

Abstract
The transcription factor 4 (TCF4) locus is a robust association finding with schizophrenia (SCZ), but little is known about the genes regulated by the encoded transcription factor. Therefore, we conducted chromatin immunoprecipitation sequencing (ChIP-seq) of TCF4 in neural-derived (SH-SY5Y) cells to identify genome-wide TCF4 binding sites, followed by data integration with SCZ association findings. We identified 11 322 TCF4 binding sites overlapping in two ChIP-seq experiments. These sites are significantly enriched for the TCF4 Ebox binding motif (>85% having ≥1 Ebox) and implicate a gene set enriched for genes downregulated in TCF4 small-interfering RNA (siRNA) knockdown experiments, indicating the validity of our findings. The TCF4 gene set was also enriched among (1) gene ontology categories such as axon/neuronal development, (2) genes preferentially expressed in brain, in particular pyramidal neurons of the somatosensory cortex and (3) genes downregulated in postmortem brain tissue from SCZ patients (odds ratio, OR = 2.8, permutation P < 4x10−5). Considering genomic alignments, TCF4 binding sites significantly overlapped those for neural DNA-binding proteins such as FOXP2 and the SCZ-associated EP300. TCF4 binding sites were modestly enriched among SCZ risk loci from the Psychiatric Genomic Consortium (OR = 1.56, P = 0.03). In total, 130 TCF4 binding sites occurred in 39 of the 108 regions published in 2014. Thirteen genes within the 108 loci had both a TCF4 binding site ±10kb and were differentially expressed in siRNA knockdown experiments of TCF4, suggesting direct TCF4 regulation. These findings confirm TCF4 as an important regulator of neural genes and point toward functional interactions with potential relevance for SCZ.
Introduction
Large-scale genome-wide assocation studies (GWAS) have converged on specific risk loci for schizophrenia (SCZ) (1). One of the most robust findings is the transcription factor 4 (TCF4) region on chromosome 18q21.2 (2). First discovered in GWAS meta-analysis (3), the finding remained significant in a follow-up study (4) and a large family-based replication study (5). Most pertinently, SNPs at TCF4 were among the top findings (P = 3.34 × 10−12) in the 2014 Psychiatric Genomics Consortium (PGC) mega-analysis of SCZ (1). Congruent with the association with SCZ, TCF4 has also been associated with SCZ endophenotypes such as neurocognition and sensorimotor gating (6–8).
The biology of TCF4 suggests a plausible role in central nervous system (CNS) disorders: (1) TCF4 encodes a transcription factor abundantly expressed in brain that has been implicated in neuronal development (9) and function (10,11); (2) mutations at TCF4 cause Pitt-Hopkins Syndrome (PHS), a rare genetic disorder characterized by neurological deficits including mental retardation (2, 12–14); (3) balanced chromosomal rearrangements in patients with neurodevelopmental disorders have encompassed TCF4 (15); (4) TCF4 is a target for transcriptional regulation by microRNA 137 (gene ID: MIR137) (16), which is also a top association finding for SCZ (1) and (5) transgenic mice that overexpress TCF4 have cognitive and sensorimotor impairments (17), which mirror deficits observed in SCZ patients. Overall, the biological rationale linking TCF4 to the CNS and SCZ is compelling (2), suggesting that further study of this gene could advance our understanding of SCZ pathogenesis.
The protein encoded by TCF4 is a basic helix-loop-helix (bHLH) transcription factor (TF) that recognizes an Ephrussi-box ('E-box') binding site ('CANNTG') (2,18). However, this motif is too small and nonspecific to accurately predict TCF4 binding computationally. A precise map of binding sites is vital for deciphering the gene regulatory networks under the influence of a TF (19). In recent years, systematic mapping of TF binding has been enabled via chromatin immunoprecipitation coupled to next-generation sequencing (ChIP-seq) (20). ChIP-seq works by precipitating the desired protein–DNA complex out of cell lysate using an antibody complementary to the protein of interest. After removal of the protein, the liberated DNA fragments are sequenced and mapped back to the reference genome to yield a map of regions bound by the protein (21).
The ENCODE Consortium, which aims to map all regulatory elements in the human genome, has used ChIP-seq extensively to map binding profiles of many TFs in human cell lines (22). TF binding is tissue specific, so separate experiments are required to characterize binding in cells from each tissue of interest. At its outset, ENCODE did not have a major CNS focus (23). The consortium attempted to map TCF4 binding sites in bone marrow-derived K562 cells, (https://www.encodeproject.org/experiments/ENCSR000FCF/; date last accessed June 20, 2018). However, these data were revoked shortly after release. Here, we describe TCF4 ChIP-seq in a CNS-derived cell line. To probe the relationship with SCZ, we tested the TCF4 gene network for overlap with SCZ risk genes from GWAS and gene expression studies.
A note on nomenclature
TCF4, located on chromosome 18, was previously known by the aliases E2–2, ITF-2, PTHS and SEF-2 (http://www.ncbi.nlm.nih.gov/gene/6925; date last accessed June 20, 2018). TCF4 and TCF7L2 (Gene ID: 6934) are often confused because they share the TCF4 alias (2,18). TCF7L2, located on chromosome 10, encodes ‘T-Cell Factor 4’, an effector of Wnt/β-catenin signaling (24) that is not an accepted risk factor for SCZ. In this study, all references to TCF4 are to the gene with co-ordinates chr18: 52889562–53332018 (hg19) and these identifiers: HUGO name=TCF4, ENTREZ = 6925, HGNC = 11634, ENSEMBL=ENSG00000196628, and UNIPROT=P15884.
Results
ChIP-seq and peak calling
At the start of the project, no validated ChIP antibodies were available for TCF4. Of eight candidate anti-TCF4 antibodies identified, three passed initial immunoblot testing according to ENCODE guidelines, whereby a single band of the appropriate mass (TCF4-B long isoform, 667 aa) accounting for >50% of the total lane intensity was observed (25) (Fig. 1A). We performed immunoprecipitation (IP) cross-reactivity studies using these antibodies, where IP with anti-TCF4 antibodies was used as the substrate for Western blot with a different anti-TCF4 antibody. Figure 1B shows that anti-TCF4 antibodies K-12 and N-16 immunoprecipitated a protein of the appropriate mass that was detected by monoclonal anti-TCF4 antibody 1G4, indicating that all three antibodies were likely detecting the same protein. (The complete blot image across the full molecular weight range is shown in Supplementary Material, Fig. S1). We also conducted mass spectrometry (MS)-based proteomics analysis of the IP of all three antibodies and successfully detected TCF4 peptides in each case (Supplementary Material, Figs S2 and S3), while we did not detect TCF4 in immunoglobulin G (IgG) control IP experiments. (Note that our proteomic output also shows common contaminants such as keratin that are typical in MS analysis of IP experiments. See for example Landt et al. (25), their Figure 2D. Following these successful validation steps, we proceeded with ChIP-seq using these antibodies.
Figure 1.
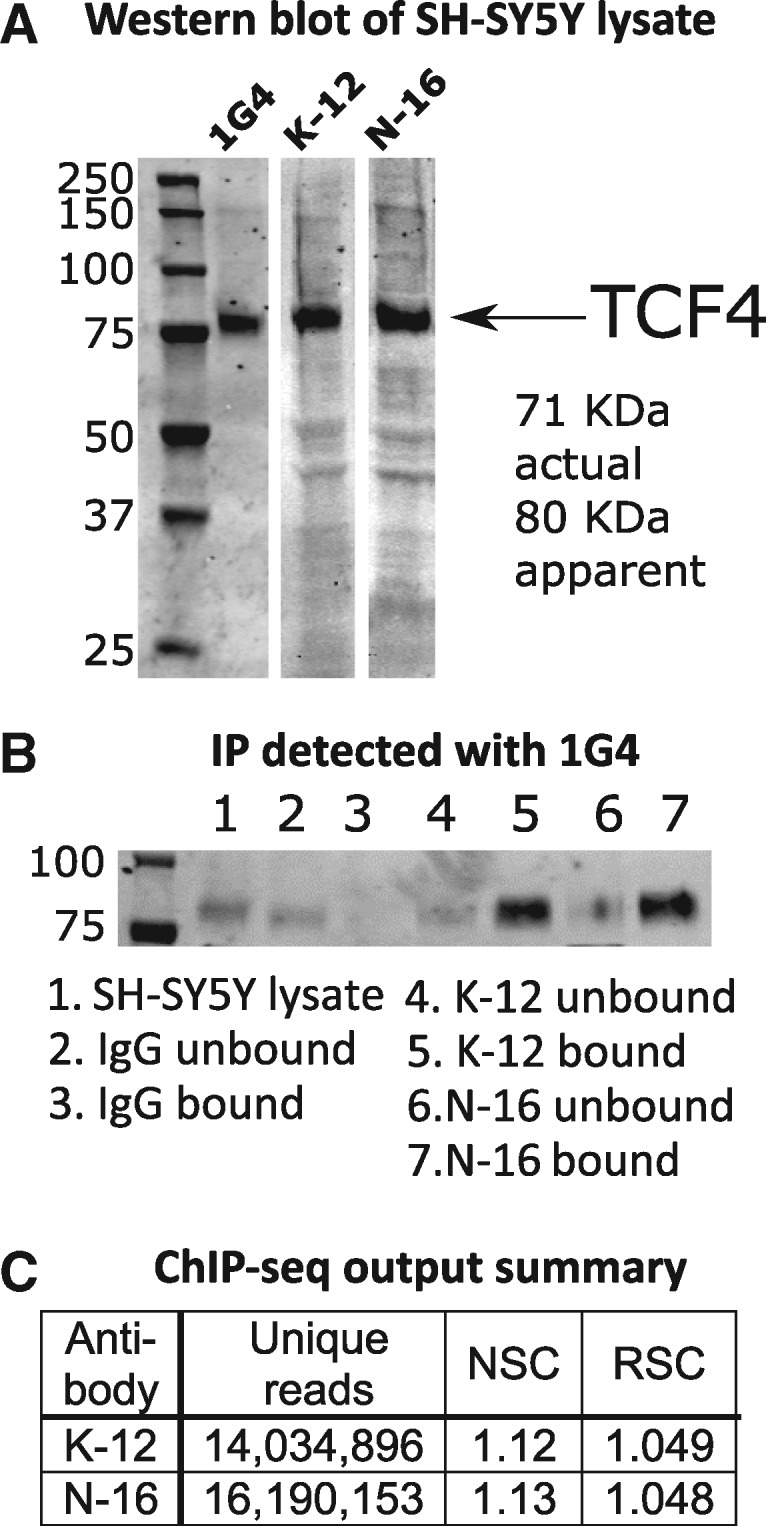
ChIP-seq outline and quality control. (A) Western blotting in SH-SY5Y cell lysate using three different anti-TCF4 antibodies (1G4, K-12 and N-16) identifies a single clear band for TCF4 (long isoform B, UniProt P15884, 667 aa). (B) Crossover IP for TCF4. In this experiment, anti-TCF4 antibodies K-12 and N-16, plus control IgG, were used to immunoprecipitate proteins that were subsequently probed in a Western blot using anti-TCF4 antibody 1G4. Using different anti-TCF4 antibodies in the IP and Western blotting increases confidence that TCF4, rather than a non-specific protein, is detected. The presence of the TCF4 band in the bound (IP) fraction, and not in the bound IgG (control) fraction, indicates that the K-12 and N-16 antibodies successfully immunoprecipitated TCF4. The full blot image is provided in Supplementary Material, Figure S1. (C) ChIP-seq output summary. The number of uniquely aligning reads for each ChIP-seq experiment are shown. The normalized strand cross-correlation coefficient (NSC) and relative strand cross-correlation coefficient (RSC) values indicate enrichment in ChIP (D), with higher values indicating more enrichment. NSC values <1.1 are relatively low and the minimum possible value is 1 (no enrichment). The minimum possible RSC value is 0 (no signal), highly enriched experiments have values >1, and values much less than 1 may indicate low quality (see genome.ucsc.edu/ENCODE/qualityMetrics.html). The values for our experiments indicate good enrichment.
Each ChIP experiment involved isolating TCF4-bound DNA from approximately 1.2 × 107 SH-SY5Y cells. Average DNA yield was within expected parameters (∼10 ng per replicate), but DNA was also recovered from ChIP using the control IgG antibodies. This indicated that nonspecific material was captured and that ChIP-seq using the IgG control antibody was the appropriate background to call peaks. After sequencing and alignment to hg19, potential polymerase chain reaction (PCR) duplicate reads were aggressively filtered (‘rmdup’ command in Samtools) to collapse all reads with the same start position to single reads. Only two of the three antibodies (K-12 and N-16) worked well. Figure 1C shows the summary statistics for these assays, and cross-correlation plots are provided in Supplementary Material, Figures S4 and S5. After filtering out ENCODE blacklisted regions, we examined overlap between peaks called for antibodies K-12 and N-16. First, we plotted the difference in starting position between peaks observed in K-12 ChIP-seq and the closest peak in N-16 ChIP-seq. Supplementary Material, Figure S6 shows that there was a clear enrichment for peaks with start sites ± 200 bp in each experiment. This is the approximate peak size (225 bp) in the ‘narrowpeak’ output format used by the SPP peak caller. Therefore, we specified that the peaks called in the two separate experiments had to overlap (at least 1bp in common) to count as ‘replicated’; neighboring, nonoverlapping peaks were not considered. Applying this criterion resulted in 11 322 TCF4 binding sites present in both experiments, using false discovery rate (FDR) threshold <1% (21). As expected, overlap was greater for peaks called with higher confidence. Sorting on SPP (21) signalValue, 65% of peaks in the top 100 in the K-12 experiment had a matching peak in the N-16 data; 50% in the top 250; 45% in the top 500 and 29% in the top 5000. To further probe data quality, we reran the overlap analysis, this time requiring peaks to overlap by almost 50% (110 bp in common). We obtained 7596 peaks showing the degree of overlap was typically high. Overall, these findings supported the consistency of our experiments.
Supplementary Material, Figure S7 shows the distribution of all 11 322 binding sites by chromosome. No binding sites were obtained for the chrY because SH-SY5Y is genetically female. The genomic distribution was nonrandom with relatively large numbers of peaks detected on chrs 7 and 17. All 11 322 binding sites are provided in Supplementary Material, Table S1.
As a further validation step, we tested for enrichment of specific sequence motifs at the TCF4 binding sites. TCF4 binds the ‘Ebox’ sequence motif (‘CANNTG’). Figure 2 shows the most significantly enriched sequence motifs at the consensus 11 322 ChIP-seq peaks. The top overall motif was the binding site for STAT1 (‘RGRAA’), while the top-ranked TCF4 Ebox (‘CAYCTG’) was fourth. The redundancy of the central 2 bp of the Ebox motif means that several different sequence combinations are possible and, for example, the ‘CACCTG’ Ebox variant was detected at E = 2.3e-016. Overall, 86.4% of the 11 322 binding sites encompassed an Ebox motif.
Figure 2.
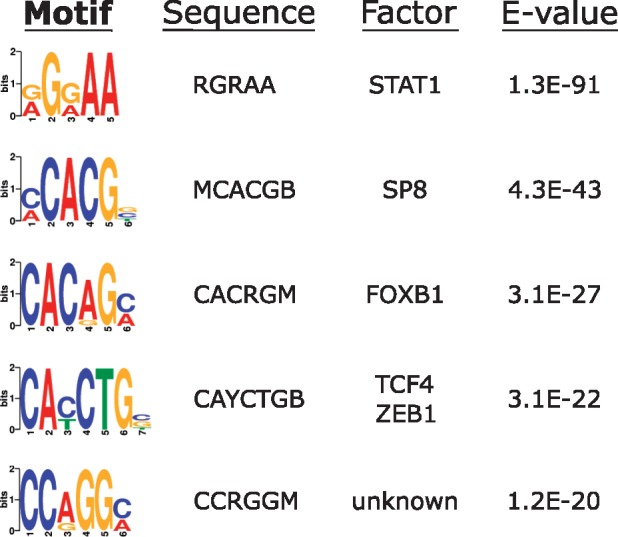
Sequence motif enrichment at TCF4 binding sites. Output from DREME shows the most enriched motifs around the TCF4 ChIP peaks. Motif sequences are shown with conventional nucleotide redundancy codes. E-values are output from DREME/MEME-ChIP and indicate the P value multiplied by the number of instances tested as a correction for multiple testing.
Genes associated with TCF4 binding sites and their tissue-specific expression
Of the 11 322 binding sites, 8923 were within 10 kb of a RefSeq gene. To obtain insight into functional role of TCF4, we used Genomic Regions Enrichment of Annotations Tool (GREAT) (26) to conduct enrichment tests for Gene Ontology (GO) categories and MSigDB pathways. GREAT first assigns genomic loci to regulatory domains associated with genes, and only 34 TCF4 binding sites could not be assigned to any gene (Supplementary Material, Fig. S8). The fact that over 2000 binding sites were not within 10 kb of a gene, yet only 34 could not be assigned to a gene suggests that a number of binding sites are at enhancers or long-range cis-regulatory elements. The results from GREAT are shown in Table 1. Significant results in the GO cellular component category had a strong neuronal theme, while insulin signaling and axon/neuronal development were notable pathway findings.
Table 1.
Functional annotation and pathway analysis of TCF4 genomic binding sites using GREAT
| Term name | Binom rank | Binom raw P value | FDR Q-Val | Fold enrichment | Observed region hits | Region set coverage |
|---|---|---|---|---|---|---|
| GO Molecular Function | ||||||
| 14–3-3 protein binding | 16 | 1.71E-31 | 3.95E-29 | 4.75 | 88 | 0.008 |
| Protein kinase A catalytic subunit binding | 92 | 1.18E-14 | 4.73E-13 | 2.82 | 75 | 0.007 |
| Ankyrin binding | 120 | 1.68E-12 | 5.17E-11 | 2.78 | 64 | 0.006 |
| GO Cellular Component | ||||||
| Cell cortex part | 24 | 2.39E-33 | 1.26E-31 | 2.40 | 243 | 0.021 |
| Axon terminus | 29 | 3.20E-27 | 1.39E-25 | 2.31 | 212 | 0.019 |
| Neuron projection terminus | 39 | 7.87E-22 | 2.55E-20 | 2.05 | 218 | 0.019 |
| Cortical cytoskeleton | 71 | 6.04E-13 | 1.08E-11 | 2.09 | 117 | 0.010 |
| GO Biological Process | ||||||
| Tooth mineralization | 69 | 2.07E-35 | 3.13E-33 | 5.27 | 90 | 0.008 |
| Insulin receptor signaling pathway | 79 | 4.36E-34 | 5.76E-32 | 2.05 | 349 | 0.031 |
| Enamel mineralization | 111 | 9.82E-30 | 9.23E-28 | 4.98 | 79 | 0.007 |
| Regulation of cell size | 171 | 2.90E-24 | 1.77E-22 | 2.00 | 260 | 0.023 |
| Spinal cord dorsal/ventral patterning | 180 | 2.74E-23 | 1.59E-21 | 2.97 | 115 | 0.010 |
| CD4-positive or CD8-positive, alpha-beta T cell lineage commitment | 196 | 3.14E-22 | 1.67E-20 | 5.36 | 54 | 0.005 |
| Spinal cord patterning | 212 | 4.64E-21 | 2.29E-19 | 2.76 | 116 | 0.010 |
| Regulation of axon extension | 225 | 1.69E-20 | 7.85E-19 | 2.16 | 182 | 0.016 |
| Negative regulation of osteoblast differentiation | 233 | 6.95E-20 | 3.12E-18 | 2.43 | 137 | 0.012 |
| Ventral spinal cord interneuron specification | 406 | 1.13E-14 | 2.92E-13 | 2.84 | 74 | 0.007 |
| MSigDB Canonical Pathways | ||||||
| Insulin signaling pathway | 4 | 9.43E-30 | 3.11E-27 | 2.04 | 309 | 0.027 |
| Sonic Hedgehog (Shh) Pathway | 6 | 7.70E-28 | 1.69E-25 | 3.74 | 101 | 0.009 |
| Genes involved in Class B/2 (Secretin family receptors) | 9 | 6.90E-23 | 1.01E-20 | 2.16 | 203 | 0.018 |
| Vibrio cholerae infection | 24 | 1.56E-17 | 8.56E-16 | 2.36 | 126 | 0.011 |
| LKB1 signaling events | 29 | 2.51E-16 | 1.14E-14 | 2.44 | 109 | 0.010 |
| Regulation of RhoA activity | 61 | 6.48E-11 | 1.40E-09 | 2.00 | 107 | 0.009 |
| p38 signaling mediated by MAPKAP kinases | 88 | 3.11E-09 | 4.67E-08 | 2.58 | 51 | 0.005 |
| Ras activation uopn Ca2+ infux through NMDA receptor | 93 | 5.53E-09 | 7.85E-08 | 2.33 | 60 | 0.005 |
| fl-arrestin-dependent Recruitment of Src Kinases in GPCR Signaling | 96 | 7.81E-09 | 1.07E-07 | 2.89 | 40 | 0.004 |
| CREB phosphorylation through the activation of CaMKII | 98 | 1.09E-08 | 1.47E-07 | 2.37 | 56 | 0.005 |
The full set of 11 322 TCF4 binding sites implicated 6528 unique genes ±10 kb of the gene body (Supplementary Material, Table S2), which is approximately one-quarter of all genes in refGene, including noncoding RNAs and genes with provisional nomenclature designations. We used FUMA (27) to test for enrichment in tissue-specific differentially expressed gene sets (Supplementary Material, Fig. S9). The TCF4 gene set was most enriched in genes upregulated in the brain and pituitary and genes downregulated in the heart and blood vessels. To further probe the expression patterns of the TCF4 gene set, we looked at single cell RNA-seq data for specific CNS cell types. We observed that the TCF4 gene set was significantly overexpressed in pyramidal neurons from the somatosensory cortex (P = 5.2 × 10−5; Fig. 3).
Figure 3.

Enrichment of TCF4 gene set expression in specific brain cell types as determined by single cell RNA-seq (51). We tested if expression of genes in the TCF4 gene set were significantly higher than for genes not in the gene set for each cell type. The bold line at ∼2.7 on the x-axis is the Bonferroni-adjusted –log10(P value) for multiple testing (α = 0.05/24). “Pyramidal SS” = pyramidal neurons of the somatosensory cortex. “Pyramidal CA1” = pyramidal neurons of the CA1 region of the hippocampus.
Differential regulation of TCF4 genes in siRNA knockdown experiments and postmortem data
We integrated our ChIP-seq data with relevant data sets to refine our findings and identify SCZ risk genes under TCF4 control. We first tested for overlap between the TCF4 gene set and gene expression data from a TCF4 knockdown experiment using small-interfering RNA (siRNA), also conducted in SH-SY5Y cells (28). We expected that genes differentially expressed following TCF4 knockdown should be enriched for those with TCF4 binding sites from ChIP-seq. A significant enrichment was observed, and this was driven by genes downregulated following TCF4 knockdown (Table 2). No significant enrichment was observed in the upregulated genes. We also tested the TCF4 gene set for enrichment in a second TCF4 siRNA knockdown experiment, this time in cortical neuron progenitor cells (29) (Table 2). Once again downregulated genes, but not upregulated genes, were significantly enriched. This consistency was encouraging. All genes overlapping between ChIP-seq and siRNA studies are provided in Supplementary Material, Tables S4 and S5.
Table 2.
Integration of TCF4 binding profile with functional and SCZ data sets
| Gene-based enrichment via permutation analysis | |||||
|---|---|---|---|---|---|
| TCF4 knockdown experiments | Set size | Overlap | Odds ratio | Fisher P value | Permutation P value |
| SH-SY5Y siRNA knockdown (all) | 921 | 267 | 1.30 | 0.0003 | 0.0005 |
| Upregulated | 396 | 100 | 1.06 | 0.3159 | n/a |
| Downregulated | 525 | 167 | 1.48 | 3.28E-05 | 0.0001 |
| Cortical precursor siRNA knockdown (all) | 502 | 138 | 1.20 | 0.0434 | 0.0438 |
| Upregulated | 240 | 47 | 0.76 | 0.9603 | n/a |
| Downregulated | 262 | 91 | 1.68 | 6.78E-05 | <4.11E-05a |
| SCZ gene expression | |||||
| Fromer et. al (2016) (all) | 623 | 190 | 1.38 | 0.0002 | 0.0001 |
| Upregulated | 296 | 37 | 0.45 | 1 | n/a |
| Downregulated | 326 | 153 | 2.77 | 2.20E-16 | <4.11E-05a |
| Genomic locus enrichment tests using LOLA | |||||
| UCSC annotations | Bed file | Overlap | Odds ratio | P value | |
| Evo CpG islands | evoCpg.bed | 3706 | 7.08 | ∼0 | |
| CpG islands | cpgIslandExt.bed | 960 | 2.76 | 2.30E-151 | |
| Switch DB TSSs | switchDbTss.bed | 584 | 3.19 | 2.46E-118 | |
| LaminBq | laminB1.bed | 4404 | 1.49 | 1.46E-94 | |
| Simple repeats | simpleRepeat.bed | 836 | 1.99 | 3.46E-68 | |
| Top 15 overlapping DNA binding protein profiles from ENCODE | |||||
| Experiment | Antibody | Overlap | Odds ratio | P value | |
| ChIP SK-N-SH | NRSF | 587 | 5.75 | 2.90E-231 | |
| ChIP GM12878 | Pol2 | 637 | 2.26 | 5.54E-71 | |
| ChIP MCF-7 | CTCF | 561 | 2.26 | 3.53E-63 | |
| ChIP H1-hESC | CTCF | 437 | 2.40 | 3.38E-56 | |
| ChIP K562 | Max | 580 | 2.09 | 3.11E-54 | |
| ChIP HEK293 | ZNF263 | 508 | 2.18 | 4.30E-53 | |
| ChIP HepG2 | Pol2 | 425 | 2.36 | 8.00E-53 | |
| ChIP PFSK-1 | FOXP2 | 305 | 2.73 | 2.83E-50 | |
| ChIP K562 | ZBTB7A | 357 | 2.48 | 2.88E-49 | |
| ChIP GM12891 | CTCF | 422 | 2.26 | 5.42E-48 | |
| ChIP A549 | CTCF | 505 | 2.02 | 3.23E-44 | |
| ChIP SK-N-SH | p300 | 494 | 2.03 | 6.72E-44 | |
| ChIP K562 | Egr-1 | 403 | 2.18 | 7.15E-43 | |
| ChIP K562 | MAZ | 450 | 2.04 | 8.06E-41 | |
| ChIP SH-SY5Y | GATA-2 | 413 | 2.09 | 9.19E-40 | |
| Psychiatric Genomics Consortium risk loci overlap | |||||
| Data file | Background | Overlap | Odds ratio | Fisher P value | Permutation P value |
| scz2.anneal.108 | ActiveDHSb | 158 | 1.38 | 8.88E-05 | 0.0816 |
| SK-N-SH DHSb | 55 | 1.56 | 0.0017 | 0.035 | |
4.11E-05 is the minimum P value obtainable when permuting over 24, 358 genes (includes non-coding RNAs and provisional IDs).
DNaseI Hypersensitivity Sites.
We next tested whether the TCF4 gene set was enriched among genes differentially expressed in postmortem brain tissue from SCZ patients. Here we used findings from Fromer et al. (30) who conducted bulk RNA-seq of dorsolateral prefrontal cortex (DLPFC) from 258 SCZ cases and 279 controls. Once again, downregulated genes were significantly enriched with almost half possessing a TCF4 binding site, compared to just 12.5% of the upregulated genes. This highly significant enrichment in downregulated genes was surprising because these are downregulated in SCZ not as a direct result of TCF4 knockdown. We hypothesized that downregulation of TCF4 causes the downregulation of genes in SCZ, analogous to the situation in the siRNA experiments. However, TCF4 expression was significantly upregulated in the SCZ patients (fold enrichment = 1.16, Fromer et al., their Supplementary data file 3 (30)). Further analysis of these gene sets showed that few genes were shared in common. That is, only seven genes with TCF4 binding sites from ChIP-seq were downregulated in the SH-SY5Y siRNA study (28) and downregulated in the SCZ expression study (30). These were ANKMY1, FAM78A, IGF2, MXRA8, NT5M, PELI3 and TNS3. Notably, IGF2 had the largest fold-change reduction of all genes in the Fromer et al. study. A further two genes with TCF4 binding sites, DBNL and PNPLA7, showed downregulation in the neural progenitor cell siRNA study (29) and the SCZ gene expression study.
Overlap of TCF4 binding sites with other TFs and SCZ risk loci
We next considered overlap of TCF4 binding sites with University of California Santa Cruz (UCSC) genome browser features. TCF4 binding sites were highly enriched (odds ratio, OR = 7.08) in CpG islands, classified according to the Weizmann Institute CpG evolution model (31) and in transcription start sites (OR = 3.19) from the SwitchGear Genomics library (www.switchgeargenomics.com; date last accessed June 20, 2018) (Table 2). We also looked at overlap with binding profiles for other TFs from ENCODE. Notable overlapping factors were FOXP2, a neural transcription factor involved in speech development (32), and p300, a chromatin remodeler encoded by the EP300 gene that is associated with SCZ (1).
Our final data integration analysis was with the 108 PGC2 SCZ risk loci published in 2014 (1). For this analysis, we used genomic locus-based enrichment rather than gene sets. This is important because many SCZ-associated genes are large and may be more likely to overlap genomic annotations by chance than a randomly selected gene set (33). Therefore, annotations can be confounded with gene size, leading to erroneous conclusions of enrichment. Testing for TCF4 binding site enrichment in the associated genomic regions, and not considering occurrence in genes, obviates this potential bias. Overall, 39 of the 108 PGC SCZ loci contained one or more TCF4 binding sites, with 130 sites in total falling within their boundaries. Permutation testing of enrichment using all known human regulatory regions as the background set revealed a nonsignificant overlap (P = 0.082). Narrowing the background set to relevant regulatory regions for the cell type used in the ChIP-seq experiments revealed a nominally significant enrichment (P = 0.035) (Table 1). We also analyzed the overlap between TCF4 binding sites and PGC SCZ risk loci using LD score regression (34, 35). We found an 8-fold SCZ h2 enrichment at the TCF4 sites, but this did not reach statistical significance (P = 0.1), possibly because of the small size of the binding sites (225bp). Accordingly, we expanded the size of the region around each peak to 1 and 2 kb, but this actually diminished the enrichment, suggesting that what signal there may be is right at the binding position (see Supplementary Material, Fig. S10). PGC SCZ genes with TCF4 binding sites ±10 kb that were also differentially expressed in either of the siRNA experiments described above were APH1A, C1orf54, CENPM, CHRNA5, DFNA5, GFOD2, GRAMD1B, LRP1, MPP6, PDCD11, SEZ6L2, TLE1 and XRCC3. Of these, both LRP1 and XRCC3 had three binding sites in our ChIP-seq data, PDCD11 had two, while the remainder had one each.
The TCF4-B long isoform is upregulated in multipotent neuronal precursors
Pathways and genes implicated in our findings suggested that TCF4-regulated genes are involved in neuronal growth and/or differentiation. To determine whether there are developmental differences in expression of the TCF4 isoform we captured in ChIP-seq, we differentiated Lund human mesencephalic cells (LUHMES) neural precursor cells using an established protocol that yields post-mitotic dopaminergic neurons in 5 days (36,37) (Fig. 4). LUHMES are a conditionally immortalized subclone of the human mesencephalic-derived cell line MESC2.10 (38). We used Western blotting with the N-16 antibody to probe for TCF4 in cell lysates and compared SH-SY5Y, undifferentiated LUHMES and differentiated LUHMES cells. Figure 4 shows a reduction in the TCF4 signal in the differentiated cells. Quantitative analysis of three duplicate experiments indicated that the TCF4 isoform recognized by N-16 was expressed in the differentiated cells at <10% of the levels in the undifferentiated, rapidly growing cells. This suggests that the specific isoform detected in our study is more abundantly expressed in multipotent neural progenitor cells than in fully differentiated neurons, at least of this dopaminergic type. This conclusion is supported by Figure 3, whereby the genes implicated by TCF4 ChIP-seq show nominally significant evidence for enrichment of expression in dopaminergic neuroblasts (P = 0.037) but no enrichment at all in adult dopaminergic neurons (P = 0.99).
Figure 4.

Differentiation of LUHMES neuronal precursor cells (A) to mature neurons (B) in culture. DAPI stain (blue) was used to identify cell bodies, while neuronal projections were stained using anti-β tubulin antibody (green). The third panel shows Western blotting using anti-TCF4 antibody N-16 and lysates from SH-SY5Y (SH), undifferentiated (A) and differentiated (B) LUHMES compared to GAPDH loading control. The Western blots (C) show that TCF4 is downregulated in the differentiated LUHMES neurons.
Discussion
We obtained data on TCF4 binding from ChIP-seq, which allowed us to probe the TCF4 gene network for association with SCZ risk genes. We validated our antibodies according to ENCODE standards. Following ChIP-seq, we further validated our findings by showing that our TCF4 binding sites were enriched for the known TCF4 Ebox binding motif. We also showed that genes with TCF4 binding sites were enriched among genes differentially expressed in TCF4 siRNA knockdown experiments. Taken together, these results strongly support the validity of the data.
A limitation of our ChIP-seq experiments was that only two of three antibodies yielded usable data. It is accepted that antibodies that pass initial characterization may still fail to yield good ChIP-seq data (25), yet the immunoblot for the antibody that failed (1G4) is arguably the cleanest (Fig. 1b). However, antibody 1G4 is monoclonal, whereas K-12 and N-16 that are polyclonal. While monoclonal antibodies have advantages in specificity and reproducibility, they are more prone to fail in ChIP-seq because the single epitope recognized in the protein may be obscured by the bound DNA. Another limitation is that we only have ChIP-seq data on the long TCF4 isoform (TCF4-B). The TCF4 locus can give rise to several distinct isoforms via alternative splicing (18). Some isoforms are exclusively nuclear while others rely on heterodimerization partners. Further work will be needed to understand the specific gene networks under their control.
The TCF4-B long isoform that we studied here possesses two transcriptional activation domains that can operate synergistically to activate transcription to a greater extent than many other isoforms of TCF4 (18). This transcriptional upregulation produced by TCF4-B can be seen in this study. Genes implicated by our TCF4 ChIP-seq were significantly enriched among those downregulated in TCF4 siRNA studies (Table 2), remembering that in RNAi knockdown the inhibition of a transcription factor will lead to a reduction in expression of genes that it activates. However, it is important to recognize that TCF4 can induce activation or repression of transcription depending on the protein complex bound to it, and there is evidence that its repressive effects may be important in the CNS (11). Indeed, we observed several genes implicated by TCF4 ChIP-seq that were upregulated in the RNAi studies (with upregulation here implying TCF4 repression under normal conditions), but the number was fewer than genes showing the opposite effect (Table 2).
Several genes that we identified as having TCF4 binding sites have been functionally linked with TCF4 in prior studies. For example, TCF4 has also been shown to affect neural excitability via regulation of potassium channel KCNQ1 (11). In our data, this gene had 19 unique TCF4 binding sites, the 16th largest number for any gene. Conversely, given the problems with “TCF4” nomenclature, delineating what is not seen in our data may be of value. Several articles describe the interaction between “TCF4”, β-catenin and p300 (39). This relates to T Cell Factor 4, encoded by TCF7L2. Beta-catenin is encoded by CTNNB1, and we did not detect a TCF4 binding site at this gene. Furthermore, although the binding profiles of p300 (encoded by EP300) and TCF4 strongly overlap (Table 2), TCF4 does not appear to regulate EP300, at least in our data. The co-occurrence of their binding sites does, however, imply that they may be involved in the regulation of a partially overlapping set of genes in CNS cells. EP300 is a PGC2 SCZ risk locus (1) and was associated with emotional processing in functional neuroimaging experiments (40). Further analysis into the overlapping pathways regulated by these two SCZ-associated DNA binding proteins may be relevant.
Insulin-like growth factor 2 (IGF2) showed evidence for TCF4 binding in our ChIP-seq experiments and showed reduced expression following TCF4 siRNA-mediated knockdown. Furthermore, IGF2 was the most downregulated gene in postmortem SCZ brain in the study by Fromer et al. (30). IGF2 may regulate neural plasticity to modulate behavior and memory (41). Furthermore, deficits in hippocampal neurogenesis in a mouse model of 22q11.2 deletion-associated SCZ can be rescued by IGF2 (42). A significant amount of work has been conducted on the role of IGF2 in the brain, yet few studies address the role of IGF2 in SCZ etiology. One issue is that IGF2 was not detected among PGC SCZ risk loci (1). This may indicate that downregulation of IGF2 in SCZ is a consequence of risk variants at other loci or environmental factors rather than as a result of risk variants at the IGF2 locus itself. It is also the case that the CommonMind Consortium sample used by Fromer et al. (30) is relatively underpowered to detect genetically driven changes to gene expression in cases versus controls, so it is unclear to what extent the differentially expressed genes in this study are driven by genetics versus secondary factors.
Several PGC SCZ risk genes contained TCF4 binding sites, and thirteen of these also showed differential expression in TCF4 siRNA experiments. The risk loci identified by the PGC may span several hundred kb and contain many genes. It is often not apparent which genes in these regions should be selected for further study. For example, there are 14 unique genes in the third most significant region identified by the PGC (chr10: 104423800–105165583, P-value = 6.12E-19) (1). Among these, PDCD11 is likely regulated by TCF4, exhibiting both TCF4 binding sites and differential expression in a TCF4 siRNA study. It is also expressed in brain (43) but is poorly characterized. Its likely regulation by TCF4 may suggest further characterization of this gene is merited.
Many of the PGC SCZ risk genes regulated by TCF4 are involved in neuronal differentiation and development. For example, APH1A encodes a subunit of gamma secretase, is crucial for Notch signaling in embryogenesis and is predominantly expressed in non-neuronal and neuronal precursor cells (44). LRP1 has diverse roles in the CNS and is a regulator of neural progenitor cell function (45), while TLE1 functions as a transcriptional repressor to regulate neuronal differentiation (46). Furthermore, we observed that the specific TCF4 isoform captured in our ChIP-seq experiments is upregulated in rapidly growing, undifferentiated neural precursors relative to differentiated neurons of a dopaminergic type. Therefore, it is plausible that TCF4 affects SCZ risk by affecting neural development.
We conclude that we have identified potential regulatory interactions between a SCZ-associated TF and several SCZ risk loci that implicate processes involved in neuronal development and function. As an “omic” study, our findings represent hypotheses to be tested in future neurobiological studies. We hope that the mapping of these interactions will stimulate further research into TCF4.
Materials and Methods
Antibody selection
At the start of this study, no ChIP-grade antibodies were available for TCF4, so we selected candidate antibodies from commercial vendors. Due to the confusion in nomenclature with T-Cell Factor 4, we discovered several mislabeled antibodies. Therefore, we limited ourselves to antibodies with published epitope sequences that could be confirmed as TCF4 via protein BLAST (NCBI). Eight antibodies were selected, of which three passed initial QC and were used for ChIP-seq: polyclonal anti-TCF4 antibodies K-12 (sc-48947) and N-16 (sc-48949) from Santa Cruz Biotechnology (Dallas, Texas, USA) and monoclonal anti-TCF4 antibody 1G4 (Novus, Littleton, Colorado, USA).
Cell culture
SH-SY5Y cells (ATCC, Manassas, Virginia) were cultured according to supplier’s standard protocols. Cell line authentication via simple tandem repeat genotyping was conducted by the University of Arizona Genomics Core Facility. LUHMES cells were cultured and differentiated according to a published protocol (36). Additional details and immunocytochemistry methods are in the Supplementary Material, Methods.
Antibody validation
We followed ENCODE guidelines for antibody validation (25). In addition to Western blotting of SH-SY5Y cell lysates, we used IP of TCF4 protein followed by Western blotting or protein mass spectrometry to characterize the proteins captured in IP (see Supplementary Material, Methods). Further validation after ChIP-seq used motif enrichment testing of binding site sequences (500 bp, centered on ChIP-seq peaks) with using DREME with default settings as implemented in MEME-ChIP (47).
ChIP-seq
Each ChIP assay used approximately 1.2 × 107 SH-SY5Y cells and was performed with the SOLiD ChIP-Seq Kit (Life Technologies, Foster City, California) according to manufacturer’s specifications, with some adjustments (Supplementary Material, Methods). ChIP-Seq libraries were validated using the BioAnalyzer high-sensitivity chip assay (Agilent, Santa Clara, California) prior to multiplexed high-throughput sequencing on the SOLiD 5500 platform. Fifty-bp single end reads were generated, with a target read number of 25 million tags per sample. In addition to ChIP samples using anti-TCF4 antibodies, their respective IgG controls and input DNA controls were sequenced.
ChIP-seq data analysis
Reads were aligned to the human genome (build hg19) using BioScope 1.2 (Life Technologies). Multi-mapping reads were discarded, and only stringent single alignments retained. Sample files were output as .bam files using BioScope. PCR duplicates were removed by dropping multiple reads with identical start positions using the Samtools (48) rmdup function and alignment files were written in tagAlign.gz format using Samtools and Bedtools (49). To call peaks and assess experiment quality, we used SPP (21) distributed with phantompeakqualtools (50). Peaks for each ChIP-seq experiment (Fig. 1c) were called against a composite IgG control comprising three replicates. This merging of control replicates was necessary to obtain sufficient material for an adequately complex control library. A false discovery rate (FDR) threshold of 1%, as implemented in SPP (21), was used to call peaks. Peaks mapping to ENCODE blacklisted regions (ftp://encodeftp.cse.ucsc.edu/users/akundaje/rawdata/blacklists/hg19/wgEncodeHg19ConsensusSignalArtifactRegions.bed.gz; date last accessed June 20, 2018) were removed. All raw and processed ChIP-seq data for this project are available through the Gene Expression Omnibus (GEO, www.ncbi.nlm.nih.gov/geo; date last accessed June 20, 2018) with accession number GSE112704.
Data integration
Bioinformatics analysis was conducted in R (www.r-project.org; date last accessed June 20, 2018). Gene lists were obtained from refGene via UCSC Genome Browser download (August 10, 2017), followed by elimination of transcripts with ambiguous mapping and pruning entries by maximum boundary to yield a single non-redundant locus per gene. Gene pathway analysis used GREAT(26) version 3.0.0, assigning proximal regulatory domains ±10 kb. Tissue-specific expression of the top TCF4 genes was evaluated using the “Gene2Func” mode in FUMA (27).
For cell-specific expression analysis, we obtained single cell RNA-seq data from five brain regions in mice (9970 single cells) that were previously clustered into 24 different cell types (51). Normalization factors were computed for each of the 9970 single cells using the scran R package (52,53) using the 50% of the genes with mean expression higher than the median. The normalization factors were computed after clustering cells using the scran quickcluster() function to account for cell type heterogeneity. We then performed 24 differential expression analyses using BPSC (54) testing each cell type against the 23 other cell types using the normalization factor as a covariate. For each differential expression analysis, the t-statistics were then transformed to a standard normal distribution. Finally, for each cell type, we used linear regression to test if the standard normalized t-statistics for genes in the TCF4 gene set were significantly higher than for genes not in the gene set.
Enrichment testing of TCF4 binding sites in significant genes from siRNA knockdown expression studies (28,29) was carried out by mapping genes ±10 kb, followed by one-sided Fisher exact tests. Permutation testing of significant findings used the shiftR package (https://github.com/andreyshabalin/shiftR; date last accessed June 20, 2018), as outlined previously (55). Test of overlap between TCF4 peaks and genomic annotations used the LOLA Bioconductor package (56), with all mappings obtained from the LOLACore annotation set (databio.org/regiondb; date last accessed June 20, 2018). The background set for this analysis was the default DNase hypersensitive sites (DHS) in multiple tissues (“activeDHS” set), which captures known human regulatory regions (56,57). Testing for overlap with PGC SCZ findings was also based on genomic locus, rather than gene. SCZ-associated loci were obtained by download of the “scz2.anneal.108” file from the PGC (https://www.med.unc.edu/pgc/results-and-downloads; date last accessed June 20, 2018). Background sets for this analysis were either “activeDHS” as above or DNase hypersensitive sites specifically for SK-N-SH cells (“wgEncodeOpenChromDnaseSknshPk” track, Duke DHS from ENCODE). No DHS data for SH-SY5Y were available, but SH-SY5Y are a subline of SK-N-SH isolated from the same donor (58). After matching to background set, enrichment testing used Fisher exact tests followed by permutation as above.
Supplementary Material
Acknowledgements
Cell Line Authentication was provided by Elizabeth Cox and the team at University of Arizona Genetics Core (via Science Exchange). Thanks to Derek Blake at the MRC Centre for Neuropsychiatric Genetics and Genomics, Cardiff University, United Kingdom for providing siRNA gene expression data, and to Jens Hjerling-Leffler, Sten Linnarsson, Ana Munoz Manchado and Amit Zeisel of the Karolinska Institute, Stockholm, Sweden for providing single-cell RNA-sequencing data. This article was submitted as a preprint to bioRxiv (11/07/2017) and assigned doi: https://doi.org/10.1101/215715.
Conflict of Interest statement. None declared.
Funding
This work was funded through grant R21 MH099419 from the US National Institute of Mental Health (NIMH) to JL McClay.
References
- 1. Schizophrenia Working Group of the Psychiatric Genomics Consortium. (2014) Biological insights from 108 schizophrenia-associated genetic loci. Nature, 511, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blake D.J., Forrest M., Chapman R.M., Tinsley C.L., O'Donovan M.C., Owen M.J. (2010) TCF4, schizophrenia, and Pitt-Hopkins Syndrome. Schizophr. Bull., 36, 443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stefansson H., Ophoff R.A., Steinberg S., Andreassen O.A., Cichon S., Rujescu D., Werge T., Pietiläinen O.P.H., Mors O., Mortensen P.B. (2009) Common variants conferring risk of schizophrenia. Nature, 460, 744–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steinberg S., de Jong S., Andreassen O.A., Werge T., Børglum A.D., Mors O., Mortensen P.B., Gustafsson O., Costas J., Pietiläinen O.P.H.. et al. (2011) Common variants at VRK2 and TCF4 conferring risk of schizophrenia. Hum. Mol. Genet., 20, 4076–4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aberg K.A., Liu Y., Bukszár J., McClay J.L., Khachane A.N., Andreassen O.A., Blackwood D., Corvin A., Djurovic S., Gurling H. (2013) A comprehensive family-based replication study of schizophrenia genes. JAMA Psychiatry, 70, 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lennertz L., Rujescu D., Wagner M., Frommann I., Schulze-Rauschenbach S., Schuhmacher A., Landsberg M.W., Franke P., Möller H.-J., Wölwer W.. et al. (2011) Novel schizophrenia risk gene TCF4 influences verbal learning and memory functioning in schizophrenia patients. Neuropsychobiology, 63, 131–136. [DOI] [PubMed] [Google Scholar]
- 7. Lennertz L., Quednow B.B., Benninghoff J., Wagner M., Maier W., Mössner R. (2011) Impact of TCF4 on the genetics of schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci., 261 (Suppl 2), S161–S165. [DOI] [PubMed] [Google Scholar]
- 8. Quednow B.B., Ettinger U., Mössner R., Rujescu D., Giegling I., Collier D.A., Schmechtig A., Kühn K.-U., Möller H.-J., Maier W.. et al. (2011) The schizophrenia risk allele C of the TCF4 rs9960767 polymorphism disrupts sensorimotor gating in schizophrenia spectrum and healthy volunteers. J. Neurosci., 31, 6684–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Flora A., Garcia J.J., Thaller C., Zoghbi H.Y. (2007) The E-protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proc. Natl. Acad. Sci. U.S.A, 104, 15382–15387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kennedy A.J., Rahn E.J., Paulukaitis B.S., Savell K.E., Kordasiewicz H.B., Wang J., Lewis J.W., Posey J., Strange S.K., Guzman-Karlsson M.C.. et al. (2016) Tcf4 Regulates Synaptic Plasticity, DNA Methylation, and Memory Function. Cell Rep., 16, 2666–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rannals M.D., Hamersky G.R., Page S.C., Campbell M.N., Briley A., Gallo R.A., Phan B.N., Hyde T.M., Kleinman J.E., Shin J.H.. et al. (2016) Psychiatric Risk Gene Transcription Factor 4 Regulates Intrinsic Excitability of Prefrontal Neurons via Repression of SCN10a and KCNQ1. Neuron, 90, 43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brockschmidt A., Todt U., Ryu S., Hoischen A., Landwehr C., Birnbaum S., Frenck W., Radlwimmer B., Lichter P., Engels H.. et al. (2007) Severe mental retardation with breathing abnormalities (Pitt-Hopkins syndrome) is caused by haploinsufficiency of the neuronal bHLH transcription factor TCF4. Hum. Mol. Genet., 16, 1488–1494. [DOI] [PubMed] [Google Scholar]
- 13. Zweier C., Peippo M.M., Hoyer J., Sousa S., Bottani A., Clayton-Smith J., Reardon W., Saraiva J., Cabral A., Gohring I.. et al. (2007) Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins syndrome). Am. J. Hum. Genet., 80, 994–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Amiel J., Rio M., de Pontual L., Redon R., Malan V., Boddaert N., Plouin P., Carter N.P., Lyonnet S., Munnich A.. et al. (2007) Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am. J. Hum. Genet., 80, 988–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Talkowski M.E., Rosenfeld J.A., Blumenthal I., Pillalamarri V., Chiang C., Heilbut A., Ernst C., Hanscom C., Rossin E., Lindgren A.M.. et al. (2012) Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell, 149, 525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kwon E., Wang W., Tsai L.-H. (2013) Validation of schizophrenia-associated genes CSMD1, C10orf26, CACNA1C and TCF4 as miR-137 targets. Mol. Psychiatry, 18, 11–12. [DOI] [PubMed] [Google Scholar]
- 17. Brzózka M.M., Radyushkin K., Wichert S.P., Ehrenreich H., Rossner M.J. (2010) Cognitive and sensorimotor gating impairments in transgenic mice overexpressing the schizophrenia susceptibility gene Tcf4 in the brain. Biol. Psychiatry, 68, 33–40. [DOI] [PubMed] [Google Scholar]
- 18. Sepp M., Kannike K., Eesmaa A., Urb M., Timmusk T. (2011) Functional diversity of human basic helix-loop-helix transcription factor TCF4 isoforms generated by alternative 5’ exon usage and splicing. PLoS One, 6, e22138.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Farnham P.J. (2009) Insights from genomic profiling of transcription factors. Nat. Rev. Genet., 10, 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Park P.J. (2009) ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet., 10, 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kharchenko P.V., Tolstorukov M.Y., Park P.J. (2008) Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat. Biotechnol., 26, 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. ENCODE Project Consortium, (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kavanagh D.H., Dwyer S., O'Donovan M.C., Owen M.J. (2013) The ENCODE project: implications for psychiatric genetics. Mol. Psychiatry, 18, 540–542. [DOI] [PubMed] [Google Scholar]
- 24. Jin T., Liu L. (2008) The Wnt signaling pathway effector TCF7L2 and type 2 diabetes mellitus. Mol. Endocrinol., 22, 2383–2392. [DOI] [PubMed] [Google Scholar]
- 25. Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B.E., Bickel P., Brown J.B., Cayting P.. et al. (2012) ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res., 22, 1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol., 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Watanabe K., Taskesen E., van Bochoven A., Posthuma D. (2017) Functional mapping and annotation of genetic associations with FUMA. Nat. Commun., 8, 1826.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Forrest M.P., Waite A.J., Martin-Rendon E., Blake D.J. (2013) Knockdown of human TCF4 affects multiple signaling pathways involved in cell survival, epithelial to mesenchymal transition and neuronal differentiation. PLoS One, 8, e73169.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hill M.J., Killick R., Navarrete K., Maruszak A., McLaughlin G.M., Williams B.P., Bray N.J. (2017) Knockdown of the schizophrenia susceptibility gene TCF4 alters gene expression and proliferation of progenitor cells from the developing human neocortex. J. Psychiatry Neurosci., 42, 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fromer M., Roussos P., Sieberts S.K., Johnson J.S., Kavanagh D.H., Perumal T.M., Ruderfer D.M., Oh E.C., Topol A., Shah H.R.. et al. (2016) Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci., 19, 1442–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cohen N.M., Kenigsberg E., Tanay A. (2011) Primate CpG islands are maintained by heterogeneous evolutionary regimes involving minimal selection. Cell, 145, 773–786. [DOI] [PubMed] [Google Scholar]
- 32. Graham S.A., Fisher S.E. (2013) Decoding the genetics of speech and language. Curr. Opin. Neurobiol., 23, 43–51. [DOI] [PubMed] [Google Scholar]
- 33. Raychaudhuri S., Korn J.M., McCarroll S.A., Consortium T.I.S., Altshuler D., Sklar P., Purcell S., Daly M.J. (2010) Accurately Assessing the Risk of Schizophrenia Conferred by Rare Copy-Number Variation Affecting Genes with Brain Function. PLoS Genet., 6, e1001097.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bulik-Sullivan B.K., Loh P.-R., Finucane H.K., Ripke S., Yang J. Schizophrenia Working Group of the Psychiatric Genomics Consortium Patterson N., Daly M.J., Price A.L., Neale B.M. (2015) LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet., 47, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Finucane H.K., Bulik-Sullivan B., Gusev A., Trynka G., Reshef Y., Loh P.-R., Anttila V., Xu H., Zang C., Farh K.. et al. (2015) Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet., 47, 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Scholz D., Pöltl D., Genewsky A., Weng M., Waldmann T., Schildknecht S., Leist M. (2011) Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J. Neurochem., 119, 957–971. [DOI] [PubMed] [Google Scholar]
- 37. Weng M.K., Natarajan K., Scholz D., Ivanova V.N., Sachinidis A., Hengstler J.G., Waldmann T., Leist M. (2014) Lineage-Specific Regulation of Epigenetic Modifier Genes in Human Liver and Brain. PLoS One, 9, e102035.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lotharius J., Barg S., Wiekop P., Lundberg C., Raymon H.K., Brundin P. (2002) Effect of mutant alpha-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J. Biol. Chem., 277, 38884–38894. [DOI] [PubMed] [Google Scholar]
- 39. Lévy L., Wei Y., Labalette C., Wu Y., Renard C.-A., Buendia M.A., Neuveut C. (2004) Acetylation of beta-catenin by p300 regulates beta-catenin-Tcf4 interaction. Mol. Cell Biol., 24, 3404–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Erk S., Mohnke S., Ripke S., Lett T.A., Veer I.M., Wackerhagen C., Grimm O., Romanczuk-Seiferth N., Degenhardt F., Tost H.. et al. (2017) Functional neuroimaging effects of recently discovered genetic risk loci for schizophrenia and polygenic risk profile in five RDoC subdomains. Transl. Psychiatry, 7, e997.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fernandez A.M., Torres-Alemán I. (2012) The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci., 13, 225–239. [DOI] [PubMed] [Google Scholar]
- 42. Ouchi Y., Banno Y., Shimizu Y., Ando S., Hasegawa H., Adachi K., Iwamoto T. (2013) Reduced adult hippocampal neurogenesis and working memory deficits in the Dgcr8-deficient mouse model of 22q11.2 deletion-associated schizophrenia can be rescued by IGF2. J. Neurosci., 33, 9408–9419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fagerberg L., Hallström B.M., Oksvold P., Kampf C., Djureinovic D., Odeberg J., Habuka M., Tahmasebpoor S., Danielsson A., Edlund K.. et al. (2014) Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteomics, 13, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jurisch-Yaksi N., Sannerud R., Annaert W. (2013) A fast growing spectrum of biological functions of γ-secretase in development and disease. Biochimica et Biophysica Acta (BBA) - Biomembranes, 1828, 2815–2827. [DOI] [PubMed] [Google Scholar]
- 45. Auderset L., Landowski L.M., Foa L., Young K.M. (2016) Low density Lipoprotein receptor related proteins as regulators of neural stem and progenitor cell function. Stem Cells Int., 2016, 1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Buscarlet M., Perin A., Laing A., Brickman J.M., Stifani S. (2008) Inhibition of cortical neuron differentiation by Groucho/TLE1 requires interaction with WRPW, but not Eh1, repressor peptides. J. Biol. Chem., 283, 24881–24888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Machanick P., Bailey T.L. (2011) MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics, 27, 1696–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. and 1000 Genome Project Data Processing Subgroup (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Quinlan A.R., Hall I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Marinov G.K., Kundaje A., Park P.J., Wold B.J. (2014) Large-scale quality analysis of published ChIP-seq data. G3 (Bethesda), 4, 209–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Skene N.G., Bryois J., Bakken T.E., Breen G., Crowley J.J., Gaspar H., Giusti-Rodriguez P., Hodge R.D., Miller J.A., Munoz-Manchado A.. et al. (2017) Genetic identification of brain cell types underlying schizophrenia. Nat. Genet., 50, 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lun A.T.L., McCarthy D.J., Marioni J.C. (2016) A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res, 5, 2122.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lun A.T.L., Bach K., Marioni J.C. (2016) Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol., 17, 75.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vu T.N., Wills Q.F., Kalari K.R., Niu N., Wang L., Rantalainen M., Pawitan Y. (2016) Beta-Poisson model for single-cell RNA-seq data analyses. Bioinformatics, 32, 2128–2135. [DOI] [PubMed] [Google Scholar]
- 55. McClay J.L., Shabalin A.A., Dozmorov M.G., Adkins D.E., Kumar G., Nerella S., Clark S.L., Bergen S.E., Hultman C.M., Magnusson P.K.E.. et al. (2015) High density methylation QTL analysis in human blood via next-generation sequencing of the methylated genomic DNA fraction. Genome Biol., 16, 291.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sheffield N.C., Bock C. (2016) LOLA: enrichment analysis for genomic region sets and regulatory elements in R and Bioconductor. Bioinformatics, 32, 587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sheffield N.C., Thurman R.E., Song L., Safi A., Stamatoyannopoulos J.A., Lenhard B., Crawford G.E., Furey T.S. (2013) Patterns of regulatory activity across diverse human cell types predict tissue identity, transcription factor binding, and long-range interactions. Genome Res., 23, 777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kovalevich J., Langford D. (2013) Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Methods Mol. Biol., 1078, 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.