

Abstract
In light of missing systematic reviews in the literature, the objective of this paper is to present the contemporary knowledge on the molecular biology of vestibular schwannomas (VS), based on a systematic literature search. In addition, current and prospected medical therapy based on molecular biology is addressed. A systematic literature search was conducted using the Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines. The systematic search was performed in the Pubmed and Embase databases. The following were the words searched: acoustic neuroma/vestibular schwannoma, molecular biology, gene, and microRNA. Specific inclusion and exclusion criteria were determined prior to search. The systematic search rendered 486 articles, ultimately yielding 69 included articles, whereas 35 were from relevant references. The occurrence of at least one mutation in the merlin gene was reported to range between 54% and 76%, whereas the loss of heterozygosity (LOH) corresponding to chromosome 22 occurs in 25% to 83% of sporadic VS. Global gene expression studies indicate that a number of genes other than merlin are at play. No high-level methylation of the merlin gene has been found. Several miRNAs are deregulated in tumor tissue, among others let-7d, miR-221, and miR-21. The acquired knowledge on molecular biology has led to several clinical implementations. Lack of the tumor suppressor merlin plays a principal role in the development of VS. Existing knowledge on the molecular biology has led to the first attempts of targeted medical treatment to prevent tumor growth. Future research is likely to introduce potential imaging markers with prognostic value and new targets for medical therapy.
Keywords: Acoustic neuroma, molecular biology, gene expression, microRNAs, review
INTRODUCTION
Vestibular schwannomas (VS) arise from the Schwann cells, sheeting the vestibular branch of the eighth (VIII) cranial nerve. Although these tumors are histologically benign, they may cause hearing loss, tinnitus, and facial palsy, and if growing rapidly to a large size, even brainstem compression and death. VS can occur unilaterally or when associated with neurofibromatosis type 2 (NF2), bilaterally. The mutual molecular hallmark of both NF2-associated VS and sporadic VS is biallelic inactivation of the merlin gene, also known as the NF2-gene, which is a tumor suppressor located at 22q12 [1]. Several genetic and epigenetic aberrations have been shown in sporadic VS, for example, various mutations in the merlin gene, loss of heterozygosity (LOH) on several chromosomes, deregulation of other genes, abnormal microRNA expression pattern, and CpG island methylation. This review summarizes the contemporary molecular biology of sporadic VS via systematic literature searches, as well as addressing current and prospected medical therapy based on molecular biology.
METHOD
Overview of Systematic Literature Review
A systematic literature review was conducted to identify published studies regarding merlin gene mutations, deregulated gene expression, LOH, DNA methylation, and deregulated microRNAs in sporadic VS.
Systematic Search Strategy and Terms
The objective of our search strategy was to identify relevant studies concerning the molecular biology of sporadic VS. First, Pubmed, Embase, and the Cochrane Center were searched for previously published reviews based on a systematic search of the literature. None were found. Second, the same sources were searched systematically for publications on the molecular biology of sporadic/unilateral VS, using both simple search strings as well as Mesh terms. No limitations besides the inclusion and exclusion criteria were employed. The search was conducted on October 1, 2017.
The search was performed in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines [2].
Pubmed
-
“Neuroma, Acoustic” AND “Genes”
○ 109 hits—36 articles of interest
-
MESH ((“Neuroma, Acoustic”[Mesh]) AND “Genes”[Mesh])
○ 311 hits—48 articles of interest
-
MESH ((“Neuroma, Acoustic”[Mesh])) AND “MicroRNAs”)
○ 6 hits—5 articles of interest
-
MESH (“Neuroma, Acoustic”[Mesh]) AND “Molecular Biology”[Mesh]
○ 13 hits—1 articles of interest
Embase
-
Acoustic Neurinoma/ AND Gene/
○ 35 hits—4 articles of interest
-
Acoustic Neurinoma/ AND microRNA/
○ 5 hits—2 articles of interest
-
Acoustic Neurinoma/ AND Molecular Biology/
○ 7 hits—0 articles of interest
Inclusion and Exclusion Criteria
Inclusion criteria were as follows: (1) analysis performed on unilateral/sporadic VS; (2) analysis on human tissue; (3) published in the English language.
Exclusion criteria were as follows: (1) not article; (2) not original research; (3) case reports.
Implementation of the Search Strategy and Study Selection Process
An initial review of the search results was conducted. All the articles were initially screened for relevance based on their titles and abstracts. Any report not excluded by this process was included for full-text analysis. The reports were then subjected to full-text analysis, using the above inclusion and exclusion criteria.
Data Extraction Process
Data were extracted by thorough examination of the included articles, on information regarding the methods used, tissue analyzed, and results. Articles containing both sporadic VS and NF2 associated VS were evaluated. If the results were divided and a clear distinction between the two groups was possible, the article was included.
Five categories were created to describe the molecular biology of sporadic VS: the merlin gene mutations; LOH; deregulated genes; DNA methylation; and microRNA.
Risk of Bias
The search and data extraction were intended to be as unbiased as possible. No attention was given to the author, source, country, or other distinctive criterion in our initial searches. The inclusion and exclusion criteria were all defined prior to the search. We received no outside funding and had no competing interests regarding the review.
Quality Assessment
All full-text articles reviewed were also informally evaluated on the quality of materials, methods, and results. No studies were excluded on the grounds of poor-quality methods or due to non-substantiated results.
RESULTS and DISCUSSION
After a systematic literature search and inclusion/exclusion of papers based on the above criteria, 33 articles were included in the review. From these 33 articles, another 35 articles were included from relevant references. A PRISMA diagram displaying the search is shown in Figure 1. Based on these 68 articles, a synopsis on the contemporary knowledge on the various aspects of the molecular biology of VS is given below. In addition, current and prospected medical therapy based on molecular biology is addressed. A meta-analysis was not possible due to the heterogeneity of methodology in the study reports.
Figure 1.
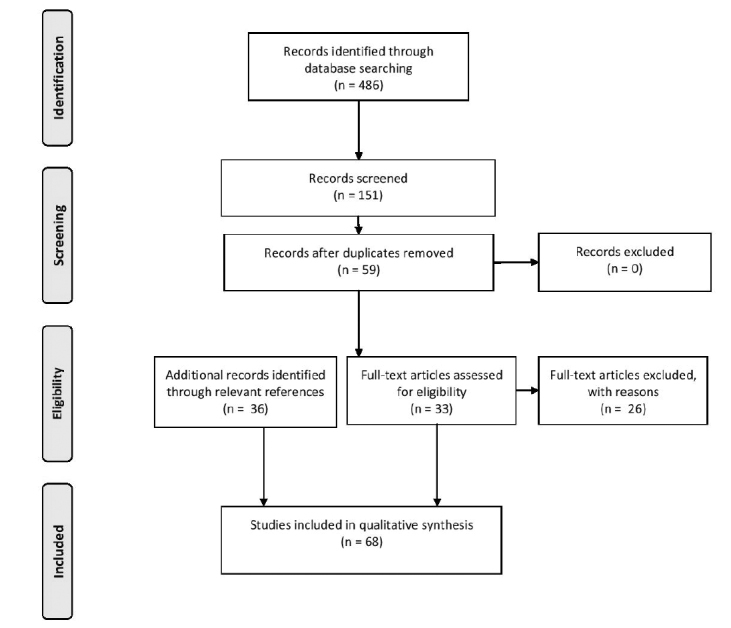
A PRISMA flowchart demonstrating the search methodology
The Merlin Gene
The protein merlin is a tumor suppressor encoded by a gene located at the chromosome 22q12.2. Silencing of the merlin gene is the mutual molecular characteristic of unilateral, sporadic vestibular schwannomas (sVS) and the bilateral VS associated with NF2. In 1993, two groups isolated the gene and its product merlin, also known as Schwannomin, by tumor deletion mapping and genetic linkage analysis[3,4].
Merlin is a member of the band 4.1-superfamily of proteins and has some sequence homology with the Ezrin, radixin, and moesin (ERM) family. Because of this sequence homology, it was first assumed that merlin was located and functioned in the cytoskeleton near the plasma membrane, equivalent to the ERM proteins. However, merlin does not contain an actin- (and thus cytoskeleton-) binding motif.
Merlin’s tumor suppressor functionality occurs when the protein is in its closed state. It is involved in a range of signaling pathways and has, as implicated, the ability to change conformation, from an open inactive state to a closed active state. The change of conformation is induced by integrin- and cadherin-mediated adhesions, as demonstrated in Figure 2. An integrin-mediated adhesion activates P21-activated kinase (PAK), which stimulates the conformation change of merlin to an open state, by phosphorylation of merlin at S518, which initiates inactivation and degradation. Conversely, cadherin-mediated adhesion inhibits PAK and thereby phosphorylation of merlin, which leads to an accumulation of the closed, active state in the nucleus [5–7]. When in closed conformation, merlin binds to DCAF1 in the cell nucleus, leading to reduction of cell proliferation through the inhibition of E3 ubiquitin ligase CRL4. The inhibited complex CRL4DCAF1 plays a role in DNA replication, where it favors the up-regulation of genes related to apoptosis and the cell-cycle stop[8]. Hence, when no functional merlin is present, the inhibition of E3 ubiquitin ligase CRL4 does not occur, and the cell continues its cell-cycle, as seen in VS. Merlin also increases the stability of p53 by inhibiting the MDM2-mediated degradation of the p53 protein [9]. P14ARF has been found to bind to the MDM2/p53 complex and inhibit the MDM2-degradation of p53. P14ARF has been found to be almost universally missing in sVS, and thus lead to p53 inactivation. Due to this inactivation of p53, a possible loss of expression of p21in sVS has been shown [10]. In addition, merlin exerts inhibitory effects on multiple mitogenic signaling pathways. The loss of functional merlin on the other hand also activates many signaling pathways. The contribution of these pathways to tumor pathogenesis and maintenance is, however, not fully understood [8, 11].
Figure 2.
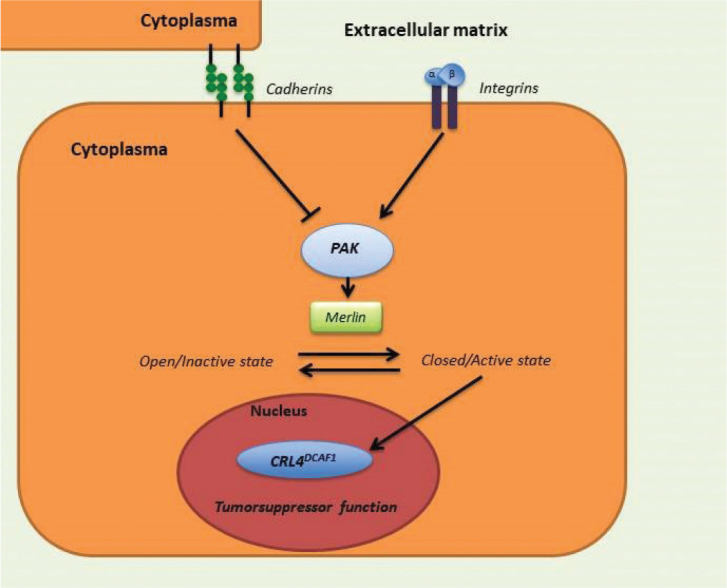
The signal pathway of merlin
Merlin Gene Mutations
In NF2-associated VS, one of the germline merlin alleles is inactivated, and the development of NF2-associated VS therefore only calls for an additional mutation, allelic loss, or silencing of the other allele [12]. In sVS, however, somatic bi-allelic merlin gene inactivation seems to be necessary for the formation of the tumor [13].
Mutations of the merlin gene consists of insertions, deletions, and single-base substitutions resulting in frameshifts and nonsense mutations [14, 15]. The occurrence of at least one mutation in the merlin gene coding sequence has been reported to range between 54% and 76% [13–23]. The consequence of a mutation is mainly the synthesis of truncated proteins (75%–93%) [14, 18]. In the studies by Torres-Martin et al. [16] and Bian et al. [17], the majority of the mutations occurred in the first half of the 17 exons in the merlin gene. Welling et al. [18] found another mutational distribution, as all mutations occurred in the exon 4–6 in the NF2-associated tumors, and nine of ten mutations occurred in exon 7–11 in sVS, which is supported by the findings of Jacoby et al. [22]. Chen et al. [24] found a higher percentage of the NF2 mutations in young patients (age under 20) with 66.7% of 12 patients compared to 34.5% of 145 adult patients. All mutations in young patients were truncated mutations. This may indicate that NF2 mutations may play a role in the formation of early sVS formation; however, the data sample was rather small, and the conclusion is that further research is needed.
Reported differences may be due to the selection bias and variable methodology. However, as mutation(s) are not occurring in almost all cases, other factors need to be at play for the development and maintenance of a tumor.
Loss of Heterozygosity
LOH is the loss of function of one allele, after the other allele has already been inactivated. Several mechanisms may account for LOH, for example, deletion resulting in a loss of a chromosome segment, mitotic recombination, translocation, and gene conversion.
Early studies showed that only 17% to 22% of sVS had a LOH on chromosome 22, whereas later studies using more sensitive methods report LOH in 25% to 83% [13, 15, 17, 23, 25–35]. The occurring differences may again be due to selected patient materials or differences in unbalanced chromosomal abnormalities versus balanced chromosomal abnormalities. Regardless of variable occurrence, the studies do however confirm that LOH is a frequent feature of sVS, and it is therefore likely to be involved in both tumor development and maintenance.
LOH at other chromosomes may also play a role in tumor pathogenesis. In six of 14 sVS (43%), Dayalan et al. [36] found LOH on chromosome 17p, which among other genes codes for tumor protein 53 (TP53). However, an overexpression of TP53 mRNA and TP53 protein was seen in all tumors. This phenomenon has been demonstrated in earlier studies of other neoplasms, for example, breast cancer [37]. However, Monoh et al. [38] found neither mutations nor LOH on chromosome 17p. They concluded that TP53 does not play an important role in the tumorigenesis or maintenance, thus contradicting Dayalan et al. [36].
Deregulation of Other Genes
Four larger studies have explored global gene deregulation in VS, whereas other studies have focused on specific, selected genes [16, 19, 39–50]. Different techniques have been used and only a few common findings exist between these studies.
Welling et al. [39] pioneered, Caye-Thomasen et al. [40] followed, and later Aarhus et al. [14] and Torres et al. [17] joined in analyzing the global gene expression in VS. They all used cDNA microarrays, although platforms, number of tumor samples, as well as number and type of control tissue differed. Welling et al. found the same 42 genes up-regulated in five VS, and eight genes significantly down-regulated in all the seven analyzed VS, compared to control nerve tissue. Five of the up-regulated and one of the down-regulated genes recur in later publications. The findings share similarities with the expression data published by Cayé-Thomasen et al., who included 16 sVS and compared them to three normal vestibular nerves. Seventy-five genes were up-regulated, and three were down-regulated. Eight of the up-regulated genes recur in other publications, whereas none of the down-regulated genes have been found by other authors. The gene Osteonectin/SPARC was found to be up-regulated by Welling et al. [39] and Aarhus et al. [19], but it was not deregulated in the study of Cayé-Thomasen et al. However, the scavenger receptor stabilin 1, which mediates targets for degradation by Osteonectin/SPARC, was up-regulated.
Aarhus et al. [19] conducted the first of the bigger studies to validate their findings with qRT-PCR. Five of the up-regulated and four of the down-regulated genes had also been identified in the other studies. Ingenuity pathway analysis was used to show that ERK was the primary network for the deregulated genes. They also found that the analyzed VS were divided into two groups regarding the mRNA expression. These findings were replicated by Torres-Martin et al. [16] who analyzed 28 sVS and compared them to nine various types of control nerve tissue. Torres-Martin et al. also found that the tyrosine-protein kinase Met (cMET) pathway, possibly enhanced by an upstream signaling of SPP1 and CAV1 among others, appears to play a principal role in the formation and preservation of VS. A possible hormonal influence was also discovered, as the androgen receptor was deregulated.
The cMET pathways and the possible cross-talk with VEGF-A was investigated by Dilawi et al. [51], who found that both cMET and VEGF-A are significantly overexpressed in sVS compared to non-neoplastic Schwann cells. A knock-down of either VEGF-A or cMET reduced the other, and inhibition of the cMET pathway reduced the proliferation in sVS cells. The cMET pathway must therefore be considered a potential target for therapeutic therapy.
Sass et al. [52] pioneered by doing a global gene expression analysis in fast-growing sVS compared to slow-growing sVS using a DNA microarray. Several notable genes were up-regulated in the fast-growing tumors, for example, the erbb2 interacting protein (Erbin), platelet-derived growth factor C, phosphatidylinositide 3-kinase, actinin alpha 1, and several toll-like receptors. Notable down-regulated genes included the brain specific protein and neural cell adhesion molecule 1. The ingenuity pathway analysis (IPA) was performed, and a number of canonical pathways were found to be related to viral infections. This supports the notion that there could be a viral etiology behind the pathogenesis and especially tumor growth of sVS. Functional molecular networks derived from the IPA demonstrated the importance of PI3K for the growth as it was in four of the five top functional molecular networks. The top network, including 23 of the total 109 deregulated genes in the study, found NF–κB and P38 MAPK to be the hubs of the network, and thus involved in several interactions with the deregulated genes. NF–κB inhibits apoptosis and is up-regulated in multiple cancers, whereas P38 MAPK may contribute to VS development and progression through an effect on Schwan cell differentiation. Dilawi et al.[53] previously completed the first functional molecular network analysis of sVS in 2015 and found that the same NF–κB was the center hub of their functional molecular network. This indicates that NF–κB is involved not only in the formation, but also the growth rate of sVS. Dilawi et al. also found that cyclin D1, Bcl2 and gene TNF encoding TNFα was overexpressed in sVS compared to control nerve in the same study.
In minor studies regarding the gene expression, the AKT1 gene, Erbb2, Neuregulin 1, and the EGF-receptor were found up-regulated and the AKT pathway to be active [44, 45, 54]. The immunoreactivity for EGFR was also found, however, deemed as not important due to low expression levels as well as a small tumor sample [46, 47]. Cayé-Thomasen et al. [48] found a positive correlation between VEGF, VEGFR-1, and tumor growth rate, while Moller et al. [55] found a positive correlation between MMP-9 and the tumor growth rate. Seol et al. [43] found a down-regulation of p27 in 67% of aggressive VS compared to 20% in non-aggressive tumors. O’Reilly et al. [41] found an overexpression of FGFR1 in growing sVS. Hence, VEGF, VEGFR-1, p27, and FGFR1 may play a role in the growth of sVS, and may be potential prognostic markers or therapeutic targets.
The Cyclin D1 expression, HIF-1alfa, Epo, EpoR, and bcl2 expression have also been found in tumor samples, as well as the silencing/hypermethylation of RASSF1A, which is associated with negative cyclin D1 expression. Hung et al.[46] found an up-regulation of the L1 cell adhesion molecule, also found in the latter three of the four larger gene expression studies [16, 19, 40]. Chen et al. [14] investigated the difference of Cyclin D1, merlin, phosphorylated merlin, and p53 expression between young patients under 18 and adults, with no significant difference.
De Vries et al. [49] analyzed 48 sVS for the 13 most frequent mutations in BRAF, EGFR, PIK3CA, and KRAS. No mutations were found, suggesting that these genes do not play a role in tumor development.
Sirén et al. [42] found an overexpression of urokinase plasminogen activator and tissue plasminogen activator (tPA) in 13 sVS samples. They also found up-regulation of the PA-PAI-I complex leading to reduced tPA activity compared to NF2-associated VS. This indicates that sVS may be more prone to hemorrhaging and thrombosis and are less likely to be invasive.
Overall, the studies on deregulated genes in sVS are relatively few, and the different methods/arrays used make comparisons difficult and overall conclusions uncertain. The MET-pathway as well as NF–κB could represent potential targets, as they appear to be principal in the development and preservation of sVS.
DNA Methylation
In sVS, the functional merlin is not present, although as many as 40% of the tumors contain an intact wild-type merlin gene. As a number of human cancers have been associated with abnormal methylation of CpG-islands located in the gene promoter regions [56], several studies have explored aberrant methylation of the merlin gene promoter regions in schwannomas, as a potential explanation for the lack of functional merlin.
Kino et al. [57] showed that a 70-base pair region of the merlin gene contained five CpG-sites playing a role in the transcriptional silencing of the gene. Fourteen of 23 sporadic schwannomas showed CpG-methylation, with an absence of a transcriptional product in eight of the 14 tumors. Gonzalez-Gomez et al. [58] found merlin gene methylation in 19% of 27 sVS. In these studies, there were no correlations with merlin gene mutations or deletions. Chen et al. [24] investigated the phosphorylation of merlin in young patients compared to adult patients and found that patients with phosphorylated merlin (3/12 patients), exhibited larger tumor size than patients with deficient merlin (9/12 patients). Kullar et al. [59] used a more sensitive pyro-sequencing technique and found no high-level methylation in any of 40 sequenced sVS. Again, a possible explanation for conflicting results may be different patient cohorts and differences in sensitivity between the methods used. Kullar et al. used the most sensitive method and did the most comprehensive study including the copy number and mutational status of each tumor. Consequently, hypermethylation of the CpG-islands of the merlin gene is likely to play little or no role in the silencing of the merlin gene.
Methylation of other genes has also been explored. Both Gonzalez-Gomez et al. [58] and Lassaletta et al. [60] examined the methylation status of a number of genes important for tumor development, including the angiogenesis inhibitor THBS1, the DNA repair protein MGMT, the extracellular matrix binding protein TIMP-3, the caspase and thus apoptosis initiator CASP8, and the apoptosis inducer and cell growth inhibitor TP73. Lassaletta et al. [61] also examined methylation of the gene RASSF1A, a tumor suppressor candidate acting downstream Ras, and found that the methylation status was inversely related to tumor growth. However, methylation status of this particular gene may be related to age, which was also found in these study [62, 63].
In conclusion, methylation of the merlin gene is likely to play little or no role in the development of VS, whereas determination of a potential role of methylation of other genes warrants additional studies.
MicroRNA
Two studies have been done on miRNAs in sVS [64, 65]. Cioffi et al. [64] used qRT-PCR and RT-PCR to analyze miR-21 overexpression in eight sporadic VS compared to nine normal vestibular nerves, and five normal greater auricular nerves. They found consistent overexpression of miR-21 and a correlating decrease in PTEN, a known target of miR-21. They also found that anti-miR-21 was related to decreased tumor growth.
Torres-Marten et al. [65] found miR-7 to be up-regulated in their study and has been shown to be deregulated in several other tumors [66, 67]. Torres-Martin et al. [64] analyzed 15 sporadic VS and one NF2-asssociated schwannoma, comparing with three controls. They found 174 microRNAs to be deregulated, including confirmation of the up-regulation of miR-221, miR-21, miR-29, miR-30a, and miR-138 and the down-regulation of miR-7, miR-638, miR-143, and miR-498. However, let-7d, miR-451, and miR-34a were not deregulated to the extent found in the studies above. They also found up-regulation of hsa-mir-363, which inhibits merlin synthesis post-transcriptional, and a global up-regulation of a cluster of miRNAs in the chromosomal region of 14q32 that might contribute to the development and maintenance of VS. In addition, a difference was found between the miRNA expression pattern in tumors with specific molecular characteristics, such as mutations in the merlin gene and/or the LOH on chromosome 22q, as compared to tumors without specific molecular characteristics.
Clinical Implications
Current treatment options are microsurgical excision, radiotherapy, and watchful waiting. However, due to the growing knowledge on the molecular biology of VS, an increasing number of potential targets for medical therapy are being identified. The PDGF and VEGF are known angiogenetic factors, and several studies have correlated tumor VEGF to either tumor growth or volume, which subsequently spawned the first medical treatment of VS, namely the anti-VEGF antibody Bevacizumab [48, 68–70]. The most recent research in this field have demonstrated that vascular biomarkers from dynamic contrast-enhanced MRI can predict the tumor response to treatment with the VEGF antibody [71]. The PDGF was first considered to be a target for therapy by Altuna et al., and later Ammoun et al. corroborated the potential by the use of a potent PDGF receptor inhibitor [72, 73].
Other therapies have targeted the EGFR/ErbB2, PI3K/AKT, HER-1/EGFR, PAK, and mTORC1, and different degrees of VS shrinkage and hearing improvement have been demonstrated, with some of them still in clinical trials [74–79]. These treatments are tested on both sporadic, as well as NF2-associated VS, with positive results. Dilawi et al. [53] used the NF–κB inhibiter Curcumin on cultures of sVS cells, resulting in a dose-dependent decrease in proliferation.
CONCLUSION
This is the first systematic review regarding the molecular biology of sVS, and the first to include studies regarding miRNA. A lack of the tumor suppressor merlin plays a principal role in the development of VS. As the merlin gene mutations are not found in all tumors, deregulation of other genes and, post-transcriptional silencing by miRNA and/or LOH, are likely factors to play a role in tumor pathogenesis and growth, whereas a role of DNA methylation appears to be unlikely. Existing knowledge on the molecular biology has led to the first attempts of targeted medical treatment to prevent tumor growth. Additional attempts are imminent, and future expansion of our knowledge on the molecular biology of these enigmatic tumors is likely to introduce potential imaging markers with prognostic value and new potential targets for medical therapy.
Footnotes
Peer-review: Externally peer-reviewed.
Author Contributions: Concept – H.S., P.Y.T.; Design - H.S., P.Y.T.; Supervision - H.S., P.Y.T.; Resource - H.S., P.Y.T.; Materials - H.S., P.Y.T.; Data Collection and/or Processing - H.S., P.Y.T.; Analysis and/or Interpretation - H.S., P.Y.T.; Literature Search - H.S., P.Y.T.; Writing - H.S., P.Y.T.; Critical Reviews - H.S., P.Y.T.
Conflict of Interest: The authors have no conflict of interest to declare.
Financial Disclosure: The authors declared that this study has received no financial support.
REFERENCES
- 1.Stemmer-Rachamimov AO, Xu L, Gonzalez-Agosti C, Burwick JA, Pinney D, Beauchamp R, et al. Universal absence of merlin, but not other ERM family members, in schwannomas. Am J Pathol. 1997;151:1649–54. [PMC free article] [PubMed] [Google Scholar]
- 2.Moher D, Liberati A, Tetzlaff J, Altman DG PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Phys Ther. 2009;89:873–80. doi: 10.1016/j.jclinepi.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 3.Trofatter JA, MacCollin MM, Rutter JL, Murrell JR, Duyao MP, Parry DM, et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;75:826. doi: 10.1016/0092-8674(93)90406-G. [DOI] [PubMed] [Google Scholar]
- 4.Rouleau GA, Merel P, Lutchman M, et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993;363:515–21. doi: 10.1038/363515a0. [DOI] [PubMed] [Google Scholar]
- 5.Okada T, Lopez-Lago M, Giancotti FG. Merlin/NF-2 mediates contact inhibition of growth by suppressing recruitment of Rac to the plasma membrane. J Cell Biol. 2005;171:361–71. doi: 10.1083/jcb.200503165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kissil JL, Johnson KC, Eckman MS, Jacks T. Merlin phosphorylation by p21-activated kinase 2 and effects of phosphorylation on merlin localization. J Biol Chem. 2002;277:10394–9. doi: 10.1074/jbc.M200083200. [DOI] [PubMed] [Google Scholar]
- 7.Xiao G-H, Beeser A, Chernoff J, Testa JR. p21-activated kinase links Rac/Cdc42 signaling to merlin. J Biol Chem. 2002;277:883–6. doi: 10.1074/jbc.C100553200. [DOI] [PubMed] [Google Scholar]
- 8.Li W, You L, Cooper J, Schiavon G, Pepe-Caprio A, Zhou L, et al. Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell. 2010;140:477–90. doi: 10.1016/j.cell.2010.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim H, Kwak NJ, Lee JY, Choi BH, Lim Y, Ko YJ, et al. Merlin neutralizes the inhibitory effect of Mdm2 on p53. J Biol Chem. 2004;279:7812–8. doi: 10.1074/jbc.M305526200. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, yan Wang Z, Wu H. P14ARF deficiency and its correlation with overexpression of p53/MDM2 in sporadic vestibular schwannomas. Eur Arch Otorhinolaryngol. 2015;272:2227–34. doi: 10.1007/s00405-014-3135-y. [DOI] [PubMed] [Google Scholar]
- 11.Okada T, You L, Giancotti FG. Shedding light on Merlin’s wizardry. Trends Cell Biol. 2007;17:222–9. doi: 10.1016/j.tcb.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 12.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–3. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bijlsma EK, Merel P, Bosch DA, Westerveld A, Delattre O, Thomas G, et al. Analysis of mutations in the SCH gene in schwannomas. Genes Chromosomes Cancer. 1994;11:7–14. doi: 10.1002/gcc.2870110103. [DOI] [PubMed] [Google Scholar]
- 14.Mohyuddin A, Neary WJ, Wallace A, Wu CL, Purcell S, Reid H, et al. Molecular genetic analysis of the NF2 gene in young patients with unilateral vestibular schwannomas. J Med Genet. 2002;39:315–22. doi: 10.1136/jmg.39.5.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hadfield KD, Smith MJ, Urquhart JE, Wallace AJ, Bowers NL, King AT, et al. Rates of loss of heterozygosity and mitotic recombination in NF2 schwannomas, sporadic vestibular schwannomas and schwannomatosis schwannomas. Oncogene. 2010;29:6216–21. doi: 10.1038/onc.2010.363. [DOI] [PubMed] [Google Scholar]
- 16.Torres-Martin M, Lassaletta L, San-Roman-Montero J, et al. Microarray analysis of gene expression in vestibular schwannomas reveals SPP1/MET signaling pathway and androgen receptor deregulation. Int J Oncol. 2013;42:848–62. doi: 10.3892/ijo.2013.1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bian LG, Tirakotai W, Sun QF, Zhao WG, Shen JK, Luo QZ. Molecular genetics alterations and tumor behavior of sporadic vestibular schwannoma from the People’s Republic of China. J Neurooncol. 2005;73:253–60. doi: 10.1007/s11060-004-5176-3. [DOI] [PubMed] [Google Scholar]
- 18.Welling DB, Guida M, Goll F, Pearl DK, Glasscock ME, Pappas DG, et al. Mutational spectrum in the neurofibromatosis type 2 gene in sporadic and familial schwannomas. Hum Genet. 1996;98:189–93. doi: 10.1007/s004390050188. [DOI] [PubMed] [Google Scholar]
- 19.Aarhus M, Bruland O, Sætran HA, Mork SJ, Lund-Johansen M, Knappskog PM. Global gene expression profiling and tissue microarray reveal novel candidate genes and down-regulation of the tumor suppressor gene CAV1 in sporadic vestibular schwannomas. Neurosurgery. 2010;67:998–1019. doi: 10.1227/NEU.0b013e3181ec7b71. discussion 1019. [DOI] [PubMed] [Google Scholar]
- 20.Den Bakker MA, Van Tilborg AAG, Kros JM, Zwarthoff EC, Nf T. Truncated NF2 proteins are not detected in meningiomas and schwannomas. Neuropathology. 2001;21:168–73. doi: 10.1046/j.1440-1789.2001.00394.x. [DOI] [PubMed] [Google Scholar]
- 21.Bruder CE, Ichimura K, Tingby O, Hirakawa K, Komatsuzaki A, Tamura A, et al. A group of schwannomas with interstitial deletions on 22q located outside the NF2 locus shows no detectable mutations in the NF2 gene. Hum Genet. 1999;104:418–24. doi: 10.1007/s004390050978. [DOI] [PubMed] [Google Scholar]
- 22.Jacoby LB1, MacCollin M, Louis DN, Mohney T, Rubio MP, Pulaski K, et al. Exon scanning for mutation of the NF2 gene in schwannomas. Hum Mol Genet. 1994;3:413–9. doi: 10.1093/hmg/3.3.413. [DOI] [PubMed] [Google Scholar]
- 23.Ikeda T, Hashimoto S, Fukushige S, Ohmori H, Horii A. Comparative genomic hybridization and mutation analyses of sporadic schwannomas. J Neurooncol. 2005;72:225–30. doi: 10.1007/s11060-004-2693-z. [DOI] [PubMed] [Google Scholar]
- 24.Chen H, Zhang X, Zhang Z, Yang T, Wang Z, Wu H. The role of NF2 gene mutations and pathogenesis-related proteins in sporadic vestibular schwannomas in young individuals. Mol Cell Biochem. 2014;392:145–52. doi: 10.1007/s11010-014-2011-9. [DOI] [PubMed] [Google Scholar]
- 25.Couturier J, Delattre O, Kujas M, Philippon J, Peter M, Rouleau G, et al. Assessment of chromosome 22 anomalies in neurinomas by combined karyotype and RFLP analyses. Cancer Genet Cytogenet. 1990;45:55–62. doi: 10.1016/0165-4608(90)90066-J. [DOI] [PubMed] [Google Scholar]
- 26.Twist EC, Ruttledge MH, Rousseau M, Sanson M, Papi L, Merel P, et al. The neurofibromatosis type 2 gene is inactivated in schwannomas. Hum Mol Genet. 1994;3:147–51. doi: 10.1093/hmg/3.1.147. [DOI] [PubMed] [Google Scholar]
- 27.Antinheimo J, Sallinen SL, Sallinen P, Haapasalo H, Helin H, Horelli-Kuitunen N, et al. Genetic aberrations in sporadic and neurofibromatosis 2 (NF2)-associated schwannomas studied by comparative genomic hybridization (CGH) Acta Neurochir (Wien) 2000;142:1099–104. doi: 10.1007/s007010070036. [DOI] [PubMed] [Google Scholar]
- 28.Mantripragada KK, Buckley PG, Jarbo C, Menzel U, Dumanski JP. Development of NF2 gene specific, strictly sequence defined diagnostic microarray for deletion detection. J Mol Med (Berl) 2003;81:443–51. doi: 10.1007/s00109-003-0458-3. [DOI] [PubMed] [Google Scholar]
- 29.Warren C, James LA, Ramsden RT, Wallace A, Baser ME, Varley JM, et al. Identification of recurrent regions of chromosome loss and gain in vestibular schwannomas using comparative genomic hybridisation. J Med Genet. 2003;40:802–6. doi: 10.1136/jmg.40.11.802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koutsimpelas D, Felmeden U, Mann WJ, Brieger J. Analysis of cytogenetic aberrations in sporadic vestibular schwannoma by comparative genomic hybridization. J Neurooncol. 2011;103:437–43. doi: 10.1007/s11060-010-0412-5. [DOI] [PubMed] [Google Scholar]
- 31.Bian LG, Sun QF, Tirakotai W, Zhao WG, Shen JK, Luo QZ, et al. Loss of heterozygosity on chromosome 22 in sporadic schwannoma and its relation to the proliferation of tumor cells. Chin Med J (Engl) 2005;118:1517–24. [PubMed] [Google Scholar]
- 32.Irving RM, Moffat DA, Hardy DG, Barton DE, Xuereb JH, Holland FJ, et al. A molecular, clinical, and immunohistochemical study of vestibular schwannoma. Otolaryngol Head Neck Surg. 1997;116:426–30. doi: 10.1016/S0194-5998(97)70289-4. [DOI] [PubMed] [Google Scholar]
- 33.Irving RM, Moffat DA, Hardy DG, Barton DE, Xuereb JH, Maher ER. Somatic NF2 gene mutations in familial and non-familial vestibular schwannoma. Hum Mol Genet. 1994;3:347–50. doi: 10.1093/hmg/3.2.347. [DOI] [PubMed] [Google Scholar]
- 34.Irving RM, Moffat DA, Hardy DG, Barton DE, Xuereb JH, Maher ER. Molecular genetic analysis of the mechanism of tumorigenesis in acoustic neuroma. Arch Otolaryngol Head Neck Surg. 1993;119:1222–8. doi: 10.1001/archotol.1993.01880230066011. [DOI] [PubMed] [Google Scholar]
- 35.Sainz J, Baser ME, Ragge NK, Nelson RA, Pulst SM. Loss of alleles in vestibular schwannomas: use of microsatellite markers on chromosome 22. Arch Otolaryngol Head Neck Surg. 1993;119:1285–8. doi: 10.1001/archotol.1993.01880240015003. [DOI] [PubMed] [Google Scholar]
- 36.Dayalan AH, Jothi M, Keshava R, Thomas R, Gope ML, Doddaballapur SK, et al. Age dependent phosphorylation and deregulation of p53 in human vestibular schwannomas. Mol Carcinog. 2006;45:38–46. doi: 10.1002/mc.20150. [DOI] [PubMed] [Google Scholar]
- 37.Singh S, Simon M, Meybohm I, Jantke I, Jonat W, Maass H, et al. Human breast cancer: frequent p53 allele loss and protein overexpression. Hum Genet. 1993;90:635–40. doi: 10.1007/BF00202481. [DOI] [PubMed] [Google Scholar]
- 38.Monoh K, Ishikawa K, Yasui N, Mineura K, Andoh H, Togawa K. P53 Tumor Suppressor Gene in Acoustic Neuromas. Acta Otolaryngol Suppl. 1998;537:11–15. doi: 10.1080/00016489850182288. [DOI] [PubMed] [Google Scholar]
- 39.Welling DB, Lasak JM, Akhmametyeva E, Ghaheri B, Chang LS. cDNA microarray analysis of vestibular schwannomas. Otol Neurotol. 2002;23:736–48. doi: 10.1097/00129492-200209000-00022. [DOI] [PubMed] [Google Scholar]
- 40.Cayé-Thomasen P, Borup R, Stangerup S-E, Thomsen J, Nielsen FC. Deregulated genes in sporadic vestibular schwannomas. Otol Neurotol. 2010;31:256–66. doi: 10.1097/MAO.0b013e3181be6478. [DOI] [PubMed] [Google Scholar]
- 41.O’Reilly BF, Kishore A, Crowther JA, Smith C. Correlation of growth factor receptor expression with clinical growth in vestibular schwannomas. Otol Neurotol. 2004;25:791–6. doi: 10.1097/00129492-200409000-00024. [DOI] [PubMed] [Google Scholar]
- 42.Sirén V, Antinheimo JP, Jääskeläinen J, Böhling T, Carpén O, Vaheri A. Plasminogen activation in neurofibromatosis 2-associated and sporadic schwannomas. Acta Neurochir (Wien) 2004;146:111–8. doi: 10.1007/s00701-003-0183-2. [DOI] [PubMed] [Google Scholar]
- 43.Seol HJ, Jung HW, Park SH, Hwang SK, Kim DG, Paek SH, et al. Aggressive vestibular schwannomas showing postoperative rapid growth - their association with decreased p27 expression. J Neurooncol. 2005;75:203–7. doi: 10.1007/s11060-005-2886-0. [DOI] [PubMed] [Google Scholar]
- 44.Jacob A, Lee TX, Neff Ba, Miller S, Welling B, Chang LS. Phosphatidylinositol 3-kinase/AKT pathway activation in human vestibular schwannoma. Otol Neurotol. 2008;29:58–68. doi: 10.1097/mao.0b013e31816021f7. [DOI] [PubMed] [Google Scholar]
- 45.Stonecypher MS, Chaudhury AR, Byer SJ, Carroll SL. Neuregulin growth factors and their ErbB receptors form a potential signaling network for schwannoma tumorigenesis. J Neuropathol Exp Neurol. 2006;65:162–75. doi: 10.1097/01.jnen.0000199575.93794.2f. [DOI] [PubMed] [Google Scholar]
- 46.Hung G, Colton J, Fisher L, Oppenheimer M, Faudoa R, Slattery W, et al. Immunohistochemistry study of human vestibular nerve schwannoma differentiation. Glia. 2002;38:363–70. doi: 10.1002/glia.10077. [DOI] [PubMed] [Google Scholar]
- 47.Sturgis EM, Woll SS, Aydin F, Marrogi AJ, Amedee RG. Epidermal growth factor receptor expression by acoustic neuromas. Laryngoscope. 1996;106:457–62. doi: 10.1097/00005537-199604000-00012. [DOI] [PubMed] [Google Scholar]
- 48.Cayé-Thomasen P, Werther K, Nalla A, B⊘g-Hansen TC, Nielsen HJ, Stangerup SE, et al. VEGF and VEGF receptor-1 concentration in vestibular schwannoma homogenates correlates to tumor growth rate. Otol Neurotol. 2005;26:98–101. doi: 10.1097/00129492-200501000-00017. [DOI] [PubMed] [Google Scholar]
- 49.de Vries M, Bruijn IB, Cleton-Jansen AM, Malessy MJ, van der Mey AGL, Hogendoorn PCW. Mutations affecting BRAF, EGFR, PIK3CA, and KRAS are not associated with sporadic vestibular schwannomas. Virchows Arch. 2013;462:211–7. doi: 10.1007/s00428-012-1342-8. [DOI] [PubMed] [Google Scholar]
- 50.Lassaletta L, Patrón M, Del Río L, Alfonso C, Roda JM, Rey JA, et al. Cyclin D1 expression and histopathologic features in vestibular schwannomas. Otol Neurotol. 2007;28:939–41. doi: 10.1097/MAO.0b013e31814b2285. [DOI] [PubMed] [Google Scholar]
- 51.Dilwali S, Roberts D, Stankovic KM. Interplay between VEGF-A and cMET signaling in human vestibular schwannomas and schwann cells. Cancer Biol Ther. 2015;16:170–5. doi: 10.4161/15384047.2014.972765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sass HCR, Borup R, Alanin M, Nielsen FC, Cayé-Thomasen P. Gene expression, signal transduction pathways and functional networks associated with growth of sporadic vestibular schwannomas. J Neurooncol. 2017;131:283–92. doi: 10.1007/s11060-016-2292-9. [DOI] [PubMed] [Google Scholar]
- 53.Dilwali S, Briët MC, Kao SY, et al. Preclinical validation of anti-nuclear factor-kappa B therapy to inhibit human vestibular schwannoma growth. Mol Oncol. 2015;9:1359–70. doi: 10.1016/j.molonc.2015.03.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Doherty JK, Ongkeko W, Crawley B, Andalibi A, Ryan AF. ErbB and Nrg: potential molecular targets for vestibular schwannoma pharmacotherapy. Otol Neurotol. 2008;29:50–7. doi: 10.1097/mao.0b013e31815d4429. [DOI] [PubMed] [Google Scholar]
- 55.M⊘ller MN, Werther K, Nalla A, et al. Angiogenesis in vestibular schwannomas: Expression of extracellular matrix factors MMP-2, MMP-9, and TIMP-1. Laryngoscope. 2010;120:657–62. doi: 10.1002/lary.20834. [DOI] [PubMed] [Google Scholar]
- 56.Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res. 1998;72:141–96. doi: 10.1016/S0065-230X(08)60702-2. [DOI] [PubMed] [Google Scholar]
- 57.Kino T, Takeshima H, Nakao M, Nishi T, Yamamoto K, Kimura T, et al. Identification of the cis-acting region in the NF2 gene promoter as a potential target for mutation and methylation-dependent silencing in schwannoma. Genes Cells. 2001;6:441–54. doi: 10.1046/j.1365-2443.2001.00432.x. [DOI] [PubMed] [Google Scholar]
- 58.Gonzalez-Gomez P, Bello MJ, Alonso ME, Lomas J, Arjona D, Campos JM, et al. CpG island methylation in sporadic and neurofibromatis type 2-associated schwannomas. Clin Cancer Res. 2003;9:5601–6. [PubMed] [Google Scholar]
- 59.Kullar PJ, Pearson DM, Malley DS, Collins VP, Ichimura K. CpG island hypermethylation of the neurofibromatosis type 2 (NF2) gene is rare in sporadic vestibular schwannomas. Neuropathol Appl Neurobiol. 2010;36:505–14. doi: 10.1111/j.1365-2990.2010.01090.x. [DOI] [PubMed] [Google Scholar]
- 60.Lassaletta L, Bello MJ, Del Río L, Alfonso C, Roda JM, Rey JA, et al. DNA methylation of multiple genes in vestibular schwannoma: Relationship with clinical and radiological findings. Otol Neurotol. 2006;27:1180–5. doi: 10.1097/01.mao.0000226291.42165.22. [DOI] [PubMed] [Google Scholar]
- 61.Lassaletta L, Patrón M, González T, Martinez-Glez V, Rey JA, Gavilan J. RASSF1A methylation and cyclin D1 expression in vestibular schwannomas. Acta Neuropathol. 2007;114:431–3. doi: 10.1007/s00401-007-0272-z. [DOI] [PubMed] [Google Scholar]
- 62.Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3(4):253–266. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- 63.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96:8681–6. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cioffi JA, Yue WY, Mendolia-loffredo S, Hansen KR, Wackym A, Hansen MR. MicroRNA-21 over-expression contributes to vestibular schwannoma cell proliferation and survival. Otol Neurotol. 2011;31:1455–62. doi: 10.1097/MAO.0b013e3181f20655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Torres-Martin M, Lassaletta L, de Campos JM, Isla A, Gavilan J, Pinto GR, et al. Global Profiling in vestibular schwannomas shows critical deregulation of MicroRNAs and upregulation in those included in chromosomal region 14q32. PLoS One. 2013;8:e65868. doi: 10.1371/journal.pone.0065868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reddy SDN, Ohshiro K, Rayala SK, Kumar R. MicroRNA-7, a homeobox D10 target, inhibits p21-activated kinase 1 and regulates its functions. Cancer Res. 2008;68:8195–200. doi: 10.1158/0008-5472.CAN-08-2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kefas B, Godlewski J, Comeau L, Li Y, Abounader R, Hawkinson M, et al. microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 2008;68:3566–72. doi: 10.1158/0008-5472.CAN-07-6639. [DOI] [PubMed] [Google Scholar]
- 68.Cayé-Thomasen P, Baandrup L, Jacobsen GK, Thomsen J, Stangerup SE. Immunohistochemical demonstration of vascular endothelial growth factor in vestibular schwannomas correlates to tumor growth rate. Laryngoscope. 2003;113:2129–34. doi: 10.1097/00005537-200312000-00014. [DOI] [PubMed] [Google Scholar]
- 69.Koutsimpelas D, Stripf T, Heinrich UR, Mann WJ, Brieger J. Expression of vascular endothelial growth factor and basic fibroblast growth factor in sporadic vestibular schwannomas correlates to growth characteristics. Otol Neurotol. 2007;28:1094–9. doi: 10.1097/MAO.0b013e31814b2787. [DOI] [PubMed] [Google Scholar]
- 70.Plotkin SR, Stemmer-Rachamimov AO, Barker FG, 2nd, Halpin C, Padera TP, Tyrrell A, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361:358–67. doi: 10.1056/NEJMoa0902579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li KL, Djoukhadar I, Zhu X, Zhao S, Lloyd S, McCabe M, et al. Vascular biomarkers derived from dynamic contrast-enhanced MRI predict response of vestibular schwannoma to antiangiogenic therapy in type 2 neurofibromatosis. Neuro Oncol. 2015;18:275–82. doi: 10.1093/neuonc/nov168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Altuna X, Lopez JP, Yu MA, Arandazi MJ, Harris JP, Wang-Rodriguez J, et al. Potential role of imatinib mesylate (Gleevec, STI-571) in the treatment of vestibular schwannoma. Otol Neurotol. 2011;32:163–70. doi: 10.1097/MAO.0b013e3182009665. [DOI] [PubMed] [Google Scholar]
- 73.Ammoun S, Schmid MC, Triner J, Manley P, Hanemann CO. Nilotinib alone or in combination with selumetinib is a drug candidate for neurofibromatosis type 2. Neuro Oncol. 2011;13:759–66. doi: 10.1093/neuonc/nor056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ahmad ErbB Expression, Activation, and Inhibition with Lapatinib and Tyrophostin (AG825) in Human Vestibular Schwannomas. Otol Neurotol. 2011;32:841–7. doi: 10.1097/MAO.0b013e31821f7d88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ammoun S, Cunliffe CH, Allen JC, Chiriboga L, Giancotti FG, Zagzag D, et al. ErbB/HER receptor activation and preclinical efficacy of lapatinib in vestibular schwannoma. Neuro Oncol. 2010;12:834–43. doi: 10.1093/neuonc/noq012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bush ML, Oblinger J, Brendel V, Santarelli G, Huang J, Akhmametyeva EM, et al. AR42, a novel histone deacetylase inhibitor, as a potential therapy for vestibular schwannomas and meningiomas. Neuro Oncol. 2011;13:983–99. doi: 10.1093/neuonc/nor072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Clark JJ, Provenzano M, Diggelmann HR, Xu N, Hansen SS, Hansen MR. The ErbB inhibitors trastuzumab and erlotinib inhibit growth of vestibular schwannoma xenografts in nude mice: a preliminary study. Otol Neurotol. 2008;29:846–53. doi: 10.1097/MAO.0b013e31817f7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Giovannini M, Bonne NX, Vitte J, Chareyre F, Tanaka K, Adams R, et al. MTORC1 inhibition delays growth of neurofibromatosis type 2 schwannoma. Neuro Oncol. 2014;16:493–504. doi: 10.1093/neuonc/not242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Licciulli S, Maksimoska J, Zhou C, Troutman S, Kota S, Liu Q, et al. FRAX597, a small molecule inhibitor of the p21-activated kinases, inhibits tumorigenesis of neurofibromatosis type 2 (NF2)-associated Schwannomas. J Biol Chem. 2013;288(40):29105–14. doi: 10.1074/jbc.M113.510933. [DOI] [PMC free article] [PubMed] [Google Scholar]