

Abstract
Background
24‐dehydrocholesterol reductase (Dhcr24) catalyzes the last step of cholesterol biosynthesis, which is required for normal development and anti‐apoptotic activities of tissues. We found that Dhcr24 expression decreased in the cTnTR 141W dilated cardiomyopathy (DCM) transgenic mice. Therefore, we tested whether rescued expression of Dhcr24 could prevent the development of DCM and its possible mechanism.
Methods
Heart tissue specific transgenic overexpression mice of Dhcr24 was generated, then was crossed to cTnTR 141W mouse to obtain the double transgenic mouse (DTG). The phenotypes were demonstrated by the survival, cardiac geometry and function analysis, as well as microstructural and ultrastructural observations based on echocardiography and histology examination. The pathway and apoptosis were analysed by western blotting and TUNEL assay in vivo and in vitro.
Results
We find that Dhcr24 decreased in hearts tissues of cTnTR 141W and LMNAE 82K DCM mice. The transgenic overexpression of Dhcr24 significantly improves DCM phenotypes in cTnTR 141W mice, and activates PI3K/Akt/HKII pathway, followed by a reduction of the translocation of Bax and release of cytochrome c, caspase‐9 and caspase‐3 activation and myocyte apoptosis. Knockdown the expression of Dhcr24 reduces the activation of PI3K/Akt/HKII pathway and inhibition of the mitochondrial‐dependent apoptosis. The anti‐apoptotic effect of Dhcr24 could be completely removed by the inhibition of PI3K pathway and partly removed by the HKII inhibitor in H9c2 cell line.
Conclusion
Compensatory expression of Dhcr24 protect against DCM through activated PI3K/Akt/HKII pathway and reduce Bax translocation. This is the first investigation for the molecular mechanism of Dhcr24 participate in development of DCM.
Keywords: apoptosis, Dhcr24, dilated cardiomyopathy, gene expression and regulation
1. INTRODUCTION
Cholesterol is an indispensable lipid involved in numerous processes that are required for the normal development and maintenance of tissues.1 It is involved in signal transduction as a component of caveolae and lipid rafts and functions as a covalent modifier of the hedgehog family of morphogenes.2, 3 24‐Dehydrocholesterol reductase (Dhcr24, also called Seladin‐1), is a member of the flavin adenine dinucleotide (FAD)‐dependent oxidoreductase family and catalyzes the last step of cholesterol biosynthesis to convert desmosterol to cholesterol.
Wechsler et al reported that DHCR24−/− mice were viable up to adulthood and displayed no gross abnormalities, and it was found that cholesterol was not obligatory for viability.4 In contrast, Mirza et al indicated that DHCR24−/− mice died within the early postnatal days and that Dhcr24 expression was essential for normal development.5 Mutations in the Dhcr24 gene result in an autosomal recessive disease called desmosterolosis, which is indicated by developmental and growth retardation.6 The reduced expression of the Dhcr24 gene is associated with increased apoptosis in adrenocortical cells as the result of caspase‐3 inhibition, which is a mediator of the response to oxidative and oncogenic stress and a reactive oxygen species scavenger.7 Furthermore, a reduced expression of Dhcr24 is also found in the temporal cortex of Alzheimer's disease patients and is associated with severe anomalies in development in which Dhcr24 is suggested to be involved in the anti‐apoptotic function in the brain,8, 9, 10 and it may be a mediator of the protective effects of estrogen in neuronal cells.11 Furthermore, Dhcr24, as a key mediator of Ras‐induced senescence following oncogenic and oxidative stress, binds the p53 amino terminus and displaces the E3 ubiquitin ligase Mdm2 from p53, thus resulting in the accumulation of p53.12 The administration of triparanol, an inhibitor of Dhcr24, into pregnant rats or mice caused the accumulation of desmosterol, zymosterol and hypocholesterolemia, and was highly teratogenic.13 Therefore, Dhcr24 is a multifunctional protein possessing both cholesterol synthesizing and anti‐apoptotic activities.
Dilated cardiomyopathy (DCM) is a primary heart muscle disease characterized by the dilation of the heart chambers with evidence of impaired systolic function. Idiopathic dilated cardiomyopathy (IDC) has a prevalence of 1:2500 and is a common cause of heart failure, especially in the young, and the disease is believed to be caused by mutations in up to 50% of the cases. Despite recent advances in pharmacological and surgical therapies, the disability and morbidity due to DCM is still high.14, 15, 16 The missense mutation R141W, in the strong tropomyosin‐binding region of cardiac troponin T (cTnT), causes familial dilated cardiomyopathy (FDC).17, 18 We reported that cTnTR141W transgenic mice manifested progressive chamber dilation and contractile dysfunction and had a pathological phenotype similar to that of human DCM.19 Dhcr24 mRNA expression was down‐regulated in the cTnTR141W transgenic mice at 3 months of age, at which time the mice developed a typical cardiomyopathy phenotype. Therefore, we generated cardiac‐specific overexpression of Dhcr24 transgenic mice to study whether compensated expression of Dhcr24 in the heart tissues could ameliorate the DCM phenotypes in the mice and uncover its possible mechanism.
2. METHODS
2.1. Animals
The α‐MHC‐cTnTR141W transgenic mice were generated in our laboratory and exhibited DCM phenotype characteristics consistent with previously reports.19 Mouse Dhcr24 cDNA (Genbank accession no. 74754) was cloned into an expression plasmid under the α‐MHC promoter. The transgenic mice were generated by the microinjection method. Genotyping of the transgenic mice was facilitated by PCR using the primers 5ʹGGGACATCCAGAAACAGGTCC3ʹ and 5ʹGACGGTGTGCAGCGCACAAAG3ʹ. A 390‐bp fragment was amplified, which represents the transgenic mouse Dhcr24 gene. The transgenic founders with high expression levels of the target gene were selected by Western blot analysis using an antibody against DHCR24 (Proteintech). The Dhcr24 and cTnTR141W double transgenic mice (DTG) were generated by crossing the Dhcr24 transgenic mice with the cTnTR141W transgenic mice. All of the mice were maintained on a C57BL/6J genetic background and bred in an AAALAC‐accredited facility. The use of the animals was approved by the Animal Care and Use Committees of the Institute of Laboratory Animal Science of Peking Union Medical College (GC08‐2001).
2.2. Cell culture
The H9c2 cells line were grown in high glucose DMEM supplemented with 10% defined fetal bovine serum (HyClone), 100 U/mL penicillin and 100 g/mL streptomycin (HyClone) in a humidified 5% CO2 incubator at 37°C. The expression construct was individually generated by cloning the full‐length Dhcr24 cDNA fragment into the pcDNA3.1(+) vector (Invitrogen). The expression construct for the Dhcr24 siRNA was a commercial product (OriGene), which corresponded to the sequence of ATGTTCGATGGCTCCTTGTACCACAAGCT in the 3ʹUTR of the Dhcr24 cDNA of mouse and rat. H9c2 cells were transfected with the constructs using Lipofectamine 2000 (Invitrogen). Stable cell lines were established by subsequent selection using 800 μg/mL G418 (Amresco). The cell lines were treated with PI3K inhibitor (LY294002, 20 μM, 24 hours, MERCK) or HexokinaseII Inhibitor, 3‐bromopyruvate (3‐BrPA, 100 μM, 4 hours, MERCK), and/or H2O2 (1 mM, 2 hour), and then the cells were harvested for Western blot analysis or TUNEL assay.
2.3. Determination of cholesterol in the heart tissues
The heart tissue was homogenized, and the lipids were extracted at once with chloroform‐methanol (2:l, v/v). The cholesterol in the extraction was analyzed by HPLC according to a modified method, as previously reported.20, 21 Briefly, the HPLC system consisted of a Varian ProStar 210 pump and a Varian ProStar 335 photodiode array detector. The analytical column was an Arcus EP‐C18 (250 × 4.6 mm, 5 mm particle size) column, and the oven temperature was set at 40°C. The mobile phase was acetonitrile‐2‐propanol (75:25, v/v), and the flow rate was 1 mL/min. The absorbance at 210 nm was recorded, and the level of cholesterol was calculated.
2.4. Echocardiography
M‐mode echocardiography was performed on mice with the small animal echocardiography analysis system (Vevo770, Canada). Briefly, mice were lightly anesthetised by intraperitoneal injection of tribromoethanol at a dose of 180 mL/kg body weight. M‐mode echocardiography of the left ventricle was recorded at the tip of the mitral valve apparatus with a 30 MHz transducer.
2.5. Survival analysis
The cumulative percent mortality was calculated every month. Upon the death of each mouse, the body was autopsied by a pathologist, and morphological and pathological changes in the heart were recorded. Kaplan‐Meier curves for survival analysis were compared using the log‐rank test (SPSS. 16.0 software).
2.6. Histological analysis
For light microscopy, cardiac tissue from mice at 5 months of age was fixed in 4% formaldehyde and mounted in paraffin blocks. The sections were used for H&E and Masson trichrome staining or immunochemical staining with an anti‐DHCR24 antibody (Proteintech). For transmission electron microscopy (TEM), the cardiac tissues were routinely fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) and postfixed in buffered 1% osmium tetroxide for 1 hour. The samples were then dehydrated using a series of ethanol immersions and embedded in Epon 812. Thin sections were stained with uranyl acetate and lead citrate and examined under a JEM‐1230 transmission electron microscope. Myocytes were analysed by an observer blinded to the genotypes of the mice.
2.7. RNA extraction and PCR analysis
Total RNA was isolated from the heart tissue using TRIzol Reagent (Invitrogen). First‐strand cDNA was synthesised from 2 μg of total RNA using random hexamer primers according to the Superscript III reverse transcriptase manufacturer's protocol (Invitrogen). Procollagen type I α1 (Col1α1), Procollagen type III α1 (Col3α1) and Integrin alpha‐8 (Igtα8) mRNA were analyzed by reverse transcriptional PCR using GAPDH for normalisation under standard conditions (primers: for Col1α1, forward, 5′CTTTGCTTCCCAGATGTCCT and reverse, 5′CGGTGTCCCTTCATTCCAG; for Col3α1, forward, 5′CTCAAGAGCGGAGAATACTGG and reverse, 5′CAATGTCATAGGGTGCGATA; for Igta8, forward, 5′CAACCACCAGTCCCATTT and reverse, 5′CGCTCAATCCCAACATAA; for GAPDH, forward, 5′CAAGGTCATCCATGACAACTTTG and reverse 5′GTCCACCACCCTGTTGCTGTAG).
2.8. Protein extraction and immunoblotting
Total protein lysates from the mouse heart tissue or the cultured cells were prepared by homogenizing with lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X‐100, 1% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, and protease inhibitor cocktail). The mitochondrial and the cytoplasmic fractions from the heart tissue and cultured cells were isolated following the Fractionation Kit manufacturer's protocol (DBI Bioscience). The total lysate samples and the fractions were separated by SDS‐PAGE and transferred to nitrocellulose (Millipore). The membranes were incubated overnight with an antibody to DHCR24 (Proteintech), HKII (Abcam), PI3K, p‐Akt, t‐Akt, Bax, caspase‐9, caspase‐3, or COX4 (Cell Signaling Technology). The primary antibody binding was detected using an HRP‐conjugated immunoglobulin G (Santa Cruz) and by a chemiluminescence detection system. GAPDH was used for normalization.
2.9. TUNEL assay
The in situ terminal dUTP nick end‐labeling (TUNEL) assay was performed on sections of the heart tissue using the In Situ Cell Death Detection Kit (Roche Diagnostics GmbH, Mannheim, Germany), principally according to the manufacturer's instructions. Eight images per heart (3 hearts per genotype group) and eight images per cell line were acquired, and the positive cells were counted individually. The results are expressed as the percentage of apoptotic cells among the total cell population.
2.10. Statistical analysis
The data were analysed with one‐way ANOVA for multiple groups followed by Tukey's post hoc analysis. The data are expressed as means ± SEMs from the individual experiments. Differences were considered significant at P < .05.
3. RESULTS
3.1. Dhcr24 expression down‐regulated in the heart tissue during aging and the development of DCM
The expression of Dhcr24 in the heart tissue of wild type (WT) mice at different ages was detected by Western blot with an anti‐Dhcr24 antibody. The results indicated that Dhcr24 expression in the heart down‐regulated during aging, with its expression level reduced by 42% until 6 months of age (Figure 1A,B). The expression of Dhcr24 in the heart tissue of the cTnTR141W mice at 3 months of age down‐regulated by 52% compared with that of the non‐transgenic (NTG) littermates (Figure 1C,D, n = 3 mice per group, P < .01 versus NTG littermates). The decreased expression was also observed by immunohistochemical staining of the heart sections of the cTnTR141W mice during the development a typical cardiomyopathy phenotype (Figure 1E). The cholesterol in the cTnTR141W hearts decreased by 42% compared to that of the NTG mice, which was a result of the down‐regulation of Dhcr24 (Figure 1F, n = 6 mice group, P < .01 versus NTG littermates).
Figure 1.
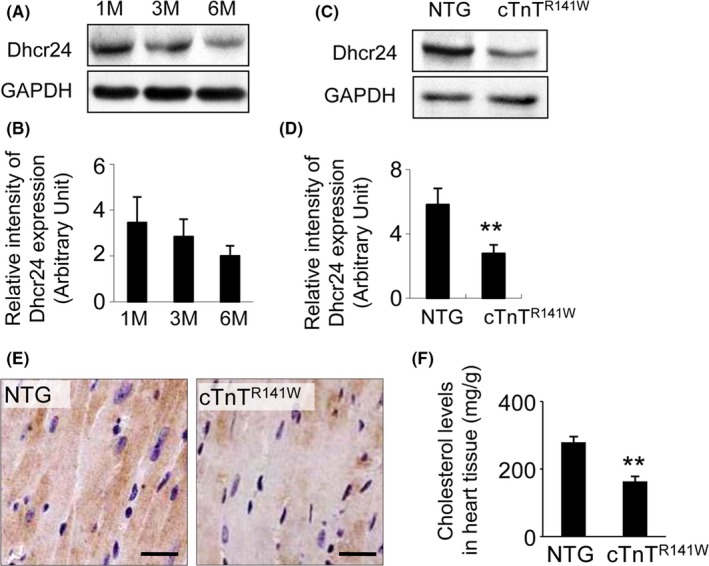
Detection of Dhcr24 expression in heart tissue. (A), The expression of Dhcr24 in the heart tissues from WT mice at 1, 3 and 6 mo of age was detected by Western blot. (B), Quantitative analysis of Dhcr24 in the WT mice used GAPDH for normalization (n = 3 mice per group). (C), Dhcr24 expression in heart tissues of the cTnTR 141W transgenic mice at 3 mo of age was detected by Western blot. (D), Quantitative analysis of Dhcr24 in the cTnTR 141W mice used GAPDH for normalization (n = 3 mice per group, **P < .01). (E), Immunodetection of Dhcr24 in the heart from the cTnTR 141W and the NTG mice at 5 mo of age. Scale bar = 20 μm. (F), The cholesterol level in the heart tissues of the cTnTR 141W and the NTG mice (n = 3 mice per group, **P < .01)
3.2. The generation of heart‐specific Dhcr24 transgenic mice
To produce the transgenic construct, the mouse Dhcr24 cDNA was inserted downstream of the α‐myosin heavy chain (MHC) cardiac‐specific promoter, and the transgenic mice were generated by the microinjection method (Figure 2A). The transgenic founders were selected by Western blot. The Dhcr24 levels in the heart tissue of the founders were increased by 24% compared with that of the NTG mice at 1 month of age (Figure 2B,C; n = 3 mice per group, P < .01 versus NTG littermates). The high level of Dhcr24 in the transgenic mice was also observed by immunohistochemical staining (Figure 2D). In addition to the increased expression of Dhcr24, cholesterol was also increased by 29% in the Dhcr24 hearts compared to those of the NTG mice (Figure 2E, n = 6 mice per group, P < .01 versus NTG littermates).
Figure 2.
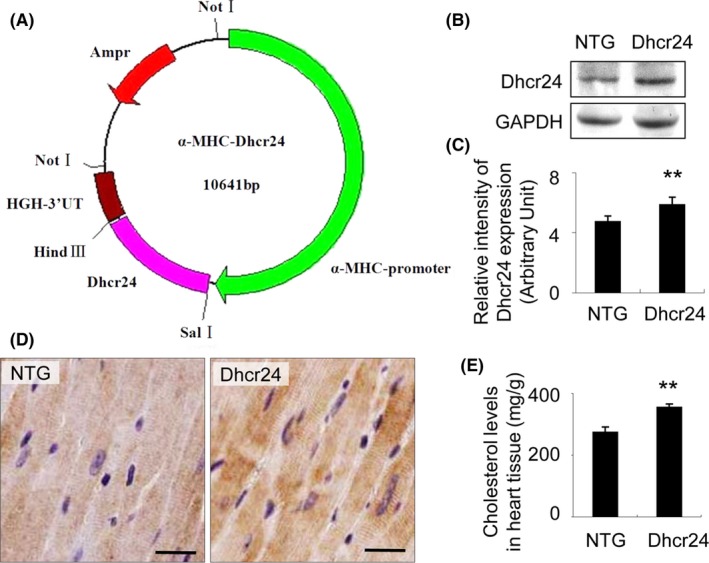
Generation of the transgenic mice. (A), The transgenic construct was generated by inserting the Dhcr24 gene under the control of the α‐MHC cardiac‐specific promoter, and the transgenic mice were created following microinjection. (B), The mouse line with Dhcr24 over‐expression was identified by Western blot. (C), Quantitative analysis of Dhcr24 in the transgenic mouse line used GAPDH for normalization. (D), Immunodetection of Dhcr24 in the heart tissue of the NTG and the Dhcr24 transgenic mice. Scale bar = 20 μm. (E), The cholesterol level in heart tissues of the NTG and the Dhcr24 transgenic mice (n = 6 mice per group, **P < .01)
3.3. The transgenic overexpression of Dhcr24 improves cardiac geometry and dysfunction and decreases mortality in cTnTR141W mice
The ventricular size and function of the NTG, Dhcr24, cTnTR141W and the Dhcr24 and cTnTR141W double transgenic mice (referred to as DTG) mice were assessed using echocardiography at 1, 3, 5, and 7 months of age. The representative M‐mode echocardiographic images at 5 months of age were shown in Figure 3A. The left ventricular (LV) posterior wall at end‐systole (LVPWS) of the DTG mice was cumulatively increased by 11% at 3 months of age and reached up to 24% at 7 months of age compared with that of the cTnTR141W transgenic mice, in which the LVPWS decreased slowly during the development of DCM (Figure 3B). The LV fractional shortening (LVFS) of the DTG mice was significantly increased by 19% to 22% compared with that of the cTnTR141W mice (Figure 3C) from 1 to 7 months of age. The DCM phenotypes were significantly improved when the decreased expression of Dhcr24 was rescued in the DTG mice. The typical M‐mode echocardiography parameters at 5 months of age are summarized in Table 1. The survival rate was 65% in the cTnTR141W mice, while the survival rate was increased to 98% in the DTG mice until 7 months of age (Figure 3D, n = 16 mice per group).
Figure 3.
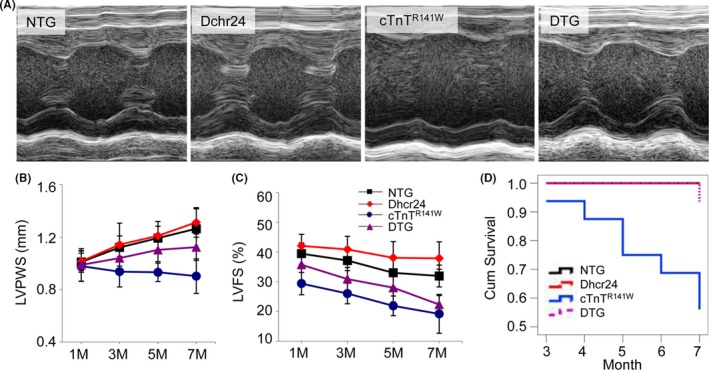
The effects of Dhcr24 on heart dimensions and survival rate. (A), The representative M‐mode echocardiographic images of the LV long‐axis from the NTG, Dhcr24, cTnTR 141W and DTG mice at 5 mo of age. (B), The left ventricle (LV) posterior wall at end‐systole (LVPW in the NTG, Dhcr24, cTnTR 141W and DTG mice at 1, 3, 5, and 7 mo of age. (C), The LV fractional shortening (LVFS) in the NTG, Dhcr24, cTnTR 141W and DTG mice at the 1, 3, 5, and 7 mo of age. (D), The cumulative survival data were recorded until 7 mo of age
Table 1.
Echocardiographic characteristics of mice at 5 mo of age
| Group Number | NTG n = 16 | Dhcr24 n = 16 | cTnTR141W n = 16 | DTG n = 28 |
|---|---|---|---|---|
| LVEDD, mm | 3.85 ± 0.17 | 3.58 ± 0.12 | 4.25 ± 0.23 | 4.12 ± 0.19 |
| LVESD, mm | 2.58 ± 0.14 | 2.21 ± 0.17 | 3.32 ± 0.25 | 2.97 ± 0.20a |
| LVPWD, mm | 0.79 ± 0.09 | 0.78 ± 0.11 | 0.70 ± 0.07 | 0.78 ± 0.09a |
| LVPWS, mm | 1.19 ± 0.09 | 1.21 ± 0.11 | 0.93 ± 0.10 | 1.10 ± 0.15a |
| LVEF, % | 61.85 ± 2.41 | 68.32 ± 4.78 | 43.31 ± 4.85 | 53.36 ± 5.08a |
| LVFS,% | 33.03 ± 4.91 | 38.08 ± 5.47 | 21.89 ± 3.30 | 27.99 ± 4.77a |
LVEDD, left ventricle (LV) end‐diastole diameter; LVESD, LV end‐systole diameter; LVPWD, LV posterior wall at end‐diastole; LVPWS, LV posterior wall at end‐systole; LVEF, LV ejection fraction; LVFS, LV fractional shortening.
P < .01 DTG vs cTnTR141W mice.
3.4. The transgenic overexpression of Dhcr24 ameliorates cardiac pathological phenotypes in the cTnTR141W mice
The morphological changes of the ventricles and the myocytes were further observed by histological examination (Figure 4A‐C). Poorly organized myofibrils with diffusion, damage and lysis were seen in the myocardium of cTnTR141W transgenic mice. The transgenic overexpression of Dhcr24 in the cTnTR141W transgenic mice significantly reduced the disorder and accumulation of collagen in the interstitial space compared with the typical histological changes observed in the heart tissues of the cTnTR141W transgenic mice. The expression levels of Col1α1, Col3α1 and Itgα8 increased by 26%, 24% and 22%, respectively, in the cTnTR141W transgenic mice (Figure 4D,E; n = 3 mice per group, P < .05 versus NTG littermates), which were all reduced to almost normal levels by the transgenic overexpression of Dhcr24 in the DTG mice (Figure 4D,E; n = 3 mice per group, P < .05 versus cTnTR141W group).
Figure 4.
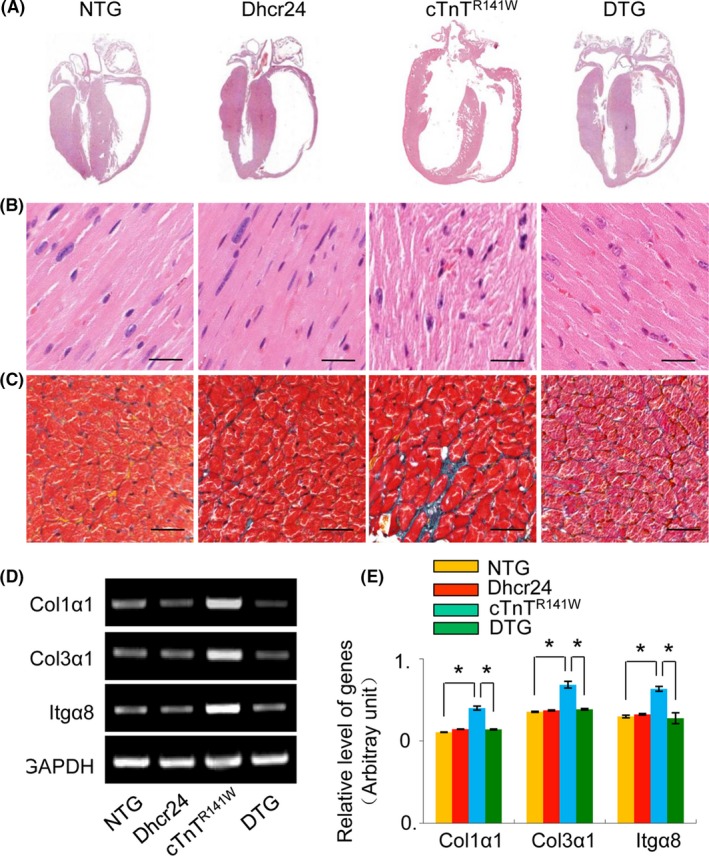
The histopathological profile of the heart tissue from mice at 5 mo of age. (A), H&E staining patterns of the whole‐heart longitudinal sections. (B), Magnification of the H&E‐stained sections of the left ventricle. (C), Magnification of the Masson trichrome stained sections of left ventricle. Scale bar = 20 μm. (D), The expression of Col1α1, Col3α1 and Itgα8 was detected by RT‐PCR. (E), Quantitative analysis of the expression of Col1α1, Col3α1 and Itgα8 using GAPDH for normalization
3.5. The transgenic overexpression of Dhcr24 activates the PI3K pathway and inhibits the mitochondrial pathway of apoptosis in cTnTR141W transgenic mice
The PI3K pathway and the mitochondrial pathway of apoptosis were analyzed in the heart tissues of the NTG, Dhcr24, cTnTR141W and DTG mice at 5 months of age (Figure 5). The levels of PI3K, HKII and phosphorylated AKT increased due to the transgenic overexpression of Dhcr24 in the Dhcr24 mice compared to that of the NTG mice, while the levels of PI3K, HKII and phosphorylated AKT were obviously reduced in the cTnTR141W transgenic mice. The reduction of those molecules was reversed by the overexpression of Dhcr24 in the DTG mice, which was indicated by the fact that PI3K expression increased by 114%, AKT phosphorylation increased by 78% and HKII expression increased by 262% compared to that of the cTnTR141W mice (Figures 5A,B; n = 3 mice per group, P < .05 or P < .01 versus cTnTR141W group). The translocation of Bax and the release of cytochrome c were significantly inhibited in the Dhcr24 transgenic mice and were increased significantly in the cTnTR141W mice compared with that of the NTG mice. The increased Bax translocation and cytochrome c release in the cTnTR141W transgenic mice reduced by 51% and 46%, respectively, due to the overexpression of Dhcr24 in the DTG mice (Figure 5C‐E, n = 3 mice per group, P < .05 or P < .01 versus cTnTR141W group). Activated caspase‐9 and ‐3 increased by 322% and 261%, respectively, in the heart tissues of the cTnTR141W transgenic mice compared with that of the NTG mice (Figures 5F,G; n = 3 mice per group, P < .01 versus NTG littermates), and the activation of caspase‐9 and ‐3 reduced by 71% and 73%, respectively, due to the overexpression of Dhcr24 in the DTG mice (Figures 5E,F; n = 3 mice per group, P < .01 versus cTnTR141W group).
Figure 5.
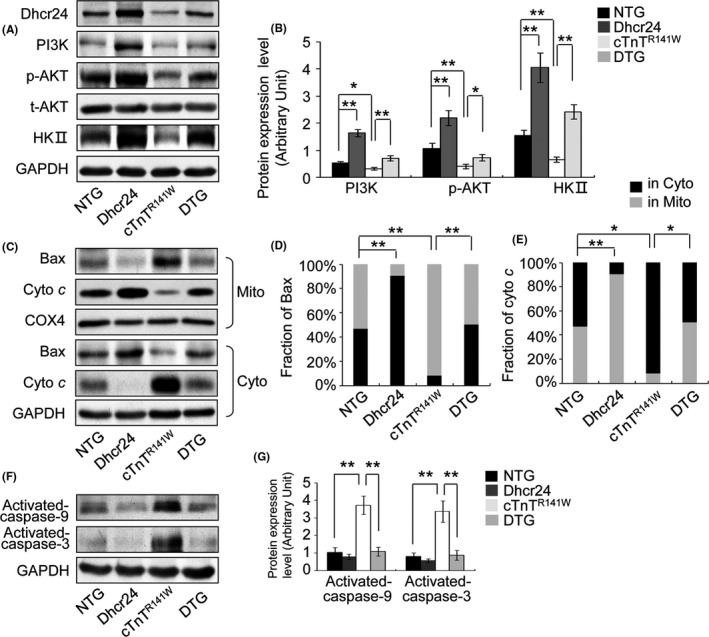
The measurement of signalling proteins of the PI3K and the mitochondrial pathways of apoptosis. The heart tissues were sampled from the NTG, Dhcr24, cTnTR 141W and DTG mice at 5 mo of age. (A), The levels of Dhcr24, PI3K, phosphorylated AKT and HKII were detected by Western blot in the heart tissues. (B), Quantitatively analyzed using GAPDH for normalization. (C), The fraction of Bax and cytochrome c in the cytoplasm and the mitochondria was detected in the heart tissues by Western blot. (D), (E), Quantitatively analysis using COX4 and GAPDH for normalization, respectively. (F), The activation of caspase‐9 and ‐3 was measured by Western blot. (G), Quantitatively analyzed using GAPDH for normalization
3.6. The transgenic overexpression of Dhcr24 improves the integrity of the mitochondrial morphology and reduces apoptosis in the myocytes of cTnTR141W transgenic mice
Disrupted ultrastructure, including swollen mitochondria with a loss of cristae and vacuolisation, was seen in the cTnTR141W mice. In contrast, the prominence of swollen mitochondria was clearly lessened by the transgenic overexpression of Dhcr24 in the DTG mice (Figure 6A). Apoptotic myocytes were rare in the NTG and Dhcr24 transgenic mice, with apoptotic indices of 0.07‰ and 0.04‰, respectively. Apoptotic myocytes increased 38.7‐fold with an apoptotic index of 2.71‰ in the cTnTR141W transgenic mice compared with that of the NTG mice (Figure 6B,C; n = 3 mice per group, P < .01 versus NTG littermates). Apoptosis was inhibited by 67.2% in the DTG mice compared with that of the cTnTR141W transgenic mice (Figures 6B,C; n = 3 mice per group, P < .05 versus cTnTR141W group) due to Dhcr24 overexpression.
Figure 6.
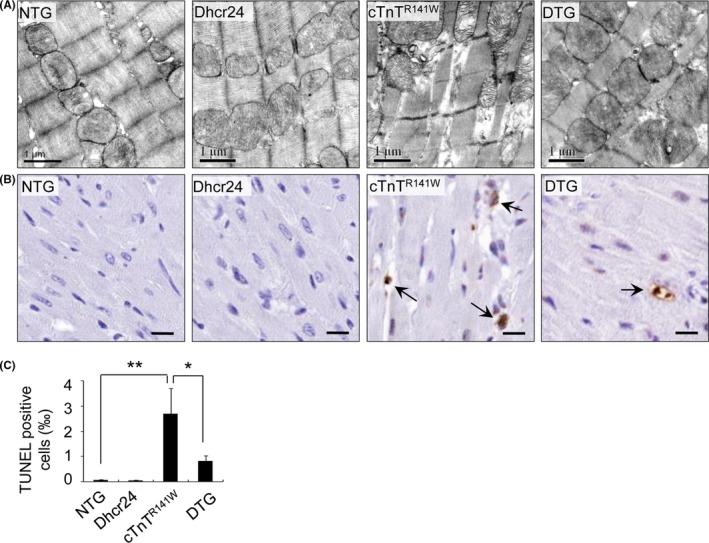
Observation of the mitochondria and TUNEL assay. (A), The morphology of the mitochondria was observed by TEM from the LV free wall in the NTG, Dhcr24, cTnTR 141W and DTG mice at 5 mo of age. (B), Photomicrographs of the heart tissue used for the TUNEL assay. The arrows indicate TUNEL‐positive cells. Scale bar = 10 μm. (C), Quantitative analysis of apoptotic cells in the hearts of mice (n = 3, *P < .05, **P < .01)
3.7. Silencing the expression of Dhcr24 inhibits the PI3K pathway and activated cytochrome c‐dependent apoptosis in H9c2 cells
H9c2 cells were transfected with the Dhcr24 overexpression construct and siRNA targeting Dhcr24. The stable cell lines with overexpressed and silenced Dhcr24 (referred to as Dhcr24‐ov and Dhcr24‐kd, respectively) were established and used for analysis of the PI3K pathway and apoptosis. The Dhcr24‐ov cells showed a 72% increased expression of Dhcr24, which resulted in increased PI3K, AKT phosphorylation and HKII by 127%, 222% and 150%, respectively, compared with that of the parental H9c2 cells (Figure 7A,B; n = 3 independent experiments, P < .01 versus control group). In contrast, the Dhcr24‐kd cells showed a 58% decreased expression of Dhcr24, which decreased the expression of PI3K, AKT phosphorylation and HKII by 21%, 41% and 60%, respectively, compared with that of the parental H9c2 cells (Figure 7A,B; n = 3 independent experiments, P < .05 or P < .01 versus control group). The translocation of Bax and the release of cytochrome c were reduced by 59% and 48%, respectively, in the Dhcr24‐ov cells and increased by 33% and 51%, respectively, in the Dhcr24‐kd cells compared with that of the parental cells (Figure 7C‐E, n = 3 independent experiments, P < 0.01 versus control group). Myocyte apoptosis induced by H2O2 treatment in the parental cells showed an apoptotic index of 11.2% (Figure 7F,G; n = 3 independent experiments, P < .05 versus control + PBS group), which reduced to 0.2% in the Dhcr24‐ov cells and increased to 33.2% in the Dhcr24‐kd cells (Figures 7F,G; n = 3 independent experiments, P < .05 versus control + H2O2 group).
Figure 7.
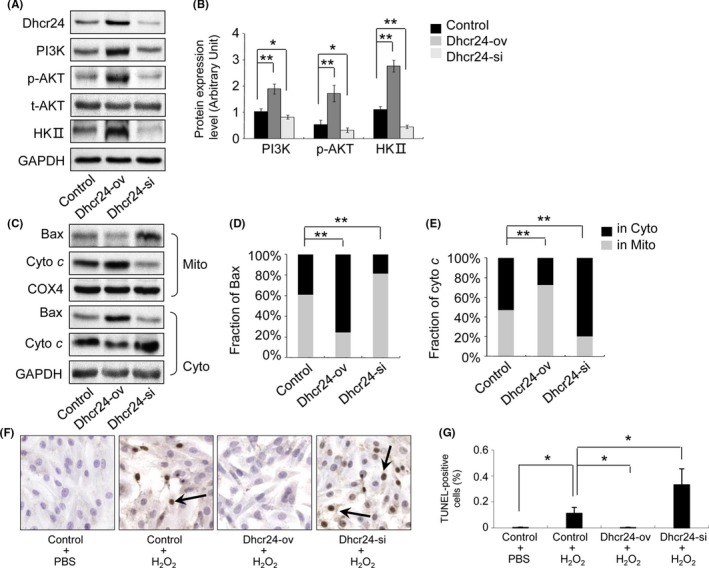
The measurement of the signalling proteins of the PI3K pathway and apoptosis in H9c2 cells. The H9c2 cells, Dhcr24 over‐expressing (Dhcr24‐ov) cells and Dhcr24 knockdown (Dhcr24‐kd) cells were sampled, and the signalling proteins of the PI3K pathway and the mitochondrial pathway of apoptosis were detected by Western blot and TUNEL assay. (A), The levels of Dhcr24, PI3K, phosphorylated AKT and HKII were measured by Western blot. (B), Quantitative analysis using GAPDH for normalization (n = 3 independent experiments, *P < .05, **P < .01). (C), The fraction of Bax and cytochrome c in the cytoplasm and the mitochondria was detected by Western blot in the heart tissues. (D), (E), Quantitative analysis of the level of Bax and cytochrome c using COX4 and GAPDH for normalization, respectively, (n = 3 independent experiments, **P < .01). (F), Photomicrographs of the H9c2 cells and the Dhcr24‐ov and Dhcr24‐kd cells used for the TUNEL assay. The arrows indicate TUNEL‐positive cells. (G), Quantitative analysis of the apoptotic cells (magnification × 400, n = 3 independent experiments, *P < .05)
3.8. AKT and HKII are required for anti‐apoptotic effects in Dhcr24 overexpressed H9c2 cells
Dhcr24 cells were treated with a PI3K inhibitor (LY294002) or an HKII inhibitor (3‐BrPA), and these inhibitors resulted in a 60% and 74% reduction of HKII expression, respectively (Figure 8A,B; n = 3 independent experiments, P < .05 or P < .01 versus PBS control). Bax translocation and cytochrome c release increased by 152% and 205% in the presence of LY294002, respectively, and by 66% and 81% in the presence of 3‐BrPA, respectively (Figure 8C‐E, n = 3 independent experiments, P < .01 versus PBS control). The apoptotic index of the Dhcr24‐ov cells significantly reduced to 0.2% compared with that of the parental cells with H2O2 treatment (Figure 7F,G; n = 3 independent experiments, P < .05 versus PBS control). However, the apoptotic index of the Dhcr24‐ov cells significantly increased by 69‐fold in the presence of LY294002 (Figure 8F,G; n = 3 independent experiments, P < .01 versus H2O2 group) and by 22‐fold in the presence of 3‐BrPA (Figure 8F,G; n = 3 independent experiments, P < .05 versus H2O2 group). These results indicated that the anti‐apoptotic effects of Dhcr24 cloud be completely inhibited by PI3K inhibition and inhibited by approximately one‐half with the HKII inhibitor.
Figure 8.
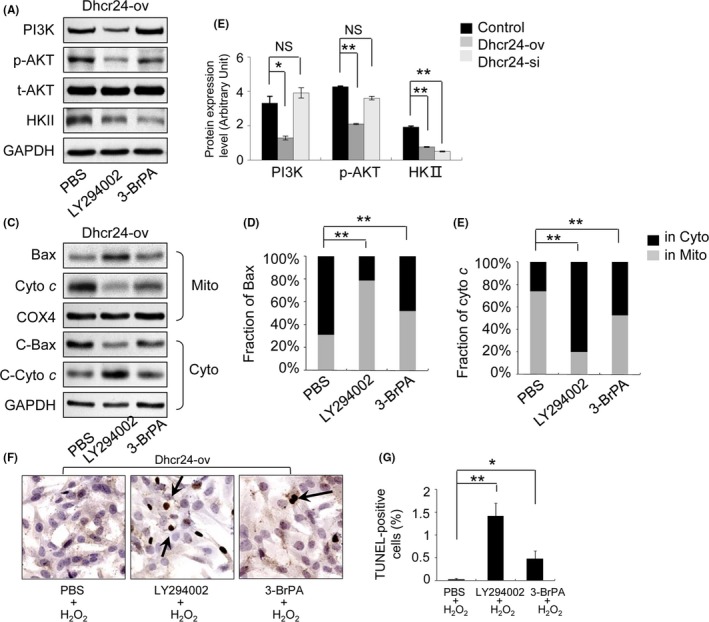
The measurement of apoptosis of the Dhcr24‐ov cells in the presence of inhibitors of PI3K and HKII. The Dhcr24‐ov cells were treated with LY294002 (PI3K inhibitor) or 3‐BrPA (HKII inhibitor), and the signalling proteins of the PI3K and the mitochondrial pathways of apoptosis were detected by Western blot and TUNEL assay. (A), The levels of PI3K, phosphorylated AKT and HKII were measured by Western blot. (B), Quantitative analysis using GAPDH for normalization. (C), The fractions of Bax and cytochrome c in the cytoplasm and the mitochondria were detected by Western blot in the heart tissues. (D), (E), Quantitative analysis using COX4 and GAPDH for normalization, respectively (n = 3 independent experiments, **P < .01). (F), Photomicrographs of Dhcr24‐ov cells treated with LY294002 (PI3K inhibitor) or 3‐BrPA (HKII inhibitor) used for the TUNEL assay. The arrows indicate TUNEL‐positive cells. (G), Quantitative analysis of apoptotic cells (magnification × 400, n = 3 independent experiments, *P < .05, **P < .01)
4. DISCUSSION
Cholesterol is an essential biological molecule in animals that is involved in signal transduction as a component of caveolae and lipid rafts and functions as a covalent modifier of the hedgehog family of morphogenes.2, 3 Dhcr24 catalyzes the last step of cholesterol biosynthesis to convert desmosterol to cholesterol, and the expression of Dhcr24 has been detected in many different organs, including the brain, adrenal thyroid gland, ovary, testis and prostate.8, 22 We found that Dhcr24 expressed in heart tissue of mouse, and its expression reduced by 42% at 6 months of age compared with 1 month of age (Figure 1A), which suggested that Dhcr24 expression down‐regulated during aging (Dhcr24 expression was also observed in human heart tissues at different ages by immunohistological staining, Figure S1). Interestingly, the expression of Dhcr24 significantly down‐regulated in the DCM heart tissues from both of the cTnTR141W (Figure 1C,D) and LMNAE82K transgenic mice (Figure S2), which resulted in the decrease of cholesterol (Figure 1F). Dhcr24 is involved in anti‐apoptotic function in the brain, and its reduced expression in the temporal cortex is involved in the pathological development of Alzheimer's disease.8, 9, 11 Our results suggested that reduced Dhcr24 expression was also associated with DCM in the mouse models. We generated a heart tissue‐specific transgenic mouse with increased Dhcr24 expression and cholesterol (Figure 2). The transgenic overexpression of Dhcr24 significantly improved cardiac geometry and dysfunction and decreased the mortality of the cTnTR141W transgenic mice (Figure 3 and Table 1). Furthermore, the transgenic overexpression of Dhcr24 ameliorated the obvious cardiac pathological phenotype in the cTnTR141W transgenic mice (Figure 4).
Cardiomyocyte cell death is a major factor that contributes to pathological heart disease. All three types of cell death (necrosis, apoptosis, and autophagy) are present in heart failure.23, 24 Based on clinical and animal studies, myocyte apoptosis plays an important role in the pathogenesis of cardiovascular diseases, such as ischemic heart disease, atherosclerosis, cardiomyopathy, and heart failure.25, 26, 27, 28, 29, 30 Both of the DCM mouse models, the cTnTR141W and the LMNAE82K transgenic mice, were generated in our laboratory and show apoptosis in the heart.31, 32
PI3K‐dependent signaling is an ubiquitous pathway involved in cell growth, proliferation, survival, migration, metabolism and several other biological responses, including cardiomyocyte growth, survival, and contractility, as well as cardiovascular inflammation.33, 34, 35 Akt is one of the primary downstream effectors in the PI3K pathway, which is phosphorylated at Thr308 and Ser473 to generate its active form and controls a variety of responses in the heart, such as inhibition of apoptosis, regulation of cell proliferation, metabolism and hypertrophy.36, 37, 38 HKII is also one of the downstream effectors in the PI3K pathway.39, 40 HKII is phosphorylated by Akt and mediates protection against Ca2+ overload in isolated heart mitochondria and against H2O2‐induced injury in neonatal rat ventricular myocytes (NRVMs), and inhibits the opening of mitochondrial permeability transition pores (MPTP) in cardiomyocytes.41 Constitutively activated Akt increases mitochondrial HK activity in rat fibroblasts and mediate cell survival. Disruption of mitochondrial HKII association impaired Akt‐mediated protection even in the absence of Bax and Bak, which are critical mediators of mitochondrial outer membrane permeabilization (MOMP).42 Akt also regulates the expression of HKII at the transcriptional level in adipose, skeletal muscle and myeloid cells.43, 44, 45 Thus, Akt and mitochondrial HKII have the ability to prevent cell death induced by mechanisms beyond those that regulate the Bcl‐2 family of proteins and MOMP.46
Our results indicated that the expression of PI3K, HKII and phosphorylated AKT reduced and followed by the release of cytochrome c, caspase‐9 and caspase‐3 activation and increased apoptosis in the cTnTR141W mouse heart. The transgenic overexpression of Dhcr24 significantly activates the PI3K pathway and inhibits the mitochondrial pathway of apoptosis and improves the integrity of the mitochondria in the DTG mouse heart (Figures 5 and 6). Furthermore, the PI3K pathway was inhibited and cytochrome c‐dependent apoptosis was activated by knockdown of the expression of Dhcr24 with siRNA. In contrast, the PI3K pathway increased and apoptosis was inhibited in the Dhcr24‐ov H9c2 cells. These results suggested that Dhcr24 regulated the PI3K signaling pathway and was involved in myocyte apoptosis in vivo and in vitro (Figure 7). Furthermore, the anti‐apoptotic effect of Dhcr24 was completely removed in the presence of a PI3K inhibitor and about half inhibited by an HKII inhibitor (Figure 8).
One of the possible mechanisms to prevent cell death is through the Akt/HKII pathway. Another is through Akt inhibition of Bax translocation from the cytosol to the mitochondria, which partly inhibits myocyte apoptosis. Our results indicated that Dhcr24 regulated the translocation of Bax from the cytosol to the mitochondria through the PI3K/Akt signaling pathway (Figures 6, 7 and 8). The Bcl‐2 family member Bax translocates from the cytosol to the mitochondria where it is oligomerized and permeabilizes into the mitochondrial outer membrane to promote apoptosis. Bax activity is counteracted by prosurvival Bcl‐2 proteins, and Akt has a direct effect on the Bcl‐2 proteins and regulates cell survival.47, 48, 49 Akt is indicated to improve myocyte survival by reducing the ratio of Bcl‐2/Bax in the cytosol during cardiac ischemia/reperfusion.50
Taken together, these results provide data supporting the idea that Dhcr24 expression reduced and is involved in the pathological development of DCM, at least in the mouse models of DCM. Compensatory expression of Dhcr24 could protect against dilated cardiomyopathy in the mouse model. Dhcr24 activated the PI3K signaling pathway and decreased myocyte apoptosis by up‐regulating the expression of HKII and reducing the translocation of Bax from the cytosol to the mitochondria.
CONFLICT OF INTEREST
None.
AUTHOR CONTRIBUTIONS
All listed authors meet the requirements for authorship. DL and LFZ conceived and designed the experiments; WD performed the experiments and wrote the main manuscript text. FFG analyzed the data of cell experiments and supervised the pathological observation. WC performed the microinjection of transgenic mice. XZ constructed the expression plasmid. SG and NL performed the genotyping and management of the transgenic mice. All authors have read and approved the manuscript.
Supporting information
ACKNOWLEDGEMENTS
The authors thank Dr James for improving the presentation of the Figures and the English.
Dong W, Guan FF, Zhang X, et al. Dhcr24 activates the PI3K/Akt/HKII pathway and protects against dilated cardiomyopathy in mice. Anim Models Exp Med. 2018;1:40–52. 10.1002/ame2.12007
Funding information
The present work was supported by the National Science and Technology Support Project (2015BAI08B01), CAMS Innovation Fund for Medical Sciences (CAMS‐I2M, 2016‐I2M‐1‐015) and Beijing Natural Science Foundation (5172027).
REFERENCES
- 1. Stevenson J, Brown AJ. How essential is cholesterol? Biochem J. 2009;420:e1–e4. [DOI] [PubMed] [Google Scholar]
- 2. Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev. 2002;54:431–467. [DOI] [PubMed] [Google Scholar]
- 3. Beachy PA, Cooper MK, Young KE, et al. Multiple roles of cholesterol in hedgehog protein biogenesis and signaling. Cold Spring Harb Symp Quant Biol. 1997;62:191–204. [PubMed] [Google Scholar]
- 4. Wechsler A, Brafman A, Shafir M, et al. Generation of viable cholesterol‐free mice. Science. 2003;302:2087. [DOI] [PubMed] [Google Scholar]
- 5. Mirza R, Hayasaka S, Kambe F, et al. Increased expression of aquaporin‐3 in the epidermis of DHCR24 knockout mice. Br J Dermatol. 2008;158:679–684. [DOI] [PubMed] [Google Scholar]
- 6. Couet J, Sargiacomo M, Lisanti MP. Interaction of a receptor tyrosine kinase, EGF‐R, with caveolins. Caveolin binding negatively regulates tyrosine and serine/threonine kinase activities. J Biol Chem. 1997;272:30429–30438. [DOI] [PubMed] [Google Scholar]
- 7. Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen‐mediated inhibition of target tissue response to Shh signaling. Science. 1998;280:1603–1607. [DOI] [PubMed] [Google Scholar]
- 8. Grubauer G, Feingold KR, Elias PM. Relationship of epidermal lipogenesis to cutaneous barrier function. J Lipid Res. 1987;28:746–752. [PubMed] [Google Scholar]
- 9. Jans R, Atanasova G, Jadot M, Poumay Y. Cholesterol depletion upregulates involucrin expression in epidermal keratinocytes through activation of p38. J Invest Dermatol. 2004;123:564–573. [DOI] [PubMed] [Google Scholar]
- 10. Crameri A, Biondi E, Kuehnle K, et al. The role of seladin‐1/DHCR24 in cholesterol biosynthesis, APP processing and Abeta generation in vivo. EMBO J. 2006;25:432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Luciani P, Deledda C, Rosati F, et al. Seladin‐1 is a fundamental mediator of the neuroprotective effects of estrogen in human neuroblast long‐term cell cultures. Endocrinology. 2008;149:4256–4266. [DOI] [PubMed] [Google Scholar]
- 12. Wu C, Miloslavskaya I, Demontis S, Maestro R, Galaktionov K. Regulation of cellular response to oncogenic and oxidative stress by Seladin‐1. Nature. 2004;432:640–645. [DOI] [PubMed] [Google Scholar]
- 13. Le Roux PJ, Coetzee JF. Tramadol today. Curr Opin Anaesthesiol. 2000;13:457–461. [DOI] [PubMed] [Google Scholar]
- 14. Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. [DOI] [PubMed] [Google Scholar]
- 15. Mogensen J, Murphy RT, Shaw T, et al. Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2004;44:2033–2040. [DOI] [PubMed] [Google Scholar]
- 16. Ho KK, Anderson KM, Kannel WB, Grossman W, Levy D. Survival after the onset of congestive heart failure in Framingham Heart Study subjects. Circulation. 1993;88:107–115. [DOI] [PubMed] [Google Scholar]
- 17. Senthil V, Chen SN, Tsybouleva N, et al. Prevention of cardiac hypertrophy by atorvastatin in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circ Res. 2005;97:285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li D, Czernuszewicz GZ, Gonzalez O, et al. Novel cardiac troponin T mutation as a cause of familial dilated cardiomyopathy. Circulation. 2001;104:2188–2193. [DOI] [PubMed] [Google Scholar]
- 19. Juan F, Wei D, Xiongzhi Q, et al. The changes of the cardiac structure and function in cTnTR141W transgenic mice. Int J Cardiol. 2008;128:83–90. [DOI] [PubMed] [Google Scholar]
- 20. Belkner J, Stender H, Kuhn H. The rabbit 15‐lipoxygenase preferentially oxygenates LDL cholesterol esters, and this reaction does not require vitamin E. J Biol Chem. 1998;273:23225–23232. [DOI] [PubMed] [Google Scholar]
- 21. Christie WW. Rapid separation and quantification of lipid classes by high performance liquid chromatography and mass (light‐scattering) detection. J Lipid Res. 1985;26:507–512. [PubMed] [Google Scholar]
- 22. Peri A, Danza G, Benvenuti S, et al. New insights on the neuroprotective role of sterols and sex steroids: the seladin‐1/DHCR24 paradigm. Front Neuroendocrinol. 2009;30:119–129. [DOI] [PubMed] [Google Scholar]
- 23. Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010;72:19–44. [DOI] [PubMed] [Google Scholar]
- 24. Konstantinidis K, Whelan RS, Kitsis RN. Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc Biol. 2012;32:1552–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Toko H, Takahashi H, Kayama Y, et al. Ca2+/calmodulin‐dependent kinase IIdelta causes heart failure by accumulation of p53 in dilated cardiomyopathy. Circulation. 2010;122:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Narula J, Pandey P, Arbustini E, et al. Apoptosis in heart failure: release of cytochrome c from mitochondria and activation of caspase‐3 in human cardiomyopathy. Proc Natl Acad Sci U S A. 1999;96:8144–8149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Frustaci A, Kajstura J, Chimenti C, et al. Myocardial cell death in human diabetes. Circ Res. 2000;87:1123–1132. [DOI] [PubMed] [Google Scholar]
- 28. Feuerstein GZ, Young PR. Apoptosis in cardiac diseases: stress‐ and mitogen‐activated signaling pathways. Cardiovasc Res. 2000;45:560–569. [DOI] [PubMed] [Google Scholar]
- 29. Tucka J, Bennett M, Littlewood T. Cell death and survival signalling in the cardiovascular system. Front Biosci (Landmark Ed). 2012;17:248–261. [DOI] [PubMed] [Google Scholar]
- 30. Latif N, Khan MA, Birks E, et al. Upregulation of the Bcl‐2 family of proteins in end stage heart failure. J Am Coll Cardiol. 2000;35:1769–1777. [DOI] [PubMed] [Google Scholar]
- 31. Lu D, Lian H, Zhang X, et al. LMNA E82K mutation activates FAS and mitochondrial pathways of apoptosis in heart tissue specific transgenic mice. PLoS ONE. 2010;5:e15167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lu D, Ma Y, Zhang W, et al. Knockdown of cytochrome P450 2E1 inhibits oxidative stress and apoptosis in the cTnT(R141W) dilated cardiomyopathy transgenic mice. Hypertension. 2012;60:81–89. [DOI] [PubMed] [Google Scholar]
- 33. Foukas LC, Okkenhaug K. Gene‐targeting reveals physiological roles and complex regulation of the phosphoinositide 3‐kinases. Arch Biochem Biophys. 2003;414:13–18. [DOI] [PubMed] [Google Scholar]
- 34. Ghigo A, Morello F, Perino A, Damilano F, Hirsch E. Specific PI3K isoform modulation in heart failure: lessons from transgenic mice. Curr Heart Fail Rep. 2011;8:168–175. [DOI] [PubMed] [Google Scholar]
- 35. Aoyagi T, Matsui T. Phosphoinositide‐3 kinase signaling in cardiac hypertrophy and heart failure. Curr Pharm Des. 2011;17:1818–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shiraishi I, Melendez J, Ahn Y, et al. Nuclear targeting of Akt enhances kinase activity and survival of cardiomyocytes. Circ Res. 2004;94:884–891. [DOI] [PubMed] [Google Scholar]
- 37. Rubio M, Avitabile D, Fischer K, et al. Cardioprotective stimuli mediate phosphoinositide 3‐kinase and phosphoinositide dependent kinase 1 nuclear accumulation in cardiomyocytes. J Mol Cell Cardiol. 2009;47:96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gude N, Muraski J, Rubio M, et al. Akt promotes increased cardiomyocyte cycling and expansion of the cardiac progenitor cell population. Circ Res. 2006;99:381–388. [DOI] [PubMed] [Google Scholar]
- 39. Pastorino JG, Hoek JB. Hexokinase II: the integration of energy metabolism and control of apoptosis. Curr Med Chem. 2003;10:1535–1551. [DOI] [PubMed] [Google Scholar]
- 40. Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, Thompson CB. Akt‐directed glucose metabolism can prevent Bax conformation change and promote growth factor‐independent survival. Mol Cell Biol. 2003;23:7315–7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase‐II. Cell Death Differ. 2008;15:521–529. [DOI] [PubMed] [Google Scholar]
- 42. Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Culbert AA, Tavare JM. Multiple signalling pathways mediate insulin‐stimulated gene expression in 3T3‐L1 adipocytes. Biochim Biophys Acta. 2002;1578:43–50. [DOI] [PubMed] [Google Scholar]
- 44. Lee AW, States DJ. Colony‐stimulating factor‐1 requires PI3‐kinase‐mediated metabolism for proliferation and survival in myeloid cells. Cell Death Differ. 2006;13:1900–1914. [DOI] [PubMed] [Google Scholar]
- 45. Printz RL, Koch S, Potter LR, et al. Hexokinase II mRNA and gene structure, regulation by insulin, and evolution. J Biol Chem. 1993;268:5209–5219. [PubMed] [Google Scholar]
- 46. Miyamoto S, Murphy AN, Brown JH. Akt mediated mitochondrial protection in the heart: metabolic and survival pathways to the rescue. J Bioenerg Biomembr. 2009;41:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell‐intrinsic death machinery. Cell. 1997;91:231–241. [DOI] [PubMed] [Google Scholar]
- 48. Del Peso L, Gonzalez‐Garcia M, Page C, Herrera R, Nunez G. Interleukin‐3‐induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. [DOI] [PubMed] [Google Scholar]
- 49. Downward J. PI 3‐kinase, Akt and cell survival. Semin Cell Dev Biol. 2004;15:177–182. [DOI] [PubMed] [Google Scholar]
- 50. Huang C, Gu H, Zhang W, Herrmann JL, Wang M. Testosterone‐down‐regulated Akt pathway during cardiac ischemia/reperfusion: a mechanism involving BAD, Bcl‐2 and FOXO3a. J Surg Res. 2010;164:e1–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials