

Abstract
The applicability of Streptomyces sp. cell lytic enzymes for devising a simple and competent biological polyhydroxyalkanoate (PHA) recovery approach from Bacillus megaterium cells was investigated. B. megaterium strain Ti3 produced 50% (w/w) PHA using glucose as carbon source. The intracellular PHA was recovered employing a non-PHA accumulating actinomycetes (Tia1) identified as Streptomyces albus, having potent lytic activity against living and heat inactivated B. megaterium. Interestingly, maximum biomass (2.53 ± 0.6 g/L by 24 h) of the lytic actinomycete was obtained in PHA production medium itself thus circumventing the prior actinomycete acclimatization just by co-inoculation with B. megaterium as an inducer. Maximum lytic activity was observed at pH 6.0, 40 °C, 220 mg of biomass and 33.3 mL of concentrated culture filtrate in a 100 mL reaction mixture. Preliminary biochemical investigations confirmed the proteolytic and caseinolytic nature of the lytic enzyme. PHA yield of 0.55 g/g by co-inoculation extraction approach was comparable with the conventional sodium hypochlorite based extraction method. Interestingly, S. albus also demonstrated a broad spectrum lytic potential against varied Gram-negative and Gram-positive PHA producers highlighting the extensive applicability of this biolytic PHA recovery approach. The lytic enzyme retained almost 100% relative activity on storage at −20 °C upto two months. 1H Nuclear magnetic resonance analysis of the extracted polymer confirmed it as a homopolymer composed of 3-hydroxybutyrate monomeric units. This is the first report on Streptomyces sp. based biological and eco-friendly, intracellular PHA recovery from Bacillus spp.
Keywords: Bacillus megaterium Ti3, Streptomyces albus Tia1, Lytic assay, Enzyme based extraction, PHA recovery, P3HB
1. Introduction
The polyhydroxyalkanoates (PHAs) have emerged as attractive and capable “Green Plastics”, due to their correlative and favourable material properties. The suitability of a bacterium for industrial PHA production is determined by diverse upstream and downstream factors including growth and PHA accumulation rates, PHA recovery or extractability, purity and molecular weight of the polymer [29], [32]. Since PHAs are intracellularly accumulated, the extraction or recovery approaches target either solubilization of the polymer itself or of the non-PHA cell mass (NPCM). PHA granules and NPCM both being in solid phase, polymer extraction and purification from the NPCM presents a technical challenge [34]. Hence, the PHA recovery (downstream processing) is a very crucial decisive step of the PHA manufacturing process [32].
PHA solubilisation with halogenated solvents (specially using chloroform) [5], [47] are the most simple, efficient and routinely employed laboratory scale polymer extraction processes, their usage is antagonistic to the concept of “Green PHA biosynthesis” from a larger environmental perspective. Diverse non-halogenated solvents have also been explored and successfully applied as an eco-friendly alternative for the same [43], [38].
Several NPCM solubilizing approaches based on chemicals (sodium hypochlorite) [20]; surfactants (sodium dodecyl sulphate (SDS), palmitoyl carnitine) [9], [37]; surfactant-chelate combination (Triton X-100 and betaine with EDTA) [8], [35]; enzymatic (commercial enzymes such as bromelain, pancreatin; microbial Cytophaga and Microbiospora sp. lysing enzymes [23], [31], [35]; ionic liquids [13]; supercritical fluids [11], [24]; γ-irradation [12]; mechanical aids [12], [17], [25], [54]; dissolved air flotation [56]; osmotic cell fragility [16], [48]; spontaneous liberation [30] etc. to name a few have been reported for PHA recovery from NPCM [34]. Though chemicals and/or surfactants recovery methods are simpler and facilitate recovery of high quality PHAs, the associated major shortcoming of these methods is the release of hazardous effluents to the environment [46]. The most conventional chemical digestion agent, sodium hypochlorite results in polymer degradation and severe molecular weight reduction under uncontrolled extraction conditions [21], [32], [34]. In contrast, the use of environmental friendly methods such as supercritical fluids, γ-irradiation, dissolved air flotation and especially mechanical aids exhibit the implementation potential at commercial levels, they necessitate high initial investments, lengthy processing time and in some cases NPCM interference also [34]. Furthermore, other eco-friendly strategies such as cell fragility accompanied with spontaneous PHA liberation usually count on either wild type osmophilic strains or development of very high polymer accumulating recombinant strains in addition to a few low polymer recovery conditions [16], [30], [48]. Very recently, a novel biological PHA recovery approach employing selective digestion of freeze-dried producer bacterial biomass in mealworms and animals was reported. The cells were consumed as feeds by both and the partially purified polymer was obtained as whitish fecal pellets. Though a new option, the strategy restricts the application potential to such methods where requirement of highly pure PHAs is undesirable. Also, the approach is still under risks and safety analysis for large scale applications [33], [42], [44].
In the light of the above, bacteriolytic PHA recovery approach is still an attractive eco-friendly process as it is operated in mild conditions and outweighs the cost factor of commercial enzymes yielding quality PHAs.
Actinomycetes play a significant role in the production of various antimicrobial agents and other industrially important substances such as enzymes. They are of enormous importance since they possess a capacity to produce and secrete a variety of extracellular hydrolytic enzymes such as amylase, lipase, and cellulases which play an important role in food, fermentation, and textile and paper industries [50]. Many actinomycetes have been isolated from various natural sources, as well as in plant tissues and rhizospheric soil. Biological functions of actinomycetes mainly depend on sources from which the bacteria are isolated. Microbial alkaline proteases for manufacturing uses are produced mostly from Streptomyces and Bacillus. Actinomycetes, particularly Streptomycetes are known to secrete multiple proteases in culture medium. The potential of actinomycetes in the discovery of novel compounds with antimicrobial activity has been realized, and hence opens exciting avenues in the field of biotechnology and biomedical research [50].
With respect to the above stated findings, the aim of the present work was to devise a simpler PHA extraction protocol using bacteriolytic actinomycetes for the recovery of PHA from Bacillus megaterium strain Ti3.
2. Materials and methods
2.1. Microorganism
PHA producer Bacillus megaterium strain Ti3 was used in the present study [26]. The strain was maintained on nutrient agar plates at 4 °C. The strain accumulates PHA during growth phase with a maximum yield of 0.6 g/L amounting to 44.1 ± 0.9% accumulation [27].
2.2. PHA production
Overnight grown Nutrient broth culture of B. megaterium strain Ti3 was used as inoculum (1% v/v) for PHA production medium (pH 7.0) containing (g/L) 6.1 glucose, 0.2 MgSO4, 0.1 NaCl, 0.75 KH2PO4, 7.5 casein hydrolysate. Production studies were carried out in 250 mL Erlenmeyer flasks containing 100 mL culture medium. Incubation was done at 30 °C, 120 rpm for 24 h [27]. After incubation, the biomass content or cell dry mass (CDM) was evaluated by gravimetry. For this, 10 mL of the culture broth was centrifuged at 10,000g for 10 min. The cell pellet obtained was washed with distilled water and dried to a constant weight at 60 °C [49].
2.3. Conventional sodium hypochlorite based PHA extraction and quantification
Ten mL culture pellet was treated with 5 mL of 40% (v/v) sodium hypochlorite solution at 30 °C for 20 min, followed by centrifugation at 10,000g for 20 min. The polymer pellet obtained was washed with distilled water, acetone, methanol and diethyl ether, respectively and dried to a constant weight at 60 °C. PHA was quantified gravimetrically and by crotonic acid estimation [36], [49]. The CDM was used as a reference for the determining the PHA yield (expressed in terms of (g/g) CDM).
2.4. Isolation, screening and identification of the potent lytic actinomycetes
Ten different rhizospheric soil samples were collected from Indian Institute of Horticulture Research (IIHR) and Gandhi Krishi Vigyan Kendra (GKVK), Bangalore. 100 μL of the 10−4 and 10−6 soil suspension serial dilution respectively was uniformly spread plated on actinomycetes isolation agar plates (Hi Media). The plates were incubated at 30 °C for 24–72 h. For screening of potent lytic Actinomycetes, well isolated, morphologically distinct colonies were plated on B. megaterium strain Ti3 agar plates and incubated at 30 °C for 24–72 h. The lytic zone diameter of different isolates was measured. The selected probable lytic isolates were subsequently stained with Sudan Black B (0.3% in 96% ethanol). Isolate exhibiting the maximum lytic potential and lacking PHA accumulation was chosen for further studies. Molecular characterization of the selected actinomycetes sp. Tia1, was done by 16SrRNA gene sequence analysis (Chromous Biotech Pvt. Ltd., Bangalore). Phylogenetic lineage analysis was done using NCBI-BLAST (Basic Local Alignment Search Tool) and Phylip tool (based on neighbour joining algorithm) [3].
2.5. Optimization of lytic culture (Streptomyces albus Tia1) biomass using different media
Inoculum of the lytic S. albus Tia1 culture was optimized by transferring 72 h old Starch casein agar culture to four different media (100 mL in 250 mL flask) viz. Nutrient broth; Starch-Casein [1% (w/v) Starch and 0.5% (w/v) Casein]; Actinomycetes Culture Media 1, pH 7.0 (g/L): 4.4 Na2HPO4·2H2O; 1.5 KH2PO4; 1 (NH4)2SO4; 0.2 MgSO4; 10 Sucrose; 1.5 Yeast extract [50] and Actinomycetes Culture Media 2 (modified PHA Production Medium) (g/L): 10 Glucose; 0.2 MgSO4; 0.1 NaCl; 0.75 KH2PO4 and 7.5 Casein. The flasks were incubated at 30 °C and 150 rpm for 24–72 h. Biomass content was gravimetrically quantified.
2.6. Production studies of the lytic principle of S. albus
To determine the nature of expression of the lytic principle of S. albus Tia1, the culture was grown in presence (induced) and absence (un-induced) of B. megaterium Ti3 cells. The culture filtrates obtained by growth of S. albus Tia1 under un-induced and induced conditions for 24 h, respectively, were tested for their efficiency to hydrolyse the PHA containing B. megaterium Ti3 cells. The time course profile of lytic principle production was evaluated by lytic assay using the culture supernatant of the actinomycetes grown for 24–72 h, respectively.
2.7. Lytic assay
Culture inoculum of S. albus Tia1 grown in ACM 2 (2.5 mg/mL) was inoculated in B. megaterium Ti3 24 h culture broth (heat inactivated at 80 °C for 10 min and then cooled), containing 2.20 mg/mL (w/v) biomass. The flasks were incubated at 120 rpm, 30 °C for 24–72 h. The supernatant was collected at every 24 h interval, by centrifugation at 10,000g for 15 min. The supernatant was 5× concentrated (reverse dialysis using 100% w/v sucrose). Lytic activity of supernatant was determined by hydrolysing B. megaterium Ti3 cells (2.2 mg dry weight) with 1 mL 5× lytic supernatant (648 µg protein) at 45 °C, pH 7 up to 4 h (Un-optimized). Optical density (O.D.) was measured at 600 nm and the activity was calculated as the amount of enzyme required to decrease the O.D. by 0.1 under standard assay conditions [35].
Optimization of various physicochemical parameters of the lytic assay was done by varying: (a) pH: 5–11 (at 40 °C for 1 h), (b) Temperature: 30–100 °C (at pH 6.0 for 1 h), (c) Lytic supernatant volume: 0.1–1.5 mL (at pH 6.0 and 40 °C). Appropriate controls (without enzyme) were also set up at the respective assay conditions [35].
2.8. Biochemical characterization of the lytic principle
To assess the biochemical nature of the lytic principle, the culture filtrate of S. albus Tia1 was subjected to two chemical and one physical pre-treatment respectively. The crude supernatant (1.0 mL) of S. albus Tia1 was treated with different concentrations of SDS (10% w/v) for 10 min at room temperature; 0.25 mL TCA (100%, w/v) for 1 h (at 4 °C and room temperature, respectively), and was incubated at 100 °C for 10 min (heat treatment) respectively. The respective preparations were subsequently assayed for residual lytic activity [55].
2.9. Elucidation of the lytic mechanism
B. megaterium Ti3 cells (1.5 mL) of the optimized reaction mixture was substituted with 25, 50 and 100 mg of bovine serum albumin (BSA) and incubated at 40 °C for 1 h. Reaction system devoid of BSA was used as control. The caseinolytic activity of the cell extract of S. albus Tia1 was determined using casein agar slide (1% w/v casein and agar respectively in 0.1 M phosphate buffer pH 7). The slide was incubated at room temperature (RT) for 4 h. Zone of hydrolysis was observed on staining with Coomassie Blue R-250 for 10 min followed by destaining [52].
2.10. Determination of Michaelis Menten constant
The lytic assay was set up with varying Bacillus biomass concentration (0.22–4.4 mg) at pH 6, 40 °C for 1 h. Non-linear hyperbolic regression was used for estimating the Km and Vmax values of the crude lytic enzyme of S. albus Tia1. Here, the starting values obtained were used by linear regression fitting of a Hanes–Woolf plot using the Hyper32 software [14], [60].
2.11. Extraction of PHA using S. albus Tia1 cells and culture filtrate
For the lytic biomass based PHA extraction, the S. albus Tia1 cell pellet was co-inoculated into B. megaterium Ti3 culture broth as mentioned above, and incubated at 30 °C for 24–72 h. After incubation, the S. albus Tia1 cells were separated by filtration using Whatman No 1. The polymer released in the broth was recovered by mixing the supernatant with chloroform in the ratio of 1:4. The dispersion was incubated on an orbital shaker at RT for 1 h. The bottom chloroform phase having PHA was dried to a constant weight at 60 °C.
For the lytic culture filtrate based polymer extraction, the induced S. albus Tia1 culture supernatant collected at every 24 h intervals (24–72 h) was used for hydrolyzing B. megaterium Ti3 cells in optimized ratio. The PHA was recovered using chloroform as mentioned earlier [35]. Efficiency of the above two extraction procedures was assessed by comparing their resultant PHA yields with the conventional sodium hypochlorite and hypochlorite-chloroform dispersion method [21], [22].
2.12. Substrate spectrum of S. albus Tia1 lytic activity
For determining the substrate specificity of the lytic enzyme, S. albus Tia1 was plated on agar plates containing different PHA producers viz. Gram-negative (Ralstonia eutropha MTCC 1954 and Pseudomonas aeruginosa P6) and Gram-positive (Bacillus sp. Ti1, B. subtilis, B. thuringiensis IAM12077, Actinomycetes Bl10, available in our Microbiology laboratory). Plates were incubated at 30 °C for 24–48 h, followed by observation of zone of lysis. Additionally, the various PHA producer cells were also subjected to enzymatic hydrolysis (in the optimized ratio) at 45 °C, pH 7 for 1 h. The O.D. was measured and the lytic activity was calculated as mentioned above.
2.13. Characterization of PHA
Structural characterization of the PHA extracted employing the biolytic method was done by 1H NMR analysis. The spectra were recorded on a Bruker Avance II 500 spectrometer at 400 MHz, 25 °C using CDCl3 as solvent. The concentration of the polymer sample used was 10 mg mL−1 (in CDCl3). Tetra methyl silane (TMS) was used as the internal standard for the chemical shifts (outsourced to IISC, Bangalore).
2.14. Statistical analysis
All the experiments were done in triplicates. One-way ANOVA analysis was performed using SPSS (IBM Statistics 19) statistical package. Duncan’s multiple range test (DMRT), was applied for the comparison of means at p < 0.05 (5% level of significance). The data sets for media optimization of lytic culture (S. albus Tia1) biomass were analyzed by Two-way ANOVA (Bonferroni Post-hoc test) using Graph Pad Prism 6 statistical package, at p < 0.05.
3. Results and discussion
3.1. Isolation, screening and identification of a potent lytic Actinomycete sp.
Twenty six morphologically distinct isolates obtained were subjected to lytic plate screening using the B. megaterium Ti3 (PHA producer) as the substrate. Of these, 17 isolates exhibited potential lytic activity with zone of clearance (ranging from 5 to 18 mm), of which eight isolates with zone size > 7.0 mm were selected (Fig. 1). Further, sudan black colony staining of the selected 8 isolates, showed Tia1 and Coa2 to be non PHA producers. Isolate Tia 1 being a non-PHA producer with maximum lytic potential was chosen for further studies.
Fig. 1.
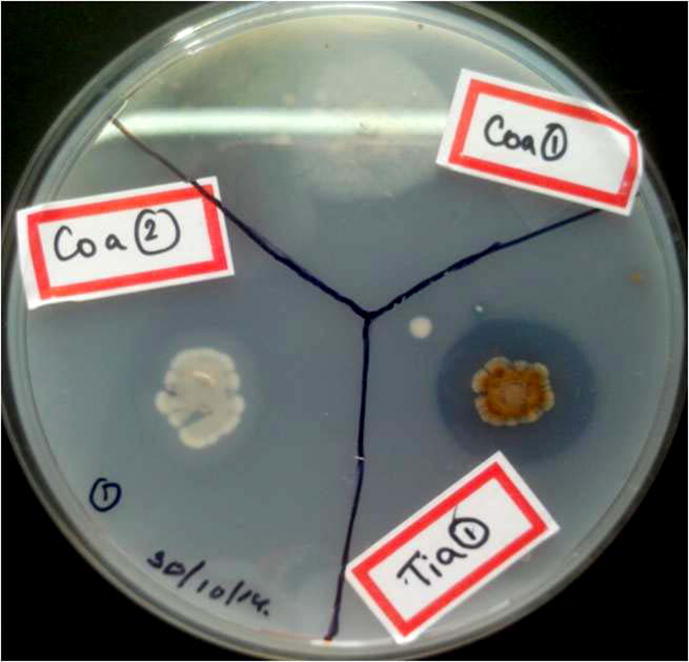
Assessment of the lytic potential of various Actinomycetes on Bacillus megaterium Ti3.
Based on its phenotypic characteristics (colorless with white aerial mycelium forming ovoidal spores on lateral branches of aerial hyphae and musty or earthy odour), the isolate Tia1 was identified as Streptomyces sp. Further, the BLAST and dendrogram analysis of the partial 16S rRNA gene sequence (740 bp) revealed Streptomyces albus strain J1074 to be the closest relative of actinomycetes sp.Tia1. The partial 16S rRNA gene sequence of strain Tia1 was submitted to NCBI database (Accession No KT003583) (Fig. 2).
Fig. 2.
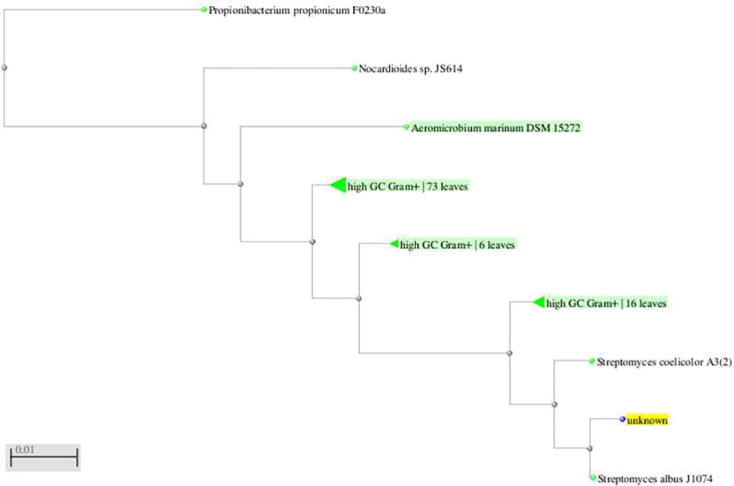
Dendrogram indicating the phylogenetic relation of the Streptomyces sp.Tia1 with other organisms. unknown: represents the isolate Actinomycetes sp. Tia1.
Actinomycetes group exhibit remarkable bacteriolytic properties through antibiotics and lytic enzymes [57]. The bacteriolytic activity is attributed to the dissolution of the heat inactivated cell wall by proteolytic enzymes [6], [40]. Hence, it was not surprising that out of 26 colonies screened almost 17 (65%) of them possessed lytic activity against bacteria.
3.2. Optimization of lytic culture (Streptomyces albus Tia1) biomass using different media
Analysis by Two-way ANOVA showed that lytic culture biomass was significantly influenced by both time of incubation and type of media (p < 0.05). As shown in Fig. 3 amongst all the four media tested, ACM 2 supported maximum cell dry mass of 2.52 ± 0.05 g/L and 2.49 ± 0.07 g/L in 24 and 48 h, respectively. Therefore, culture media ACM 2 and time period of 24 h for growth was considered for all the future studies. The growth and nutrient conditions may influence the production of lytic principle in the organism which may be explored for designing an optimum media and growth condition for mass cultivation of the lytic organism to enable efficient extraction of PHA. Since the ACM 2 medium is the PHA production medium for B. megaterium Ti3, the lytic actinomycete which showed highest biomass in ACM 2 itself does not require additional adaptation in any new media for lytic activity production.
Fig. 3.
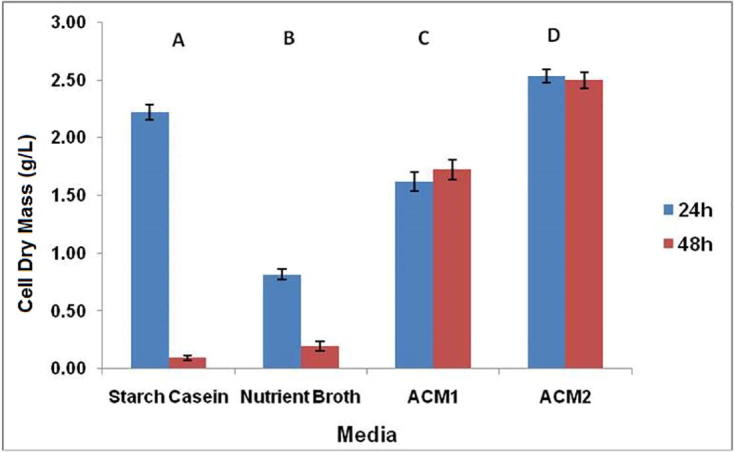
Optimization of S. albus Tia1 biomass using different media. Alphabets A, B, C, and D indicate that means of different media are significantly different at p < 0.05. Two way ANOVA showed that all the values were significant except starch casein vs nutrient broth (48 h).
3.3. Determination of the inducible nature of the lytic principle of S. albus
The culture filtrate obtained by 24 h growth of S. albus Tia1 in presence and absence of B. megaterium Ti3 as substrate, were tested for its efficiency to hydrolyse the PHA containing B. megaterium Ti3 cells. In comparison to the un-induced (13.3 ± 4.71 U), the lytic activity for induced culture filtrate was observed to increase by 2 folds (26.6 ± 4.71 U), indicative of a inducible lytic mechanism possessed by S. albus Tia1. Literature reports have depicted the lytic enzymes of various actinomycetes viz. Microbispora sp. [35] and Streptomyces globisporus 1829 strain [61] to be of inducible and constitutive nature, respectively.
3.4. Time course induction of lytic activity of S. albus Tia1
Time course evaluation of lytic activity by S. albus Tia1 grown with B. megaterium Ti3 in the PHA production media showed time dependent induction of the lytic potential. The lytic activity gradually increased from 8 h to 120 h with peak activity at 72 h of induction (50 ± 8.16 U) (Fig. 4). The time course induction profile of S. albus enzyme is in concordance with earlier studies on lytic organisms which reported maximum lytic activity between 60 and 72 h of growth [1], [12], [35].
Fig. 4.
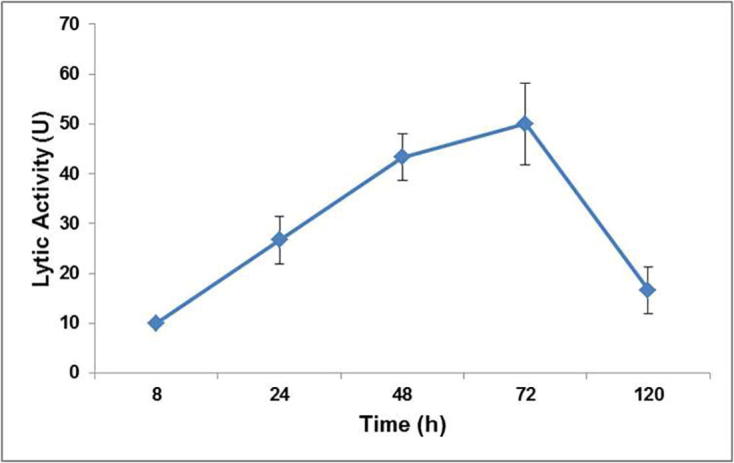
Time course induction of lytic activity of S. albus Tia1.
3.5. Optimization of various physicochemical parameters of the lytic assay
Contact time between the B. megaterium Ti3 cells and the lytic principle is crucial for efficient lysis and release of the polymer for recovery. pH and temperature of an assay system are also known to influence the enzyme activity significantly. Standardization of assay conditions by one factor approach resulted in maximum cell lytic activity (223 ± 4.7 U) at pH 6, 40 °C by 1 h (Fig. 5a–c). Many bacteriolytic enzymes have optimum pH in acidic range which is similar to pH ranges of cell wall proteins [10].
Fig. 5.
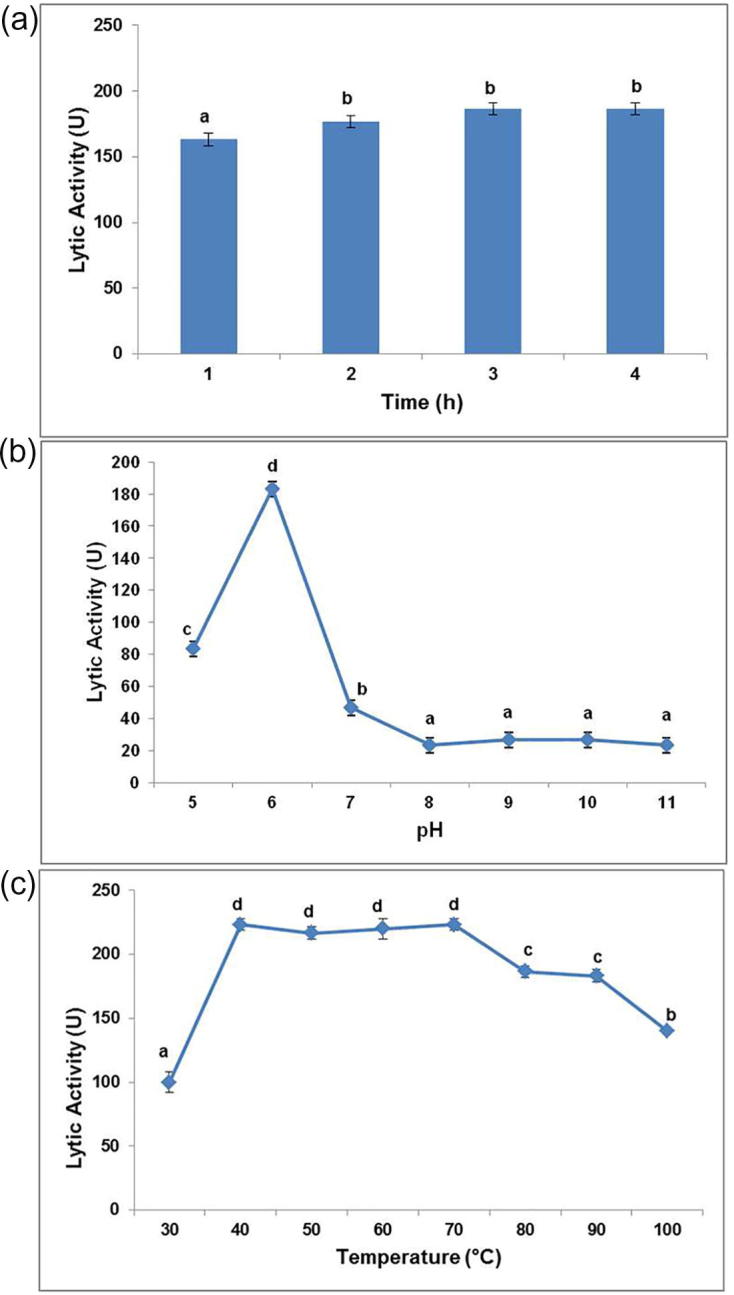
Optimization of (a) incubation time, (b) pH and (c) temperature for lytic activity of S. albus Tia1 on B. megaterium Ti3. Different alphabets (a, b, c and d) indicate the significant difference amongst the means, at p < 0.05.
3.6. Determination of mechanism of lysis
The significant decrease in the lytic activity due to the three pre-treatments (SDS, TCA and heat), respectively, suggested a proteinic lytic mechanism of S. albus Tia1. Further, this was also confirmed by inhibition of lytic activity using Bovine Serum Albumin (BSA) as a competitive substrate. Furthermore, the zone of clearance on casein agar plate demonstrated the caseinolytic nature of the crude extract. Streptomyces strains have been reported to be producers of a complex system of lytic enzymes [18]. The proteolytic and caseinolytic nature of the actinomycetes lytic enzymes has been reported earlier studies too [15], [18], [19], [35], [45], [53], [58], [59], [62].
3.7. Determination of Michaelis Menten constants of the lytic enzyme of S. albus Tia1
Optimum stoichiometry of substrate cells with lytic enzyme is needed to achieve efficient lysis of PHA producer cells for increased PHA yield. The highest lytic activity (270 U) was observed at pH 6, 40 °C, 2.2 mg/mL of biomass and concentrated culture filtrate containing 324 µg of protein. The apparent Km and Vmax values were 1.227 ± 0.1692 mg/mL and 346.6 ± 28.49 U/mL, respectively (Fig. 6a and b). Thus, the optimal ratio of substrate cells to lytic enzyme obtained was used for future enzyme based PHA extraction experiments.
Fig. 6.
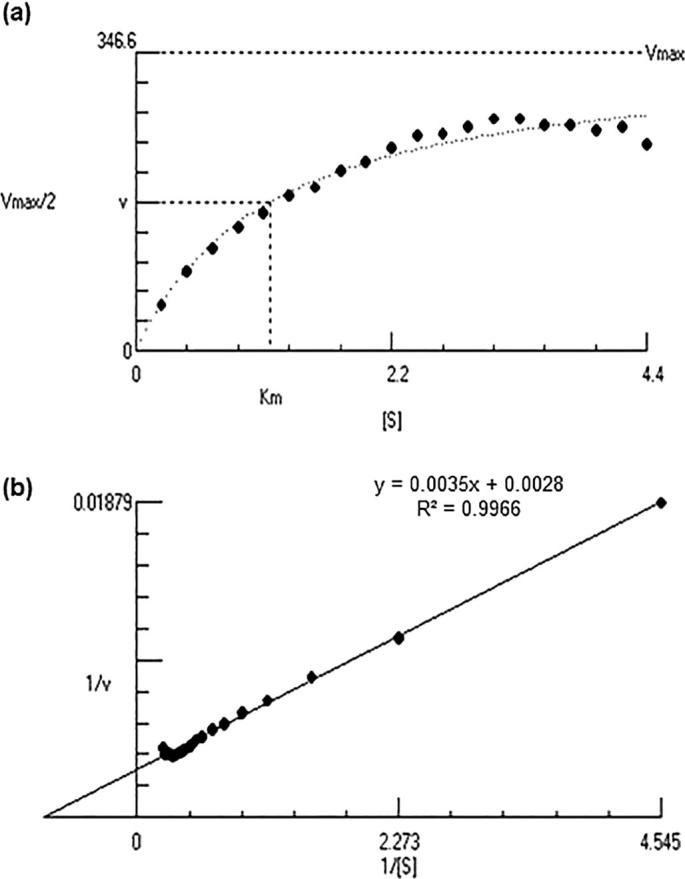
Effect of varying concentration of biomass on the lytic activity of S. albus Tia1 on B. megaterium Ti3. (a) Michaelis-Menten Hyperbola, (b) Lineweaver-Burk Plot.
3.8. Extraction of PHA using S. albus Tia1 cells and culture filtrate
The Co-inoculation studies had clearly demonstrated the appreciable growth of S. albus Tia1 in the fermented broth of B. megaterium Ti3, resulting in lysis of producer cells and release of the polymer (Fig. 7). Time of contact between the producer and lytic actinomycetes significantly influenced the polymer recovery, which increased from 24 h (0.27 g/g CDM), to a maximum by 48 h (0.32 g/g CDM) followed by decline further by 72 h (0.22 g/g CDM).
Fig. 7.
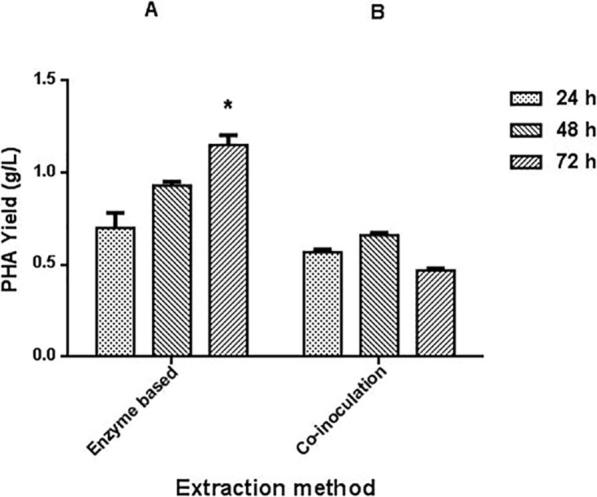
Influence of induction time period on the PHA yields recovered from B. megaterium Ti3 by the different biolytic strategies. Alphabets A and B indicated means of PHA yields recovered using the enzyme based and co-inoculation strategy being significantly different at p < 0.05. * indicates the respective mean was overall statistically significant. Two way ANOVA showed that all the values were significant except co-inoculation (24 h vs 72 h) and enzyme-24 h vs co-inoculation 48 h.
For the culture filtrate based extraction, the co-induced culture filtrate collected at every 24 h interval (up to 72 h) was used for the polymer from B. megaterium Ti3 cells [100 mL extraction mixture containing 220 mg cells with 50 mL of crude enzyme (≈16.2 mg protein)]. PHA was successfully recovered using all the co-induced culture filtrate samples with 72 h induced filtrate supporting the highest PHA recovery yield of 0.55 g/g CDM at 40 °C for 2 h (Fig. 7).
3.9. Comparison of PHA extraction by different methods
Significantly different PHA yields were obtained from B. megaterium Ti3 cells using the four different extraction methods (p < 0.05), respectively. Interestingly, the enzyme based extraction yielded 0.55 g/g CDM PHA amounting to 100% as obtained by the conventional sodium hypochlorite extraction method (Fig. 8). The enzyme based extraction led to a 1.74 fold increase in the PHA yield as compared to co-inoculation approach attributable to the utilization of the released polymer by the growing S. albus Tia1 cells.
Fig. 8.
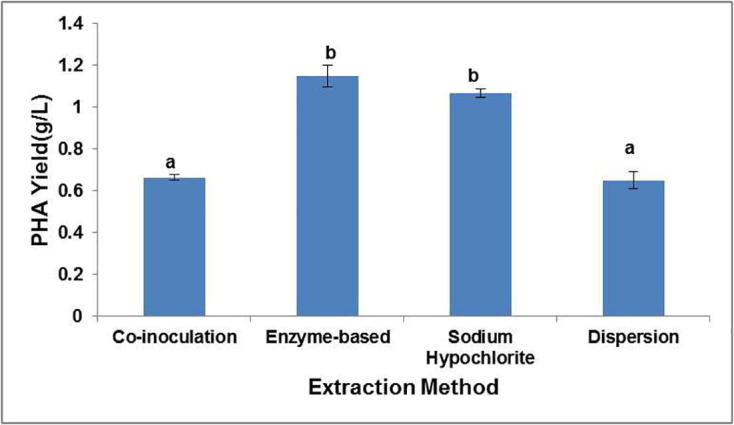
Comparison of PHA yields by different extraction methods. Alphabets a and b indicate that means within columns are significantly different at p < 0.05.
Release of the intracellular PHA using lytic cultures have been reported earlier, to quote a few complete lysis of Ralstonia eutropha cells upon treatment with Cytophaga sp. lytic enzymes was reported by Harrison et al. [23]. A Microbispora sp. protease induced hydrolysis in the gram –ve cells of Sinorhizobium meliloti cells accumulating 50% of PHA [35]. A similar study, reported use of Microbispora sp. enzymes for lysing of gram +ve PHA producing B. flexus cells followed polymer separation by aqueous two phase system (ATPS) resulting in 92% pure PHA [12].
Thus, the enzymatic approaches are quite attractive due to their mild operation conditions, specificity and an expectation of good quality PHAs being recovered. The main shortcoming presented by this approach is the high cost of enzymes for large scale recovery processes [34]. However, the lytic bacteria co-culturing with the PHA producer culture at the appropriate harvest time can be explored as an appreciably economical and environmental friendly polymer recovery strategy.
3.10. Substrate spectrum of S. albus Tia1 lytic activity
Interestingly, the S. albus Tia1 lytic enzyme exhibited broad substrate specificity, capable of lysing both Gram- positive and Gram- negative bacteria. The extent of lysis varied significantly with respect to different bacteria, with the highest lytic activity (280 U) against B. megaterium Ti3 (p < 0.05) (Fig. 9). Additionally, the enzyme stored up to a period of two months at −20 °C retained almost 100% activity as assessed by lytic assay. The diverse enzymatic susceptibility towards different bacterial cells may be due the variations in their mucopeptide structures, its chemistry and thickness, and the secondary effects of components present [18], [51]. Similar observations were obtained in case of Micromonospora sp. Strain 152-enzyme depicting lytic activity towards both Gram- positive and Gram- negative bacteria [53]. On the contrary studies have shown the lytic enzymes of Sorangium α-, β- enzyme [58], MyxobacterAL-1 enzyme [15], MR-enzymes from Streptomyces albus G [19], [45], and Fl-enzyme from Streptomyces griseus [58] to be inactive toward Gram-negative bacteria such as Serratia marcescens, P. aeruginosa, E. coli and P. vulgaris. Hence, the present study S. albus Tia1 lytic enzymatic potential paves way for a generalized enzyme based PHA extraction from various bacterial PHA producers. However, the extraction procedure needs to be optimized for different PHA producers.
Fig. 9.
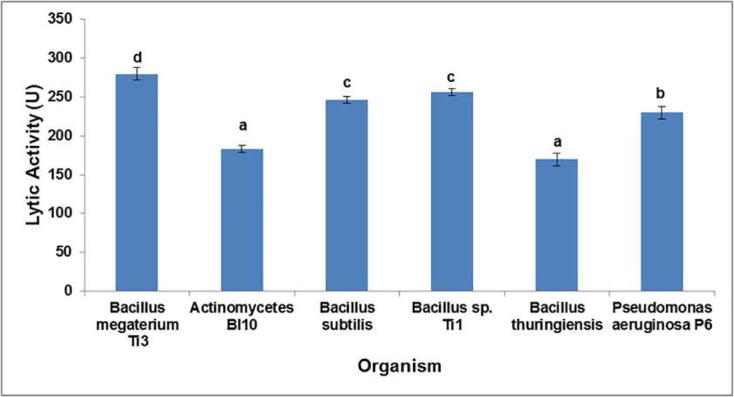
Spectrum of S. albus Tia1 lytic activity on different PHA producers. Alphabets a, b, c and d indicate that means within columns are significantly different at p < 0.05.
3.11. Characterization of PHA
The assignments of the peaks for the 1H NMR spectrum of the enzymatically extracted PHA are given in Fig. 10. A doublet at around 1.2 ppm, multiplet at 2.4–2.6 and 5.2 ppm were observed respectively. The ratio of integrated peak values was 3:2:1. The 1.2 ppm shift represented the three protons of the C4 (methyl group or -CH3 side chain. The 2.4–2.6 ppm shift was corresponding to the two protons of C2 (methylene group; -CH2 group) and 5.2 ppm represented the single proton bonded to C3 (methine group; -CH group), respectively. The spectrum was well in correlation with the P(3HB) 1H NMR spectra reported in literature (Table 1) [2], [4], [7]. Thus, the monomers present in the polymer were of 3HB type only. Also, absence of the 1.6 and 0.9 shifts representing C4 (-CH2 group of 3HV) and C5 (-CH3 group of 3HV), respectively [39], [41] confirmed the nature of the enzymatically extracted polymer to be a pure homopolymer of poly (3-hyroxybutyrate) or P3HB. The 1H NMR spectrum obtained was also well in agreement with the earlier reports of homopolymer P3HB accumulation by B. megaterium Ti3 on glucose as a carbon source [27], [28] thus validating the extracted material as a polyhydroxyalkanoate of P3HB type.
Fig. 10.
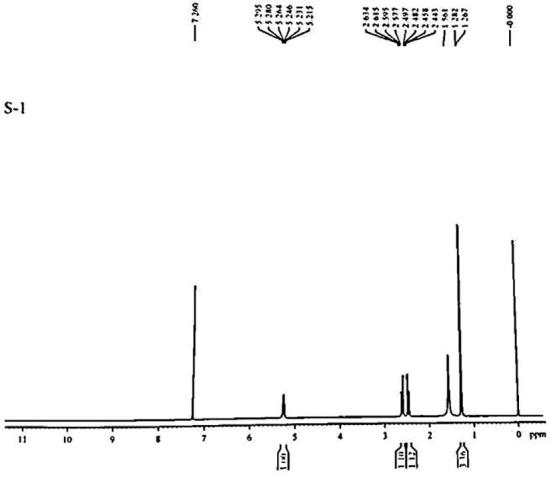
1H NMR spectrum of the polymer extracted from B. megaterium Ti3.
Table 1.
1H NMR peak assignments for the PHA extracted from B. megaterium Ti3 by the enzymatic approach.
4. Conclusion
The biolytic method using the lytic potential of S. albus, devised in this study, utilized a broad spectrum biological hydrolysis mechanism for different PHA producers, wherein the lytic culture grew on producer cells as a sole carbon and nitrogen source, without buffering, centrifugation or re-suspension of the cells. The present proposed biolytic P3HB extraction strategy can be a viable eco-friendly alternative evading or minimizing the need of chemicals (sodium hypochlorite), solvents and expensive mechanical aids. Furthermore, development of an immobilized bacteriolytic system could be explored as a more effective and inexpensive PHA recovery strategy.
Acknowledgments
Acknowledgements
The authors express their sincere thanks to the management of Jain University and Council of Scientific and Industrial Research, New Delhi for providing the necessary infrastructure and financial support (Direct-Senior Research Fellowship, Grant No. 9/1115(0002)2K14 EMR-I to Ms. Neetu Israni), respectively, for carrying out this work.
Conflict of interest
None.
Footnotes
Peer review under responsibility of National Research Center, Egypt.
References
- 1.Aksnes L., Grov A. Acta Chem Scand. 1974;28:185–192. doi: 10.3891/acta.chem.scand.28b-0185. [DOI] [PubMed] [Google Scholar]
- 2.Alarfaj A.A., Arshad M., Sholkamy E.N., Munusamy M.A. Braz Arch Biol Tech. 2015;58:781–788. [Google Scholar]
- 3.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 4.Ansari S., Fatma T. PLoS One. 2016;11:e0158168. doi: 10.1371/journal.pone.0158168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berger E., Ramsay B.A., Ramsay J.A., Chavarie C., Braunegg G. Biotechnol Tech. 1989;3:227–232. [Google Scholar]
- 6.Born V.R. J Gen Microbiol. 1952;6:344. doi: 10.1099/00221287-6-3-4-344. [DOI] [PubMed] [Google Scholar]
- 7.Brinda A.D., Nachiyar C.V., Kaviyarasi T., Samrot A.V. Int J Pharm Sci. 2014;7:140–144. [Google Scholar]
- 8.Chen G.Q., Zhang G., Park S.J., Lee S.Y. Appl Microbiol Biotechnol. 2001;57:50–55. doi: 10.1007/s002530100755. [DOI] [PubMed] [Google Scholar]
- 9.Choi J.I., Lee S.Y. Biotechnol Bioeng. 1999;62:546–553. doi: 10.1002/(sici)1097-0290(19990305)62:5<546::aid-bit6>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 10.Croux C., Canard B., Goma G., Soucaille P. J Gen Microbiol. 1992;138:861–869. doi: 10.1099/00221287-138-5-861. [DOI] [PubMed] [Google Scholar]
- 11.Daly S.R., Fathi A., Bahramian B., Manavitehrani I., McClure D.D., Valtchev P. J Supercrit Fluids. 2018;135:84–90. [Google Scholar]
- 12.Divyashree M.S., Shamala T.R., Rastogi N.K. Biotechnol Bioprocess Eng. 2009;14:482–489. [Google Scholar]
- 13.Dubey S., Bharmoria P., Gehlot P.S., Agrawal V., Kumar A., Mishra S. ACS Sustain Chem Eng. 2017 [Google Scholar]
- 14.Duggleby R.G. Design and analysis of enzyme and pharmacokinetic experiments. In: Endrenyi L., editor. Kinetic data analysis. Plenum Press; New York and London: 1981. pp. 1–24. [Google Scholar]
- 15.Ensign J.C., Wolfe R.S. J Bacteriol. 1966;91:524–534. doi: 10.1128/jb.91.2.524-534.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.A.M. Escalona, F.R. Varela, A.M. Gomis. US Patent 5536419, 1996.
- 17.Ghatnekar M.S., Pai J.S., Ganesh M. J Chem Technol Biotechnol. 2002;77:444–448. [Google Scholar]
- 18.Ghuysen J.M., Tipper D.J., Strominger J.L. Methods Enzymol. 1966;8:685–699. [Google Scholar]
- 19.Ghuysen J.M., Dierickx L., Coyette J., Leyh-Bouille M., Guinand M., Campbell J.N. Biochemistry. 1969;8:13–222. doi: 10.1021/bi00829a031. [DOI] [PubMed] [Google Scholar]
- 20.Hahn S.K., Chang Y.K., Lee S.Y. Appl Environ Microbiol. 1995;61:34–39. doi: 10.1128/aem.61.1.34-39.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hahn S.K., Chang Y.K., Kim B.S., Lee K.M., Chang H.N. Biotechnol Tech. 1993;7:209–212. [Google Scholar]
- 22.Hahn S.K., Chang Y.K., Kim B.S., Chang H.N. Biotechnol Bioeng. 1994;44:256–261. doi: 10.1002/bit.260440215. [DOI] [PubMed] [Google Scholar]
- 23.Harrison S.T.L. Biotechnol Adv. 1991;9:217–240. doi: 10.1016/0734-9750(91)90005-g. [DOI] [PubMed] [Google Scholar]
- 24.Hejazi P., Vasheghani-Farahani E., Yamini Y. Biotechnol Prog. 2003;19:1519–1523. doi: 10.1021/bp034010q. [DOI] [PubMed] [Google Scholar]
- 25.Hwang K.J., You S.F., Don T.M. J Chin Inst Chem Eng. 2006;37:209–216. [Google Scholar]
- 26.Israni N., Shivakumar S. Int J Pharm Biol Sci. 2013;4(3B):934–945. [Google Scholar]
- 27.Israni N., Shivakumar S. J Sci Ind Res. 2015;74:290–295. [Google Scholar]
- 28.Israni N., Shivakumar S. J Bionanosci. 2016;10:381–389. [Google Scholar]
- 29.Jacquel N., Lo C.W., Wei Y.H., Wu H.S., Wang S.S. Biochem Eng J. 2008;39:15–27. [Google Scholar]
- 30.Jung I.L., Phyo K.H., Kim K.C., Park H.K., Kim I.G. Res Microbiol. 2005;156:865–873. doi: 10.1016/j.resmic.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 31.Kapritchkoff F.M., Viotti A.P., Alli R.C.P., Zuccolo M., Pradella J.G.C., Maiorano A.E. J Biotechnol. 2006;122:453–462. doi: 10.1016/j.jbiotec.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 32.Koller M., Niebelschütz H., Braunegg G. Eng Life Sci. 2013;13:549–562. [Google Scholar]
- 33.Kunasundari B., Arza C.R., Maurer F.H.J., Murugaiyah V., Kaur G., Sudesh K. Sep Purif Technol. 2017;172:1–6. [Google Scholar]
- 34.Kunasundari B., Sudesh K. eXPRESS Polym Lett. 2011;5:620–634. [Google Scholar]
- 35.Lakshman K., Shamala T.R. Enzyme Microb Technol. 2006;39:1471–1475. [Google Scholar]
- 36.Law J.H., Slepecky R.A. J Bacteriol. 1961;82:33–36. doi: 10.1128/jb.82.1.33-36.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee K.M., Chang H.N., Chang Y.K., Kim B.S., Hahn S.K. Biotechnol Tech. 1993;7:295–300. [Google Scholar]
- 38.P.E. Mantellato, N.A.S. Durao. US Patent No. 20080193987 A1, 2008.
- 39.Moorkoth D., Nampoothiri K.M. Bioresour Technol. 2016;201:253–260. doi: 10.1016/j.biortech.2015.11.046. [DOI] [PubMed] [Google Scholar]
- 40.Muggleton P.W., Webb M. Biochim Biophys Acta. 1952;8:431. doi: 10.1016/0006-3002(52)90069-3. [DOI] [PubMed] [Google Scholar]
- 41.Mumtaz T., Abd-Aziz S., Lai Yee P., Wan Yunus W.M.Z., Shirai Y., Hassan M.A. Int J Polym Anal Ch. 2010;15:329–340. [Google Scholar]
- 42.Murugan P., Han L., Gan C.Y., Maurer F.H., Sudesh K.J. J Biotechnol. 2016;239:98–105. doi: 10.1016/j.jbiotec.2016.10.012. [DOI] [PubMed] [Google Scholar]
- 43.Nonato R., Mantelatto P., Rossell C. Appl Microbiol Biotechnol. 2001;57:1–5. doi: 10.1007/s002530100732. [DOI] [PubMed] [Google Scholar]
- 44.Ong S.Y., Kho H.P., Riedel S.L., Kim S.W., Gan C.Y., Taylor T.D. J Biotechnol. 2018;265:31–39. doi: 10.1016/j.jbiotec.2017.10.017. [DOI] [PubMed] [Google Scholar]
- 45.Petit J.F., Munoz E., Ghuysen M. Biochemistry. 1966;5:2764–2776. doi: 10.1021/bi00872a037. [DOI] [PubMed] [Google Scholar]
- 46.Rameshwari R., Meenakshisundaram M. Int J Pure App Biosci. 2014;2:68–80. [Google Scholar]
- 47.Ramsay J.A., Berger E., Voyer R., Chavarie C., Ramsay B.A. Biotechnol Tech. 1994;8:589–594. [Google Scholar]
- 48.Rodriguez-Valera F., Lillo J.A.G. FEMS Microbiol Rev. 1992;103:181–186. [Google Scholar]
- 49.Senthil K.B., Prabakaran G. Ind J Biotechnol. 2006;5:76–79. [Google Scholar]
- 50.Sharma M. Int J Curr Microbiol App Sci. 2014;3:801–832. [Google Scholar]
- 51.Strominger J.L., Izaki K., Matsuhashi M., Tipper D.J. Federation Proc. 1967;26:9–22. [PubMed] [Google Scholar]
- 52.Sumantha A., Sandhya C., Szakacs G., Soccol C.R., Pandey A. Food Technol Biotechnol. 2005;43:313–319. [Google Scholar]
- 53.Suzuki K., Uyeda Masaru, Shibata Motoo. Agric Biol Chem. 1985;49:1719–1726. [PubMed] [Google Scholar]
- 54.Tamer I.M., Moo-Young M., Chisti Y. Ind Eng Chem Res. 1998;37:1807–1814. [Google Scholar]
- 55.Tendulkar S.R., Saikumari Y.K., Patel V., Raghotama S., Munshi T.K., Balaram P. J Appl Microbiol. 2007;103:2331–2339. doi: 10.1111/j.1365-2672.2007.03501.x. [DOI] [PubMed] [Google Scholar]
- 56.van Hee P., Elumbaring A.C.M.R., van der Lans R.G.J.M., Van der Wielen L.A.M. J Coloid Interf Sci. 2006;297:595–606. doi: 10.1016/j.jcis.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 57.Welsch M. J Gen Microbiol. 1947;18:491–497. doi: 10.1099/00221287-18-2-491. [DOI] [PubMed] [Google Scholar]
- 58.Whitaker D.R., Jurasek L., Roy C. Biochem Biophys Res Commun. 1966;24:173–178. doi: 10.1016/0006-291x(66)90715-7. [DOI] [PubMed] [Google Scholar]
- 59.Whitaker D.R. Academic Press; New York: 1970. Methods in enzymology; pp. 599–613. [Google Scholar]
- 60.Wilkinson G.N. Biochem J. 1961;80:324–332. doi: 10.1042/bj0800324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yokogawa K., Kawata S., Yoshimura Y. Agric Biol Chem. 1972;37:799–808. [Google Scholar]
- 62.Yoshimoto T., Tsuru D. J Biochem. 1972;72:379. doi: 10.1093/oxfordjournals.jbchem.a129913. [DOI] [PubMed] [Google Scholar]