

Abstract
We use deep sequencing to identify sources of variation in mRNA splicing in the dorsolateral prefrontal cortex (DLFPC) of 450 subjects from two aging cohorts. Hundreds of aberrant pre-mRNA splicing events are reproducibly associated with Alzheimer’s disease. We also generate a catalog of splicing quantitative trait loci (sQTL) effects: splicing of 3,006 genes is influenced by genetic variation. We report that altered splicing is the mechanism for the effects of the PICALM, CLU, and PTK2B susceptibility alleles. Further, we performed a transcriptome-wide association study and identified 21 genes with significant associations to Alzheimer’s disease, many of which are found in known loci, but 8 are in novel loci. This highlights the convergence of old and new Alzheimer’s disease genes in autophagy-lysosomal-related pathways. Overall, this study of the aging brain’s transcriptome provides evidence that dysregulation of mRNA splicing is a feature of Alzheimer’s disease and is, in some cases, genetically driven.
INTRODUCTION
Alternative splicing is an important post-transcriptional regulatory mechanism through which pre-mRNA molecules can produce multiple distinct mRNAs. Alternative splicing affects over 95% of human genes1, contributing significantly to the functional diversity and complexity of proteins expressed in tissues2. Alternative splicing is abundant in human nervous system tissues3 and contributes to phenotypic differences within and between individuals: at least 20% of disease-causing mutations may affect pre-mRNA splicing4. Mutations in RNA-binding proteins (RBPs) involved in splicing regulation and aberrant splicing have been linked to Amyotrophic lateral sclerosis (ALS)5 and Autism6. Further, disruptions in RNA metabolism, including mRNA splicing, are associated with age-related disorders, such as Frontotemporal lobar dementia (FTD)7, Parkinson’s disease8 and Alzheimer’s disease9,10. These studies have largely focused on alternative splicing of selected candidate genes, including the amyloid precursor protein (APP) 8 and microtubule associated protein Tau (MAPT)8,9,11. However, proteomic profiles of Alzheimer’s disease brains identified an increased aggregation of insoluble U1 snRNP, a small nuclear RNA (snRNA) component of the spliceosomal complex, suggesting that the core splicing machinery may be altered in Alzheimer’s disease12. Apart from these studies, there have been few investigations of the possibility of more widespread splicing disruption affecting brain transcriptomes in Alzheimer’s disease13. However, a comprehensive study of cis- and trans- acting genetic factors that regulate alternative splicing in aging brains is lacking.
Over twenty-four genetic loci have now been associated with Alzheimer’s disease susceptibility by Genome-wide Association Studies (GWAS)14, and these variants are enriched for associations with gene expression levels in peripheral myeloid cells and often lie within cis-regulatory elements15. For example, we reported that one of these variants influences splicing of CD3316. Given the high abundance of alternative splicing in the brain, we hypothesized that other Alzheimer’s disease-associated genetic variants might also affect pre-mRNA splicing, possibly by disrupting efficient binding of splicing factors.
Here, by applying state-of-the-art analytic methods, we generated a comprehensive genome-wide map of splicing variation in the aging prefrontal cortex. We use this map to identify: (1) aberrant mRNA splicing events related to Alzheimer’s disease; (2) a new reference of thousands of genetic variants influencing local mRNA splicing in the brain; (3) trans acting splicing factors that are involved in intron excision; and (4) association of GWAS findings to specific genes within each Alzheimer’s disease susceptibility locus. Overall, we deepen our understanding of genetic regulation in the aging brain’s transcriptome and provide a foundation for the formulation of mechanistic hypotheses in Alzheimer’s disease and other neurodegenerative diseases.
RESULTS
Aberrant mRNA splicing in Alzheimer’s disease
We deeply sequenced RNA from frozen DLFPC samples obtained at autopsy from 450 participants in either the Religious Order Study (ROS) or the Memory and Aging Project (MAP), two prospective cohort studies of aging that include brain donation. All subjects were without known dementia at study entry. During the study, some subjects experienced cognitive decline, and, at autopsy, they displayed a range of amyloid-β and Tau pathology, with 60% of subjects having a pathologic diagnosis of Alzheimer’s disease17,18 (Supplementary Table 1). We have previously reported changes in RNA expression level in relation to Alzheimer’s disease in these data19.
Following alignment and quantification of RNA-Seq reads, LeafCutter20 was applied to estimate “percent spliced in” values (PSI) for local alternative splicing events (Fig. 1). We identified 53,251 alternatively spliced intronic excision clusters in 16,557 genes. We report more alternatively spliced intron clusters in the cortex than any other tissues or brain regions previously analyzed21. To identify aberrant splicing events, we analyzed the association between the PSI of each intron excision event and a pathologic diagnosis of Alzheimer’s disease or quantitative measures of neuropathology including neuritic plaques (NP), neurofibrillary tangles (NFT), and amyloid-β burden, while accounting for confounding factors. At a False Discovery Rate (FDR) < 0.05, we identified a total of 82 differentially spliced introns in 67 genes associated with different neuropathologies including 5 with NP, 20 with amyloid, and 48 with NFT (Supplementary Table 2). A heat map of the top differentially spliced introns associated with NFT is shown in Fig. 2a. On average, these differentially excised introns explain ~2–13% of total variation in neuropathologic burden after accounting for biological and technical covariates (Fig. 2b; Supplementary Fig. 1).
Figure 1: Overview of the study.
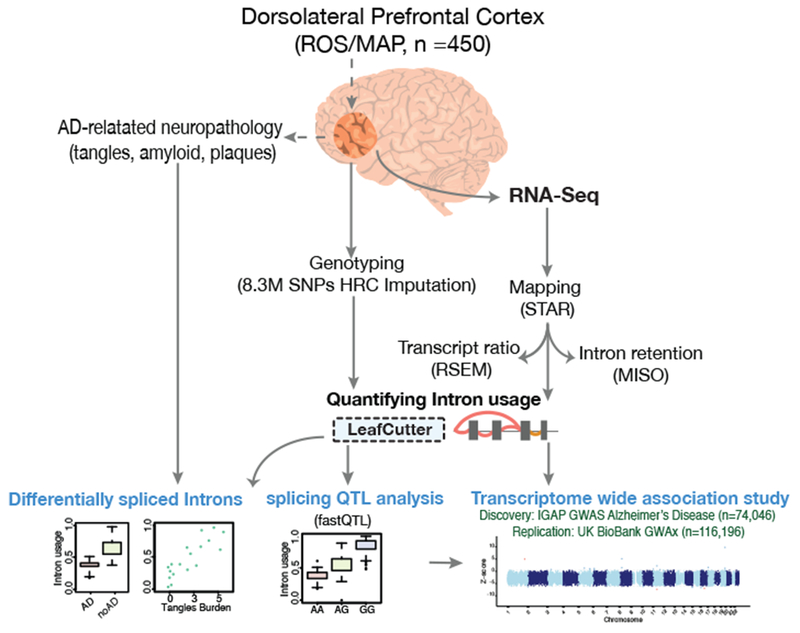
RNA was sequenced from the gray matter of the dorsal lateral prefrontal cortex (DLPFC) of 542 samples (450 remained after QC and matching for genotype data) from the ROS/MAP cohort. RNA-Seq data were processed, aligned and quantified by our parallelized pipeline. The intronic usage ratios for each cluster were then computed using LeafCutter20, standardized (across individuals) and quantile normalized. The intronic usage ratios were used for differential splicing analysis, for calling splicing QTLs, and for transcriptome-wide association studies (TWAS). TWAS was performed on summary statistics from IGAP Alzheimer’s disease GWAS of 74,046 individuals14.
Figure 2: Differential splicing analysis in relation to Alzheimer’s disease diagnosis and neuropathology.
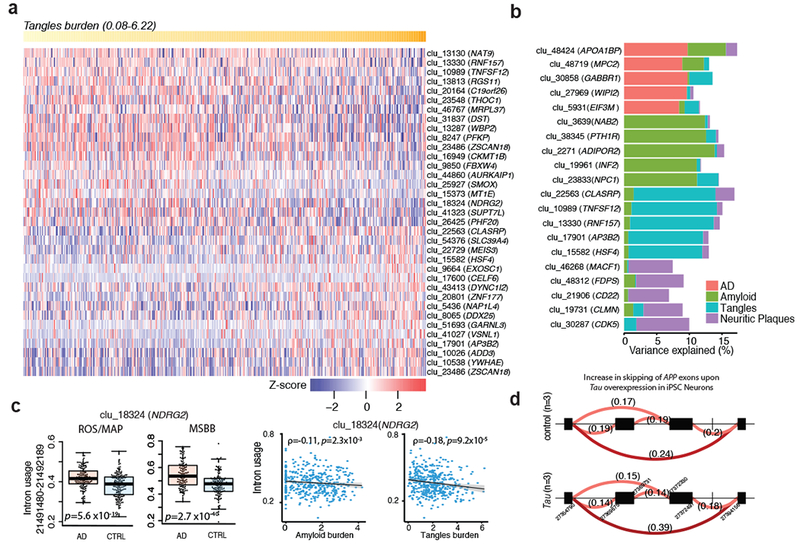
(a) Heat map of top 35 differently excised intron association with burden of tangles in ROSMAP. Each column is one subject, who are ordered by their tangles burden (yellow row at the top of the panel). The association’s Z-score strength and direction are denoted using the key at the bottom of the panel. (b) Variance explained (%) of top 5 differently excised introns association for four different traits. (c) The left two panels present the mean and distribution of intron usage for differently excised introns in NDRG2 in relation to a clinical diagnosis of Alzheimer’s disease in ROSMAP and in MSBB. The right two panels display the association of amyloid or tangle burden to intron usage in NDRG2. (d) Differentially excised intron in APP upon Tau overexpression in iPSC Neurons.
To test for association with the clinical diagnosis of Alzheimer’s disease, we used LeafCutter20 to identify differentially spliced introns by jointly modeling intron clusters using a Dirichlet-multinomial GLM (Online Methods). At a Bonferroni-corrected P < 0.05, we identified a total of 87 intron clusters (corresponding to 84 genes) that displayed altered splicing in relation to Alzheimer’s disease (Supplementary Table 3). Of these, 11 genes are also differentially expressed, suggesting that our splicing analysis is identifying novel associations that had been missed in conventional approaches evaluating gene expression levels alone. For example, the most significant differentially excised intron (chr10: 3147351–3147585) is found in the phosphofructokinase gene (PFKP): the frequency of this splicing event was associated with Alzheimer’s disease (P < 4.9×10−24; β=−0.27) and all pathologic measures tested in this study. Similarly, the next most differentially excised intron (chr14: 21490656–21491400) associated with Alzheimer’s disease is found in the alpha/beta-hydrolase fold protein gene NDRG family member 2 (NDRG2) (P < 5.6 × 10−19; β=−0.058) and is also associated with measures of both amyloid and Tau pathology (Fig. 2c). Differential splicing of both PFKP and NDRG2 in human brains has been previously shown to be associated with Alzheimer’s disease pathogenesis22,23, offering a measure of replication. Other genes with differentially excised introns associated with Alzheimer’s disease at a Bonferroni-corrected P < 0.05 include APP (P < 1.6 × 10−3; β=−0.003) and genes in known GWAS loci including PICALM (P < 0.02; β=0.005) and CLU (P < 3.2 × 10−4; β=−0.019). These differential splicing genes are not necessarily expressed in a single cell type (e.g., neurons) but are expressed across many cell types including astrocytes (Supplementary Figs. 2 and 3; Supplementary Table 4). Moreover, co-splicing network analysis using WGCNA24 suggests that the differentially spliced genes are enriched in specific functional modules and are part of a coherent biological process (Supplementary Figs. 2 and 3; Supplementary Tables 5–7; Supplementary Note).
Next, to assess the robustness of our results, we performed a replication analysis using RNA-Seq data from the Mount Sinai Brain Bank (MSSB)25 involving 301 samples from Alzheimer’s disease and control brains (Supplementary Note). Of the 84 genes with differentially spliced intron clusters in ROSMAP, 52 (including APP, PFKP, and NDRG2) were significant at a Bonferroni-corrected P < 0.05 thresholds in the MSBB data and the effect sizes are highly correlated between the two datasets (Pearson’s r=0.35; P <6.75× 10−14) (Fig. 2c; Supplementary Fig. 4; Supplementary Table 8). This constitutes an independent replication of specific, aberrant splicing alterations in Alzheimer’s disease brains. Finally, to further validate and explore the mechanism of our observations, we analyzed RNA-Seq data derived from 3 control induced pluripotent stem cells (iPSC)-derived neurons (iN) and the same iN line overexpressing Tau: differential intron excision was noted for 42 genes (FDR < 0.05), of which 11 genes overlap with the Alzheimer’s disease-associated splicing in the cortex including APP, PICALM, and NDRG2 (Fig. 2d; Supplementary Table 9). Despite the small sample size, these in vitro data suggest that tau accumulation in neurons – at a stage in which neurons are accumulating phospho-tau but are not apoptotic – may be sufficient to induce some of the splicing alterations that we observed in cortical tissue of human subjects; this in vitro validation of disease-related splicing changes suggests that this Alzheimer’s disease-altered splicing (1) is unlikely to be related to confounding factors from autopsy or the agonal state and (2) has specific target RNAs that can be modeled in vitro.
Genetic effects on pre-mRNA splicing in aging brains
We next performed a sQTL study to identify local genetic effects that drive variation in RNA splicing in the DLFPC. First, we assessed the splice events from the LeafCutter20 algorithm (Fig. 1); 30% of these 53,251 intron excision clusters are novel splicing events, not previously reported in other sQTL studies. The PSI values were adjusted for known and hidden factors (15 principal components) and then fit to imputed SNP data using an additive linear model implemented in fastQTL26 (Online Methods; Supplementary Fig. 5). At FDR < 0.05, we found 9,028 sQTLs in 3,006 genes (Supplementary Table 10). As expected, splicing was most strongly affected by variants in the splice region itself (59.8%): 20.2% of variants are mapped to splice acceptor sites and 16.4% to splice donor sites. The remaining (23.2%) mapped to other splice regions or are found within an intron (Supplementary Fig. 6). Further, sQTLs are mapped to distinct regulatory features as defined by 15 chromatin states in DLPFC27: sQTLs were significantly enriched in actively transcribed regions and enhancers. They are depleted in repressed chromatin marked with polycomb, heterochromatin, and quiescent regions (Fig. 3a), consistent with the diminished transcription noted in these regions.
Figure 3: Enrichment of splicing QTLs in epigenomic marks and in Alzheimer’s disease GWAS.
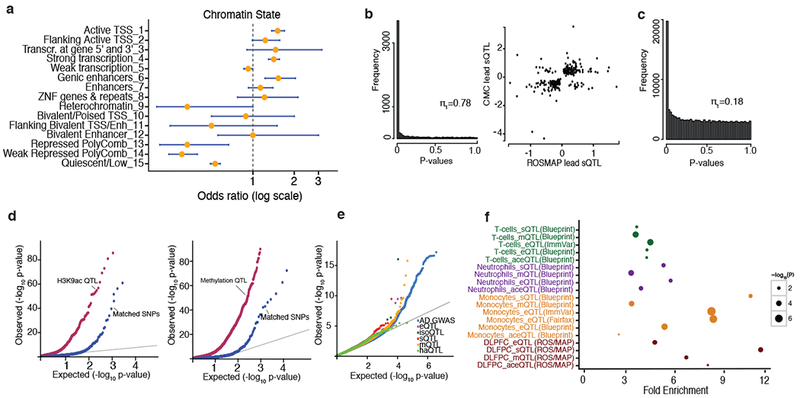
(a) Splicing QTLs are enriched in regions (or chromatin states) associated with active transcription and genic enhancers, and they are depleted in polycomb regions that are transcriptionally repressed in the DLPFC. (b) Left: P-value distribution of ROSMAP sQTLs that are significant in CMC (FDR < 0.05). The majority (78%) of sQTLs in ROSMAP are also discovered in CMC. Right: The direction of effect is consistent for the majority (93%) of the significant (FDR < 0.05) lead sQTLs in CMC and in ROSMAP. (c) P-value distribution of ROSMAP eQTLs that are significant sQTLs (FDR < 0.05). (d) SNPs that drive QTLs in H3K9ac and DNA methylation data in the same ROSMAP brains are more likely to be sQTLs than matched SNPs within H3K9ac domains (left) and near DNA methylated CG (right). (e) QQ-plot for Alzheimer’s disease GWAS suggests that sQTLs are enriched among Alzheimer’s disease GWAS (IGAP study14) compared to other types of QTLs. (f) Fold-enrichment of Alzheimer’s disease GWAS SNPs (GWAS P < 10−5) among QTL SNPs driving variation in gene expression, splicing, histone acetylation, and DNA methylation in primary monocytes15,31,50, T-cells15,31, or DLFPC29.
To assess the extent of sQTL replication, we compared our sQTLs to the recently published dataset from the CommonMind Consortium (CMC), consisting of DLFPC profiles from 258 persons with schizophrenia and 279 control subjects28 (Supplementary Note). Our sQTLs yield a Storey’s π1 = 0.78 in the CMC data, suggesting substantial sharing of sQTLs between these two different brain collections (Fig. 3b). Moreover, 93% of shared sQTLs showed the same direction of effect (Fig. 3b). The fraction of sQTLs that are novel deserves further evaluation to assess the extent to which they may be context-specific given that the average age at death of our participants (88 years) is significantly older than that of the CMC dataset.
In agreement with recent findings in lymphoblastoid cell lines (LCLs)21, we found that a majority of sQTLs act independent of gene expression effects, as evident by the low degree of sharing between sQTLs and eQTLs29 from the same brains (π1 = 0.18) (Fig. 3c). Of the 9,045 lead sQTL SNPs, only 42 are also a lead eQTL, suggesting that a substantial fraction of sQTLs are unique and are not detected by standard eQTL analysis.
To further understand the mechanisms underlying sQTLs, we assessed the overlap of sQTLs with SNPs influencing epigenomic marks (xQTLs) such as DNA methylation (mQTL) and histone H3 acetylation on lysine 9 (H3K9Ac, haQTL)29 that are available from the same DLPFC samples. Indeed, we found that such xQTLs29 are significantly enriched among sQTLs when compared to randomly selected, matched SNPs (Kolmogorov–Smirnov test P < 0.001): of the lead sQTL, 9% (578) were also associated with an haQTL, and 19% (1246) were also an mQTL (Fig. 3d). This suggests that there an important subset of genetic variants co-influences splicing, methylation levels, and histone modifications. In a complementary analysis, we found significant sharing of sQTL SNPs among SNPs that also influence histone (π1 = 0.74) or methylation (π1 = 0.82). These overlaps suggest that there is a contribution of epigenomic regulation in splicing.
Given prior reports21,30, we evaluated whether our sQTLs from the aging brain were enriched for Alzheimer’s disease susceptibility variants (Figs. 3e and 3f). We also assessed enrichment of Alzheimer’s disease SNPs (GWAS P < 1 × 10−5) in splicing, methylation or expression QTLs from DLFPC29, monocytes15,31, neutrophils31 and T-cells15,31. We found that DLFPC sQTLs are more likely to be enriched for Alzheimer’s disease GWAS SNPs, followed by sQTL and eQTL from monocytes (Fig. 3f). These findings highlight (1) the important role of RNA splicing on variation in Alzheimer’s disease susceptibility, (2) the prominent role of myeloid cells in Alzheimer’s disease susceptibility15 but also (3) the fact that a number of Alzheimer’s disease variants have mechanisms that may be mediated through non-myeloid effects.
Some of these effects of Alzheimer’s disease variants on splicing are known, such as the 8-fold increase in full-length CD33 isoform16,32, but several of these - in CLU, PICALM, and PTK2B - have not been previously reported (Supplementary Table 10). These results delineate the initial events along the cascade of functional consequences for these three Alzheimer’s disease variants and provide important mechanistic insights into their development as potential therapeutic targets.
Splicing regulators associated with alternative splicing
Splicing of pre-mRNA is catalyzed by a large ribonucleoprotein complex called the spliceosome, which consists of five snRNAs and numerous splicing factors33. To identify brain splicing factors that regulate sQTL events in trans, we evaluated whether the lead sQTL SNPs identified in our study are enriched in RBP binding sites using publicly available cross-linking immunoprecipitation (CLIP)-Seq datasets from 76 RBPs in CLIPdb34. We found that binding targets of 18 RBPs are significantly enriched among lead sQTLs (Fig. 4a). The most enriched RBP is PTBP1, followed by HNRNPC, CPSF7, and ELAVL1 (P < 0.05, Fisher’s exact test with Benjamini-Hochberg correction). Notably, the enrichment for neuronal ELAVL1 RBP target sites is consistent with a recent report that, upon neuronal ELAVL1 depletion, BIN1 and PICALM transcripts were found to have lower exon inclusion for those sites in which ELAVL binding sites directly overlapped with SNPs associated with Alzheimer’s disease35.
Figure 4: Enrichment of RNA-binding protein (RBP) binding sites among sQTLs.
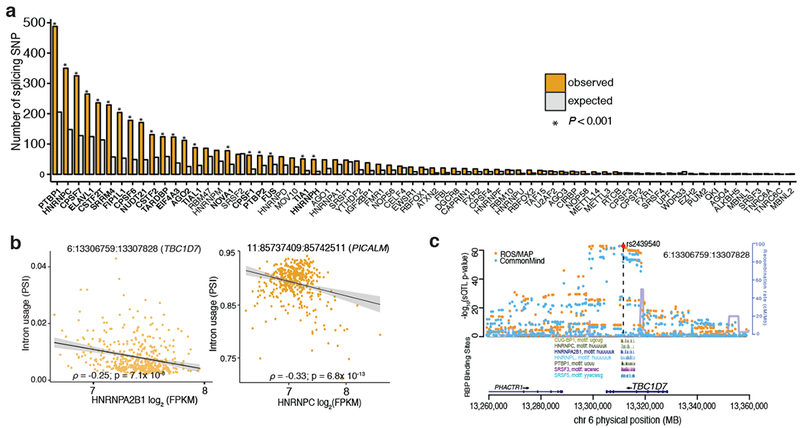
(a) RBP enrichment (expected vs. observed) among the lead sQTLs. Significant RBSs are in bold and shown with an “*”.(b) Association of hnRNPA2B1 (left) and hnRNPC (right) gene expression levels with differential intron usage in TBC1D7 (left) and in PICALM (right). (c) Regional plot of sQTL results for SNPs in the vicinity of TBC1D7 (6:13306759:13307828). SNPs driving splicing QTLs for TBC1D7 overlap CLIP binding sites (from CLIPdb34) for several splicing factors. The top SNP (rs2439540, red) overlaps motifs for a number of RBPs. Splicing QTL results are highly consistent between ROSMAP (orange) and CommonMind (blue) data.
On the other hand, we also observed significant enrichment for the lead sQTL SNPs within the binding sites for a number of heterogeneous nuclear ribonucleoproteins (hnRNP), including hnRNP C (P < 0.009). Further, we find that the expression levels of hnRNP splicing factors are correlated with intronic excision levels of hundreds of genes, many of which are in Alzheimer’s disease susceptibility loci including BIN1, PICALM, APP, and CLU (Fig. 4b; Supplementary Figs. 7 and 8). The hnRNP C factor has been linked to Alzheimer’s disease in previous studies, including in a recent biochemical study reporting the translational regulation of APP mRNA by hnRNP C36. This observation goes towards the mechanism of the sQTL: consistent with the assumption that, altering the sequence of a binding site changes the likelihood that a splicing event occurs in vivo. In one example of an sQTL affecting intron usage, a SNP within intron of TBC1D7 is found within CLIP-defined binding sites for hnRNP C as well as other RBPs (Fig. 4c). Thus, incorporating RBP binding sites as a functional annotation allows for improving our accuracy in selecting plausible variants that may disrupt binding of splicing factors to cause the alternative-splicing event. Further biochemical studies will be required to understand the full regulatory program that orchestrates the disease-related splicing changes.
TWAS prioritizes Alzheimer’s disease genes in autophagy pathways
To identify genes whose mRNA expression or alternative splicing is associated with Alzheimer’s disease and mediated by genetic variation, we performed two Transcriptome-wide association studies (TWAS)37 by using either the ROSMAP expression data or its intronic excision levels as reference panels to re-analyze summary level data from the International Genomics of Alzheimer’s Project (IGAP) GWAS14. A total of 4,746 genes and 15,013 differentially spliced introns could be analyzed, and we identified 21 genes at FDR < 0.05 whose imputed gene expression or intronic excision levels were significantly associated with Alzheimer’s disease status (Fig. 5a; Supplementary Table 11). Among these, there were genes in known Alzheimer’s disease loci including SPI1, CR1, PTK2B, CLU, MTCH2, and PICALM. These results help to pinpoint the likely gene that is the target of the known susceptibility variant in each locus, particularly at the MTCH2 locus in which the functional consequence of the risk allele was unclear. However, the new Alzheimer’s disease genes are even more interesting, and 8 of these associations are found in loci that harbored only suggestive evidence of association in the IGAP study. These genes include AP2A1, AP2A2, FUS, MAP1B, TBC1D7 and others that are now significant at a threshold adjusted for genome-wide testing. This analysis therefore helps to prioritize the long list of suggestive IGAP associations (Figs. 5a; Supplementary Figs. 9–17). Interestingly, both AP2A1 and MAP1B were recently identified as hub proteins in Alzheimer’s disease proteome networks and had lower protein expression levels in Alzheimer’s disease brains compared to ALS, PD, or control brains38.
Figure 5: Transcriptome-wide association study of Alzheimer’s Disease.
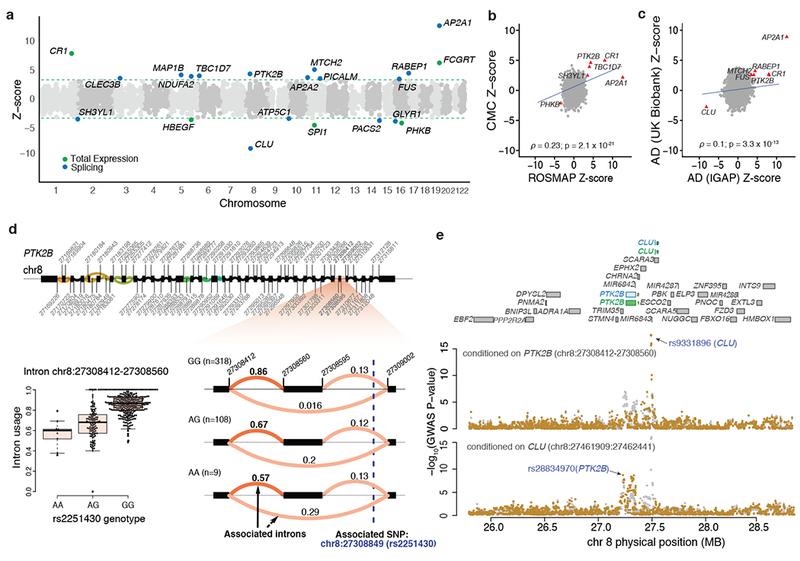
(a) Transcriptome-wide results using the IGAP GWAS summary statistics; each dot is one gene. The dotted green line denotes the threshold of significance (FDR 0.05). Genes for which there is evidence of significant differential intron usage are highlighted in blue. In green, we highlight those genes where the TWAS using total gene expression results are significant. (b) Replication of ROSMAP TWAS in CMC DLFPC data. The red triangles denote genes where the replication analysis is significant. (c) Replication of IGAP Alzheimer’s disease TWAS using the UK BioBank GWAS based on an independent set of subjects. (d) PTK2B gene structure (top): clusters of differential splicing events are noted with the colored curves. The panel then zooms to highlight differential intronic usage for chr8:27308412–27308560 stratified by rs2251430 genotypes (right). On the left, we show the same data use a box plot. (e) Conditional analysis of IGAP GWAS results for two splicing effects for PTK2B and CLU in Alzheimer’s disease GWAS data. As noted in the top aspect of the panel, these two Alzheimer’s disease genes are located close to one another. The intronic excision events for PTK2B and CLU are present in both ROSMAP (blue) and in CMC (green) dataset. When the Alzheimer’s disease GWAS is conditioned on the PTK2B (chr8:27308412–27308560) splicing effect, the CLU effect remained significant, demonstrating its independence from the PTK2B association. The reciprocal analysis conditioning on the CLU (chr8:27461909:27462441) effect, the PTK2B association remained significant.
To replicate these results, we first assessed whether an expression imputation model built using the CMC dataset28 that was then deployed in the IGAP GWAS yields similar results. We focused on the 21 significantly associated genes above: four genes (CR1, PTK2B, TBC1D7, and SH3YL1) replicated at FDR < 0.05 and two genes (AP2A1 and PHKB) were suggestive at P < 0.05 with the expression and splicing inference from CMC (Fig. 5b). The directions of effect for all six associations were consistent in both datasets (Fig. 5b). Thus, we see replication of our results: they are not due to the unique properties of the ROSMAP dataset. Second, we used a different Alzheimer’s disease GWAS - the UK BioBank (UKBB) GWAS by proxy39 - to replicate the IGAP TWAS results. We note that, despite analyzing data from 116,196 subjects, the UKBB GWAS is underpowered since the GWAS does not use Alzheimer’s disease cases but, rather, subjects who have a first-degree relative with Alzheimer’s disease as “cases”. Nevertheless, we were able to replicate (at a nominal P < 0.05) seven of our IGAP TWAS associations in the UKBB TWAS (Fig. 5c). These two complementary replication efforts demonstrate the robustness of our results. Finally, we performed a TWAS using the summary statistics of a meta-analysis of IGAP and UKBB GWAS, and identified three additional genes (ABCA7, RHBDF1, and VPS53) that meet a genome-wide significant threshold in the meta-analysis, with ABCA7 being one of the well-validated Alzheimer’s disease loci (Supplementary Table 12).
Most of the TWAS associations are the result of differential intron usage, suggesting the importance of pre-mRNA splicing in Alzheimer’s disease (Fig. 5a). An example of TWAS association with intron usage at PTK2B, a known Alzheimer’s disease susceptibility locus, is shown in Fig. 5d. We often observed multiple TWAS-associated genes in the same locus, likely due to co-expression of genes in close physical proximity or allelic heterogeneity within the susceptibility locus40. To account for multiple associations in the same locus, we applied conditional and joint association methods that rely on summary statistics40,41 to identify genes that had significant TWAS associations when analyzed jointly (Figs. 5e and 6b). A region with multiple TWAS association includes the PTK2B/CLU locus, which shows independent co-localized association for both GWAS14 and splicing effects (Fig. 5e).
Figure 6: TWAS prioritizes Alzheimer’s disease genes in endocytosis and autophagy-related pathway.
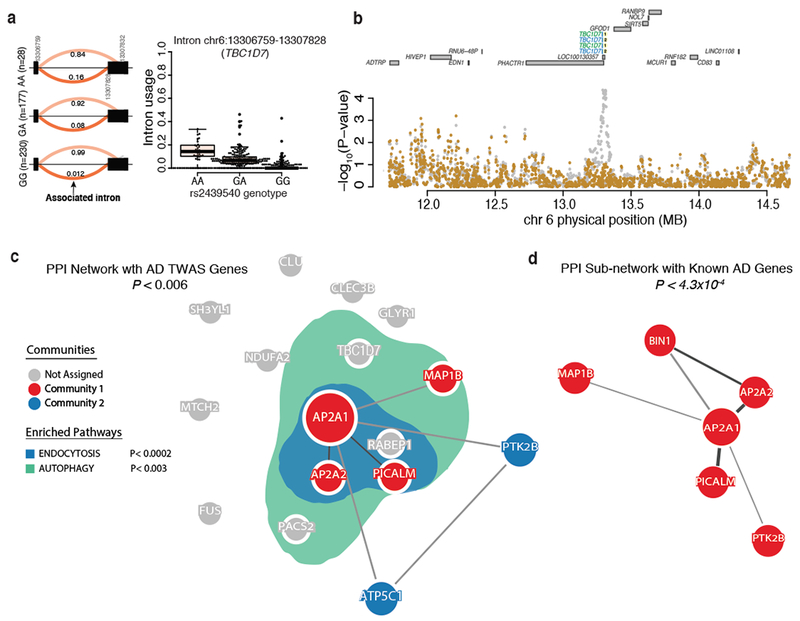
(a) Differential intronic usage for chr6: 13306759:13307828 (TBC1D7) stratified by rs2439540 genotypes (left). Box plot for the same data (right). (b) Regional plot showing the IGAP P-values in TBC1D7 locus. Two intronic excision events at TBC1D7 are present in both ROSMAP (blue) and in CMC (green) dataset. The Alzheimer’s disease GWAS effect is mostly explained by intronic usage of chr6:13306759:13307828. The AD GWAS at TBC1D7 is suggestive in the original IGAP study (p<10−5). (c) The product of three of the novel Alzheimer’s disease genes (AP2A2, AP2A1, and MAP1B) are members of the same PPI network (P < 0.006). The genes in this network and others not in the network (i.e., TBC1D7, PACS2, and RABEP1) are significantly enriched in genes annotated as being involved in endocytosis (blue; P < 0.0002) and autophagy-related pathways (green; P < 0.003). (d) The novel Alzheimer’s disease genes (AP2A2, AP2A1, and MAP1B) form a significant PPI sub-network (P < 4.3 ×10−4) with known Alzheimer’s disease genes (i.e., PICALM, BIN1, and PTK2B).
Refining known associations is important to translate results into functional studies, but the newly validated Alzheimer’s disease genes (Figs. 6a and 6b) also offer new insights into disease: we used GeNets42 to evaluate the connectivity of our new Alzheimer’s disease genes with the network of known susceptibility genes that are interconnected by protein-protein interaction (PPI)43. These new and known Alzheimer’s disease susceptibility genes are directly connected (i.e., they form shared ‘communities’) (P < 0.006) (Fig. 6c). Further, this joint network is enriched for endocytosis pathways (P < 0.0002), highlighting the existing narrative of endocytosis pathways as being preferentially targeted in Alzheimer’s disease. The enrichment for autophagy-lysosomal related pathway (P < 0.003) is more interesting (Fig. 6c). The genes in the autophagy-lysosomal related pathway (AP2A2, AP2A1, and MAP1B) form a statistically significant (P < 4.3×10−4) PPI sub-network with known Alzheimer’s disease genes (PTK2B, PICALM and BIN1) (Fig. 6d). Protein degradation pathways have been implicated previously in Alzheimer’s disease44. Overall, these PPI analyses suggest that our new TWAS-derived genes are not a random set of genes but are part of an Alzheimer’s disease network.
DISCUSSION
In this study, we directly examined alternative splicing events in a large dataset of aging brains, which led to both the observation that specific alternative splicing events are reproducibly associated with Alzheimer’s disease and that certain validated genetic associations affect splicing of a nearby gene as the proximal functional consequence of the susceptibility allele. Our replication efforts demonstrate that the observed Alzheimer’s disease-related perturbations in splicing are not simply due to spliceosomal failure: specific genes are reproducibly affected in a specific manner. Further, results from our in vitro model of tau overexpression in iPSC-derived neurons suggest that perturbation of MAPT may be sufficient to cause at least a subset of these disease-related splicing changes that are observed in the human cortex at autopsy.
We used the powerful TWAS approach, which leverages our splicing map and common genetic variants to test the hypothesis that the effect of such variants in Alzheimer’s disease is mediated, by altering splicing levels. These analyses confirmed many of the known Alzheimer’s disease genes (i.e., CLU and PTK2B), which supports the role of regulation of splicing levels as key mechanisms in certain loci, but also found several new loci: TBC1D7, AP2A1, AP2A2, and MAP1B (Figs. 5a and 6b; Supplementary Figs. 7, 9, and 13). These new genes reinforce the association of the Clathrin/AP2 adaptor complex with Alzheimer’s disease susceptibility45. Both AP2A2 and AP2A1, which are components of the AP2 adaptor complex that serves as a cargo receptor, selectively sorting membrane proteins involved in receptor-mediated endocytosis46. The AP2 complex and PICALM interact with APP, directing it to degradation and autophagy46.
Our study also offers insights for several well-known Alzheimer’s disease loci in which the gene was known but the functional mechanism remained unclear. Similar to our work in CD3316, the careful analysis of these cortical data highlights a specific splicing mechanism for the Alzheimer’s disease risk alleles at CLU, PICALM, and PTK2B. All three are complex proteins with a large number of exons, so our results prioritize specific domains in these proteins as harboring the functional domain that influences Alzheimer’s disease risk. Further, our analyses of RBP involved in splicing regulation of Alzheimer’s disease susceptibility genes including PICALM, and RNA binding site analysis of HNRNPC (Fig. 4c; Supplementary Fig. 6) and ELAVL helps to prioritize the variant that may be driving the genetic association and to elaborate the series of events upstream of the susceptibility variant that enable its expression. Thus, our catalog of splicing variants made available with this study provides a starting point for further focused molecular and biochemical experimental validation to fully elucidate the role of these splicing variants in the etiology of Alzheimer’s disease.
This study has several limitations. We only characterize splicing events in one region (the DLPFC) of the aging brain. The DLPFC is a region that is affected by amyloid pathology relatively early as it spreads throughout the neocortex47. The accumulation of Tau pathology progresses in a stereotypic manner captured by the Braak stages48, and the DLPFC displays accumulation of NFTs containing Tau typically when individuals begin to be symptomatic. Thus, both amyloid and Tau pathology accumulate in the DLPFC in Alzheimer’s disease, and we use quantitative measures of these pathologies to enhance our power in discovering the molecular features that are associated with these pathologies. Some of these splicing changes may contribute to the accumulation of these pathologies while others may be a reaction to the presence of pathology or may be the result of indirect effects of the pathology in other brain regions. Currently, we cannot differentiate these three sources of variation in our results. Expression datasets from multiple brain regions exist in the GTEx project49, but the sample size is small (n=88–136) to build a robust transcriptome model for TWAS. The MSBB has RNA-seq data across three brain regions but due to the lack of availability for individual level genotypes we are unable to build reference models from those data. Another limitation of this study is the small sample size of the in vitro experiment; thus, these intriguing results will require testing in a much larger number of iPSC lines to confirm that this effect of MAPT overexpression is generalizable. We note that these MAPT overexpressing iPSC-derived neurons are functioning normally at the time when they were sampled; thus, these in vitro data suggest that at least some of the disease-associated splicing changes that we report may occur very early in the series of molecular events that are caused by perturbation in MAPT expression.
This transcriptome-wide reference map of RNA splicing in the aging cortex is a new resource that highlights strong effects of neuropathology and genetic variation on splicing. It will be useful in annotating the results of genetic and epigenomic studies of neurologic and psychiatric diseases, but it has an immediate impact in (1) identifying the functional consequences of several Alzheimer’s disease susceptibility alleles, (2) extending the list of loci involved in Alzheimer’s disease, and (3) implicating the protein degradation machinery in the pathology of Alzheimer’s disease.
ONLINE METHODS
Study Cohorts
Religious Orders Study (ROS):
From January 1994 through June of 2010, 1,148 persons agreed to annual detailed clinical evaluation and brain donation at the time of death. Of these, 1,139 have completed their baseline clinical evaluation: 68.9% were women; 88.0% were white, non-Hispanic; their mean age was 75.6 years; and mean education was 18.1 years. There were 287 cases of incident dementia and 273 cases of incident Alzheimer’s disease with or without a coexisting condition. Details of the clinical and pathologic methods have been previously reported 17.
Memory and Aging Project (MAP):
From October 1997 through June 2010, 1,403 persons agreed to annual detailed clinical evaluation and donation of the brain, spinal cord, nerve, and muscle at the time of death. Of these, 1,372 completed their baseline clinical evaluation: 72.7% were women; 86.9% were white, non-Hispanic; their mean age was 80.0 years; and mean education was 14.3 years with 34.0% with 12 or fewer years of education. There were 250 cases of incident dementia and 238 cases of incident Alzheimer’s disease with or without a coexisting condition. Details of the clinical and pathologic methods have been previously reported 51. To avoid population stratification artifacts in the genetic analyses, the study was limited to non-Latino whites.
See Supplementary Notes for the details of CommonMind Consortium (CMC) and Mount Sinai Brain Bank (MSBB) datasets.
Data acquisition, quality control, and normalization
Genotyping.
DNA from ROS and MAP subjects was extracted from whole blood, lymphocytes or frozen post-mortem brain tissue and genotyped on the Affymetrix GeneChip 6.0 platform at the Broad Institute’s Center for Genotyping. Only self-declared non-Latino Caucasians were genotyped to minimize population heterogeneity. PLINK software52 was used to implement our QC pipeline. We applied standard QC measures for subjects (genotype success rate >95%, genotype-derived gender concordant with reported gender, excess inter/intra-heterozygosity) and for single nucleotide polymorphisms (SNPs) (HWE P > 0.001; MAF > 0.01, genotype call rate > 0.95; misshap test > 1×10−9) to these data. Subsequently, EIGENSTRAT53 was used to identify and remove population outliers using default parameters. Imputation was performed using Michigan Imputation Server with Minimac354 using Haplotype Reference Consortium (HRC version r1.1, 2016)55 panel consisting of 64,940 haplotypes of predominantly European ancestry. Imputation filtering of r2 > 0.3 was used for quality control. After QC, 450 individuals and 8,383,662 genotyped or imputed markers were used for sQTL analysis.
RNA-Seq data.
RNA was sequenced from the gray matter of dorsal lateral prefrontal cortex (DLPFC) of 542 samples, corresponding to 540 unique brains. These samples were extracted using Qiagen’s miRNeasey mini kit and the RNase free DNase Set. RNA was quantified using Nanodrop. The quality of RNA was evaluated by the Agilent Bioanalyzer. All samples were chosen to pass two initial quality filters: RNA integrity (RIN) score >5 and quantity threshold of 5 μg (and were selected from a larger set of 724 samples). RNA-Seq library preparation was performed using the strand specific dUTP method14 with poly-A selection. Sequencing was performed on the Illumina HiSeq with 101bp paired-end reads and achieved coverage of 150M reads of the first 12 samples. These 12 samples served as a deep coverage reference and included 2 males and 2 females of non-impaired, mild cognitive impaired, and Alzheimer’s cases. The remaining samples were sequenced with target coverage of 50M reads; the mean coverage for the samples passing QC is 95 million reads (median 90 million reads). The libraries were constructed and pooled according to the RIN scores such that similar RIN scores would be pooled together. Varying RIN scores result in a larger spread of insert sizes during library construction and leads to uneven coverage distribution throughout the pool.
The RNA-Seq data were processed by a parallelized pipeline. This pipeline includes trimming the beginning and ending bases from each read, identifying and trimming adapter sequences from reads, detecting and removing rRNA reads, and aligning reads to reference genome. Specifically, RNA-Seq reads in FASTQ format were inspected using FASTQC program. Barcode and adapter contamination, low-quality regions (8bp at beginning and 7bp at ending of each FASTQ reads) were trimmed using FASTX-toolkit. To remove rRNA contamination, we aligned trimmed reads to rRNA reference (rRNA genes were downloaded from UCSC genome browser selecting the RepeatMask table) by BWA then extracted only paired unmapped reads for transcriptome alignment. STAR (v2.5)56 (was used to align reads to the transcriptome reference, and RSEM (v1.3.0)57 was used to estimate expression levels for all transcripts. To quantify the contribution of experimental and other confounding factors to the overall expression profiles, we used the COMBAT algorithm58 to account for the effect of batch and linear regression to remove the effects of RIN, post-mortem interval (PMI), sequencing depth, study index (ROS sample or MAP sample), genotyping PCs, age at death, and sex. Finally, only highly expressed genes were kept (mean expression >2 log2-FPKM), resulting in 13,484 expressed genes for eQTL analysis. The details for cis-eQTL analysis are in Ng et al.29.
Intron usage mapping and quantification.
We used LeafCutter20 to obtain clusters of variably spliced introns. Leafcutter allows the identification of splicing events without relying on existing annotations, which are typically incomplete, especially in the setting of large genes or individual/population-specific isoforms. Leafcutter defines “clusters” of introns that represent alternative splicing choices. To do this, it first groups together overlapping introns (defined by spliced reads). For each of these groups, Leafcutter constructs a graph where nodes are introns and edges represent overlapping introns. The connected components of this graph define the intron clusters. Singleton nodes (introns) are discarded. For each intron cluster, it iteratively (1) removed introns that were supported with fewer than 100 reads or fewer than 5% of the total number of intronic read counts for the entire cluster, and (2) re-clustered introns according to the procedure above. The intron usage ratio for each clusters was next computed and standardized (across individuals) and quantile normalized (across sample) as in Li et al.20. LeafCutter was carefully benchmarked against other methods (see Li et al.20), and was able, for example, to identify as many or more differentially spliced events d than compared to other methods.
Association of intron usage with Alzheimer’s disease and neuropathology traits.
The association analysis with neuropathology traits and intron usage was performed using a linear model, adjusting for experimental batch, RNA integrity number (RIN), sex, age at death, and post-mortem interval (PMI). To test for association with Alzheimer’s disease, we limited the comparison to those participants clinical diagnosis of Alzheimer’s disease and those who have neither diagnosis (Supplementary Table 1). We used Leafcutter20 to identify intron clusters with at least one differentially excised intron by jointly modeling intron clusters using a Dirichlet-multinomial GLM20. To account for neuronal loss and cell type proportion in each brain sample, we used gene expression level of cell type specific genes as an additional covariate. However, these measures did not affect our association analysis. We report differentially spliced introns at Bonferroni-corrected P < 0.05 to correct for multiple hypothesis testing.
We used variancePartition59 to estimate the proportion of variance explained of differently excised introns association with Alzheimer’s disease, burden of amyloid, burden of tangles, and neuritic plaques.
Splicing QTL mapping.
We used Leafcutter20 to obtain the proportion of intron defining reads to the total number of reads from the intron cluster it belongs to. This intron ratio describes how often an intron is used relative to other introns in the same cluster. We used WASP60 to remove read-mapping biases caused by allele-specific reads. This is particularly significant when a variant is covered by reads that also span intron junctions as it can lead to a spurious association between the variant and intron excision level estimates. We standardized the intron ratio values across individuals for each intron and quantile normalize across introns61 and used this as our phenotype matrix. We used linear regression (as implemented in fastQTL)26 to test for associations between SNP dosages (MAF ≥ 0.01) within 100kb of intron clusters and the rows of our phenotype matrix that correspond to the intron ratio within each cluster. As covariate, we used the first 3 principal components of the genotype matrix to account for the effect of ancestry plus the first 15 principal components of the phenotype matrix (PSI) to regress out the effect of known and hidden factors. The principal components regress out the technical and biological covariates such as experimental batch, RNA integrity number (RIN), sex, age at death, and post-mortem interval (PMI). To estimate the number of sQTLs at any given false discovery rate (FDR), we ran an adaptive permutation scheme26, which maintains a reasonable computational load by tailoring the number of permutations to the significance of the association. We computed the empirical gene-level p-value for the most significant QTL for each gene. Finally, we applied Benjamini-Hochberg correction on the permutation p-values to extract all significant splicing QTL pairs with an FDR < 0.05.
Transcriptome-wide Association Study.
We used RNA-seq data and genotypes from ROSMAP to impute the cis genetic component of expression/intron usage37,40 into large-scale late-onset Alzheimer’s disease GWAS of 74,046 individuals from the International Genomics of Alzheimer’s Project (IGAP)14. The complete TWAS pipeline is implemented in FUSION suite of tools40. The details steps implemented in FUSION are: (1) estimate heritability of gene expression or intron usage unit and stop if not significant. We estimated using a robust version of GCTA-GREML62, which generates heritability estimates per feature as well as the as well as the likelihood ratio test (LRT) P-value. Only features that have a heritability of Bonferroni-corrected P < 0.05 were retained for TWAS analysis. (2) The expression or intron usage weights were computed by modeling all cis-SNPs (1MB +/− from TSS) using best linear unbiased prediction (BLUP), or modeling SNPs and effect sizes (BSLMM), LASSO, Elastic Net and top SNPs37,40. A cross-validation for each of the desired models are performed; (3) Perform a final estimate of weights for each of the desired models and store results. The imputed unit is treated as a linear model of genotypes with weights based on the correlation between SNPs and expression in the training data while accounting for LD among SNPs. To account for multiple hypotheses, we applied an FDR < 0.05 within each expression and splicing reference panel that was used.
We used the same TWAS pipeline to process the CMC datasets (see Supplementary Notes).
Joint and conditional analysis.
Joint and conditional analysis of TWAS results was performed using the summary statistic-based method described in Yang et al.41, which we applied to genes instead of SNPs. We used TWAS statistics from the main results and a correlation matrix to evaluate the joint/conditional model. The correlation matrix was estimated by predicting the cis-genetic component of expression for each TWAS gene and computing Pearson correlations across all pairs of genes. We used FUSION tool to perform the joint/conditional analysis, generate conditional outputs, and generate plots.
Gene Expression, DNA Methylation, Histone Modification QTL Mapping.
The details of ROSMAP gene expression, DNA methylation, and histone modification data are described in Supplementary Notes. The quantitative trait locus (xQTL) analysis on a multi-omic dataset is described in Ng et al.29. The xQTL results and analysis scripts can be accessed through online portal, xQTL Serve (see URLs).
QTL Sharing.
We used the Storey’s π1 statistics63 also described in Nica et al.64, QTL sharing was estimated as the proportion of true associations π1 among the top SNP in each QTLs in the second QTL.
Enrichment of sQTLs within epigenomic marks and splicing factor binding sites.
We selected a set of 71 human curated RNA-binding proteins (RBP) splicing regulatory proteins from the SpliceAid-F database65 to analyze the relationship between gene expression levels of RBP and intron usage patterns across all samples. To test for enrichment of sQTLs in RBP binding sites, we downloaded human CLIP data in BED format from ClipDB34. We used GREGOR66 (Genomic Regulatory Elements and Gwas Overlap algoRithm) to evaluate global enrichment of trait-associated variants in splicing factor binding sites. GREGOR66 evaluates the significance of the observed overlap (of sQTL and splicing factor binding sites) by estimating the probability of the observed overlap of the lead sQTL relative to expectation using a set of matched control variants (random control SNPs are selected across the genome that match the index SNP for a number of variants in LD, minor allele frequency, and distance to nearest intron). We used Fisher’s Exact Test in combination with Benjamini-Hochberg False Discovery Rate (FDR) correction for multiple testing.
Enrichment of sQTLs in Chromatin States.
We downloaded chromatin states from the Roadmap Epigenomics Project. The 15 chromatin states were generated from 5 chromatin marks in DLPFC of a cognitively non-impaired MAP subject with minimal pathology as part of the Roadmap Epigenomics Consortium27. A ChromHMM model applicable to brain epigenome was learned by virtually concatenating consolidated data corresponding to the core set of 5 chromatin marks assayed (H3K4me3, H3K4me1, H3K36me3, H3K27me3, H3K9me3). BED files downloaded from Washington University in St. Louis Roadmap Epigenome Browser. To test for enrichment for sQTLs among the 15 chromatin states, we used GREGOR66 to evaluate global enrichment of trait-associated variants in splicing factor binding sites.
GWAS Enrichment Analyses.
We used GARFIELD (unpublished; http://www.ebi.ac.uk/birney-srv/GARFIELD/) to test for enrichment of IGAP Alzheimer’s disease GWAS SNPs among sQTLs and other publicly available QTL datasets. GARFIELD performs greedy pruning of GWAS SNPs (LD r2 >0.1) and then annotates them based on functional information overlap. It quantifies fold enrichment at GWAS P <10−5 significant cutoff and assesses them by permutation testing, while matching for minor allele frequency, distance to nearest transcription start site and a number of LD proxies (r2 > 0.8).
Q-Q plots show quantiles of one dataset against quantiles of a second dataset and are commonly used in GWAS to show a departure from an expected P-value distribution. We generated Q-Q plots for LD-pruned GWAS SNPs (PLINK with the settings “-- indep- pairwise 100 5 0.8”). We compared the sQTLs overlapping with LD-pruned GWAS SNPs and compared the distribution to a random set of SNPs with similar MAF.
GWAS Datasets.
We performed transcriptome-wide association study using GWAS summary statistics from: (1) Alzheimer’s disease GWAS from the International Genomics of Alzheimer’s Project (stage 1 data)14; (2) Alzheimer’s disease genome-wide association study by proxy (GWAx) in 116,196 individuals from the UK Biobank39.
Protein-protein Interaction Network and Pathway Analysis.
We constructed a protein-protein interaction (PPI) network using the GeNets42 to determine whether the Alzheimer’s disease TWAS genes significantly interact with each other and with known Alzheimer’s disease associated proteins. GeNets create networks of connected proteins using evidence of physical interaction from the InWeb database, which contains 420,000 high-confidence pair-wise interactions involving 12,793 proteins67. Community structures of the underlying genes are displayed in GeNets. These “communities” are also called modules or clusters. This feature highlights genes that are more connected to one another than they are to other genes in other modules. To assess the statistical significance of PPI networks, GeNets applies a within-degree node-label permutation strategy to build random networks that mimic the structure of the original network and evaluates network connectivity parameters on these random networks to generate empirical distributions for comparison to the original network. In addition to PPI network analysis, GeNets allows for gene set enrichment analysis on genes within the PPI network. We used Molecular Signatures Database (MSigDB) Curated Gene Sets (C2), curated from various sources such as online pathway databases, the biomedical literature, and knowledge of domain experts and Canonical Pathways (CP), curated from pathway databases such KEGG, BioCarta, and Reactome to test for gene set enrichment within the PPI network. Then a hypergeometric testing is applied to get P-value for gene set enrichment. We used Bonferroni-corrected P < 0.05 to correct for multiple hypothesis testing.
Life Sciences Reporting Summary.
Further information on experimental design is available in the Life Sciences Reporting Summary.
Data availability.
The ROSMAP splicing QTL visualization (Shiny App) browser is made available at https://rajlab.shinyapps.io/sQTLviz_ROSMAP/. The ROSMAP data are available at the RADC Research Resource Sharing Hub at www.radc.rush.edu. The ROSMAP and MSBB mapped RNA-seq data that support the findings of this study are available in AMP-AD Knowledge Portal (https://www.synapse.org/#!Synapse:syn2580853) upon authentication by the Consortium. The CommonMind Consortium data are available in CMC Knowledge Portal: https://www.synapse.org/#!Synapse:syn4923029.
Supplementary Material
Acknowledgments
We thank the participants of ROS and MAP for their essential contributions and gift to these projects. We thank A. Gusev for helpful discussions and for sharing the source code and scripts for TWAS. We thank the International Genomics of Alzheimer’s Project (IGAP) for providing summary results data for these analyses. T.R. is supported by grants from the NIH National Institute on Aging (R01AG054005) and the Alzheimer’s Association. P.L.D. is supported by NIH R01AG036836. D.AB. is supported by NIH P30AG10161, R01AG015819, R01AG017917. P.L.D and D.A.B are supported by NIH U01AG046152.
We thank the patients and families who donated material for CommonMind Consortium data. The CommonMind Consortium data are available in CMC Knowledge Portal: https://www.synapse.org/#!Synapse:syn4923029. Data were generated as part of the CMC supported by funding from Takeda Pharmaceuticals Company Limited, F. Hoffman-La Roche Ltd and NIH grants R01MH085542, R01MH093725, P50MH066392, P50MH080405, R01MH097276, RO1-MH-075916, P50M096891, P50MH084053S1, R37MH057881 and R37MH057881S1, HHSN271201300031C, AG02219, AG05138 and MH06692. Brain tissue for the study was obtained from the following brain bank collections: the Mount Sinai NIH Brain and Tissue Repository, the University of Pennsylvania Alzheimer’s Disease Core Center, the University of Pittsburgh NeuroBioBank and Brain and Tissue Repositories and the NIMH Human Brain Collection Core. CMC Leadership: Pamela Sklar, Joseph Buxbaum (Icahn School of Medicine at Mount Sinai), Bernie Devlin, David Lewis (University of Pittsburgh), Raquel Gur, Chang-Gyu Hahn (University of Pennsylvania), Keisuke Hirai, Hiroyoshi Toyoshiba (Takeda Pharmaceuticals Company Limited), Enrico Domenici, Laurent Essioux (F. Hoffman-La Roche Ltd), Lara Mangravite, Mette Peters (Sage Bionetworks), Thomas Lehner, Barbara Lipska (NIMH).
Footnotes
URLs.
ROSMAP sQTL browser, https://rajlab.shinyapps.io/sQTLviz_ROSMAP/
LeafCutter, https://github.com/davidaknowles/leafcutter;
xQTL Browser, http://mostafavilab.stat.ubc.ca/xQTLServe;
FUSION, http://gusevlab.org/projects/fusion/
MISO, http://genes.mit.edu/burgelab/miso/;
SpliceAid-F, http://srv00.recas.ba.infn.it/SpliceAidF/;
Roadmap Epigenomics Project, http://egg2.wustl.edu/roadmap/web_portal/chr_state_learning.html;
GREGOR, http://genome.sph.umich.edu/wiki/GREGOR;
GARFIELD, http://www.ebi.ac.uk/birney-srv/GARFIELD;
GeNets, https://apps.broadinstitute.org/genets;
Michigan Imputation Server, https://imputationserver.sph.umich.edu/index.html;
The RUSH Alzheimer’s Disease Research Center Research Resource Sharing Hub, https://www.radc.rush.edu;
AMP-AD Synapse Portal, https://www.synapse.org/#!Synapse:syn2580853/wiki/409844;
CommonMind Consortium Knowledge Portal, https://www.synapse.org/#!Synapse:syn2759792/wiki/69613;
IGAP GWAS summary statistics, http://web.pasteur-lille.fr/en/recherche/u744/igap/igap_download.php;
UK Biobank summary statistics, http://gwas-browser.nygenome.org/downloads/gwas-browser/.
Competing financial interests
The authors declare no competing financial interests.
REFERENCES
- 1.Wang ET et al. Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–6 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kornblihtt AR et al. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol 14, 153–65 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Barbosa-Morais NL et al. The evolutionary landscape of alternative splicing in vertebrate species. Science 338, 1587–93 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Wang GS & Cooper TA Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet 8, 749–61 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Dredge BK, Polydorides AD & Darnell RB The splice of life: alternative splicing and neurological disease. Nat Rev Neurosci 2, 43–50 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Parikshak NN et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540, 423–427 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arai T et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351, 602–11 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Trabzuni D et al. MAPT expression and splicing is differentially regulated by brain region: relation to genotype and implication for tauopathies. Hum Mol Genet 21, 4094–103 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rockenstein EM et al. Levels and alternative splicing of amyloid beta protein precursor (APP) transcripts in brains of APP transgenic mice and humans with Alzheimer’s disease. J Biol Chem 270, 28257–67 (1995). [DOI] [PubMed] [Google Scholar]
- 10.Buee L, Bussiere T, Buee-Scherrer V, Delacourte A & Hof PR Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 33, 95–130 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Valenca GT et al. The Role of MAPT Haplotype H2 and Isoform 1N/4R in Parkinsonism of Older Adults. PLoS One 11, e0157452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bai B et al. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc Natl Acad Sci U S A 110, 16562–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaquero-Garcia J et al. A new view of transcriptome complexity and regulation through the lens of local splicing variations. Elife 5, e11752 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lambert JC et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45, 1452–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raj T et al. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science 344, 519–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raj T et al. CD33: increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum Mol Genet 23, 2729–36 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennett DA, Schneider JA, Arvanitakis Z & Wilson RS Overview and findings from the religious orders study. Curr Alzheimer Res 9, 628–45 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bennett DA et al. Selected findings from the Religious Orders Study and Rush Memory and Aging Project. J Alzheimers Dis 33 Suppl 1, S397–403 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mostafavi S et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat Neurosci 21, 811–819 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li YI et al. Annotation-free quantification of RNA splicing using LeafCutter. Nat Genet 50, 151–158 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li YI et al. RNA splicing is a primary link between genetic variation and disease. Science 352, 600–4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tollervey JR et al. Analysis of alternative splicing associated with aging and neurodegeneration in the human brain. Genome Res 21, 1572–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitchelmore C et al. NDRG2: a novel Alzheimer’s disease associated protein. Neurobiol Dis 16, 48–58 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Zhang B & Horvath S A General Framework for Weighted Gene Co-Expression Network Analysis. Statistical Applications in Genetics and Molecular Biology 4, Article 17 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Wang M et al. Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer’s disease. Genome Med 8, 104 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ongen H, Buil A, Brown AA, Dermitzakis ET & Delaneau O Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics 32, 1479–85 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernstein BE et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol 28, 1045–8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fromer M et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci 19, 1442–1453 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ng B et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat Neurosci 20, 1418–1426 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicolae DL et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet 6, e1000888 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen L et al. Genetic Drivers of Epigenetic and Transcriptional Variation in Human Immune Cells. Cell 167, 1398–1414 e24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malik M et al. CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J Neurosci 33, 13320–5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sibley CR, Blazquez L & Ule J Lessons from non-canonical splicing. Nat Rev Genet 17, 407–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang YC et al. CLIPdb: a CLIP-seq database for protein-RNA interactions. BMC Genomics 16, 51 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scheckel C et al. Regulatory consequences of neuronal ELAV-like protein binding to coding and non-coding RNAs in human brain. Elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Borreca A, Gironi K, Amadoro G & Ammassari-Teule M Opposite Dysregulation of Fragile-X Mental Retardation Protein and Heteronuclear Ribonucleoprotein C Protein Associates with Enhanced APP Translation in Alzheimer Disease. Mol Neurobiol 53, 3227–3234 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Gusev A et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet 48, 245–52 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seyfried NT et al. A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease. Cell Syst 4, 60–72 e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu JZ, Erlich Y & Pickrell JK Case-control association mapping by proxy using family history of disease. Nat Genet 49, 325–331 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Gusev A et al. Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat Genet 50, 538–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang J et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet 44, 369–75, S1–3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li T et al. GeNets: a unified web platform for network-based genomic analyses. Nat Methods 15, 543–546 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raj T et al. Alzheimer disease susceptibility loci: evidence for a protein network under natural selection. Am J Hum Genet 90, 720–6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nixon RA New perspectives on lysosomes in ageing and neurodegenerative disease. Ageing Res Rev 32, 1 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Emmett MJ et al. Histone deacetylase 3 prepares brown adipose tissue for acute thermogenic challenge. Nature 546, 544–548 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tian Y, Chang JC, Fan EY, Flajolet M & Greengard P Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer’s APP-CTF for terminal degradation via autophagy. Proc Natl Acad Sci U S A 110, 17071–6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ingelsson M et al. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 62, 925–31 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Guillozet AL, Weintraub S, Mash DC & Mesulam MM Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol 60, 729–36 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Consortium GT Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–60 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fairfax BP et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 343, 1246949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bennett DA et al. Overview and findings from the rush Memory and Aging Project. Curr Alzheimer Res 9, 646–63 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Purcell S et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–75 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patterson N, Price AL & Reich D Population structure and eigenanalysis. PLoS Genet 2, e190 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Das S et al. Next-generation genotype imputation service and methods. Nat Genet 48, 1284–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCarthy S et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 48, 1279–83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnson WE, Li C & Rabinovic A Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–27 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Hoffman GE & Schadt EE variancePartition: interpreting drivers of variation in complex gene expression studies. BMC Bioinformatics 17, 483 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van de Geijn B, McVicker G, Gilad Y & Pritchard JK WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat Methods 12, 1061–3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Degner JF et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature 482, 390–4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang J, Lee SH, Goddard ME & Visscher PM GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88, 76–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Storey JD & Tibshirani R Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100, 9440–5 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nica AC et al. The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet 7, e1002003 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Giulietti M et al. SpliceAid-F: a database of human splicing factors and their RNA-binding sites. Nucleic Acids Res 41, D125–31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schmidt EM et al. GREGOR: evaluating global enrichment of trait-associated variants in epigenomic features using a systematic, data-driven approach. Bioinformatics 31, 2601–6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lage K et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat Biotechnol 25, 309–16 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ROSMAP splicing QTL visualization (Shiny App) browser is made available at https://rajlab.shinyapps.io/sQTLviz_ROSMAP/. The ROSMAP data are available at the RADC Research Resource Sharing Hub at www.radc.rush.edu. The ROSMAP and MSBB mapped RNA-seq data that support the findings of this study are available in AMP-AD Knowledge Portal (https://www.synapse.org/#!Synapse:syn2580853) upon authentication by the Consortium. The CommonMind Consortium data are available in CMC Knowledge Portal: https://www.synapse.org/#!Synapse:syn4923029.