoculate 10 mL of Luria-Bertani (LB) medium containing 100 μg/mL ampicillin and grown overnight at 37 °C, 200 rpm. The overnight culture were diluted 1:100 into fresh medium containing appropriate antibiotic. Bacterial cells were grown to approximately OD600 = 0.5 at 37 °C, 200 rpm. The expression of the recombinant proteins were induced with 1 mM IPTG and left for 3 h at 37 °C. For optimization of inducer concentration, after bacterial cells were grown at OD600 = 0.5 at 37 °C, 200 rpm, IPTG concentration between 0 and 1 mM, (0.05, 0.1, 0.2, 0.5 or 1 mM) were added into bacterial culture, followed by cultivation for 3 h at 37 °C. For optimization of induction time, after bacterial cells were grown at OD600 = 0.5 at 37 °C, 200 rpm and induced with optimal concentration IPTG, the bacterial culture were grown for 1, 2, 3, 4, 5 or 6 h before harvesting. An amount of bacterial samples equivalent to OD600 = 0.5 (approximately 1.5 × 107 CFU/mL) was collected by centrifugation (3000 rpm, 10 min) and stored at -80 °C until SDS-PAGE and immunoblotting analysis with a rabbit GST-tag antibody (Genscript, USA).
2.4. Protein purification
Bacterial cell pellets were suspended in 50 mL of PBS buffer plus 1 mg/mL lysozyme and 1 mM PMSF and were incubated for 30 min on ice. The cells were disrupted by sonication, and the soluble fraction was collected. The soluble supernatant was passed through a 0.45 μM pore-size filter.
Glutathione-Agarose resins (Genscript, USA) were washed three times with PBS. After that bacterial clear lysate from strain overexpressing GST-LipL32 was combined with slurry resins. Glutathione beads bound with GST-LipL32 protein were mixed and incubated for 30 min at 4 °C. The mixer was washed three times with PBS plus 0.1% Tween 20. The purified GST-LipL32 recombinant protein was eluted using 10 mM glutathione. Eluted proteins were analyzed by SDS-PAGE and immunoblotting. The total protein concentration was determined using Bradford protein assay.
2.5. Sensitization of latex bead with GST-LipL32 recombinant proteins
Latex polystyrene beads, 0.8 μM diameter (Sigma, USA) were sensitized with recombinant GST-LipL32 protein according to standard method. Briefly, latex bead suspension was washed twice with carbonate-bicarbonate buffer. After that, latex suspension was incubated with 400 μg of GST-LipL32 recombinant protein at 37 °C for 6 h. The sensitized beads were washed and suspended in PBS containing 5 mg/mL of BSA to final concentration as 2%. The latex mixers were left overnight at 37 °C. Finally, latex suspension was washed again and suspended with PBS containing 0.5 mg/mL of BSA and 0.1% sodium azide to a 1% latex suspension. The sensitized latex beads were stored at 4 °C until examination.
2.6. Human sera samples
For evaluation of latex agglutination tests were performed using 3 groups of human serum samples. First group was 30 healthy donor’s serum samples from Thailand National Red Cross, Thailand. Second group consisted of 10 samples of dengue fever, 10 samples of scrub typhus and 10 melioidosis specimens at Nakonratchaseema regional medical science center, Thailand. Both sample groups showed negative result for indirect immunofluorescences assay (IFA) with leptospiral antigen. Third group was thirty serum samples from leptospirosis patients at Nakonratchaseema regional medical science center, Thailand. This group presented positive result for testing with IFA.
2.7. Latex agglutination test (LAT)
The LAT was performed by mixing 25 μL of sensitized latex bead and 25 μL of sample serum on a glass slide. The mixture was gently mixed and read agglutination result within 2 min. For positive result, agglutination was observed as a formation of fine granular particles at the edge of mixtures. For positive results, latex suspension reacted weakly, moderately or strongly clumping, LAT was graded for 1+, 2+ or 3+, respectively. For negative result, the mixture remains homogenous suspension. PBS was used as negative control.
2.8. Statistical analysis
The comparisons were made on assumption that the specimens tested with IFA were authentic positive or negative. For this study, sensitivity, specificity, accuracy and positive and negative predictive value were obtained when results from LAT were compared with results from IFA in same sample. In addition, agreement between results from LAT and IFA was measured by Kappa (κ) coefficient.
3. Results
3.1. Amplification and cloning of lipl32 gene
Genomic DNA of Leptospira interrogans serovar Pyrogenes was used as template for amplification with l32F and l32R primers. The PCR products showed approximately 800 bp (Fig. 1A). The PCR product was digested with appropriate restriction enzymes and ligated with pGEX-2T plasmid. After pGEX-2T containing DNA fragment, pGRK32 was digested with BamHI and EcoRI for releasing any insert, the result showed the insertion of the expected size for the amplified DNA fragment encoding LipL32Δ1-20 (approximately 800 bp.) (Fig. 1B).
Fig. 1.
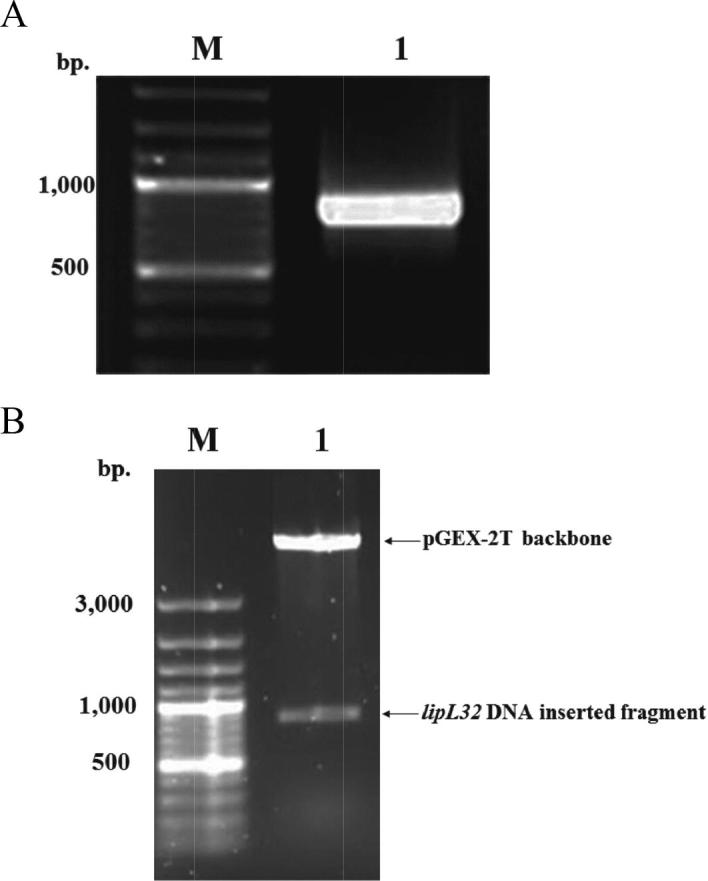
EtBr stained agarose gel of PCR product amplified from the gene encoding LipL32Δ1-20. (A) Lane M: 1 kb ladder DNA marker, lane 1: PCR product of DNA fragment encoding LipL32Δ1-20, approximately 800 bp. EtBr stained agarose gel of the restriction analysis of pGRK32. (B) Lane M: 1 kb ladder DNA marker, lane 1: pGRK32 plasmid was digested with BamHI and EcoRI restriction enzymes. The top band represents pGEX-2T plasmid backbone. The bottom band represents the DNA inserted fragment.
3.2. Expression and condition optimization for production of GST-LipL32 recombinant protein
E. coli BL21(DE3) strain harboring pGRK32 recombinant plasmid was induced with 1 mM of IPTG for 3 h. Whole cell protein lysates were analyzed by SDS-PAGE and western blot with specific GST antibody. Results from SDS-PAGE and immunoblotting indicated that E. coli strain containing pGRK32 expressed the GST-LipL32 fusion proteins (approximately 50 kDa) (Fig. 2A and B).
Fig. 2.

SDS-PAGE (A) and western blot analysis (B) of the expression of pGEX-2T or pGRK32 in E. coli. Lane M: Prestained protein marker, Lane 1: E. coli containing pGEX-2T before IPTG induction, Lane 2: bacteria containing pGEX-2T after IPTG induction, Lane 3: bacteria containing pGRK32 before IPTG induction, Lane 4: bacteria containing pGRK32 after IPTG induction.
For optimal condition experiment, the optimal IPTG concentration for GST-LipL32 fusion proteins expression were determined by inducing with IPTG concentration 0.05, 0.1, 0.2, 0.5 and 1 mM at 37 °C for 3 h. The cell pellets were analyzed by SDS-PAGE. Result showed that bacterial strain harboring pGRK32 recombinant plasmid inducing with 0.5 mM of IPTG was able to express maximum amounts of GST-LipL32 protein (Fig. 3A). The optimal incubation time for GST-LipL32 protein expression were determined by inducing with 0.5 mM IPTG with various incubation time from 1 to 6 h. at 37 °C. Result from SDS-PAGE demonstrated that GST-LipL32 proteins were expressed with the highest amount at 5 h (Fig. 3B). In this study, the optimal condition for GST-LipL32 production was induced with 0.5 mM of IPTG for 5 h at 37 °C.
Fig. 3.

Optimization condition for GST-LipL32 recombinant protein expression. (A) SDS-PAGE analysis of expression level in different concentration of IPTG at 37 °C for 3 h. (B) SDS-PAGE analysis of GST-LipL32 fusion protein production in different time course after induction with 0.5 mM IPTG at 37 °C. Lane M: Prestained protein marker. Arrow indicates GST-LipL32 protein.
3.3. Purification of GST-LipL32 fusion proteins and development of latex agglutination test
E. coli BL21(DE3) strain harboring pGRK32 recombinant plasmid were cultured under optimal condition (0.5 mM IPTG for 5 h). Bacterial cell were lysed by sonication. Properties of recombinant protein were analyzed by SDS-PAGE and immunoblotting. The SDS-PAGE results illustrated that GST-LipL32 fusion protein was expressed in soluble and insoluble form (Fig. 4A). The soluble GST-LipL32 fusion protein were purified with Glutathione-Agarose resins, then elution step was performed with 10 mM glutathione and analyzed by SDS-PAGE. Results from protein electrophoresis indicated that elution fractions contain the marginally GST-LipL32 recombinant proteins (approximately 50 kDa) (Fig. 4B). The concentration of protein in eluted fractions was 3.1 mg/mL. Additionally, immunoblotting result showed that anti-sera from leptospirosis patient was able recognition for elution fraction containing GST-LipL32 fusion protein (Fig. 4C). Finally, we developed latex agglutination test (LAT) by sensitization of 1% of suspension latex beads with 400 μg of fusion proteins.
Fig. 4.

Purification and immunoblotting analysis of soluble GST-LipL32 fusion protein. (A) SDS-PAGE analysis of GST-LipL32 in E. coli; IPTG induced total cell lysate (W), the insoluble form (I) and the soluble form (S). (B) Purification of soluble GST-LipL32 from bacterial cell lysates by Glutathione-Agarose resins column. Lane M: Prestained protein marker, Lane 1: bacterial cell lysate, Lane 2: flow through, Lane 3 to 5: washing fraction, Lane 6 to 9: elution fractions. (C) Western blot analysis of elution fraction containing purified GST-LipL32 fusion protein with anti-sera from leptospirosis patient. Lane M: Prestained protein marker, Lane 1: elution fraction. Arrow indicates GST-LipL32 protein.
3.4. Evaluation of the efficacy of our LAT
Comparison of our LAT and IFA was performed on three groups of sample as described in Materials and Methods. In group of 30 healthy donor’s serum samples with negative results from IFA testing produced all negative (0/30) results in LAT. For second group of specimens giving negative result for IFA, one specimen from dengue fever patients reacted with LAT. Moreover, there were 5 and 2 samples from scrub typhus and melioidosis specimens had agglutination reaction with LAT, respectively. In last group, thirty sera samples from leptospirosis patients with positive results from IFA testing showed 20 (20/30) agglutination with LAT. All results are shown in Table 2.
Table 2.
Summary results of LAT.
| Serum samples (no.) | LAT results |
|
|---|---|---|
| Positive | Negative | |
| Healthy blood donor (N = 30) | 0 | 30 |
| Human dengue fever, (N = 10) | 1 | 9 |
| Human scrub typhus (N = 10) | 5 | 5 |
| Melioidosis (N = 10) | 2 | 8 |
| Human leptospirosis (N = 30) | 20 | 10 |
Overall of positive and negative results is given in Table 3. The diagnostic sensitivity, specificity, accuracy and positive and negative predictive value were calculated to be 66.7%, 86.7%, 80%, 71.42% and 83.9% respectively. The agreement between two assays was estimated by kappa coefficient as 0.42.
Table 3.
Concordance of results obtained between IFA and LAT.
| LAT results | IFA results |
|
|---|---|---|
| Positive | Negative | |
| Positive | 20 | 8 |
| Negative | 10 | 52 |
| Total | 30 | 60 |
Kappa coefficient, κ = 0.421.
4. Discussion
Leptospirosis is an emerging disease in Thailand, which has dramatic increased since 1996 [1]. Leptospirosis is diagnosed in the laboratory either by isolating the causal organism or serological assay. Recombinant antigen-based serological test achieve higher sensitivity and specificity than using whole-bacterial cells because of the purity of antigen and the lack of non-specific moieties presenting in whole-cell preparations [9].
In this study was to develop and evaluate a latex agglutination test with GST-LipL32 recombinant proteins for serodiagnosis in human leptospirosis serum. The latex agglutination test is simple and inexpensive. Moreover, the sensitized latex beads were highly stable. The agglutination reaction of latex particle is not difficult to examine, the examiner is not required for special training and equipment [16]. However, in this study showed that limitation for latex agglutination in this study is not suitable for plasma sample.
In this study, the recombinant GST-LipL32 proteins were developed from the genome of L. interogans serovar Pyrogenes. This bacterial strain was found to be associated with disease in Thailand [17]. Leptospiral LipL32 protein is found in only pathogenic Leptospira species [12]. The LipL32 protein is strongly effective in inducing specific antibody responses during infection of pathogenic Leptospira species in humans. In addition, LipL32 protein is major antigens in the humoral immune response to leptospirosis and immunology-based diagnostics [18].
After overexpression, recombinant protein was determined characteristic by SDS-PAGE. The results showed that GST-LipL32 protein was partially solubilized in the cytoplasm. Chalayon P, et al. demonstrated that same characteristic of rLipL32 protein occurred [19]. After purification and determination of protein concentration of GST-LipL32 protein was 3,140 µg/mL.
Although, Microscopic Agglutination Test (MAT) is the standard serological test for diagnosis of leptospirosis but it is only performed in reference labs and requires paired sera for confirmation [6]. In the previous reports, a comparative evaluation of different method such as MAT, Indirect Hemagglutination (IHA), Microcapsule Agglutination test (MCAT), Dipstick, ELISA and IFA serological diagnosis of leptospirosis were performed by Naigowit P et al. The results showed that the IFA method was significantly more sensitivity (92.5%) and specificity (95.0%) than other methods. They concluded that IFA is suitable for a reference laboratory [20]. In this study, we preferred IFA method as definitive test and compared with our latex agglutination test. The result of latex agglutination test showed diagnostic sensitivity, diagnostic specificity, positive predictive value, negative predictive value and accuracy were 66.7%, 86.7%, 71.42%, 83.87% and 80.00% respectively when compared with IFA method.
Based on static calculation, our developed LAT test showed simple, rapid and a high specificity test comparing with IFA method. It suggested that this method cloud be usefulness for ruling-in of leptospirosis from positive samples [21]. It is recommended for field study and some situations, such as disease outbreak. However, its low sensitivity may make it unsuitable for routine using [22]. Moreover, the agreement rate when the two methods were compared was, 0.421 by kappa analysis. This value demonstrated a moderate agreement.
Eight false-positive latex agglutination results were from 1 dengue fever’s patient, 5 scrub typhus’s patient and 2 melioidosis’s patient (Table 2). These results are similar to previous studies. Study of Hull-Jackson, et al. [23] showed that false-positive latex agglutination results were from 8.5% dengue fever’s patient and 20% melioidosis’s patient. From study of Phadutkanchana and Nakarin illustrated that false-positive latex agglutination results were from 50% scrub typhus’s patient [16]. Scrub typhus is a major problem in a false positive result for immunodiagnostic test of leptospirosis [5]. Reasons of false positive are unclear, but two organisms may probably share a common antigen or previous exposure to leptospries in patient with scrub typhus may occur [16]. For leptospirosis, the rising antibody levels between pair sera is another way to confirm leptospirosis infection [19].
5. Conclusions
In conclusion, our LAT is easy to perform and interpret. This test does not require special equipment for detection and reporting results in a short time. It showed an acceptable relationship to standard method. In addition, our LAT illustrated a high degree of specificity. It is a suitable for screening leptospiral infection. Even though, more samples should be studied to obtain the most accurate statistical results before using routinely.
Acknowledgments
Acknowledgements
The financial support provided by Naresuan University, Phitsanulok, Thailand. We are grateful to thank Thai Red Cross and Nakonratchaseema regional medical science center, Thailand for their support for samples.
Conflict of interest
The authors declare that there are no conflict of interest.
Ethical approval
The use of stored human serum samples in this study was approved by Human Ethics Committee of Naresuan University (approval number 55 01 02 0010).
Footnotes
Peer review under responsibility of National Research Center, Egypt.
References
- 1.Tangkanakul W., Smits H.L., Jatanasen S. Southeast Asian J Trop Med Public Health. 2005;36(2):281–288. [PubMed] [Google Scholar]
- 2.McBride A.J., Athanazio D.A., Reis M.G. Curr Opin Infect Dis. 2005;18(5):376–386. doi: 10.1097/01.qco.0000178824.05715.2c. [DOI] [PubMed] [Google Scholar]
- 3.Silva J.J., Dalston M.O., Carvalho J.E. Rev Soc Bras Med Trop. 2002;35(4):395–399. doi: 10.1590/s0037-86822002000400017. [DOI] [PubMed] [Google Scholar]
- 4.Bajani M.D., Ashford D.A., Bragg S.L. J Clin Microbiol. 2003;41(2):803–809. doi: 10.1128/JCM.41.2.803-809.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pradutkanchana J., Pradutkanchana S., Kemapanmanus M. Southeast Asian J Trop Med Public Health. 2003;34(1):175–178. [PubMed] [Google Scholar]
- 6.Bharti A.R., Nally J.E., Ricaldi J.N. Lancet Infect Dis. 2003;3(12):757–771. doi: 10.1016/s1473-3099(03)00830-2. [DOI] [PubMed] [Google Scholar]
- 7.Fernandes C.P., Seixas F.K., Coutinho M.L. Hybridoma (Larchmt) 2008;27(5):381–386. doi: 10.1089/hyb.2008.0029. [DOI] [PubMed] [Google Scholar]
- 8.Senthilkumar T., Subathra M., Phil M. Indian J Med Microbiol. 2008;26(1):45–49. doi: 10.4103/0255-0857.38857. [DOI] [PubMed] [Google Scholar]
- 9.Lin X., Chen Y., Yan J. Clin Vaccine Immunol. 2008;15(11):1711–1714. doi: 10.1128/CVI.00189-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cullen P.A., Xu X., Matsunaga J. Infect Immun. 2005;73(8):4853–4863. doi: 10.1128/IAI.73.8.4853-4863.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cullen P.A., Haake D.A., Bulach D.M. Infect Immun. 2003;71(5):2414–2421. doi: 10.1128/IAI.71.5.2414-2421.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo D., Xue F., Ojcius D.M. Vaccine. 2009;28(1):243–255. doi: 10.1016/j.vaccine.2009.09.089. [DOI] [PubMed] [Google Scholar]
- 13.Murray G.L., Srikram A., Hoke D.E. Infect Immun. 2009;77(3):952–958. doi: 10.1128/IAI.01370-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Subathra M., Senthilkumar T.M., Ramadass P. Appl Biochem Biotechnol. 2013;169(2):431–437. doi: 10.1007/s12010-012-9973-4. [DOI] [PubMed] [Google Scholar]
- 15.Sambrook J., Russell D.W., Cold Spring Harbor Laboratory . 3rd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor; N.Y: 2001. Molecular cloning: a laboratory manual. [Google Scholar]
- 16.Pradutkanchana S., Nakarin J. J Med Assoc Thai. 2005;88(10):1395–1400. [PubMed] [Google Scholar]
- 17.Tansuphasiri U., Deepradit S., Phulsuksombati D. J Med Assoc Thai. 2005;88(3):391–398. [PubMed] [Google Scholar]
- 18.Guerreiro H., Croda J., Flannery B. Infect Immun. 2001;69(8):4958–4968. doi: 10.1128/IAI.69.8.4958-4968.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chalayon P., Chanket P., Boonchawalit T. Trans R Soc Trop Med Hyg. 2011;105(5):289–297. doi: 10.1016/j.trstmh.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 20.Pimjai N., Wimol P., Ornnalin L. J Trop Med Parasitolo. 2000;23(2):59–63. [Google Scholar]
- 21.Akobeng A.K. Acta Paediatr. 2007;96(3):338–341. doi: 10.1111/j.1651-2227.2006.00180.x. [DOI] [PubMed] [Google Scholar]
- 22.Raj P., Bhan M.K., Prasad A.K. Indian J Med Res. 1989;89:165–169. [PubMed] [Google Scholar]
- 23.Hull-Jackson C., Glass M.B., Ari M.D. J Clin Microbiol. 2006;44(5):1853–1855. doi: 10.1128/JCM.44.5.1853-1855.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]