

Plasmodiophora brassicae stimulates phloem differentiation and expression of phloem-specific SWEET transporters to hijack host metabolism.
Abstract
Successful biotrophic plant pathogens can divert host nutrition toward infection sites. Here we describe how the protist Plasmodiophora brassicae establishes a long-term feeding relationship with its host by stimulating phloem differentiation and phloem-specific expression of sugar transporters within developing galls. Development of galls in infected Arabidopsis (Arabidopsis thaliana) plants is accompanied by stimulation of host BREVIS RADIX, COTYLEDON VASCULAR PATTERN, and OCTOPUS gene expression leading to an increase in phloem complexity. We characterized how the arrest of this developmental reprogramming influences both the host and the invading pathogen. Furthermore, we found that infection leads to phloem-specific accumulation of SUGARS WILL EVENTUALLY BE EXPORTED TRANSPORTERS11 and 12 facilitating local distribution of sugars toward the pathogen. Utilizing Fourier-transform infrared microspectroscopy to monitor spatial distribution of carbohydrates, we found that infection leads to the formation of a strong physiological sink at the site of infection. High resolution metabolic and structural imaging of sucrose distributions revealed that sweet11 sweet12 double mutants are impaired in sugar transport toward the pathogen, delaying disease progression. This work highlights the importance of precise regulation of sugar partitioning for plant–pathogen interactions and the dependence of P. brassicae’s performance on its capacity to induce a phloem sink at the feeding site.
INTRODUCTION
Plasmodiophora brassicae is a soil-borne biotrophic pathogen. Each year infection of oilseed rape (Brassica napus) by this protist leads to extensive crop losses (Strehlow et al., 2014). It infects plants through root hair cells; this primary infection leads to production of secondary zoospores and eventually their release from root hair cells into the soil. Released zoospores are the source of secondary infection of cortical tissues leading to gall formation (Supplemental Figure 1; Kageyama and Asano, 2009). The pathogen colonizes the underground parts of plants, reprogramming existing meristematic activities to form feeding sites as well as creating favorable conditions for resting spore formation (Malinowski et al., 2012). Galls in the root and hypocotyl are characteristic symptoms of infection caused by P. brassicae, hence its common name: clubroot. This disease affects most members of the Brassicaceae including the model species Arabidopsis, which can conveniently be used to study the biological basis of P. brassicae-plant interactions.
Investigation of this pathosystem revealed that, although gall formation in P. brassicae-infected plants is not a prerequisite for completion of the pathogen lifecycle, galls are critical structures—securing space for pathogen growth and nutrient availability, thereby influencing the number of resting spores released into the soil (Malinowski et al., 2012). Galls are sites of intense sugar consumption, therefore understanding how this strong physiological sink is created by the pathogen is crucial for development of new strategies for tackling clubroot disease. It has been suggested that, in plants, establishment of a strong carbohydrate sink is preceded by increased cytokinin accumulation, which stimulates a local increase of sucrose invertases (Roitsch and Ehneß, 2000; Walters and McRoberts, 2006). During clubroot infection, there is increased expression of extracellular sugar invertases (Siemens et al., 2011). Infected ipt1;3;5;7 quadruple mutant plants, defective in cytokinin biosynthesis, displayed significantly delayed resting spore formation that compromised their subsequent infectivity (Malinowski et al., 2016). The exact role of cytokinins, their synthesis and turnover during clubroot disease is still unclear because infection of wild-type Col-0 plants did not induce any significant changes in cytokinin levels (Malinowski et al., 2016).
In this work we investigate the role of phloem-specific changes in sucrose transport toward the infection site. The phloem bundle is composed of sieve elements (SE), companion cells (CCs), and phloem parenchyma cells (PP). SEs play the major conductive role within the phloem, whereas CC are involved in SE support and sugar transport (Lucas et al., 2013). As phloem comprises only a small fraction of cells within plant organs and its differentiation requires precisely regulated events within single cells, our knowledge of the molecular networks governing phloem development is limited. The molecular regulation of SE differentiation can be divided into three modules: (1) regulation of time and rate of differentiation; (2) nucleus and organellar breakdown; and (3) SE pore formation (Rodriguez-Villalon, 2016). The timing and rate of protophloem differentiation is regulated by two plasma membrane-associated proteins, BREVIS RADIX (BRX) and OCTOPUS (OPS). Their activity antagonizes the BARELY ANY MERISTEM (BAM3) receptor-like kinase, which is a part of a negative regulation module with its peptide ligand, CLAVATA/EMBRYO SURROUNDING REGION45 (CLE45) (Depuydt et al., 2013). The inositol polyphosphate 5-phosphatase COTYLEDON VASCULAR PATTERN2 (CVP2) and its homolog CVP2-LIKE1 (CVL1) are also involved in early steps of phloem differentiation (Rodriguez-Villalon, 2016). An expression marker for later differentiation steps such as sieve cell enucleation is the ALTERED PHLOEM DEVELOPMENT (APL) gene whose activity is also linked to the regulatory balance between cell differentiation and proliferation (Bonke et al., 2003). Downstream of APL, two transcription factors, NAC DOMAIN CONTAINING PROTEIN45 (NAC45) and NAC86, control enucleation events through the exonucleases NAC 45/86-DEPENDENT EXONUCLEASE-DOMAIN PROTEIN1/2/4 (Furuta et al., 2014). The last steps related to SE differentiation depend on callose synthesis and are regulated by CALLOSE SYNTHASE3/7 (CALS3/7) enzymes (Vatén et al., 2011).
In Arabidopsis, the sucrose produced in leaves during photosynthesis in mesophyll cells is transported through the plasmodesmata to adjacent cells until it reaches the PP. Then, with the help of SUGARS WILL EVENTUALLY BE EXPORTED TRANSPORTERS (SWEET) family proteins, it is exported into the phloem apoplasm. Subsequently, sucrose is transported to phloem CCs by ATPase-dependent H+ sucrose symporters (SUT/SUC). Actively loaded sugar is transported and unloaded in appropriate sink locations; this process also involves SWEET proteins. In the Arabidopsis genome, 17 members of the SWEET family have been identified. Based on their sequence similarity, they were grouped into four different clades: clades I, II, and IV working as glucose, galactose, and fructose efflux factors and clade III members involved in sucrose transport (Chandran, 2015).
Pathogens can influence assimilate destination via modification of loading mechanisms, phloem transport, unloading, and the availability of phloem-delivered sugar (Chen et al., 2012; Chen, 2014). We observed previously that within the clubroot gall there is an increased formation of phloem cells and an arrest of xylogenesis (Malinowski et al., 2012). Additionally, we found that cle41 mutants, where the phloem/xylem differentiation process was disrupted, produced smaller galls and that their underground parts disintegrated prematurely compared with infected non-mutant plants. These findings stimulated our interest in understanding the patterns of phloem architecture remodeling elicited by P. brassicae and its subsequent consequences for sugar distribution. We found that P. brassicae infection leads to reprogramming of the molecular network regulating early steps of phloem cell differentiation. The consequence of this is an increased number of cells within phloem bundles. This developmental reprogramming is amplified by an increase in the potential of phloem for sugar unloading at the site of infection. We found increased levels of SWEET11 and 12 sugar permeases in phloem cells within the hypocotyls of infected plants. Infected sweet11;12 double mutants gave rise to smaller galls accumulating lower amounts of sucrose compared with infected Col-0 controls; this change resulted in a disruption of pathogen development. Collectively, our data indicate how P. brassicae increases sucrose delivery and its diffusion into the intracellular spaces of nonvascular tissues surrounding the pathogen. This may represent a critical aspect of the pathogen-driven host reprogramming influencing disease progression.
RESULTS
All Phloem Lineage Cells are Present in Higher Numbers in P. brassicae–Infected Tissue
In our previous work we found that P. brassicae infection does not increase the number of phloem bundles but, instead, leads to an increase in their cellular complexity (Malinowski et al., 2012). To further understand the anatomical and structural changes within the phloem at the site of infection, we adopted an electron microscopy approach. Because we found previously that differences in phloem bundle complexity observed between the hypocotyls of infected and non-infected plants were most evident toward the later stages of disease progression, 26 d after inoculation (DAI), this time-point was selected for further analysis.
Typically, each SE is accompanied by a single CC whose main role is to maintain the metabolic activity of SE cells. Through the careful examination of sections we found that the number of all phloem cells within phloem bundles increased after infection (Figures 1A and 1D); however, in most cases the SE/CC proportion was not changed (Figure 1B). Within 30 independent infected hypocotyls we found only three examples of bundles with increased SE/CC ratios (see example in Figure 1E). In some cases clusters of proliferating cells, presumably the source of future phloem bundles, were observed (Figure 1D, red stars)—a phenomenon not seen in non-infected tissue (Figure 1C). The uniform increase of all phloem cell types in response to infection indicates that the clubroot-induced reprogramming of phloem development must occur early in the differentiation process, raising the number of precursor cells that then differentiate into appropriate functional cells.
Figure 1.

P. Brassicae-Induced Phloem Anatomical Changes in Hypocotyls of the Infected Host.
(A) The average number of se, CCs, and parenchyma cells per bundle in control and infected plants 26 DAI, determined from inspection of electron micrographs. Asterisks denote a significant difference in cell type counts between infected and uninfected samples (*P < 0.05, **P < 0.01, ***P < 0.001); error bars indicate the se.
(B) The proportion of each cell type within the phloem bundles of uninfected and infected plants calculated from (A), no significant differences were detected upon infection.
(C) to (E) Electron microscopy micrographs showing representative phloem bundles in the Col-0 hypocotyls of non-infected plants (C) and P. brassicae infected Col-0 at 26 DAI (D) and (E).
(E) A rare example of a phloem bundle with altered ratios of phloem cell types.
Yellow stars in (D) and (E) indicate host cells colonized by the pathogen. Red stars in (D) denote proliferating cells near differentiating phloem bundles. Scale bars represent 10 µm. Images were acquired from radial sections of hypocotyls.
P. brassicae Infection Increases Expression of Phloem Identity Genes
To determine which genes associated with phloem cell lineage might be implicated in the pathogen-driven changes to phloem differentiation, we analyzed the expression patterns of key regulators of phloem cell fate. Results from previous gene expression profiling by RNA-Seq (Malinowski et al., 2016) were checked independently by RT-qPCR to confirm the behavior of phloem-related genes during clubroot disease. The emerging picture shows that P. brassicae infection leads to extensive reprogramming of phloem development (Supplemental Figure 2). In particular, at 16 DAI the expression of the cell fate regulators BRX and CVL1 and enucleation factors NEN1,2,3 are significantly induced; the up-regulation of some factors determined to be significant in the RNA-Seq data-set were not significant at the P value < 0.05 threshold with RT-qPCR but the trend for induction upon infection was consistent for APL (P = 0.097), BAM3 (P = 0.064), and CVP2 (P = 0.072).
To further understand the reprogramming of phloem cell fate, we have investigated spatial changes in the cell-specific expression patterns of factors involved in early steps of phloem differentiation (OPS, CVP2, BRX, BAM3, CLE45) and the master regulatory gene (APL) involved both in phloem cell fate specification and the enucleation process. With the use of appropriate promoter:GUS transgenic lines, we observed that infection led to phloem-specific accumulation of OPS and CVP2 genes during the proliferative stage of gall formation 16 DAI (Figure 2). At this time-point the expression levels in control, mock-inoculated hypocotyls were very low, such that staining was not visible in 10-µm thin sections (Figure 2). At 26 DAI, expression of these factors was lower in P. brassicae-infected plants than in controls. Expression of BRX was higher in infected hypocotyls during both the proliferative (16 DAI) and expansive (26 DAI) stages of gall formation. At 16 DAI in uninfected hypocotyls, BAM3 signals were present not only in phloem cells but also in cambium and its progeny. Only phloem-specific signals were observed in infected hypocotyls at 16 DAI. The spatial expression patterns of CLE45 were not altered significantly following infection.
Figure 2.

Promoter Activity of Genes Involved in Phloem Differentiation.
Comparison of the APL:GUS; BAM3:GUS; BRX:GUS; CLE45:GUS; OPS:GUS; CVP2:GUS promoter activity at indicated time (16 DAI or 26 DAI) in mock inoculated plants (Mock) or after P. brassicae infection (Inf). Signals were observed as a blue product of β-Glucuronidase activity in 10-µm Technovit embedded and Safranin-O counterstained hypocotyl sections. Due to the large size of the galls at 26 DAI, a quarter segment of the representative section for each hypocotyl is presented. Insets show representative phloem bundle images from regions marked with squares on the whole section images. Each inset is duplicated in the upper grayscale panel; the phloem cells are highlighted for the corresponding GUS staining image beneath it. Those phloem cells stained for promoter activity are marked in red and other phloem cells, not showing GUS staining, are marked in green. Scale bars represent 200 µm for radial sections and 20 µm for the insets provided. The experiment was performed on 30 plants for each combination and repeated three times. After visual inspection, the 10 most representative hypocotyls from each combination were used for sectioning.
As the phloem bundle complexity increases in infected hypocotyls at 16 DAI, it is possible that more cells would exhibit APL promoter activity; however, the pattern of this SE differentiation marker did not change upon infection and was still expressed specifically in cells differentiating into SEs 16 DAI, decreasing markedly 26 DAI (Figure 2). Our results show that clubroot-induced gall formation involves activation of the OPS/CVP2/BRX positive regulatory module governing the early steps of phloem formation. Different patterns of accumulation observed between OPS and BRX likely reflects the fact that they are elements of independent parallel regulatory pathways (Rodriguez-Villalon, 2016). Taken together, our observations show that the activation of phloem differentiation occurs during the proliferative stage of gall formation and decreases in the expansive growth phase previously described by Malinowski et al. (2012).
Decreased Phloem Transport Capacity Restricts Gall Formation and Results in Premature Death of the Host
To gain insight into the importance of phloem reprogramming for the host-pathogen interaction, we examined disease progress in plants harboring mutations in genes involved in the early steps of phloem differentiation (ops-2, cvp2-1 cvl1-1, and brx-2 loss of function mutants). The anatomy of galls from infected mutant lines differed substantially from those of Col-0 controls (Figure 3). At 16 DAI, hypocotyls of infected brx plants were smaller than in corresponding infected Col-0 and they produced clusters of small undifferentiated cells (Figure 3A). In the ops mutant, the dramatic increase in cell number within phloem bundles typically seen in infected wild-type hypocotyls was not observed (Figure 3A). The 30-d-old (16 DAI) and 40-d-old (26 DAI) double mutant cvp2 cvl1 plants exhibited a very strong developmental phenotype even in the absence of infection (Figure 3). They also failed to respond to P. brassicae infection with increased phloem cellular complexity, but to a much lower extent than ops (Figure 3). The hypocotyl width of infected brx and cvp2 cvl1 mutants was significantly reduced at both 16 DAI and 26 DAI (Figure 3B). The uninfected hypocotyls of ops mutants were larger than wild type but the P. brassicae–induced expansion of hypocotyl size was significantly reduced in ops compared with Col-0 at 26 DAI (Figure 3C).
Figure 3.

Disease Development In Galls Of Phloem Differentiation Mutant Plants.
(A) Representative 10-µm sections across hypocotyls of Col-0 and selected phloem differentiation mutants: brx, ops, and cvp2 cvl1 at 16 DAI and 26 DAI. Sections were stained using toluidine blue. Scale bars represent 200 µm for radial sections and 50 µm for the insets provided. Insets show higher magnification of regions containing phloem cells. Each inset is duplicated in the upper grayscale panel; the phloem cells are highlighted in green. Clusters of undifferentiated cells observed in hypocotyls of infected brx and cvp2 cvl1 mutants are colored magenta.
MP, multinuclear plasmodia; RS, resting spores.
(B) The influence of phloem differentiation mutants on P. brassicae gall development 16 DAI and 26 DAI. Hypocotyl widths were measured and the means and ses of 15 replicates were calculated using a general linear model, different letters indicate a significant difference between means (Benjamini-Hochburg adjusted P < 0.05).
(C) The P. brassicae-induced increase in hypocotyl width calculated from (B) is plotted; asterisks denote a significant difference between mutant and wild type (*P < 0.05, **P < 0.01, ***P < 0.001).
(D) The number of phloem cells per bundle based on calculations performed on three randomly chosen bundles from 10 independent hypocotyls for each combination (30 bundles in total).
(E) The number of phloem bundles per hypocotyl observed on 10 independent hypocotyls for each combination.
In all three mutant lines the P. brassicae lifecycle was accelerated; this was already visible at 16 DAI but the difference at 26 DAI was more pronounced, with resting spores formed in each mutant line whereas in Col-0 plants the pathogen had yet to undergo sporogenesis and remained as multicellular plasmodia with few resting spores yet formed (Figure 3A). Hypocotyls of all three mutant lines started disintegrating at 26 DAI, a process not typically observed in Col-0 until more than 32 DAI. In later stages (32 DAI), a dramatic increase in the number of dead plants with disintegrated underground parts was observed in all phloem development mutants examined (Supplemental Figures 3B and 3C). Observation of the aboveground parts of plants during infection revealed earlier onset and exacerbated symptoms of stress in the phloem mutants compared with Col-0 controls (Supplemental Figure 3A).
P. brassicae Infection Stimulates Expression of SWEET11 and SWEET12 in the Vicinity of the Pathogen
It has been reported that P. brassicae infection leads to changes in sugar distribution and metabolism within the host (Siemens et al., 2011). The successful formation of P. brassicae feeding sites largely depends on the host potential for soluble sugar delivery. In fact, P. brassicae frequently locates near the phloem cells; therefore it is of interest whether this pathogen also manipulates the expression of host genes facilitating access to soluble sugars at the site of infection. Our transcriptional profiling showed that infection had a profound effect on enzymes associated with soluble carbohydrate metabolism and transport. The expression of most invertase genes (cytosolic and vacuolar) declined upon gall formation although expression of CWINV1, an apoplastic invertase, increased (Supplemental Figure 4A). This was previously observed by Siemens et al. (2011). By contrast, the expression of the sucrose synthase genes SUS2 and SUS3 increased markedly upon infection (Supplemental Figure 4A).
Previous studies have shown that cytosolic invertases CINV1 and CINV2 are critical for the catabolism of sucrose and growth of non-photosynthetic tissue under normal conditions, while the combined loss of soluble sucrose synthases in the sus1;2;3;4 quadruple mutant have little effect (Barratt et al., 2009). However, low oxygen conditions can shift the balance of sucrose catabolism pathways and sus1 sus4 mutants do exhibit reduced root growth under hypoxic conditions (Bieniawska et al., 2007). Because P. brassicae infection induces hypoxia responses (Gravot et al., 2016), we wanted to examine the role of both these scenarios on gall development. To determine the impact of CINV and SUS activity on clubroot disease, the quadruple sucrosesynthase mutant sus1;2;3;4 and the double cytosolic invertase mutant cinv1 cinv2 were infected with P. brassicae. In both cases, the hypocotyls of mutant plants were significantly smaller than wild type, 26 DAI, whether infected or not (Supplemental Figure 5). However, microscopic analysis of infected mutants revealed typical plasmodial development (Supplemental Figure 5A). The increase in hypocotyl size in response to P. brassicae was indistinguishable from that of wild type (Supplemental Figure 5B), thus the sink limitations in these mutants must be sufficiently alleviated by clubroot infection to allow normal gall formation.
RNA-Seq profiling of transcriptional responses of genes associated with sucrose and hexose sugar transport revealed an increase in the expression of the sugar permeases SWEET11 and SWEET12 during gall formation, whereas SWEET16 and SWEET17 genes were strongly repressed (Supplemental Figure 4A.) In addition, expression of the monosaccharide transporters STP8 and STP13 was also up-regulated upon infection (Supplemental Figure 4A). SWEET16 and 17 transport fructose out of vacuoles (Guo et al., 2014) while STP8 and STP13 transport hexose and galactose, respectively, and are associated with programmed cell death in pathogen defense and cell wall remodeling (Büttner, 2007). SWEET11 and 12 are sugar permeases, capable of bidirectional transport of both sucrose and hexose sugars (Chen et al., 2012), and thus were attractive candidates for the delivery of carbohydrate to intracytoplasmic secondary plasmodia within developing galls.
Studies recently performed for two near-isogenic lines of Brassica rapa differing in the clubroot resistance CRb locus showed that several BrSWEET genes, which encode sugar permeases, were up-regulated in susceptible plants subjected to P. brassicae infection (Li et al., 2018). We interrogated the behavior of SWEET11 and 12 at the site of P. brassicae infection through microscopy studies. Cross sections were made of the hypocotyls of control and infected plants bearing SWEET11:SWEET11-GUS and SWEET12:SWEET12-GUS transgenes to localize their spatial accumulation. We found that infection leads to increased accumulation of both tested transporters (Figure 4). In non-infected lines, no signals for SWEET11 and 12 were observed at 16 DAI. At 26 DAI in control SWEET11:SWEET11-GUS and SWEET12:SWEET12-GUS plants, specific signals were observed in the cambial area; however, signals in SWEET11 plants were very weak and were visible only in hand-cut thick sections or whole mount tissue. Sixteen-DAI–infected plants showed a large increase in SWEET11 accumulation in xylem parenchyma, phloem, and cambial cells whereas SWEET12 accumulated in the region of the phloem only. The phloem-specific up-regulation of both SWEET11 and 12 proteins was still evident at 26 DAI.
Figure 4.

SWEET11 and 12 Protein Colocalization in Response to P. Brassicae Infection.
The presence of SWEET11-GUS and SWEET12-GUS fusion proteins in hypocotyls was studied by β-Glucuronidase activity assay followed by embedding in Technovit and sectioning. Shown are 10-μm Technovit embedded and Safranin-O counterstained sections. A and B, SWEET11:SWEET11-GUS (A) and SWEET12:SWEET12-GUS (B), both 16 DAI and 26 DAI with (Inf) and without (Mock) P. brassicae infection. Insets show representative phloem bundles from regions marked with squares on larger images of radial hypocotyl sections. Scale bars = 200 μm for radial sections and 20 μm for insets.
SWEET11 and SWEET12 Transporters are Involved in the Increase of Sugar Availability at the Site of Infection.
To understand the role of observed phloem-specific SWEET11 and SWEET12 accumulation for P. brassicae life-cycle progression, we inspected anatomical changes in developing galls of the sweet11-1 sweet12-1 double mutant (sweet11;12). We found that the pathogen development in sweet11;12 plants was delayed at 26 DAI, with no resting spores deposited at this time and fewer, smaller multinucleate plasmodia formed than in Col-0 (Figure 5). The hypocotyls of infected sweet11;12 double mutants were smaller than Col-0, and the P. brassicae–induced increase in hypocotyl size was significantly reduced (Figures 5B and 5C). Complementation of sweet11;12 with either SWEET11 or SWEET12 resulted in plants that were indistinguishable from wild type in terms of both pathogen development and gall size (Figure 5), indicating that SWEET11 and SWEET12 compensate effectively for the loss of either in the delivery of carbohydrate toward the pathogen.
Figure 5.

P. brassicae Disease Development Progression in the sweet11;12 Double Mutant and Single Gene Complementation Lines.
(A)Toluidine blue stained 10-μm hypocotyl sections of Col-0, sweet11;12 and sweet11;12 SWEET11, sweet11;12 SWEET12 complementation lines at 16 DAI and 26 DAI. Scale bar =200 μm. RS, resting spores; MP, multinuclear plasmodia.
(B) Gall development in sweet11;12 mutants. Mean hypocotyl widths of 14 or 28 replicates calculated using a mixed linear model are plotted along with the se. Different letters indicate significant differences between means (Benjamini-Hochburg adjusted P value < 0.05).
(C) Hypocotyl width increase induced by P. brassicae infectioncalculated from (B). Asterisks denote a significant difference between mutant and wild type (P < 0.05).
The phloem in sweet11;12 plants exhibited the typical pattern of cell proliferation in response to P. brassicae infection with no significant differences in the number of phloem bundles or cells per bundle to those of wild-type plants (Supplemental Figure 6). Both Col-0 and sweet11;12 plants responded to P. brassicae infection with an increase in phloem complexity (Supplemental Figures 6A to 6D). This pattern was also reflected by increased expression of CVP2 and BRX factors, positively regulating phloem development (Supplemental Figures 6E and 6F). The decreased gall size in sweet11;12 plants was not associated with any change in the mortality of infected plants, as had been observed in mutants with impaired phloem differentiation (Supplemental Figure 3).
Delay in pathogen development in sweet11;12 mutants supports the hypothesis that, in addition to reprogramming host cell development to favor phloem proliferation, the pathogen stimulates host sugar permeases to deliver carbohydrate to developing plasmodia. To confirm the impact of sweet11;12 mutations on the pattern of sugar redistribution we used Fourier-transform infrared (FTIR) microspectroscopy. Such an approach allowed us to construct a topographical map where representative color-coded FTIR images visualize carbohydrate distribution within tissues (Figures 6A to 6D). In mock treated Col-0 plants we could observe characteristic strong accumulation of carbohydrates in regions containing conductive tissue, but the maximum levels were observed in the cambial region. This was particularly striking at 26 DAI, when intense secondary growth occurs in the hypocotyl (Figure 6A). Similar to Col-0 plants strong accumulation of carbohydrates in the cambial area, increasing in later developmental stages, was observed in hypocotyls of uninfected sweet11;12 mutants; however, the degree of accumulation was much stronger (Figure 6).
Figure 6.

Changes in Carbohydrate and Sucrose Distribution Observed in Col-0 and the sweet11;12 Double Mutant.
(A) to (D) FTIR visualization of carbohydrate levels within the hypocotyls of Col-0 (A and B) and sweet11;12 (C and D) plants at indicated time (16 DAI and 26 DAI) with (Inf) and without (Mock) P. brassicae infection. Scale bar represents 400 μm.
(E) and (F) Differences in carbohydrate distribution changes that occurred over 10 d (from 16 DAI to 26 DAI) in Col-0 (E) and sweet 11;12 (F) plants. Values were calculated for whole radial hypocotyl sections or extracted from digitally selected cambium, phloem, and xylem regions of Mock and Inf plants at 16 DAI and 26 DAI.
(G) Quantification of changes in sucrose concentration observed with FTIR imaging between two time-points (16 DAI and 26 DAI) after P. brassicae infection, averaged for three individual plants of Col-0 and sweet11;12. Boxplot graphs show median values as a point and inter-quartile range (IQR) as a box, with whiskers representing 1.5× IQR. The error bar represents the variation between the two distributions as quantile absolute difference (QAD). Significant differences with Mann-Whitney P values below 0.05 are marked * and ** when the Divergence Effect Size > 0.2. Spectral features of carbohydrates or sucrose fingerprints along with baseline features were extracted using an extended multiplicative signal correction model (see “Methods”).
Upon P. brassicae infection both Col-0 and sweet11;12 plants form a strong carbohydrate sink and accumulate more carbohydrates in the inner central xylem region, ring of cambial cells, and their closest progeny as well as phloem bundle areas with adjacent parenchymatic cells. The main difference between the two host genotypes lies in the magnitude of response to infection: The accumulation of carbohydrates in infected sweet11;12 mutant is strongly reduced compared with the Col-0 plants (Figure 6). This is particularly visible in the cambial region.
To further understand the importance of the phloem-specific increase in SWEET11 and SWEET12 transporters observed after P. brassicae infection for facilitating delivery of sugars, we quantified sucrose accumulation in corresponding regions (Figure 6G). We found that across 10 d of disease progression (16 DAI to 26 DAI), a significantly lower accumulation of sucrose occurs in each of these regions (Figure 6G). The largest disparity in sucrose accumulation between infected Col-0 and sweet11;12 mutant was observed in the phloem and proximal tissue. These results support the involvement of SWEET11 and SWEET12 transporters in pathogen-driven manipulation of sucrose distribution within hypocotyls of infected plants. Analysis of disease progress in galls shows that compromising this route negatively impacts pathogen development (Figure 5; Supplemental Figure 3).
DISCUSSION
P. brassicae–Induced Galls are Complex Structures with Internal Anatomical Changes Facilitating Host Nutrient Uptake
The development of special structures facilitating nutrient uptake is a common mechanism used by biotrophic pathogens to create feeding sites within the host. Organisms such as nematodes, leaf gall insects, and microorganisms like Rhizobium or pathogenic Agrobacterium strains use particular strategies to create an appropriate environment for efficient nutrient uptake, ensuring successful multiplication for future generations (Damiani et al., 2012; Ji et al., 2013; Isaias et al., 2014). Frequently, pathogen feeding sites can be observed as groups of proliferated or hypertrophied cells called galls. The complexity of these structures can differ greatly depending on the degree of reprogramming of host plant morphogenesis. This includes differentiation of new vasculature facilitating nutrient transport to the gall (Isaias et al., 2014). Our previous work showed that infection of Arabidopsis plants by the soil-borne pathogen P. brassicae leads to gall development characterized by inhibition of xylogenesis and an increase in phloem cell number (Malinowski et al., 2012). So far, the molecular basis of this phloem reprogramming and its importance for the plant–pathogen interaction has not been described. In our previous work we showed that the conserved molecular mechanism involving the TDIF-PXY/TDR-WOX4 module (Hirakawa et al., 2010), which regulates the balance between phloem and xylem formation, is not modified by the pathogen. Here, we decided to perform a detailed anatomical study followed by observation of changes in expression pattern of genes that were recently identified as being involved in phloem tissue differentiation. Electron microscopy showed increased numbers for all phloem cell types in P. brassicae-induced galls formed on Arabidopsis hypocotyls—however, we did not observe aberrant phloem differentiation. As with the hypocotyls of mock-inoculated control plants in clubroot galls, typically each SE was accompanied by a CC cell. There was no significant change in the SE:CC ratio, suggesting that P. brassicae–driven phloem reprogramming occurs during the early steps of differentiation and that further steps such as cell enucleation were not perturbed.
To study the molecular basis of observed anatomical changes we examined the expression of regulators of phloem lineage. Most functional studies of these genes have been performed during protophloem formation and phloem differentiation in the early developmental stages, therefore our work attempted to visualize their action during formation of phloem from the vascular cambium progeny cells in secondary thickening.
Gene expression profiling within whole hypocotyls showed that infection triggers wholesale changes in the regulatory pathways governing phloem development. The increased expression of CVL1, CVP2, and BRX genes observed 16 DAI confirmed our anatomical studies, suggesting that pathogen-driven reprogramming starts at the earliest steps of phloem differentiation. Further verification of the spatial patterns of promoter activity showed that induction of OPS, CVP2, and BRX expression occurred at 16 DAI. These factors regulate vascular cell identity acquirement and entry to the phloem cell differentiation pathway. Based on existing data, it is clear that their action is at least partially independent. Overexpression of the OPS gene in Arabidopsis plants did not result in ectopic phloem formation (Truernit et al., 2012). Both ops and cvp2 cvl1mutants do not show any disruption in the initiation of phloem bundles, but have altered vascular bundle organization (Carland et al., 1999; Truernit et al., 2012).
BRX is a factor positively regulating entry to phloem differentiation and early steps of transition to SE identity (Scacchi et al., 2010). Its action seems to be crucial for mediating auxin and brassinosteroid mediated vascular tissue development (Mouchel et al., 2006). Recent studies of P. brassicae disease clearly support a scenario in which these phytohormones are crucial for cell fate reprogramming within developing galls (Ludwig-Müller, 2014; Schuller et al., 2014). Our results show that P. brassicae infection manipulates the phloem cell identity program by influencing these factors. Despite elevated levels of transcript accumulation, the spatial expression pattern of the MYB transcription factor APL, which is known to be involved in regulation of xylem/phloem ratio (Bonke et al., 2003) and enucleation during the SE differentiation process (Furuta et al., 2014), was not changed in hypocotyls of infected plants at 16 DAI. We suggest that changes in transcript level observed in the whole hypocotyl can be attributed to the higher number of cells within the phloem bundle resulting from reprogramming that had taken place at earlier developmental stages.
Taken together, our studies of anatomy, transcriptomics, and promoter activity showed that P. brassicae phloem reprogramming occurs during early steps of phloem formation during the proliferative stage of gall formation (Supplemental Figure 6). This overlaps with an increase of existing meristematic activity (16 DAI) and precedes accumulation of multinuclear plasmodia.
Changes in Early Steps Regulating Phloem Differentiation are Important for the Host and the Pathogen During Infection
Pathogens interfere with the host source-sink balance to acquire the nutrients necessary for their successful multiplication. P. brassicae is an example of a pathogen colonizing sink organs; therefore we can assume that increasing phloem bundle complexity in the hypocotyl may act as an important element for the strengthening of a pathogen-oriented sink. To test this hypothesis we used mutants of phloem-related genes whose activity increased during the proliferative stage of gall formation. We tested the previously described ops-2 (Truernit et al., 2012) mutant, the cvp2-1 cvl1-1 double mutant (Carland and Nelson, 2009), and the brx-2 mutant (Rodrigues et al., 2009). In all of these mutants, defects in phloem development were reported. However, in our study, where later stages of plant development were studied, anatomical changes within the hypocotyl of uninfected plants were only clearly visible in the cvp2 cvl1 double mutant. By contrast, the effect of these mutations on gall development and clubroot disease progression was striking. Infection led to the development of different cellular patterns that can be attributed to the different functions of the mutated genes. All mutants were unable to react to P. brassicae infection with respect to increased phloem complexity.
Another notable observation was that, in all phloem mutants examined, pathogen development was accelerated. P. brassicae–infecting phloem mutants developed resting spores more quickly than in the Col-0 genetic background and at 32 DAI, a higher number of roots and hypocotyls of mutant plants had disintegrated. The acceleration of the infection process in the mutants led to premature death and disintegration of plants. Disease progress acceleration in the mutants was accompanied by reduced growth and increased pigmentation and senescence in the leaf rosettes. Taken together, these results demonstrate that, during infection, the reprogramming of phloem development within galls facilitates maintenance of host tissue integrity and is important for pathogen development.
Inability to respond to infection with increased phloem complexity results in disease acceleration and premature death of plants. When P. brassicae fails to stimulate phloem complexity in the regulatory mutants, it can adapt to the lower nutrient supply and limits imposed on gall formation by hastening its lifecycle to release spores into the soil that may encounter more favorable hosts. Infected wild-type Arabidopsis will typically survive long enough to form viable seed, but not mutants incapable of forming phloem architecture that satisfies the requirements of P. brassicae. The triggering of phloem developmental reprogramming therefore represents an interesting point of balance in the evolving host-parasite relationship.
P. brassicae Uses the Host Sugar Efflux Machinery for Nutrient Supply at the Site of Infection
It has been known for many years that clubroot-induced galls are strong sinks for carbohydrates. Keen and Williams (1969) demonstrated that 14C-labeled assimilates were translocated more rapidly to the hypocotyls of clubroot-infected cabbage plants and that export from infected areas was reduced. Our FTIR microspectroscopy measurements support this view because higher accumulation of carbohydrates was visible in the hypocotyls of infected plants. Understanding how this strong physiological sink is created by the pathogen is crucial for development of new strategies for tackling clubroot disease. The formation of the sink is governed by both developmental and biochemical processes. Assimilates are delivered to the site of infection via the phloem, production of which is maintained in infected tissue despite the extensive reprogramming of host development and repression of xylogenesis that occurs during gall formation. The metabolism of proliferating host and pathogen cells contributes to increased sink strength. Starch and sucrose accumulate in infected tissue, and plasmodial development is associated with accumulation of trehalose and lipid droplets within the pathogen (Evans and Scholes, 1995; Brodmann et al., 2002; Bi et al., 2016).
Sucrose is transported across membranes (both host and pathogen) by specific sugar transport proteins including ATPase-dependent H+ sucrose transporters (SUT/SUC) and sugar permeases (SWEET family members). A unique set of potential sugar transporters has also been identified in the P. brassicae genome (Rolfe et al., 2016). Here we report finding of increased levels of the SWEET11 and SWEET12 transcripts. Observations made using the SWEET11:SWEET11-GUS and SWEET12:SWEET12-GUS lines show very pronounced increase in SWEET11 and SWEET12 protein accumulation in phloem bundles within hypocotyls of infected plants. It has been proposed that the SWEET transporters mediate the facilitative diffusion of sugars, by allowing solute permeation across biological membranes down a concentration gradient (Eom et al., 2015). Members of this group of proteins are known to be involved in loading of the sucrose to phloem; however, there are also reports showing their importance for diffusion of sugars toward the intercellular space at the infection site for a variety of pathogens (Chen et al., 2010; Chen, 2014). SWEET transporters and apoplastic invertases may also have a protective role as hydrolysis of apoplastic sucrose and subsequent uptake of hexose sugars into the cytoplasm will limit the availability of carbohydrate to pathogens located in the apoplast. Clubroot, as an intracytoplasmic pathogen, may have evolved to exploit these defense mechanisms—processes that restrict carbohydrate supply to the apoplast may favor nutrient supply to the plasmodia. Because P. brassicae inhabits both symplastic and apoplastic spaces, these two scenarios may be relevant at different stages of the pathogen’s lifecycle.
Our studies on the sweet11;12 double mutant confirm the specific importance of these proteins for clubroot infection. Li et al. (2018) showed that SWEET transporters are up-regulated in Brassica rapa upon P. brassicae infection. The authors also reported that clubroot disease progress is delayed in the Arabidopsis sweet11 mutant; however, in our experiments performed on sweet11;12 double mutants complemented with either SWEET11 or SWEET12, no phenotypic differences with respect to gall development were observed, indicating that these transporters are capable of effectively compensating for each other in supplying P. brassicae with adequate sucrose for gall formation. After infection, the double mutant plants accumulate less sucrose in phloem bundle areas within hypocotyls, indicating that the availability of this compound is lower than in infected Col-0. Also the total carbohydrate levels in the cambial area of infected hypocotyls observed by FTIR are lower than in Col-0. Lower levels of carbohydrates within the cambial area may be translated into less pronounced stimulation of meristematic activity in this region, resulting in the smaller galls of sweet11;12 mutants during infection; the promotive role of sugars on cell proliferation has previously been reported (Skylar et al., 2011).
Interestingly, loss of SWEET11 and SWEET12 permeases does not affect the host as strongly as mutations compromising phloem proliferation. The cues that trigger P. brassicae to accelerate its lifecycle in the phloem mutants where gall formation is impaired were clearly not present in sweet11;12 plants, though they represent a similarly limited environment. The sweet11;12 mutants did not die prematurely as the phloem regulatory network mutants did. This shows that the role of these proteins is more restricted to targeting of the nutrient supply toward developing plasmodia rather than the systemic response necessary for the integrity of P. brassicae–infected plants. Plants incapable of pathogen-driven phloem remodeling can host the pathogen for a more restricted time only. This may reflect the fact that phloem acts as a long-distance conduit for a wide range of vital substances including other nutrients, particularly nitrogen-containing metabolites or signaling molecules regulating biotic interactions (Lucas et al., 2013). Figure 7 summarizes, schematically, the changes in host networks regulating phloem development and possible involvement of SWEET transporters during gall development and subsequent P. brassicae spore maturation. Our work shows that both levels of reprogramming are crucial for disease progression.
Figure 7.

Schematic Representation of the P. brassicae-Driven Developmental Phloem Reprogramming and Stimulation of Sucrose Transport to Pathogen-Colonized Cells.
P. brassicae infection triggers critical reprogramming of the early phloem differentiation related genes (OPS, BRX, and CVP2). Transcriptional changes lead to an increase of phloem bundle complexity influencing the host–pathogen interaction. Proliferated phloem bundles are the site of intense SWEET11 and SWEET12 sugar permease expression, facilitating efficient redirection of sugars toward the pathogen site. Accumulation of SWEETs enable the pathogen to usurp the host assimilate. PPC, phloem precursor cells; SE, sieve element cells; CC, companion cells; SC, sink cells; Pb, P. brassicae.
METHODS
Plant Material
Arabidopsis (Arabidopsis thaliana) accession Columbia-0 (Col-0) was used as the genetic background for all mutants and transgenic lines, except for the OPS:GUS line, which was in the Wassilewskaja (Ws-0) accession. The reporter lines OPS:GUS (Truernit et al., 2012), BRX:GUS (Mouchel et al., 2006), CVP2:GUS (Carland and Nelson, 2004), CLE45:GUS, BAM3:GUS (Depuydt et al., 2013), and pSWEET11:SWEET11-GUS-11, pSWEET12:SWEET12-GUS-6 (Chen et al., 2012) have been previously described, as have the cvp2-1 cvl1-1 double mutant (cvl1-1 = SALK_029945) (Carland and Nelson, 2009), ops-2 (SALK_139316) (Truernit et al., 2012), and brx-2 (Rodrigues et al., 2009). The pSWEET11:SWEET11-GUS and pSWEET12:SWEET12-GUS lines were obtained from the Nottingham Arabidopsis Stock Centre (http://arabidopsis.info) with IDs N68825 and N68828, respectively. The sweet11-1 sweet12-1 double mutant used is a cross of SALK_073269 and SALK_031696 T-DNA lines and the revertant sweet11 and sweet12 lines, derived from complementation of the sweet11;12 double mutant, which were also obtained from the Nottingham Arabidopsis Stock Centre (accessions N68831 and N68833, respectively); all three lines of SWEET gene mutants used in this study were previously characterized in Chen et al. (2015).
Growth Conditions and Inoculation
In all experiments plants were grown under controlled conditions with a growth irradiance of 120 μmol m−2 s−1 provided by FluorA L 36W/77 fluorescent bulbs (Osram) with a 9-h light/15-h dark photoperiod and temperatures of 22°C day/20°C night. Seeds were sterilized by 1% sodium hypochlorite (commercial bleach), kept at 4°C for 2 d, and sown on 0.7% agar medium (BioShop) supplemented by 0.5 Murashige & Skoog salts (MS; Sigma-Aldrich) and 1% (w/v) sucrose, pH- adjusted to 5.7 with KOH. Seven d after germination, seedlings were transferred to soil mix (Klasmann No. 11 and perlite, 5:1). Plants were inoculated 14 d after germination using 2 mL of 106 mL−1 spores suspended in water. Control plants were mock inoculated with 2 mL of water. The Plasmodiophora brassicae isolate used in this study was classified by the European Clubroot Differential set as 16/2/12 (Devos et al., 2005). Brassica rapa L. ssp pekinensis cv Amager was used for pathogen propagation; 8 weeks after inoculation, well-developed large galls were collected and homogenized. Resting spores were extracted according to Mithen and Magrath (1992) and suspended in deionized water. Spore viability was assessed according to Siemens et al. (2002). Before inoculation, spores were diluted to a concentration of 106 mL−1. At defined time points (16 DAI and 26 DAI), hypocotyls were harvested. Each experiment used 30 infected and 30 control plants and was repeated three times for each combination. For the experiment assessing plant mortality, three biological repeats each containing 15 infected plants were scored at 32 DAI and those with totally disintegrated underground organs and desiccated aboveground parts were scored as dead.
Histochemical GUS Detection and Light Microscopy
Histochemical analyses of GUS expression was performed as described by Jefferson et al. (1986). For better specificity, plant material was preincubated with 90% (v/v) ice cold acetone for 5 min, washed in 100 mM phosphate buffer at pH 7.0, and infiltrated with substrate solution under vacuum for 5 min. After 16 h incubation at room temperature in substrate solution containing 0.1 mm 5-bromo-4-chloro-3-indolyl-β-glucuronic acid cyclohexylammonium salt (X-GLUC DIRECT), materials were dehydrated in 70% then 99.98% ethanol, preinfiltrated in 1:1 EtOH/Technovit 7100 (v/v) and finally infiltrated with 100% Technovit 7100. Embedded hypocotyls were sectioned on a Leica RM2135 microtome (Leica) and 10µm sections were placed on glass slides in a drop of water, and dried on a heating plate (62°C). After staining with 0.05% (w/v) Safranin-O solution for GUS observations or 0.05% Toluidine Blue (Sigma-Aldrich) solution for anatomical observation, sections were mounted in 50% (w/v) glycerol solution, and then examined and photographed in bright field under an AXIO Image M2 microscope (Zeiss) equipped with an AxioCamICc5 camera. For GUS assay after visual inspection, the 10 most representative hypocotyls from each time-point were sectioned. Hypocotyl measurements were analyzed by either general or mixed effect linear models using the “lm” and “lmer” functions of the lme4 package in the R environment (Bates et al., 2015).
Transmission Electron Microscopy
Uninfected and P. brassicae–infected Col-0 plants were grown and inoculated as described above. At 16 DAI and 26 DAI hypocotyls were dissected and immediately immersed in a fixative composed of 2% (w/v) paraformaldehyde (Sigma-Aldrich) and 2% (v/v) glutaraldehyde (Sigma-Aldrich) dissolved in 0.05 M cacodylic buffer (pH 7.2) for 2 h. Thereafter they were rinsed in the same buffer for 2 h and post-fixed in 2% osmium tetroxide (Roth) in 0.05 M cacodylic buffer for 2 h. After rinsing with 0.05 M cacodylic buffer for 2 h they were dehydrated in an ascending graded ethanol series, which was replaced with pure propylene oxide for 1 h. The samples were infiltrated with an ascending graded series of a medium grade epoxy resin (Sigma-Aldrich) for 24 h in total. Then they were transferred into flat embedding molds and the resin was polymerized at 65°C for 16 h (Piszczek et al., 2011). Ultrathin (90-nm-thick) sections were taken on a UCT ultramicrotome (Leica Microsystems) and collected on Formvar-coated single slot copper grids. They were stained with a saturated ethanolic (70% v/v) solution of uranyl acetate and an aqueous solution of lead citrate for 10 and 15 min, respectively, and examined under an 268M “Morgagni” (FEI) transmission electron microscope operating at 80 kV. The images were acquired with a SIS “Morada” (Olympus) digital camera at 10 MPx resolution. They were equalized for similar brightness and contrast, and cropped and resized using Adobe Photoshop software. Fifteen uninfected and infected hypocotyls per time-point were sectioned and examined.
FTIR Imaging
Cryo-sections were prepared from frozen plant materials of non-infected and infected Col-0 and sweet11;12 mutant plants at 16 d and 26 d after infection. Samples taken from hypocotyls were embedded in Tissue-Tek cryomolds using Tissue-Tek O.C.T. (Sakura Finetek) at −20°C. Embedded tissue blocks were trimmed and cross-sectioned (16-μm-thick) with a cryotome (CryoStar NX7; Thermo Fisher Scientific, and Microm International). Lyophilized cryo-sections from three biological replicates were subsequently used for imaging on a Hyperion 3000 FTIR microscope (Bruker Optics) coupled to a Tensor 27 FTIR spectrometer (Bruker Optics) with an internal mid-infrared source. The system is equipped with a focal plane array detector (64×64 pixel), which was used in transmission mode. The imaging system was purged with dry air continuously. FTIR images were recorded in the spectral range of 3900 cm−1 to 800 cm−1 at a spatial resolution of 5.5 µm (OPUS: binning 2×2 pixels) and a spectral resolution of 6 cm−1 using 15× infrared magnification objectives (Bruker Optics). Each spectrum comprised 64 coadded scans. A reference of a single focal plane array window of the empty light path was acquired before image acquisition and automatically subtracted from the recorded image by the software OPUS (Bruker Optics).
FTIR Data Processing
Atmospheric absorptions of water vapor and CO2 were partially corrected by OPUS (Bruker Optics) during image acquisition. OPUS files were imported into MatLab (The MathWorks) as ENVI files using the multiband-read function or by the “irootlab” toolbox (Trevisan et al., 2013). Image data was reduced to the desired areas of interest and cropped to a spectral range (wave number) of 2200 cm−1 to 850 cm−1. Spectral features like carbohydrates and sucrose fingerprints along with baseline features were extracted using an extended multiplicative signal correction model (Bassan et al., 2010; Baker et al., 2014; Nguyen et al., 2016). This feature extraction is an iterative approach by which the combined spectral features of the tissue spectra are modeled from pure component spectra to elucidate the spectral composition. Using a partial least-squares algorithm, a library of chemical components common in plant tissue along with spectral features of scattering artifacts as a principle component matrix are fit to each pixel spectrum. Baseline features are subtracted at the end of each iteration until the model converges toward a purely chemical spectral data set. Standardization of the component-specific images derived from unique fingerprint features of single (sucrose) or multiple compounds (carbohydrates) enables the quantitative assessment (Gündel et al., 2018). Images were digitally dissected into phloem, cambium, and xylem regions of the hypocotyl cross section. Extracted features from these regions were compared by quantile absolute distance (QAD), and signal distributions for each feature across both developmental stages (16 DAI and 26 DAI) were tested for significance using the non-parametric Mann-Whitney test and comparisons were deemed to be significant if the P value was <0.05 and the divergence effect size (D) was >0.2. QAD compares the difference of quantiles of two distributions over the entire range of probabilities and is calculated from the mean difference of the quantile functions A−1 and B−1 of two populations as described in Equation 1:
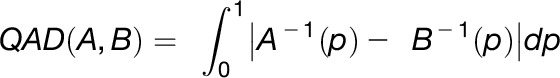 |
(1) |
QAD values are always positive, and will only be 0 if A and B are identical. Divergence effect size is a probability distance between two cumulative distribution functions A and B regardless of their context, according to Equation 2:
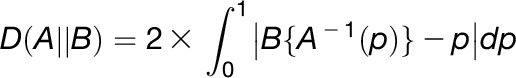 |
(2) |
Since it is not a symmetric measure D(A|B) ≠ D(B|A), a symmetric measure of this parameter can be achieved by Equation 3:
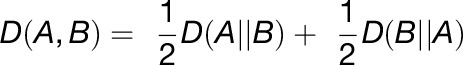 |
(3) |
This effect size is a bounded parameter that can take values between 0 (identical) and 1 (no commonalities).
Transcript Sequencing
RNA-Seq was performed at the Applied Genomics and Analytical Technologies Department, National Research Council of Canada using the Illumina Hi-Seq 2500 system. This data has been previously published by Malinowski et al. (2016) and raw data from this experiment can be obtained from the European Nucleotide Archive (https://www.ebi.ac.uk/ena) under the identifier PRJEB12261.
Real-Time PCR
To perform quantitative determination of changes in the expression of P. brassicae genes, RNA from hypocotyls was isolated using the Trizol method (Chomczynski and Sacchi, 1987) and ethanol precipitated in the presence of 1 µg of RNase free glycogen (Ambion). RNA was quantified using Nano-Drop and Qubit equipment (Thermo Fisher Scientific). First-strand cDNA synthesis was performed on 1 µg of DNase (Ambion) treated total RNA with the MMLV reverse transcriptase (Promega) according to the manufacturer’s protocol. A quantity of 1 μL of a 4-fold dilution of this cDNA was used as a template for PCR amplification. Primers were designed using the QuantPrime tool for RT-qPCR (http://quantprime.mpimp-golm.mpg.de/). The reactions were performed using the LightCycler 480 instrument (Roche) and the SensiMix SYBR No-ROX Kit (Bioline). Each amplification was performed with gene-specific oligonucleotide primers (Supplemental Table ), with three technical replicates combined, to give an average value for each biological replicate and three independent biological replicates were analyzed for each condition. Expression levels were calculated relative to the geometric mean of three normalization genes (LOS1-At1g56070, VAB1-At1g76030, and YLS8-At5g08290) according to the method described by Pfaffl (2001).
Accession Numbers
Sequence data from this article can be found in the Arabidopsis Genome Initiative or GenBank/EMBL databases under the following accession numbers: ANAC020: AT1G54330, APL: AT1G79430, BAM3: AT4G20270, BRX: AT1G31880, CALS3: AT5G13000, CALS7: AT1G06490, CHER1: AT3G15380, CLE45: AT1G69588, CRN: AT5G13290, CVL1: AT2G32010, CVP2: AT1G05470, MAKR5: AT5G52870, MP: AT1G19850, NAC45: AT3G03200, NAC86: AT5G17260, NEN1: AT5G07710, NEN2: AT5G61390, NEN3: AT1G74390, NEN4: AT4G39810, OPS: AT3G09070, OPL2: AT2G38070, CINV1 A/N-INVG: AT1G35580, CINV2 A/N-INVI: AT4G09510, A/N-INVA: AT1G56560, A/N-INVC: AT3G06500, A/N-INVD: AT1G22650, A/N-INVE: AT5G22510, A/N-INVF: AT1G72000, A/N-INVH: AT3G05820, CWINV1: AT3G13790, CWINV2: AT3G52600, CWINV5: AT3G13784, VAC-INV1: AT1G62660, VAC-INV2: AT1G12240, SUS1: AT5G20830, SUS2: AT5G49190, SUS3: AT4G02280, SUS4: AT3G43190, SUS5: AT5G37180, SUS6: AT1G73370, SUC1: AT1G71880, SUC2: AT1G22710, SUC3: AT2G02860, SUC4: AT1G09960, SUC5: AT1G71890, SUC6: AT5G43610, SWEET1: AT1G21460, SWEET2: AT3G14770, SWEET4: AT3G28007, SWEET11: AT3G48740, SWEET12: AT5G23660, SWEET13: AT5G50800, SWEET15: AT5G13170, SWEET16: AT3G16690, SWEET17: AT4G15920, STP1: AT1G11260, STP3: AT5G61520, STP4: AT3G19930, STP7: AT4G02050, STP8: AT5G26250, STP9: AT1G50310, STP10: AT3G19940, STP11: AT5G23270, STP12: AT4G21480, STP13: AT5G26340, STP14: AT1G77210, LOS1: AT1G56070, YLS8: AT5G08290, VAB1: AT1G76030.
Supplemental Data
Supplemental Figure 1. Introduction to the experimental system. (A) Plasmodiophora brassicae life cycle in Arabidopsis, adapted from Kageyama and Asano (2009).
Supplemental Figure 2. Expression of key phloem cell fate regulators in P. brassicae infected hypocotyls
Supplemental Figure 3. Disease progression in phloem differentiation mutants and the sweet11;12 double mutant
Supplemental Figure 4. P. brassicae-driven changes in the expression of sucrose metabolism and transport genes in Arabidopsis hypocotyls
Supplemental Figure 5. Mutations in sus1;2;3;4 and cinv1 cinv2 lead to reduced hypocotyl size but have no effect on P. brassicae disease progression
Supplemental Figure 6. No effect of sweet11;12 mutation on phloem differentiation response to infection was observed
Supplemental Table. List of primer sequences
Dive Curated Terms
The following phenotypic, genotypic, and functional terms are of significance to the work described in this paper:
Acknowledgments
Research work by R.M. and W.T. has been supported by the European Union’s Seventh Framework Programme for research, technological development, and demonstration under grant agreement 621321. R.M. is the BIO-TALENT ERA-CHAIR team leader. This work was also financed from Polish financial sources for education in the years 2015–2019 allocated to an international cofinanced project. P.W., M.O., and R.M. were supported by the National Science Centre Poland SONATA BIS2 grant 2012/07/E/NZ3/00510 “Integrated Approach for Deciphering the Mechanism Leading to Shift in Cell Proliferation/Differentiation Balance Accompanying Clubroot Infection.” L.B. and A.G. acknowledge Deutsche Forschungsgemeinschaft for the Project grant BO1917/5-1 as well as the contribution of Steffen Wagner (Leibniz Institute of Plant Genetics and Crop Plant Research) to FTIR work. L.B. and L.G. acknowledge the contribution of Steffen Wagner (Leibniz Institute of Plant Genetics and Crop Plant Research) to FTIR work.
We thank Prof. Christian Hardtke (Department of Plant Molecular Biology, University of Lausanne, Switzerland) for sharing materials, making insightful comments, and the critical reading of this manuscript.
AUTHOR CONTRIBUTIONS
P.W. performed all experiments aimed at understanding of the pathogen-driven phloem development reprogramming as well as experiments with SWEET11 and SWEET12 protein fusions and the sweet11;12 double mutant; he was also involved in all data mining and interpretation as well as the manuscript writing. A.G. and L.B. performed FTIR-based metabolite imaging and analyzed data. N.Y. and S.R. performed experiments on the infection of sus1;2;3;4, cinv 1;2 and sweet11;12 mutants. M.S. performed electron microscopy studies on the pathogen-driven phloem anatomy changes. W.T. performed bioinformatics analyses and was involved in the manuscript preparation. M.O. was involved in embedding and sectioning of some materials as well as performing one experiment on the sus1;2;3;4 and cinv1;2 mutants. R.M. supervised and designed experiments and wrote the final version of the manuscript. The authors declare no conflict of interest.
Footnotes
[CC-BY]: Article free via Creative Commons CC-BY 4.0 license.
References
- Baker M.J., et al. (2014). Using Fourier transform IR spectroscopy to analyze biological materials. Nat. Protoc. 9: 1771–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barratt D.H.P., Derbyshire P., Findlay K., Pike M., Wellner N., Lunn J., Feil R., Simpson C., Maule A.J., Smith A.M. (2009). Normal growth of Arabidopsis requires cytosolic invertase but not sucrose synthase. Proc. Natl. Acad. Sci. USA 106: 13124–13129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassan P., Kohler A., Martens H., Lee J., Jackson E., Lockyer N., Dumas P., Brown M., Clarke N., Gardner P. (2010). RMieS-EMSC correction for infrared spectra of biological cells: Extension using full Mie theory and GPU computing. J. Biophotonics 3: 609–620. [DOI] [PubMed] [Google Scholar]
- Bates D., Mächler M., Bolker B., Walker S. (2015). Fitting Linear Mixed-Effects Models Using lme4. Journal of Statistical Software 67: 1–48. [Google Scholar]
- Bi K., He Z., Gao Z., Zhao Y., Fu Y., Cheng J., Xie J., Jiang D., Chen T. (2016). Integrated omics study of lipid droplets from Plasmodiophora brassicae. Sci. Rep. 6: 36965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniawska Z., Paul Barratt D.H., Garlick A.P., Thole V., Kruger N.J., Martin C., Zrenner R., Smith A.M. (2007). Analysis of the sucrose synthase gene family in Arabidopsis. Plant J. 49: 810–828. [DOI] [PubMed] [Google Scholar]
- Bonke M., Thitamadee S., Mähönen A.P., Hauser M.-T., Helariutta Y. (2003). APL regulates vascular tissue identity in Arabidopsis. Nature 426: 181–186. [DOI] [PubMed] [Google Scholar]
- Brodmann A., Schuller A., Ludwig-Müller J., Aeschbacher R.A., Wiemken A., Boller T., Wingler A. (2002). Induction of trehalase in Arabidopsis plants infected with the trehalose-producing pathogen Plasmodiophora brassicae. Mol. Plant Microbe Interact. 15: 693–700. [DOI] [PubMed] [Google Scholar]
- Büttner M. (2007). The monosaccharide transporter(-like) gene family in Arabidopsis. FEBS Lett. 581: 2318–2324. [DOI] [PubMed] [Google Scholar]
- Carland F.M., Nelson T. (2004). COTYLEDON VASCULAR PATTERN2-mediated inositol (1,4,5) triphosphate signal transduction is essential for closed venation patterns of Arabidopsis foliar organs. Plant Cell 16: 1263–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carland F.M., Nelson T. (2009). CVP2- and CVL1-mediated phosphoinositide signaling as a regulator of the ARF GAP SFC/VAN3 in establishment of foliar vein patterns. Plant J. 59: 895–907. [DOI] [PubMed] [Google Scholar]
- Carland F.M., Berg B.L., FitzGerald J.N., Jinamornphongs S., Nelson T., Keith B. (1999). Genetic regulation of vascular tissue patterning in Arabidopsis. Plant Cell 11: 2123–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran D. (2015). Co-option of developmentally regulated plant SWEET transporters for pathogen nutrition and abiotic stress tolerance. IUBMB Life 67: 461–471. [DOI] [PubMed] [Google Scholar]
- Chen L.-Q., et al. (2010). Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468: 527–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.-Q. (2014). SWEET sugar transporters for phloem transport and pathogen nutrition. New Phytol. 201: 1150–1155. [DOI] [PubMed] [Google Scholar]
- Chen L.-Q., Qu X.-Q., Hou B.-H., Sosso D., Osorio S., Fernie A.R., Frommer W.B. (2012). Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science 335: 207–211. [DOI] [PubMed] [Google Scholar]
- Chen L.-Q., Lin I.W., Qu X.-Q., Sosso D., McFarlane H.E., Londoño A., Samuels A.L., Frommer W.B. (2015). A cascade of sequentially expressed sucrose transporters in the seed coat and endosperm provides nutrition for the Arabidopsis embryo. Plant Cell 27: 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P., Sacchi N. (1987). Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162: 156–159. [DOI] [PubMed] [Google Scholar]
- Damiani I., Baldacci-Cresp F., Hopkins J., Andrio E., Balzergue S., Lecomte P., Puppo A., Abad P., Favery B., Hérouart D. (2012). Plant genes involved in harbouring symbiotic rhizobia or pathogenic nematodes. New Phytol. 194: 511–522. [DOI] [PubMed] [Google Scholar]
- Depuydt S., Rodriguez-Villalon A., Santuari L., Wyser-Rmili C., Ragni L., Hardtke C.S. (2013). Suppression of Arabidopsis protophloem differentiation and root meristem growth by CLE45 requires the receptor-like kinase BAM3. Proc. Natl. Acad. Sci. USA 110: 7074–7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos S., Vissenberg K., Verbelen J.P., Prinsen E. (2005). Infection of Chinese cabbage by Plasmodiophora brassicae leads to a stimulation of plant growth: impacts on cell wall metabolism and hormone balance. New Phytol. 166: 241–250. [DOI] [PubMed] [Google Scholar]
- Eom J.-S., Chen L.-Q., Sosso D., Julius B.T., Lin I.W., Qu X.-Q., Braun D.M., Frommer W.B. (2015). SWEETs, transporters for intracellular and intercellular sugar translocation. Curr. Opin. Plant Biol. 25: 53–62. [DOI] [PubMed] [Google Scholar]
- Evans J.L., Scholes J.D. (1995). How does clubroot alter the regulation of carbon metabolism in its host? Asp. Appl. Biol. 42: 125–132. [Google Scholar]
- Furuta K.M., et al. (2014). Plant development. Arabidopsis NAC45/86 direct sieve element morphogenesis culminating in enucleation. Science 345: 933–937. [DOI] [PubMed] [Google Scholar]
- Gravot A., Richard G., Lime T., Lemarié S., Jubault M., Lariagon C., Lemoine J., Vicente J., Robert-Seilaniantz A., Holdsworth M.J., Manzanares-Dauleux M.J. (2016). Hypoxia response in Arabidopsis roots infected by Plasmodiophora brassicae supports the development of clubroot. BMC Plant Biol. 16: 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gündel A., Rolletschek H., Wagner S., Muszynska A., Borisjuk L. (2018). Micro imaging displays the sucrose landscape within and along its allocation pathways. Plant Physiol. pp.00947.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W.-J., Nagy R., Chen H.-Y., Pfrunder S., Yu Y.-C., Santelia D., Frommer W.B., Martinoia E. (2014). SWEET17, a facilitative transporter, mediates fructose transport across the tonoplast of Arabidopsis roots and leaves. Plant Physiol. 164: 777–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa Y., Kondo Y., Fukuda H. (2010). TDIF peptide signaling regulates vascular stem cell proliferation via the WOX4 homeobox gene in Arabidopsis. Plant Cell 22: 2618–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaias R.M.S., de Oliveira D.C., da Silva C.R.G., Kraus J.E. (2014). Developmental anatomy of galls in the neotropics: Arthropods stimuli versus host plant constraints. In Neotropical Insect Galls, Santos G.W.F.J.C., ed (New York: Springer; ), pp. 15–34. [Google Scholar]
- Jefferson R.A., Burgess S.M., Hirsh D. (1986). beta-Glucuronidase from Escherichia coli as a gene-fusion marker. Proc. Natl. Acad. Sci. USA 83: 8447–8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H., Gheysen G., Denil S., Lindsey K., Topping J.F., Nahar K., Haegeman A., De Vos W.H., Trooskens G., Van Criekinge W., De Meyer T., Kyndt T. (2013). Transcriptional analysis through RNA sequencing of giant cells induced by Meloidogyne graminicola in rice roots. J. Exp. Bot. 64: 3885–3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama K., Asano T. (2009). Life cycle of Plasmodiophora brassicae. J. Plant Growth Regul. 28: 203–211. [Google Scholar]
- Keen N.T., Williams P.H. (1969). Translocation of sugars into infected cabbage tissues during clubroot development. Plant Physiol. 44: 748–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Li X., Xuan Y., Jiang J., Wei Y., Piao Z. (2018). Genome wide identification and expression profiling of SWEET genes family reveals its role during Plasmodiophora brassicae-induced formation of clubroot in Brassica rapa. Front. Plant Sci. 9: 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas W.J., et al. (2013). The plant vascular system: Evolution, development and functions. J. Integr. Plant Biol. 55: 294–388. [DOI] [PubMed] [Google Scholar]
- Ludwig-Müller J. (2014). Auxin homeostasis, signaling, and interaction with other growth hormones during the clubroot disease of Brassicaceae. Plant Signal. Behav. 9: e28593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinowski R., Smith J.A., Fleming A.J., Scholes J.D., Rolfe S.A. (2012). Gall formation in clubroot-infected Arabidopsis results from an increase in existing meristematic activities of the host but is not essential for the completion of the pathogen life cycle. Plant J. 71: 226–238. [DOI] [PubMed] [Google Scholar]
- Malinowski R., Novák O., Borhan M.H., Spíchal L., Strnad M., Rolfe S.A. (2016). The role of cytokinins in clubroot disease. Eur. J. Plant Pathol. 145: 543–557. [Google Scholar]
- Mithen R., Magrath R. (1992). A contribution to the life history of Plasmodiophora brassicae: Secondary plasmodia development in root galls of Arabidopsis thaliana. Mycol. Res. 96: 877–885. [Google Scholar]
- Mouchel C.F., Osmont K.S., Hardtke C.S. (2006). BRX mediates feedback between brassinosteroid levels and auxin signalling in root growth. Nature 443: 458–461. [DOI] [PubMed] [Google Scholar]
- Nguyen T.N.Q., Jeannesson P., Groh A., Piot O., Guenot D., Gobinet C. (2016). Fully unsupervised inter-individual IR spectral histology of paraffinized tissue sections of normal colon. J. Biophotonics 9: 521–532. [DOI] [PubMed] [Google Scholar]
- Pfaffl M.W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piszczek E., Dudkiewicz M., Sobczak M. (2011). Molecular cloning and phylogenetic analysis of cereal type II metacaspase cDNA from wheat. Biol. Plant. 55: 614. [Google Scholar]
- Rodrigues A., Santiago J., Rubio S., Saez A., Osmont K.S., Gadea J., Hardtke C.S., Rodriguez P.L. (2009). The short-rooted phenotype of the Brevis radix mutant partly reflects root abscisic acid hypersensitivity. Plant Physiol. 149: 1917–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Villalon A. (2016). Wiring a plant: Genetic networks for phloem formation in Arabidopsis thaliana roots. New Phytol. 210: 45–50. [DOI] [PubMed] [Google Scholar]
- Roitsch T., Ehneß R. (2000). Regulation of source/sink relations by cytokinins. Plant Growth Regul. 32: 359–367. [Google Scholar]
- Rolfe S.A., Strelkov S.E., Links M.G., Clarke W.E., Robinson S.J., Djavaheri M., Malinowski R., Haddadi P., Kagale S., Parkin I.A.P., Taheri A., Borhan M.H. (2016). The compact genome of the plant pathogen Plasmodiophora brassicae is adapted to intracellular interactions with host Brassica spp. BMC Genomics 17: 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scacchi E., Salinas P., Gujas B., Santuari L., Krogan N., Ragni L., Berleth T., Hardtke C.S. (2010). Spatio-temporal sequence of cross-regulatory events in root meristem growth. Proc. Natl. Acad. Sci. USA 107: 22734–22739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller A., Kehr J., Ludwig-Müller J. (2014). Laser microdissection coupled to transcriptional profiling of Arabidopsis roots inoculated by Plasmodiophora brassicae indicates a role for brassinosteroids in clubroot formation. Plant Cell Physiol. 55: 392–411. [DOI] [PubMed] [Google Scholar]
- Siemens J., Nagel M., Ludwig-Müller J., Sacristán M.D. (2002). The interaction of Plasmodiophora brassicae and Arabidopsis thaliana: Parameters for disease quantification and screening of mutant lines. J. Phytopathol. 150: 592–605. [Google Scholar]
- Siemens J., González M.-C., Wolf S., Hofmann C., Greiner S., Du Y., Rausch T., Roitsch T., Ludwig-Müller J. (2011). Extracellular invertase is involved in the regulation of clubroot disease in Arabidopsis thaliana. Mol. Plant Pathol. 12: 247–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skylar A., Sung F., Hong F., Chory J., Wu X. (2011). Metabolic sugar signal promotes Arabidopsis meristematic proliferation via G2. Dev. Biol. 351: 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strehlow B., de Mol F., Struck C. (2014). Risk potential of clubroot disease on winter oilseed rape. Plant Dis. 99: 667–675. [DOI] [PubMed] [Google Scholar]
- Trevisan J., Angelov P.P., Scott A.D., Carmichael P.L., Martin F.L. (2013). IRootLab: A free and open-source MATLAB toolbox for vibrational biospectroscopy data analysis. Bioinformatics 29: 1095–1097. [DOI] [PubMed] [Google Scholar]
- Truernit E., Bauby H., Belcram K., Barthélémy J., Palauqui J.C. (2012). OCTOPUS, a polarly localised membrane-associated protein, regulates phloem differentiation entry in Arabidopsis thaliana. Development 139: 1306–1315. [DOI] [PubMed] [Google Scholar]
- Vatén A., et al. (2011). Callose biosynthesis regulates symplastic trafficking during root development. Dev. Cell 21: 1144–1155. [DOI] [PubMed] [Google Scholar]
- Walters D.R., McRoberts N. (2006). Plants and biotrophs: A pivotal role for cytokinins? Trends Plant Sci. 11: 581–586. [DOI] [PubMed] [Google Scholar]