

Abstract
Ubiquitin-specific protease 44 (USP44) has been implicated in tumor progression and metastasis across various tumors. However, the function of USP44 in prostate cancers and regulatory mechanism of histone-modifying enzymes by USP44 in tumors is not well-understood. Here, we found that enhancer of zeste homolog 2 (EZH2), a histone H3 lysine 27 methyltransferase, is regulated by USP44. We showed that EZH2 is a novel target of USP44 and that the protein stability of EZH2 is upregulated by USP44-mediated deubiquitination. In USP44 knockdown prostate cancer cells, the EZH2 protein level and its gene silencing activity were decreased. Furthermore, USP44 knockdown inhibited the tumorigenic characteristics and cancer stem cell-like behaviors of prostate cancer cells. Inhibition of tumorigenesis caused by USP44 knockdown was recovered by ectopic introduction of EZH2. Additionally, USP44 regulates the protein stability of oncogenic EZH2 mutants. Taken together, our results suggest that USP44 promotes the tumorigenesis of prostate cancer cells partly by stabilizing EZH2 and that USP44 is a viable therapeutic target for treating EZH2-dependent cancers.
Keywords: EZH2, prostate cancer cells, protein stability, USP44
INTRODUCTION
Ubiquitin-specific protease 44 (USP44) has been known as a tumor suppressor that prevents chromosome missegregation by preventing premature anaphase onset (Holland and Cleveland, 2012; Stegmeier et al., 2007). USP44 directly deubiquitinates the anaphase-promoting complex (APC) coactivator Cdc20 to stabilize the APC-inhibitory Mad2–Cdc20 complex. However, recent studies showed that the function of USP44 in cancer is complex and context-dependent. In colorectal neoplasia, epigenetic inactivation of USP44 by CpG island promoter methylation was verified as a frequent and early event (Sloane et al., 2014). In breast cancer, USP44 was upregulated in cancer stem cell (CSC) subpopulations and contributed to aggressive behaviors by promoting vasculogenic mimicry (Liu et al., 2015). Moreover, USP44 promoted the malignant progression of gliomas by stabilizing the oncoprotein securin (Zou et al., 2017). Interestingly, USP44 was shown to regulate histones, and Sowa et al. reported its interaction with histones H2A and H2B (Sowa et al., 2009). Additionally, USP44 contributes to gene repression by H2B deubiquitination and is required for the invasiveness of triple-negative breast cancer cells (Lan et al., 2016).
Enhancer of zeste homolog 2 (EZH2) is a histone-modifying enzyme that promotes transcriptional repression by methylating lysine 27 of histone H3 (H3K27) as a component of the polycomb repressive complex 2 (PRC2) (Margueron and Reinberg, 2011). Accumulating evidence indicates that EZH2 is involved in the development and progression of various cancers. By acting as a core component of PRC2, EZH2 modulates the expression of many genes involved in cell cycle regulation and DNA repair, thereby promoting tumorigenesis (Kim and Roberts, 2016). Overexpression and functional changes in EZH2 are frequently observed in various types of cancer (Hu et al., 2010; Melling et al., 2015; Varambally et al., 2002; Zingg et al., 2015). PRC2-independent transcriptional activity of EZH2 has also been reported, promoting the transcription of AR in prostate cancer and nuclear factor-κB target genes in breast cancer (Lee et al., 2011; Xu et al., 2012). Recently, a tumor-promoting function independent of the catalytic activity of EZH2 was reported in SWI-SNF mutant cancers (Kim et al., 2015).
Based on the regulation of USP44 in histone function, we examined whether USP44 participates in regulating histone modifying enzymes. To identify the histone-modifying enzymes regulated by USP44, we screened a panel of several histone-modifying enzymes for their interactions with USP44 and identified that EZH2 is a novel target of USP44. USP44 deubiquitinates and stabilizes EZH2 protein. Furthermore, USP44 promotes the malignancies of prostate cancer cells partly by stabilizing EZH2. Collectively, we found that the cellular protein levels of EZH2, which is important for its oncogenic function, can be controlled by modulating USP44 levels and activity.
MATERIALS AND METHODS
Cell culture and cell lines
PC3 and HEK293T cells were maintained in Dulbecco’s modified Eagle medium (DMEM), DU145 cells were maintained in RPMI 1640. All media were supplemented with 10% FBS, an antibiotic-antimycotic solution (100 U/ml penicillin, 0.1 mg/ml streptomycin, and 0.25 mg/ml amphotericin B) and grown at 37°C under standard cell culture conditions (5% CO2, 95% humidity).
Antibodies
The following antibodies were used: USP44 (SC-377203), β-catenin (SC-7963), vimentin (SC-32322), Nanog (SC-134218), BMI1 (SC-390443), Sox2 (SC-17320) (all from Santa Cruz Biotechnology), EZH2 (#4905), Oct4 (#2750) (both from Cell Signaling Technology), H3K27me3 (EDM Millipore, #07-449), E-cadherin (BD Transduction Laboratories, #610181), Klf4 (Abcam, Cambridge, ab151733), HA (Covance, MMS-101R), Xpress (Invitrogen, P/N 46-0528), Flag M2 (Sigma-Aldrich, F3165), and β-actin (Sigma-Aldrich, A1978)
Generation of USP44 knockdown stable cell lines
Target sequences for small hairpin RNA (shRNA) for USP44 are 5′-AAGCAGUCAUCCUGUUGCAUU-3′ (shRNA1) and 5′-AGGUGGUCAGGACGUAAUAAC-3′ (shRNA2). Stable USP44 shRNA transfectants were generated as previously described (Jang et al., 2011).
Real-time RT-PCR and primers
The quantity of mRNA of specific genes was calculated using the ΔΔCt method and normalized to GAPDH. All measurements were performed in triplicate. The sequences of the primer pairs were as follows: USP44 5′-TGAGTACAACTG GTTTGGAGGA-3′ and 5′-CAGCCATGTCTGGTTACTGAAA-3′ (Sloane et al., 2014), EZH2 5′-TTCATGCAACACCCAACAC TT-3′ and 5′-GGTGGGGTCTTTATCCGCTC-3′ (Peng et al., 2015), E-cadherin 5′-GTCACTGACACCAACGATAATCCT-3′ and 5′-TTTCAGTGTGGTGATTACGACGTTA-3′ (Ye et al., 2010), ADRB2 5′-TTCCTCTTTGCATGGAATTTG-3′ and 5′-AGAGGAGTGGGGGAA GAGTC-3′ (Yu et al., 2007), SLIT2 5′-GCGGCGGGGAAAGATGC-3′ and 5′-AGCGCCAGCCCGT GACAG-3′ (Yu et al., 2010), DAB2IP 5′-TGGACGATGTGCT CTATGCC-3′ and 5′-GGATGGTGATGGTTTGGTAG-3′ (Chen et al., 2005). CD44 5′-GACAAGTTTTGGTGGCACG-3′ and 5′-CACGTGGAATACACCTGCAA-3′ (Swarts et al., 2013), CD133 5′-CACTACCAAGGACAAGGCGT-3′ and 5′-TCCTTG ATCGCTGTTGCCAT-3′ (Le et al., 2013).
Wound healing, transwell migration, and matrigel invasion assays
Wound healing, migration, and matrigel invasion assays were conducted as previously described (Jang et al., 2011).
Sphere formation assay
Stable cells were dissociated into single cells and seeded into 24-well Ultra-low Attachment plates (Corning Incorporated) at a density of 200 cells/well and cultured in serum-free DMEM/F12K media supplemented with 4 μg/ml insulin, B27, and 20 ng/ml EGF and bFGF. Sphere formation capacity was assessed as the number of spheres with a diameter exceeding 200 μm counted after 14 days under a microscope at 10× magnification.
Drug resistance assay
A total of 5 × 104 PC3 or DU145 stable cells was added to a 6-well plate. Twenty-four hours after seeding, the cells were treated with different concentrations of doxorubicin or etoposide. After treatment for 24 h, viable cells were counted by the trypan blue-exclusion assay.
Immunocytochemistry
The cells plated on PLL-coated glass coverslips were fixed with 2% formaldehyde in phosphate-buffered saline (PBS) for 30 min at room temperature, followed by permeabilization with 0.5% Triton X-100 in PBS. All subsequent dilutions and washes were carried out with PBS containing 0.1% Triton X-100 (PBST). Nonspecific binding sites were saturated by incubation with 3% horse serum and 10% gelatin in PBST for 30 min. The cells were incubated with primary antibody overnight and washed with PBST four times at 10-min intervals. Fluorescein isothiocyanate-or tetramethylrhodamine isothiocyanate-conjugated secondary antibody (Jackson Laboratories) were incubated with the cells for 1 h and washed with PBST four times at 10-min intervals. The coverslips were mounted in Vectashield with DAPI (Vector Laboratories) and the cells were visualized with a Zeiss Axio-vision/LSM 510 META inverted confocal microscope.
RESULTS
EZH2 is a new binding partner of USP44
To identify the histone-modifying enzymes regulated by USP44, we screened a panel of several histone-modifying enzymes for their interactions with USP44 by immunoprecipitation assay (Supplementary Fig. S1). We found that USP44 interacted with EZH2 and the interaction between USP44 and EZH2 was dependent on USP44 catalytic activity (Figs. 1A and 1B). EZH2 binding to USP44 was only detected for wild-type USP44, but not for the USP44 catalytic mutant (C282A) with disabled deubiquitinating activity. In the metastatic prostate cancer cell line DU145, we verified the endogenous interaction between USP44 and EZH2 (Fig. 1C). We next confirmed the nuclear co-localization of USP44 and EZH2 in PC3 and DU145 cells by immunocytochemistry (Fig. 1D). In DU145 cells, the ectopically expressed wild-type and USP44 catalytic mutant resided in the nucleus, indicating that the lack of an interaction between USP44 catalytic mutant and EZH2 was not due to a difference in cellular localization (Fig. 1E).
Fig. 1. EZH2 interacts with USP44.
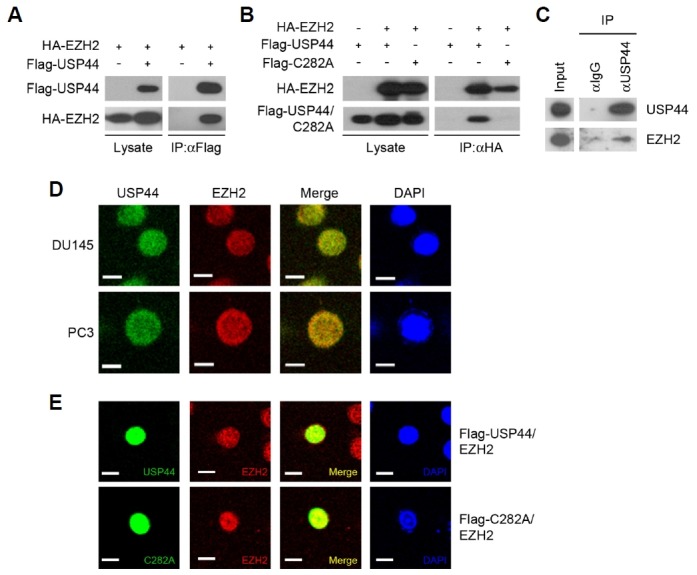
(A) HEK293T cells were transfected as indicated. Each cell lysate was immunoprecipitated with a Flag antibody followed by immunoblotting with Flag and HA antibodies. (B) HEK293T cells were transfected as indicated. Each cell lysate was immunoprecipitated with HA antibody followed by immunoblotting with Flag and HA antibodies. (C) Immunoprecipitation of USP44 from DU145 cell extract using an USP44 antibody followed by immunoblotting with USP44 and EZH2 antibodies. (D) Immunofluorescent staining of USP44 and EZH2 in DU145 and PC3 cells. USP44 was stained green and EZH2 was stained red. (E) DU145 cells were transfected with Flag-USP44 or Flag-USP44 C282A. Flag-USP44 or Flag-USP44 C282A was stained green and EZH2 was stained red. The blue signal represents nuclear DNA stained by DAPI. The bar indicates 10 μm.
USP44 deubiquitinates and stabilizes EZH2 protein
To characterize the functional interaction between USP44 and EZH2, we investigated the regulation of EZH2 protein stability by USP44. Ectopically expressed EZH2 was stabilized by wild-type USP44, but not by the USP44 catalytic mutant (Fig. 2A). Moreover, knockdown of USP44 by specific shRNAs destabilized endogenous EZH2 protein (Fig. 2B). To further confirm the increase in EZH2 protein stability by USP44, the EZH2 protein level was determined after inhibiting protein synthesis with cycloheximide (CHX) in the presence of the wild-type or USP44 catalytic mutant. USP44 overexpression strongly increased the stability of EZH2 protein compared to the control (Fig. 2C). However, the USP44 catalytic mutant did not stabilize EZH2 after CHX treatment. To further examine the regulatory effects of USP44 on EZH2 protein stability, we investigated whether EZH2 is deubiquitinated by USP44. As shown in Fig. 2D, EZH2 was deubiquitinated by USP44. In contrast, knockdown of USP44 by shRNA increased the ubiquitination of EZH2 (Fig. 2E). These results indicate that USP44 regulates the ubiquitination status of EZH2 and thereby EZH2 protein stability.
Fig. 2. EZH2 protein is stabilized by USP44.
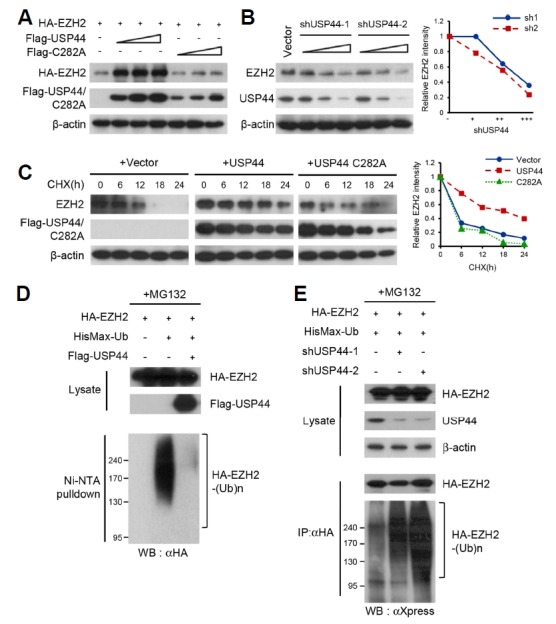
(A) HEK293T cells were transfected as indicated and each cell lysate was immunoblotted to detect HA-EZH2 and Flag-USP44. (B) USP44 shRNA was transfected as indicated and protein from each cell lysate was immunoblotted to detect EZH2 and USP44. Line graph representing the relative protein levels of EZH2 normalized to β-actin. (C) Mock or Flag-USP44/USP44 C282A-expressing HEK293T cells were treated with cycloheximide (CHX) for the indicated times. Equal amounts of protein were used to detect EZH2 by immunoblotting. Line graph representing the relative protein levels of EZH2 normalized to β-actin using a densitometer and expressed as the relative intensity compared to the non-treated control. (D) HEK293T cells were transfected as indicated and treated with MG132 for 12 h before harvesting. Each cell lysate was subjected to Ni-NTA pulldown followed by immunoblotting with HA antibody. (E) HEK293T cells were transfected as indicated and treated with MG132 for 12 h before harvesting. Each cell lysate was subjected to immunoprecipitation with HA antibody followed by immunoblotting with Xpress and HA antibodies.
USP44 knockdown activates EZH2 repressive target genes in prostate cancer cells
A study showed that EZH2 overexpression was associated with the worsening of hormone-refractory, metastatic prostate cancer (Varambally et al., 2002). To gain insight into the biological importance of EZH2 stabilization by USP44, we investigated the USP44 expression level in prostate cancer cell lines. In cells with more malignant and metastatic properties such as PC3 and DU145, USP44 protein expression was higher than in benign and weakly metastatic cell lines including RWPE1, RWPE2, and LNCaP (Fig. 3A). To characterize the physiological function of the USP44-mediated increase of EZH2 protein stability, we generated USP44 knockdown PC3 and DU145 stable cell lines. In these USP44 knockdown cells, the level of EZH2 protein and H3K27me3 was decreased (Fig. 3B). However, USP44 knockdown did not decrease the EZH2 mRNA level, indicating that USP44 regulates EZH2 at the protein level (Fig. 3C). In prostate cancer, the role of EZH2 in tumor progression is indicated by its transcriptional repression of multiple genes, including Ecadherin, SLIT2, DAB2IP, and ADRB2 (Cao et al., 2008; Chen et al., 2005; Yu et al., 2007; 2010). Given that EZH2 protein and its catalytic product H3K27me3 are decreased in USP44 knockdown cells, we investigated the expression of EZH2 repressive target genes following USP44 knockdown. In USP44 knockdown cells, the expression of E-cadherin, DAB2IP, ADRB2, and SLIT2 was upregulated as compared to the control (Fig. 3D). These results indicate that USP44 increases both the protein level and gene silencing activity of EZH2 in prostate cancer cells.
Fig. 3. USP44 knockdown activates the expression of EZH2 repressive target genes.
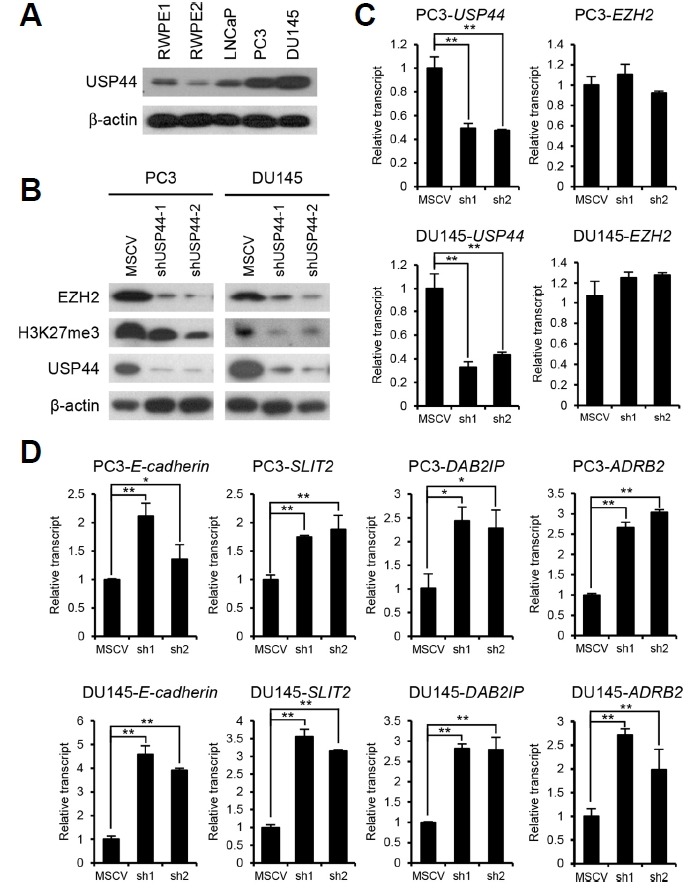
(A) Equal amounts of protein from each prostate cancer cell line were immunoblotted with USP44 antibody. (B) Equal amounts of protein from each USP44 knockdown stable cell line were immunoblotted with EZH2, H3K27me3, and USP44 antibodies. (C) Real-time RT-PCR analysis of USP44 and EZH2 mRNAs in each of the USP44 knockdown stable cell lines. Values are expressed as the mean ± SD of three independent experiments. (D) Real-time RT-PCR analysis on the transcript levels of E-cadherin, ADRB2, SLIT2, and DAB2IP in USP44 knockdown stable cell lines. Values are expressed as the mean ± SD of three independent experiments. The p value is shown from a Student’s t-test. *p < 0.05, **p < 0.01
USP44 increases the tumorigenic abilities of prostate cancer cells
USP44 was initially identified as a tumor suppressor that regulates the spindle checkpoint by preventing premature anaphase onset. According to recent studies, the function of USP44 in tumorigenesis is diverse depending on the cell type. However, the function of USP44 in prostate cancer progression has not been reported. The activation of EZH2 repressive target genes in USP44 knockdown cells raised the possibility that USP44 affects the tumorigenic abilities of prostate cancer cells. During tumor progression, epithelial-to-mesenchymal transition (EMT) is important for cancer cells to acquire invasive and metastatic capabilities (Thiery, 2002). To determine whether USP44 affects the EMT in prostate cancer cells, we examined the expression of EMT markers in USP44 knockdown cells. The expression of E-cadherin and β-catenin, epithelial markers, was increased, while the mesenchymal marker vimentin was decreased in USP44 knockdown cells (Fig. 4A). Next, we evaluated whether USP44 knockdown affects the cell migration and invasiveness of prostate cancer cells. Wound healing was inhibited in USP44 knockdown cells (Fig. 4B, Supplementary Fig. S2). Moreover, transwell migration assays indicated that USP44 knockdown resulted in decreased cell migration through the filter (Fig. 4C). We also conducted quantitative in vitro invasion assays to assess the effect of USP44 on the ability of prostate cancer cells to invade matrigel (Fig. 4D). Knockdown of USP44 decreased the invasive potential of metastatic PC3 and DU145 cells. Overall, USP44 knockdown was successful in decreasing the wound healing, migration, and invasion of prostate cancer cells.
Fig. 4. USP44 knockdown decreases the tumorigenic abilities of prostate cancer cell lines.

(A) Expression of E-cadherin, β-catenin, and vimentin in USP44 knockdown stable cell lines was examined by immunoblotting. (B) Wound healing assays of USP44 knockdown stable cell lines. Monolayers of stable cell lines were scratched with a yellow micropipette tip. Values are expressed as the mean ± SD of three independent experiments. (C) Migration assays of USP44 knockdown stable cell lines. Equal amounts of cells from the stable cell lines were seeded into the transwell migration chamber. After 22 h, the number of cells that had migrated to the lower chamber was counted. Values are expressed as the mean ± SD of three independent experiments. (D) Matrigel invasion assays of USP44 knockdown stable cell lines. Equal numbers of cells from the stable cell lines were seeded into the matrigel-coated chamber. After 22 h, the invaded cells on the lower surface of the membrane were counted. Values are expressed as the mean ± SD of three independent experiments. The p value is shown from a Student’s t-test. *p < 0.05, **p < 0.01
USP44 upregulates the CSC characteristics of prostate cancer cells
USP44 has been suggested as a marker of breast cancer stem cells and was implicated in the regulation of embryonic stem cell differentiation by H2Bub deubiquitination (Fuchs et al., 2012; Liu et al., 2015). To investigate whether USP44 regulates the CSC characteristics of prostate cancer cells, we assessed the effects of USP44 knockdown on CSC-like behaviors such as pluripotent stem cell marker expression, self-renewal ability, and drug resistance. In USP44 knockdown cells, the expression of pluripotent stem cell markers including Oct4, Nanog, and BMI1 was decreased. However, the expression of Klf4 and Sox2 was not changed by USP44 knockdown (Fig. 5A). Cell surface markers are often used to verify and analyze CSC populations. USP44 knockdown reduced the expression of CD44 and CD133, which are known to be highly expressed in prostate CSCs (Fig. 5B). We next performed sphere formation assays to determine the effect of USP44 on the self-renewal capacity of prostate cancer cells. USP44 knockdown clearly decreased the size and number of spheres formed (Fig. 5C). CSCs typically enable tumors to survive exposure to chemotherapeutic agents. Thus, we assessed the susceptibility of prostate cancer cells to chemotherapeutic drugs following USP44 knockdown. Decreased USP44 expression sensitized prostate cancer cells to exposure to doxorubicin or etoposide (Fig. 5D). These observations indicate that USP44 plays key roles in upregulating the CSC-like properties of prostate cancer cells.
Fig. 5. USP44 knockdown inhibits the CSC-like properties of prostate cancer cells.
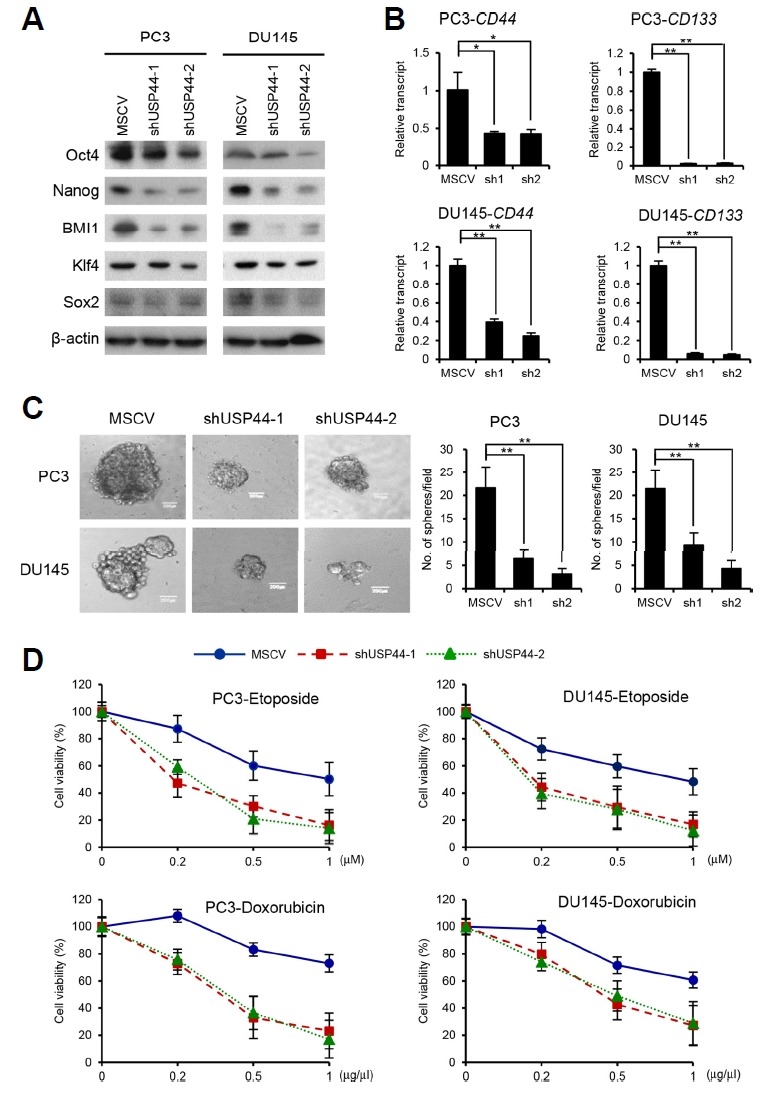
(A) Expression of pluripotent CSC markers including Oct4, Nanog, Bmi1, Klf4, and Sox2 were examined by immunoblotting in USP44 knockdown stable cell lines. (B) Real-time RT-PCR analysis on the transcript levels of CD44 and CD133 in USP44 knockdown stable cell lines. Values are expressed as the mean ± SD of three independent experiments. (C) Self-renewal potency was evaluated by the formation of tumor spheres in USP44 knockdown stable cell lines. Stable cells were seeded in 24-well Ultra-low Attachment plates and cultured in serum-free media. The number of spheres larger than 200 μm in diameter was counted. The figure shows the representative images of each stable cell line and each scale bar is 200 μm. Values are expressed as the mean ± SD of three independent experiments. (D) Equal numbers of cells from USP44 knockdown stable cell lines were seeded into 6-well plates. Twenty-four hours after seeding, the cells were treated with varying concentrations of doxorubicin or etoposide. Twenty-four hours after treatment, viable cells were counted by the trypan blue-exclusion assay. The p value is shown from a Student’s t-test. *p < 0.05, **p < 0.01
USP44 promotes prostate cancer cell tumorigenesis through EZH2 stabilization
We demonstrated that USP44 increases the tumorigenic abilities and CSC-like behaviors of prostate cancer cells. To assess whether USP44’s effect on oncogenesis are attributed to EZH2 protein stabilization, EZH2 was introduced in USP44 knockdown prostate cancer cells (Supplementary Fig. S3). Introduction of EZH2 significantly rescued the USP44 knockdown-induced suppression of wound healing, migration, and invasion activity both in PC3 and DU145 stable cell lines (Figs. 6A–6C, Supplementary Figs. S4A–S4C). Furthermore, the decrease in the size and number of spheres by USP44 knockdown was restored to the control level by EZH2 introduction (Fig. 6D, Supplementary Fig. S4D). In summary, overexpression of EZH2 restored the tumorigenic abilities that were decreased following USP44 knockdown in prostate cancer cells. These results demonstrate that USP44 promotes prostate cancer cell tumorigenesis, in part by stabilizing EZH2.
Fig. 6. USP44 promotes the malignancies of prostate cancer cells through EZH2 stabilization.
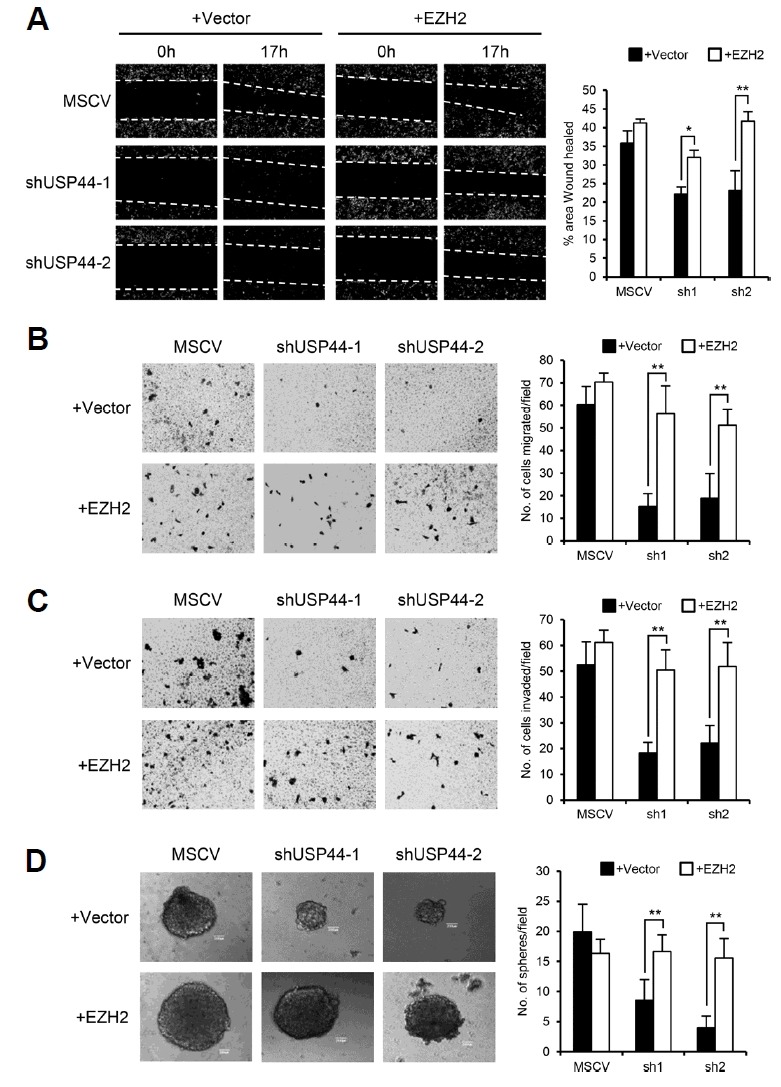
(A) Wound healing assays of USP44 knockdown PC3 stable cell lines after vector or EZH2 overexpression. Each cell monolayer was scratched with a yellow micropipette tip and cell images were acquired at the time points indicated. Values are expressed as the mean ± SD of three independent experiments. (B) Migration assays of USP44 knockdown PC3 stable cell lines after vector or EZH2 overexpression. Equal numbers of each cell were seeded in the transwell migration chamber. After 22 h, the number of cells that had migrated to the lower chamber was counted. Values are expressed as the mean ± SD of three independent experiments. (C) Matrigel invasion assays of USP44 knockdown PC3 stable cell lines after vector or EZH2 overexpression. Equal numbers of each cell were seeded onto the matrigel-coated chamber. After 22 h, the invaded cells on the lower surface of the membrane were counted. Values are expressed as the mean ± SD of three independent experiments. (D) Sphere formation assays of the USP44 knockdown PC3 stable cell lines after vector or EZH2 overexpression. Equal numbers of each cell were seeded into 24-well Ultra-low Attachment plates and cultured in serum-free media. The number of spheres larger than 200 μm in diameter were counted. The figure shows representative images from each cell and the scale bar is 200 μm. Values are expressed as the mean ± SD of three independent experiments. The p value is shown from a Student’s t-test analysis. *p < 0.05, **p < 0.01
USP44 stabilizes oncogenic EZH2 mutant protein
Next, we investigated the USP44-mediated regulation of the protein stability of several EZH2 mutants. Gain-of-function mutations within the EZH2 catalytic SET domain have been recurrently found in some lymphomas. For instance, replacement of tyrosine 641 occurs in 21.7% of diffuse large B-cell lymphomas and 7.2% of follicular lymphomas (Morin et al., 2010). Mutation of EZH2 Y641 (to Y641F, Y641N, Y641S, or Y641H) contributes to augmented conversion of H3K27 to the trimethylated form by cooperation with wild-type EZH2 (Sneeringer et al., 2010). The EZH2 mutations A677G and A687V have also been identified in non-Hodgkin’s lymphomas and they promote the hypertrimethylation of H3K27 (Majer et al., 2012; McCabe et al., 2012). Given that these EZH2 mutants contribute to lymphoma pathogenesis, we investigated the effects of USP44 on the protein stability of oncogenic EZH2 mutants. All gain-of-function EZH2 mutants tested in this study bound to USP44 with variations in affinity (Fig. 7A). Binding affinity to USP44 was strong for the Y641N, Y641S, and A677G mutants compared to the wild-type or A687V mutant. USP44 overexpression increased the protein level of each EZH2 mutant (Fig. 7B, Supplementary Fig. S5A). In contrast, USP44 knockdown reduced the EZH2 mutant protein level (Fig. 7C, Supplementary Fig. S5B). Prolonged treatment with the highly potent EZH2 enzymatic inhibitor EI1 in diffuse large B-cell lymphoma, which contains a heterozygous EZH2 Y641N mutation, caused the development of two novel secondary EZH2 mutations (Y111L and Y661D) in resistant cells (Gibaja et al., 2016). Given that Y661D occurred cis to the EZH2 Y641N allele and Y111L occurred on the EZH2 wild-type allele, we generated the Y111L and Y641N/Y661D EZH2 mutants. These two therapy-resistant EZH2 mutants bound to USP44 (Fig. 7D). USP44 overexpression increased the protein level of Y111L and Y641N/Y661D EZH2 mutant (Fig. 7E, Supplementary Fig. S5C). Additionally, USP44 knockdown decreased the protein stability of these two therapy-resistant EZH2 mutants (Fig. 7F, Supplementary Fig. S5D). Collectively, USP44 stabilizes both wild-type and oncogenic mutant EZH2 protein. These results suggest that USP44 can be used as a new therapeutic strategy for treating EZH2-dependent cancers.
Fig. 7. USP44 stabilizes oncogenic EZH2 mutants.
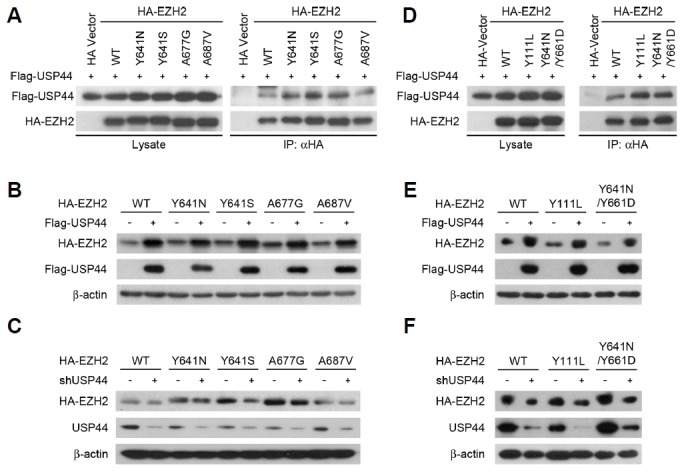
(A) Interaction of USP44 and gain-of-function EZH2 mutants was verified by immunoprecipitation. (B) HEK293T cells were transfected with Flag-USP44 and HA-tagged gain-of-function EZH2 mutants. Equal amounts of protein from each cell lysate were immunoblotted to detect HA-EZH2 mutant and Flag-USP44. (C) HEK293T cells were transfected with shUSP44 and HA-tagged gain-of-function EZH2 mutants. Equal amounts of protein from each cell lysate were immunoblotted to detect HA-EZH2 mutant and USP44. (D) Interaction of USP44 and therapy-resistant EZH2 mutants was verified by immunoprecipitation. (E) HEK293T cells were transfected with Flag-USP44 and therapy-resistant HA-EZH2 mutants. Equal amounts of protein from each cell lysate were immunoblotted to detect HA-EZH2 mutant and Flag-USP44. (F) HEK293T cells were transfected with shUSP44 and HA-tagged therapy-resistant EZH2 mutants. Equal amounts of protein from each cell lysate were immunoblotted to detect HA-EZH2 mutant and USP44.
DISCUSSION
The distinct role of USP44 in tumorigenesis has been reported in various tissue types. However, few studies have examined the role of USP44 in prostate cancer. In this study, we found that USP44 is involved in the progression of prostate cancer in part by increasing EZH2 protein stability. EZH2 overexpression and its association with worsening of prostate cancer were reported previously (Varambally et al., 2002). Interestingly, USP44 is upregulated in the highly metastatic prostate cancer cell lines DU145 and PC3 compared to the benign and weakly metastatic cells. To gain insight into the physiological function of EZH2 stabilization by USP44, we constructed the USP44 knockdown DU145 and PC3 cell lines. In USP44 knockdown cells, EZH2 protein and H3K27me3 levels were reduced. Moreover, USP44 knockdown activated EZH2 repressive target genes. Furthermore, USP44 knockdown inhibited the tumorigenic abilities and CSC-like properties of prostate cancer cells. The inhibition of tumor-promoting activities following USP44 knockdown were rescued by ectopic introduction of EZH2. These results demonstrate that the tumor-promoting activity of USP44 in prostate cancer cells is facilitated in part by EZH2 protein stabilization. However, these analyses were conducted only in vitro studies using prostate cancer cell lines. The function of USP44 identified in this study should be confirmed using in vivo models in further studies.
Several E3 ubiquitin ligases that ubiquitinate EZH2 and facilitate subsequent EZH2 degradation have been reported. For instance, EZH2 phosphorylation by CDK5 kinase is a prerequisite for FBW7-mediated EZH2 ubiquitination and degradation in pancreatic cancer cells, which is consistent with the negative correlation between EZH2 and FBW7 protein levels in human pancreatic cancer specimens (Jin et al., 2017). In prostate cancer, TRAF6 catalyzes the K63-linked polyubiquitination of EZH2, which is blocked upon SKP2 elevation (Lu et al., 2017). Moreover, Jak2-mediated phosphorylation of EZH2 on Y641 directs β-TrCP (FBXW1)-mediated EZH2 ubiquitination and degradation (Sahasrabuddhe et al., 2015). Smurf2-mediated degradation of EZH2 derepresses the peroxisome proliferator-activated receptor-γ expression that is required for neuron differentiation (Yu et al., 2013). Additionally, Praja1 E3 ubiquitin ligase promotes skeletal myogenesis by degrading EZH2 upon p38α activation (Zoabi et al., 2011). Considering the number of E3 ligases found to modulate EZH2, several deubiquitinating enzymes (DUBs) acting in distinct situations are likely to be identified. However, the deubiquitination of EZH2, which reverses the activity of E3 ligases, has not been widely studied. In bladder cancer, USP21 was demonstrated to be an EZH2-stabilizing protein through its DUB activity (Chen et al., 2017). Recently, Zhang et al. screened six DUBs including USP44 as EZH2-interacting DUBs (Zhang et al., 2018). Although USP21 and USP7 were found to bind to EZH2, no binding was identified by Zhang et al. (Chen et al., 2017; de Bie et al., 2010). Among six EZH2-interacting DUBs, ZRANB1 was only demonstrated to deubiquitinate and stabilize the EZH2 protein. However, we verified the deubiquitination and protein stabilization activity of USP44 toward EZH2. The distinct results for deubiquitination and protein stabilization of EZH2 by USP44 between Zhang et al. and our group may be because of differences in the experimental settings and conditions.
As a master epigenetic regulator of cancer development, EZH2 has become an attractive therapeutic target (Kim and Roberts, 2016). Because studies of the involvement of EZH2 in carcinogenesis have mainly been focused on its catalytic activity, EZH2 inhibitors developed for cancer treatments are directed against its methyltransferase activity. However, a recent study demonstrated that the tumor-promoting activity of EZH2 is diverse and complex. Kim et al. demonstrated that SWI/SNF-mutant cancer cells are primarily dependent on the non-catalytic role of EZH2 in stabilizing the PRC2 complex (Kim et al., 2015). Additionally, two novel EZH2 mutations (Y111L and Y661D) that are resistant to multiple EZH2 enzymatic inhibitors were identified in cell line model (Gibaja et al., 2016). These findings raise the concern that enzymatic inhibitors may not fully suppress the oncogenic function of EZH2. As an alternative, the regulatory mechanism mediating EZH2 protein stability may provide a reasonable cancer therapeutic target for efficient inhibition of the various oncogenic activities of EZH2 and overcome the emergence of resistant EZH2 mutations.
In this study, USP44 knockdown reduced both the EZH2 protein level and oncogenic activity of prostate cancer cells. Further, our study demonstrated that USP44 regulates the protein stability of both gain-of-function EZH2 mutants and drug-resistant EZH2 mutants. These findings strongly suggest that inhibition of USP44 is an efficient anticancer strategy regardless of the enzyme activity of EZH2 in EZH2-dependent cancers. USP44-specific inhibitors can be used to overcome not only the dysregulation of EZH2 in cancers but also the emergence of acquired resistant mutations. Unfortunately, specific inhibitors for USP44 are currently unavailable.
In conclusion, we verified the oncogenic function of USP44 in prostate cancer cells, which is mediated by EZH2 stabilization. The cellular levels of EZH2 and its methyltransferase activity, which are important for maintaining the tumorigenic phenotype, can be controlled by modulating USP44 levels and activity. Therefore, USP44 may be a powerful therapeutic target for treating EZH2-dependent cancers.
Supplementary data
ACKNOWLEDGEMENTS
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2013R1A1A2005972 and NRF-2015R1D1A1A01057558).
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Cao Q., Yu J., Dhanasekaran S.M., Kim J.H., Mani R.S., Tomlins S.A., Mehra R., Laxman B., Cao X., Yu J., et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene. 2008;27:7274–7284. doi: 10.1038/onc.2008.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Tu S.W., Hsieh J.T. Down-regulation of human DAB2IP gene expression mediated by polycomb Ezh2 complex and histone deacetylase in prostate cancer. J Biol Chem. 2005;280:22437–22444. doi: 10.1074/jbc.M501379200. [DOI] [PubMed] [Google Scholar]
- Chen Y., Zhou B., Chen D. USP21 promotes cell proliferation and metastasis through suppressing EZH2 ubiquitination in bladder carcinoma. Onco Targets Ther. 2017;10:681–689. doi: 10.2147/OTT.S124795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bie P., Zaaroor-Regev D., Ciechanover A. Regulation of the Polycomb protein RING1B ubiquitination by USP7. Biochem Biophys Res Commun. 2010;400:389–395. doi: 10.1016/j.bbrc.2010.08.082. [DOI] [PubMed] [Google Scholar]
- Fuchs G., Shema E., Vesterman R., Kotler E., Wolchinsky Z., Wilder S., Golomb L., Pribluda A., Zhang F., Haj-Yahya M., et al. RNF20 and USP44 regulate stem cell differentiation by modulating H2B monoubiquitylation. Mol Cell. 2012;46:662–673. doi: 10.1016/j.molcel.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibaja V., Shen F., Harari J., Korn J., Ruddy D., Saenz-Vash V., Zhai H., Rejtar T., Paris C.G., Yu Z., et al. Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene. 2016;35:558–566. doi: 10.1038/onc.2015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland A.J., Cleveland D.W. The deubiquitinase USP44 is a tumor suppressor that protects against chromosome missegregation. J Clin Invest. 2012;122:4325–4328. doi: 10.1172/JCI66420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S., Yu L., Li Z., Shen Y., Wang J., Cai J., Xiao L., Wang Z. Overexpression of EZH2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer Biol Ther. 2010;10:788–795. doi: 10.4161/cbt.10.8.12913. [DOI] [PubMed] [Google Scholar]
- Jang M.J., Baek S.H., Kim J.H. UCH-L1 promotes cancer metastasis in prostate cancer cells through EMT induction. Cancer Lett. 2011;302:128–135. doi: 10.1016/j.canlet.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Jin X., Yang C., Fan P., Xiao J., Zhang W., Zhan S., Liu T., Wang D., Wu H. CDK5/FBW7-dependent ubiquitination and degradation of EZH2 inhibits pancreatic cancer cell migration and invasion. J Biol Chem. 2017;292:6269–6280. doi: 10.1074/jbc.M116.764407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.H., Kim W., Howard T.P., Vazquez F., Tsherniak A., Wu J.N., Wang W., Haswell J.R., Walensky L.D., Hahn W.C., et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat Med. 2015;21:1491–1496. doi: 10.1038/nm.3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.H., Roberts C.W. Targeting EZH2 in cancer. Nat Med. 2016;22:128–134. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan X., Atanassov B.S., Li W., Zhang Y., Florens L., Mohan R.D., Galardy P.J., Washburn M.P., Workman J.L., Dent S.Y.R. USP44 Is an Integral Component of N-CoR that Contributes to Gene Repression by Deubiquitinating Histone H2B. Cell Rep. 2016;17:2382–2393. doi: 10.1016/j.celrep.2016.10.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le H., Zeng F., Xu L., Liu X., Huang Y. The role of CD133 expression in the carcinogenesis and prognosis of patients with lung cancer. Molecular Medicine Reports. 2013;8:1511–1518. doi: 10.3892/mmr.2013.1667. [DOI] [PubMed] [Google Scholar]
- Lee S.T., Li Z., Wu Z., Aau M., Guan P., Karuturi R.K., Liou Y.C., Yu Q. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Mol Cell. 2011;43:798–810. doi: 10.1016/j.molcel.2011.08.011. [DOI] [PubMed] [Google Scholar]
- Liu T., Sun B., Zhao X., Li Y., Zhao X., Liu Y., Yao Z., Gu Q., Dong X., Shao B., et al. USP44+ Cancer Stem Cell Subclones Contribute to Breast Cancer Aggressiveness by Promoting Vasculogenic Mimicry. Mol Cancer Ther. 2015;14:2121–2131. doi: 10.1158/1535-7163.MCT-15-0114-T. [DOI] [PubMed] [Google Scholar]
- Lu W., Liu S., Li B., Xie Y., Izban M.G., Ballard B.R., Sathyanarayana S.A., Adunyah S.E., Matusik R.J., Chen Z. SKP2 loss destabilizes EZH2 by promoting TRAF6-mediated ubiquitination to suppress prostate cancer. Oncogene. 2017;36:1364–1373. doi: 10.1038/onc.2016.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majer C.R., Jin L., Scott M.P., Knutson S.K., Kuntz K.W., Keilhack H., Smith J.J., Moyer M.P., Richon V.M., Copeland R.A., et al. A687V EZH2 is a gain-of-function mutation found in lymphoma patients. FEBS Lett. 2012;586:3448–3451. doi: 10.1016/j.febslet.2012.07.066. [DOI] [PubMed] [Google Scholar]
- Margueron R., Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe M.T., Graves A.P., Ganji G., Diaz E., Halsey W.S., Jiang Y., Smitheman K.N., Ott H.M., Pappalardi M.B., Allen K.E., et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27) Proc Natl Acad Sci USA. 2012;109:2989–2994. doi: 10.1073/pnas.1116418109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melling N., Thomsen E., Tsourlakis M.C., Kluth M., Hube-Magg C., Minner S., Koop C., Graefen M., Heinzer H., Wittmer C., et al. Overexpression of enhancer of zeste homolog 2 (EZH2) characterizes an aggressive subset of prostate cancers and predicts patient prognosis independently from pre- and postoperatively assessed clinicopathological parameters. Carcinogenesis. 2015;36:1333–1340. doi: 10.1093/carcin/bgv137. [DOI] [PubMed] [Google Scholar]
- Morin R.D., Johnson N.A., Severson T.M., Mungall A.J., An J., Goya R., Paul J.E., Boyle M., Woolcock B.W., Kuchenbauer F., et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D., Kryczek I., Nagarsheth N., Zhao L., Wei S., Wang W., Sun Y., Zhao E., Vatan L., Szeliga W., et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249–253. doi: 10.1038/nature15520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahasrabuddhe A.A., Chen X., Chung F., Velusamy T., Lim M.S., Elenitoba-Johnson K.S. Oncogenic Y641 mutations in EZH2 prevent Jak2/beta-TrCP-mediated degradation. Oncogene. 2015;34:445–454. doi: 10.1038/onc.2013.571. [DOI] [PubMed] [Google Scholar]
- Sloane M.A., Wong J.W., Perera D., Nunez A.C., Pimanda J.E., Hawkins N.J., Sieber O.M., Bourke M.J., Hesson L.B., Ward R.L. Epigenetic inactivation of the candidate tumor suppressor USP44 is a frequent and early event in colorectal neoplasia. Epigenetics. 2014;9:1092–1100. doi: 10.4161/epi.29222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneeringer C.J., Scott M.P., Kuntz K.W., Knutson S.K., Pollock R.M., Richon V.M., Copeland R.A. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human Bcell lymphomas. Proc Natl Acad Sci USA. 2010;107:20980–20985. doi: 10.1073/pnas.1012525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa M.E., Bennett E.J., Gygi S.P., Harper J.W. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmeier F., Rape M., Draviam V.M., Nalepa G., Sowa M.E., Ang X.L., McDonald E.R., 3rd, Li M.Z., Hannon G.J., Sorger P.K., et al. Anaphase initiation is regulated by antagonistic ubiquitination and deubiquitination activities. Nature. 2007;446:876–881. doi: 10.1038/nature05694. [DOI] [PubMed] [Google Scholar]
- Swarts D.R., Henfling M.E., Van Neste L., van Suylen R.J., Dingemans A.M., Dinjens W.N., Haesevoets A., Rudelius M., Thunnissen E., Volante M., et al. CD44 and OTP are strong prognostic markers for pulmonary carcinoids. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research. 2013;19:2197–2207. doi: 10.1158/1078-0432.CCR-12-3078. [DOI] [PubMed] [Google Scholar]
- Thiery J.P. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- Varambally S., Dhanasekaran S.M., Zhou M., Barrette T.R., Kumar-Sinha C., Sanda M.G., Ghosh D., Pienta K.J., Sewalt R.G., Otte A.P., et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- Xu K., Wu Z.J., Groner A.C., He H.H., Cai C., Lis R.T., Wu X., Stack E.C., Loda M., Liu T., et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y., Xiao Y., Wang W., Yearsley K., Gao J.X., Shetuni B., Barsky S.H. ERalpha signaling through slug regulates Ecadherin and EMT. Oncogene. 2010;29:1451–1462. doi: 10.1038/onc.2009.433. [DOI] [PubMed] [Google Scholar]
- Yu J., Cao Q., Mehra R., Laxman B., Yu J., Tomlins S.A., Creighton C.J., Dhanasekaran S.M., Shen R., Chen G., et al. Integrative genomics analysis reveals silencing of beta-adrenergic signaling by polycomb in prostate cancer. Cancer Cell. 2007;12:419–431. doi: 10.1016/j.ccr.2007.10.016. [DOI] [PubMed] [Google Scholar]
- Yu J., Cao Q., Yu J., Wu L., Dallol A., Li J., Chen G., Grasso C., Cao X., Lonigro R.J., et al. The neuronal repellent SLIT2 is a target for repression by EZH2 in prostate cancer. Oncogene. 2010;29:5370–5380. doi: 10.1038/onc.2010.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.L., Chou R.H., Shyu W.C., Hsieh S.C., Wu C.S., Chiang S.Y., Chang W.J., Chen J.N., Tseng Y.J., Lin Y.H., et al. Smurf2-mediated degradation of EZH2 enhances neuron differentiation and improves functional recovery after ischaemic stroke. EMBO Mol Med. 2013;5:531–547. doi: 10.1002/emmm.201201783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P., Xiao Z., Wang S., Zhang M., Wei Y., Hang Q., Kim J., Yao F., Rodriguez-Aguayo C., Ton B.N., et al. ZRANB1 Is an EZH2 Deubiquitinase and a Potential Therapeutic Target in Breast Cancer. Cell Rep. 2018;23:823–837. doi: 10.1016/j.celrep.2018.03.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingg D., Debbache J., Schaefer S.M., Tuncer E., Frommel S.C., Cheng P., Arenas-Ramirez N., Haeusel J., Zhang Y., Bonalli M., et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat Commun. 2015;6:6051. doi: 10.1038/ncomms7051. [DOI] [PubMed] [Google Scholar]
- Zoabi M., Sadeh R., de Bie P., Marquez V.E., Ciechanover A. PRAJA1 is a ubiquitin ligase for the polycomb repressive complex 2 proteins. Biochem Biophys Res Commun. 2011;408:393–398. doi: 10.1016/j.bbrc.2011.04.025. [DOI] [PubMed] [Google Scholar]
- Zou Y., Qiu G., Jiang L., Cai Z., Sun W., Hu H., Lu C., Jin W., Hu G. Overexpression of ubiquitin specific proteases 44 promotes the malignancy of glioma by stabilizing tumor-promoter securin. Oncotarget. 2017;8:58231–58246. doi: 10.18632/oncotarget.16447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.