

Summary
Context
Cryptorchidism is common in Prader‐Willi syndrome (PWS) males, but the testicular histology in childhood remains uncertain. The association between testicular histology and long‐term gonadal function in PWS males is also unknown.
Objectives
To evaluate the relationship between testicular histology in childhood and long‐term gonadal function in PWS males.
Patients and Methods
Forty men with PWS were assessed longitudinally at our institute over the past 24 years. Clinical examinations and blood tests for LH, FSH and testosterone levels were compared with normal reference values. Tissue specimens were collected during orchiopexy and analyzed based on Nistal categories.
Results
Of nine testes available for pathological assessments, two showed favourable histology (Nistal I) and seven showed unfavourable histology (Nistal II or III). Of five postpubertal males with histology available, four reached puberty spontaneously, but only one reached Tanner stage 5. In a male with favourable histology, LH and FSH were high, but testosterone was normal, though below the average of the reference range. In three males with unfavourable histology, LH was normal, but FSH was highly elevated, and testosterone was at the lower limit of normal. One patient took hCG treatment to induce puberty; this patient showed favourable histology, but LH, FSH and testosterone were not elevated in adolescence.
Conclusions
Testicular histology of PWS men in childhood varies from normal to Sertoli Cell‐Only Syndrome. Regardless of the testicular histology in childhood, hypogonadism in PWS adults arises as a consequence of primary testicular dysfunction with highly elevated FSH and insufficient testosterone levels.
Keywords: gonadal function, hypogonadism, longitudinal studies, Prader‐Willi syndrome, testicular histology
1. INTRODUCTION
Prader‐Willi syndrome (PWS) is a genetic disorder resulting from the lack of expression of the paternally derived chromosome 15q11‐q13.1, 2 It is caused by a deletion (70%), uniparental disomy (25%), imprinting centre defect (5%), or balanced translocation.1, 2 PWS is characterized by hypotonia, hyperphagia, obesity, short stature, behaviour disorder and hypogonadism. Clinical signs of hypogonadism in PWS males include cryptorchidism, scrotal hypoplasia, small testicular volume, delayed or incomplete pubertal development and infertility. Because most symptoms of PWS are considered to result from hypothalamic dysfunction, hypogonadism in PWS was hypothesized to be hypogonadotropic.3, 4, 5 However, recent studies of hypothalamic, pituitary and testicular function in PWS have shown that all forms of hypogonadism (central origin, peripheral and combined) are found in males with PWS.6, 7, 8, 9 Cryptorchidism is found in a majority of PWS males, but only two reports have evaluated the testicular histology of PWS males.10, 11 These studies demonstrated that testicular histology ranged from normal seminiferous tubule tissue with germ cells to Sertoli Cell‐Only Syndrome (SCOS), but the details remain uncertain because they were small case series. 10, 11 Further, the association between testicular histology in childhood and the long‐term prognosis for gonadal function in PWS males remain unclear, because no data are available.
The aim of this study was to evaluate the testicular histology of PWS males in childhood and determine the association between testicular histology in childhood and long‐term gonadal function.
2. MATERIALS AND METHODS
2.1. Patients
After receiving Institutional Review Board approval, the charts of 40 male children and adults (mean 12.1 ± 8.3 years; range 1.0‐28.0 years) consecutively diagnosed with PWS at our institute from July 1991 to August 2015 were retrospectively reviewed. The mean follow‐up was 10.6 ± 7.8 years (range, 0.6‐25.0 years). Thirty‐seven patients (92.5%) were longitudinally assessed until the present study, but three patients were followed by another hospital. Children were followed every 3 months by clinical examinations and at least once a year by blood sampling. After 18 years, they were followed every 6 months. Pubertal development was evaluated using the Tanner classification12 by a paediatric endocrinologist (SI). Puberty was defined as Tanner pubertal stage 2 (G2).
2.2. Hormone assays
Blood samples were periodically taken to measure LH, FSH and testosterone levels. Serum FSH and LH levels were determined using a two‐site enzyme immune‐assay (TOSOH I; TOSOH, Tokyo, Japan). From March 2000, serum FSH and LH levels were determined using TOSOH II. Total serum testosterone was determined by chemiluminescent immunoassay (TOSOH I, TOSOH), up to June 2013. Since July 2013, total serum testosterone was determined by enzyme immunoassay (TOSOH II, TOSOH). Values for LH and FSH obtained in the newer TOSOH II assay comparable to those measured in the older TOSOH I assay.13, 14 Values for testosterone were comparable in the older and newer assays.15 Detectable levels were 0.2‐200 mIU/mL for LH, 0.1‐200 mIU/mL for FSH and 7‐2000 ng/dL for testosterone. For ethical reasons, no age‐matched control group consisting of healthy children and men was available. The hormonal data were compared with previously published age‐dependent reference values in Japan.16, 17 As shown in Figure 1, we divided the hormonal data into puberty related six groups: 2 years before puberty onset, 1 year before puberty onset, puberty onset, 1 year after puberty onset, 2 years after puberty onset, and adults (>18 years).
Figure 1.
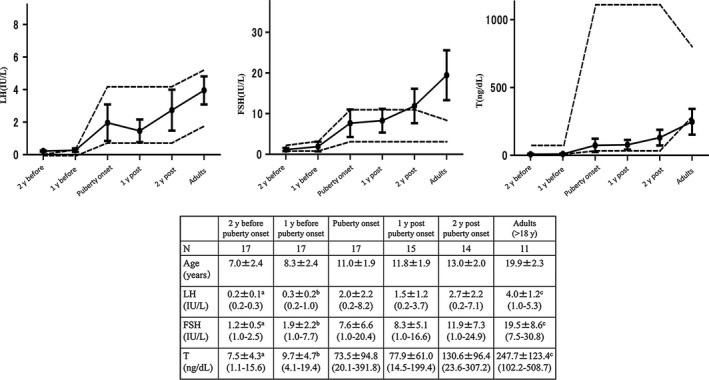
Longitudinal changes of the average values of serum LH, FSH and testosterone (T) levels in PWS males. The black dots represent the mean values of each age with the error bars representing 95% confidence intervals for the mean. The 5th to 95th percentiles of normal reference values for LH, FSH and testosterone levels are indicated by dotted lines. aThe values in the group at 2 y before puberty onset differed significantly from the values in the group at puberty onset for LH, P < 0.009; FSH, P < 0.001; and T, P < 0.0001. bThe values in the group at 1 y before puberty onset differed significantly from the values in the group at puberty onset for LH, P < 0.005; FSH, P < 0.001; and T, P < 0.0001. cThe values in the group at adults differed significantly from the values in the group at puberty onset for LH, P < 0.005; FSH, P < 0.001; and T, P < 0.0007
2.3. hCG stimulation test
Three consecutive daily intramuscular injections of hCG (3000 IU hCG/m2/d) were given, and serum testosterone levels were measured pre‐injection, 24 hours later and 48 hours later. A maximal value higher than 150 ng/dL was considered a normal response.18, 19
2.4. Surgery/Histology
During surgery, testicular location (abdominal, intra‐canalicular, extra‐canalicular) and size measured with a ruler were recorded by paediatric urologists (F Matsui and F Matsumoto). A testicular biopsy was taken (if possible), immediately fixed in formalin for 24 hours, and embedded in paraffin. All biopsies showed at least 30 seminiferous tubules and were, therefore, considered representative of the remaining testis. Four‐micrometre‐thick sections were prepared and stained with haematoxylin and eosin. To identify germ cells, additional specific immunohistochemical markers were used, including testis‐specific protein Y‐encoded (TSPY). Testicular histology was classified into 1 of the 3 modified Nistal categories20, 21 based mainly on TFI (tubular fertility index), which is the per cent of tubules containing spermatogonia. In category I, the testis has minimal alterations, TFI is higher than 60%, or mean tubular diameter is normal or slightly decreased. In category II, the testis has marked germinal hypoplasia, TFI is between 30% and 60%, or the tubules are slightly to markedly hypoplastic. In category III, the testis has severe germinal hypoplasia, TFI is below 30%, or the tubules are severely hypoplastic. All histological slides were reviewed by two experienced pathologists (KM and MT).
2.5. Statistics
Data were analyzed using Statistical Package for the social sciences statistical software version 15.0 (SPSS, Chicago, IL). Results are presented as means ± SD for continuous variables. The clinical data were compared with normal reference data from published reports.16, 17 Comparison of hormonal levels between age groups was evaluated with the Mann‐Whitney U test. A value of P < 0.05 was considered significant.
3. RESULTS
3.1. Patient characteristics
Table 1 shows the baseline characteristics of all 40 subjects. The main genetic defect was a deletion type abnormality (52.5%). Cryptorchidism (bilateral in 27 and unilateral in 8) was noted in 35 of 40 males (87.5%) with PWS, and all of them underwent orchiopexy at 1.9 ± 1.1 years (0.8‐6.3 years) at our institute. Forty‐four testes (71%) were extra‐canalicular testes, 10 (16.1%) were intra‐abdominal testes, and 8 (12.9%) were intra‐canalicular testes. The remaining five males presented bilaterally descended testis. The mean long diameter of the testes was 11.0 ± 1.9 mm (6.0‐15.0 mm) (Table 1). The mean follow‐up after orchiopexy was 10.7 ± 7.7 years (0.2‐23.5 years). Among patients who did not undergo orchiopexy, the mean follow‐up was 9.3 ± 8.3 years (0.9‐23.1 years). Twenty‐nine patients were on GH treatment.
Table 1.
Baseline characteristics of 40 Prader‐Willi syndrome males
| Genetic defect, n (%) | |
| Deletion | 21 (52.5) |
| Uniparental disomy | 6 (15.0) |
| Imprinting centre defect | 3 (7.5) |
| Translocation | 1 (2.5) |
| Not known | 9 (22.5) |
| Cryptorchidism at presentation, n (%) | 35 (87.5) |
| None | 5 (12.5) |
| Bilateral cryptorchid | 27 (67.5) |
| Lt cryptorchid | 2 (5.0) |
| Rt cryptorchid | 6 (15.0) |
| Age (y) at orchiopexy (range) | 1.9 ± 1.1 (0.8‐6.3) |
| Testis position at surgery, n (%) | |
| Extra‐canalicular | 44 (71.0) |
| Intra‐canalicular | 8 (12.9) |
| Intra‐abdominal | 10 (16.1) |
| Long diameter of testis (mm), (range) | 11 ± 1.9 (6‐15) |
| Age (y) at endocrinological sampling, (range) | 1.9 ± 1.1 (0.8‐6.3) |
| LH (mIU/mL) | 0.3 ± 0.4 (0.2‐1.7) |
| FSH (mIU/mL) | 1.9 ± 1.3 (1.0‐5.0) |
| Testosterone (ng/dL) | 7.6 ± 5.5 (3.0‐20.1) |
| Follow‐up after orchiopexy (y), (range) | 10.7 ± 7.7 (0.2‐23.5) |
| Follow‐up in patients who did not undergo orchiopexy (y), (range) | 9.3 ± 8.3 (0.9‐23.1) |
Normal range for LH: 0‐1 y, 0.3‐1.9 mIU/mL; 2‐5 y, 0.3‐1.2 mIU/mL. Normal range for FSH: 0‐1 y, 0.8‐3.0 mIU/mL; 2‐5 y, 0.8‐2.8 mIU/mL. Normal range for testosterone: <1 y, 12‐21 ng/dL; 1‐6 y, 3‐32 ng/dL.
3.2. Pubertal development
Of 14 patients older than 15 years during this study (Table 2), 13 (92.8%) spontaneously reached puberty during the study period. The onset of puberty was at a mean age of 11 ± 1.1 years (10.0‐13.0 years). The remaining patient (Case 14 in Table 2) started hCG treatment to induce puberty at 14 years, reached Tanner stage 2 after 6 months, and stopped the replacement therapy at 15 years. Thirteen patients of 14 patients underwent orchiopexy. Of 11 adults (>18 years) with PWS, only 2 (18.2%) reached Tanner stage 4‐5. Four adults reached Tanner stage 3, and 5 patients reached Tanner stage 2. Mean testicular volume was small (6.1 ± 2.5 mL) in adults.
Table 2.
Pubertal development of 14 Prader‐Willi syndrome males older than 15 y
| Case (case in Table 3) | Genetic defect | Testis position | Age at puberty onset | Pubertal development | Pubertal stage (>18 y) | |
|---|---|---|---|---|---|---|
| Rt | Lt | |||||
| 1 (case 3) | DEL | Extra‐canalicular | Extra‐canalicular | 10 | Spontaneously | 3 |
| 2 (case 4) | DEL | Extra‐canalicular | Scrotum | 11 | Spontaneously | 5 |
| 3 (case 5) | DEL | Extra‐canalicular | Extra‐canalicular | 11 | Spontaneously | 2 |
| 4 | DEL | Extra‐canalicular | Scrotum | 10 | Spontaneously | 2 |
| 5 | Not known | Scrotum | Extra‐canalicular | 13 | Spontaneously | 3 |
| 6 | Not known | Extra‐canalicular | Extra‐canalicular | 12 | Spontaneously | 4 |
| 7 | DEL | Extra‐canalicular | Extra‐canalicular | 10 | Spontaneously | 2 |
| 8 | ICD | Extra‐canalicular | Extra‐canalicular | 12 | Spontaneously | 2 |
| 9 | DEL | Extra‐canalicular | Extra‐canalicular | 12 | Spontaneously | 2 |
| 10 | Not known | Extra‐canalicular | Extra‐canalicular | 12 | Spontaneously | 3 |
| 11 | Not known | Scrotum | Scrotum | 13 | Spontaneously | 3 |
| 12 | DEL | Extra‐canalicular | Extra‐canalicular | 11 | Spontaneously | Not reached |
| 13 | UPD | Intra‐abdominal | Extra‐canalicular | 10 | Spontaneously | Not reached |
| 14 (case 2) | Not known | Extra‐canalicular | Scrotum | 14 | hCG treatment | Not reached |
DEL, deletion; ICD, imprinting centre defect; Lt, left; Rt, right; UPD, uniparental disomy.
On the other hand, three patients had a prior history of early pubertal development. Patient 1 was 8.0 years boy. Height was 122.1 cm (−1 SD), pubic hair (Ph) 2 and testis 3 mL. We found increased growth velocity (7 cm/y), and advanced bone age (9.9 years). Serum levels of LH were normal (LH 2.2 mIU/mL), but FSH and testosterone levels were high (FSH 18.2 mIU/mL and testosterone 216.5 ng/dL, respectively), Pituitary MRI was normal. At 9.7 years, the patient presented bone age of 12.3 years. Patient 2 was 8.0 years boy, height was 119.5 cm (−1 SD), and Ph 2. We found increased growth velocity (9 cm/y), and advanced bone age (7.9 years) at 7.0 years. Serum levels of LH and FSH were normal (LH 0.2 mIU/mL and FSH 1.0 mIU/mL, respectively), but testosterone levels were high (testosterone 37.3 ng/dL). Pituitary MRI was normal. At 9.9 years, the patient presented bone age of 11.6 years. Patient 3 was 9.0 years boy, height was 132.9 cm (+1 SD), Ph 2 and testis 5 mL. We found increased growth velocity (7 cm/y), high testosterone levels (testosterone 48.7 ng/dL) and advanced bone age (12.3 years). Serum levels of LH and FSH were high (LH 3.5 mIU/mL and FSH 8.3 mIU/mL, respectively). Pituitary MRI was normal. LHRH analogue therapy was started at 9.3 years and discontinued at 12.5 years, when the patient had bone age of 13.8 years. After discontinuing LHRH therapy, the patient demonstrated spontaneous pubertal progression with pubertal gonadotropin and testosterone. At 13.5 years, Ph 3, testis 6 mL. Serum levels of LH, FSH and testosterone increased (LH 8.2 mIU/mL, FSH 20.4 mIU/mL and testosterone 391.8 ng/dL, respectively).
3.3. Longitudinal studies of reproductive hormones
Prader‐Willi syndrome males in early childhood (1.9 ± 1.1 years) showed normal LH (0.3 ± 0.4 mIU/mL), FSH (1.9 ± 1.3 mIU/mL), and testosterone (7.6 ± 5.5 ng/dL) levels compared with the reference range (Table 1); hCG stimulation tests were performed at 1.2 ± 0.5 years in 11 cases, and 10 (90.9%) showed normal reactions. The testosterone peak level was 345.1 ± 300.9 ng/dL (59.4‐1000 ng/dL). Serum levels of LH and FSH were within the normal range prior to the onset of puberty (8.3 ± 2.4 years). After the onset of puberty (11.0 ± 1.9 years), LH and FSH levels significantly increased (2.0 ± 2.2 mIU/mL and 7.6 ± 6.6 mIU/mL, respectively), but they were within the normal range (Figure 1). Two years after the onset of puberty (13.0 ± 2.0 years) and in adults, the FSH level was persistently highly elevated compared with the reference range (19.5 ± 8.6 mIU/mL), but the LH level remained normal (4.0 ± 1.2 mIU/mL). Testosterone levels significantly increased after the onset of puberty, but they were low (247.7 ± 123.4 ng/dL) in adults; two patients (11.7%) continued to have low LH, FSH and testosterone levels until the age of 15 years.
3.4. Testicular histology in cryptorchidism with PWS
Of nine testes in nine patients available for pathological assessments (Table 3), 2 (22.2%) showed a Nistal score of I (a favourable histology), 1 (11.1%) showed a Nistal score of II and 6 (66.7%) showed a Nistal score of III. In category III, a total of three testes showed SCOS (Figure 2). Six (66.6%) showed spermatogonia. The mean age at testicular biopsy was 2.2 ± 1.6 years.
Table 3.
Clinical, anatomical, cytogenetic, hormonal and histological data of 9 Prader‐Willi syndrome males who underwent testicular biopsy
| Case | Age (y) | Genetic defect | Biopsy side | Cryptorchidism | Site of testis | Length (mm) | Nistal score | hCG stimulation test | Post Puberty | Pubertal stage | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T (ng/dL) | Age (y) | LH (mIU/mL) | FSH (mIU/mL) | T (ng/dL) | |||||||||
| Peak | |||||||||||||
| 1 | 1.3 | DEL | Lt | Lt | Intra‐abdominal | 16 | I | 300.4 | 13 | 8.2 | 20.4 | 391.8 | 3 |
| 2 | 1.4 | Not known | Rt | Rt | Extra‐canalicular | 10 | I | 59.4 | 15 | 0.2 | 1.0 | 27.8 | 2 |
| 3 | 1.4 | DEL | Rt | Bil | Extra‐canalicular | Unknown | III | Unknown | 21 | 4.9 | 29.6 | 508.7 | 3 |
| 4 | 2.6 | DEL | Rt | Rt | Extra‐canalicular | Unknown | III(SCOS) | Unknown | 21 | 4.9 | 30.3 | 202.6 | 5 |
| 5 | 6.3 | DEL | Rt | Bil | Extra‐canalicular | 12 | III(SCOS) | Unknown | 18 | 5.3 | 18.8 | 162.5 | 2 |
| 6 | 1.6 | UPD | Lt | Bil | Extra‐canalicular | 9 | II | 867.9 | Puberty not reached | ||||
| 7 | 1.2 | UPD | Lt | Bil | Intra‐canalicular | 9 | III | 249.3 | Puberty not reached | ||||
| 8 | 1.6 | DEL | Rt | Bil | Extra‐canalicular | 10 | III | Unknown | Puberty not reached | ||||
| 9 | 2.5 | DEL | Rt | Bil | Extra‐canalicular | 11 | III(SCOS) | Unknown | Puberty not reached | ||||
Bil, bilateral; DEL, deletion; Lt, left; Rt, right; SCOS, Sertoli Cell‐Only Syndrome; T, testosterone; UPD, uniparental disomy.
Figure 2.
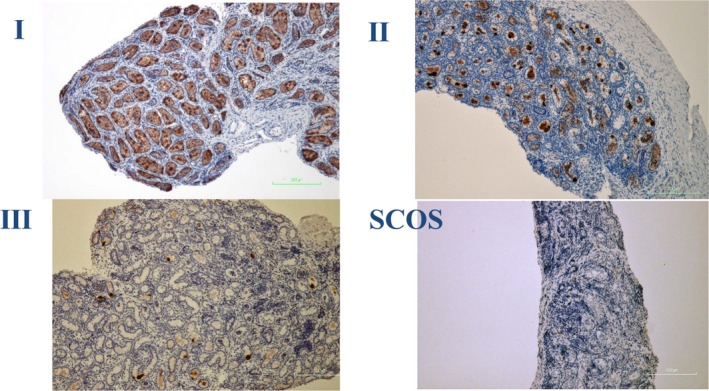
Histology cuts of testicular biopsies from boys with PWS. Germ cells (TSPY) are stained red. Nistal score of I in Case 1, Nistal score of II in Case 6 and Nistal score of III in Case 7. Sertoli Cell‐Only Syndrome (SCOS) in Case 9
The relationship between the age at surgery and testicular histology was examined to clarify why favourable histology (Nistal I) tended to occur in younger patients (mean age 1.4 years), while unfavourable histology (Nistal II and III) tended to occur in older patients (mean age 2.5 years). The lower position of the testis did not tend to have a favourable histology, because one intra‐abdominal testis was Nistal I, while six of seven extra‐canalicular testes were Nistal II or III. There were severely decreased numbers or total absence of spermatogonia even in two cases with a normal testosterone response to hCG stimulation in early childhood.
Furthermore, the association between testicular histology in childhood and gonadal function in adolescence and adults with PWS was assessed. Of five males above postpubertal age with testicular histology available, four spontaneously reached puberty. In a male with favourable histology (Case 1), LH and FSH levels were high (8.2 mIU/mL, 20.4 mIU/mL, respectively), but the testosterone level was normal, though below the average (391.8 ng/dL) of the reference range. In three males with unfavourable histology (Cases 3, 4, 5), the LH levels were normal, but testosterone levels were at the lower limit of normal (5.0 ± 0.23 mIU/mL, 291.3 ± 189.4 ng/dL, respectively). The FSH levels were highly elevated (26.2 ± 6.4 mIU/mL). One patient (Case 2) took hCG treatment to induce puberty. The patient showed favourable histology (Nistal I), but LH, FSH and testosterone levels were not elevated in adolescence, which may indicate hypogonadotropic hypogonadism of central origin for LH and/or FSH. The present data demonstrate that the FSH level was highly elevated and high in all PWS adolescents and adults regardless of the testicular histology in childhood, with the exception of one patient with hypogonadotropic hypogonadism.
4. DISCUSSION
In this study, the longitudinal data related to the clinical signs of pubertal development and reproductive hormones were reported in PWS males from infancy to adulthood. Furthermore, testicular histology in childhood was assessed, and the association between testicular histology and gonadal function was evaluated in adolescents and adult males with PWS.
It was found that the LH level was normal compared with a reference population from early in childhood to adulthood. On the other hand, the FSH level was normal compared with a reference population from early in childhood to 1 year after puberty onset, but the FSH level increased to above the 95th percentile 2 years after puberty onset. The testosterone level increased after the onset of puberty but remained below the 5th percentile in adulthood. These results were consistent with the longitudinal studies by Hirsch et al22 and Siemensma et al6 Inhibin B is secreted by Sertoli cells and is responsible for the function of Sertoli cells. Recent studies showed that inhibin B levels were mostly within the normal range in prepubertal males with PWS. After the onset of puberty, most PWS males showed decreasing inhibin B levels to below the 5th percentile.6, 23 Elevated FSH levels were associated with decreasing inhibin B levels. In the present study, most PWS children showed normal reactions to the hCG stimulation test, which indicated that the function of Leydig cells was normal before puberty onset. It has been reported that normal increases of LH, FSH and testosterone levels occur in PWS infants within a few months after birth, so‐called mini‐puberty,23, 24 whereas patients with isolated hypogonadotropic hypogonadism (IHH) show no testosterone rise during infancy.25 In the present study, approximately 10% of PWS males had low LH, FSH and testosterone levels after the onset of puberty. These results are similar to the finding of Radicioni et al,7 who found hypogonadotropic hypogonadism in 5 of 24 PWS males older than 13.5 years.
Similar to other reports, the present results confirmed that most PWS males start puberty spontaneously but do not reach Tanner stage 4‐5.7, 22 Although cryptorchidism is common in PWS males, testicular volume was below the 5th percentile compared with age‐matched normal testes during early in childhood. The testes grew less than in normal males after puberty (>14 mL in healthy adults). In this study, penile length was not confirmed, but there have been conflicting reports about penile length. Micropenis was considered to be a common feature of PWS males.26 In contrast, Hirsch et al22 reported normal penile length during infancy and early in childhood, but most adults had penile length below the lower limit of normal.
Precocious puberty in PWS males is rare, but several cases have been reported in the literature.24, 27, 28, 29 The mechanism of precocious puberty in PWS is uncertain. It has been reported that the mutation in MKRN3 gene, an imprinted gene located in the PWS critical region (chromosome 15q11‐q13) is associate with familial central precocious puberty.30 Ischaemic damage of the brain has been implicated in precocious puberty in PWS.27
In summary, most PWS males show a normal pituitary‐gonadal axis and normal testosterone secretion from early in childhood to prepubertal ages. After puberty onset, FSH levels are highly elevated, and hypogonadism arises as a consequence of primary testicular dysfunction. Furthermore, the testosterone level is insufficient for mature sexual organ growth.
The present study demonstrated that testicular histology of PWS men in childhood showed various findings, from normal seminiferous tubule tissue with germ cells to SCOS. Of the nine biopsies obtained, 22.2% showed favourable testicular histology, while 77.8% showed unfavourable histology. Vogels et al11 reported that six of eight prepubertal boys showed decreased numbers or total absence of spermatogonia, while two boys showed normal testicular histology with spermatogonia. Vogels et al found no correlation between testicular histology and the number of spermatogonia with the molecular diagnosis. Bakker et al10 reported that, of the 15 testicular biopsies obtained during orchiopexy, two had TFI >60%, three had TFI 30%‐60%, seven had TFI <30% and three had SCOS. They demonstrated that younger age and greater testosterone increases or higher inhibin B levels during hCG stimulation were correlated with favourable TFI.10 In the present study, younger age at surgery tended to be associated with favourable testicular histology, but a lower position of the testis did not tend to be associated with a favourable histology. Further, a normal reaction was observed on the hCG stimulation test, even in cases with severely decreased numbers or total absence of spermatogonia.
No previous reports have evaluated the association between testicular histology in childhood and long‐term gonadal function in PWS males. In the present study, regardless of the testicular histology in childhood, four of five cases who reached puberty showed FSH levels that were persistently elevated after puberty, which indicated that hypogonadism in PWS arises after puberty as a consequence of primary testicular dysfunction. The other case had favourable testicular histology (Nistal score 1), but gonadotropin and testosterone levels remained low until the age of 15 years. The presence of low gonadotropin levels in a 15‐year‐old PWS boy does not necessarily prove hypogonadotropic hypogonadism, because previous studies demonstrate that FSH levels may not rise to high levels until after age 20 years.6, 22
Klinefelter's syndrome (KS) adults are characterized by hypergonadotropic hypogonadism as a consequence of primary testicular dysfunction with highly elevated FSH and LH levels.31 In childhood, KS boys show normal concentrations of testosterone, LH, FSH, AMH and inhibin B. The testicular architecture in KS seems normal except for the decrease in the number of germ cells until puberty, but germ cell degeneration progresses rapidly during puberty and adolescence, eventually resulting in extensive fibrosis and hyalinization of the seminiferous tubules and hyperplasia of the interstitium in the adults.31 On the other hand, there has been one report of the testicular tissue of PWS in adulthood, and it showed Sertoli cell hyperplasia with the formation of Sertoli cell nodules, markedly vacuolized Leydig cells, and atrophic tubules with peritubular hyalinization.11 In childhood, the testicular histology of PWS males seems to be different from that of KS males in the heterogeneity that is seen in PWS, with normal testicular histology to SCOS. It has been reported that C15orf2, of which biallelic transcription is observed entirely in the testis, has an important role in spermatogenesis.32 It is suspected that genes located in the PWS critical region 15q11‐13 might have a role directly or indirectly in this process of early gonadal failure.32 In PWS boys, the disorder of testicular tissue may progress prenatally, unlike KS. Cryptorchidism was reported in only 14% of KS cases, and scrotal hypoplasia was not seen.31 This suggests that the foetal gonadotropin and androgen environment differs between PWS and KS. The difference in the foetal hormonal environment between PWS and KS might be correlated with the observed differences in testicular histological findings.
Some limitations of the present study must be acknowledged. This investigation was retrospective and limited to follow‐up from infancy to adulthood, and about half of the patients did not reach puberty. Unfortunately, histology was available only in a limited number of cases with cryptorchidism, and histology data were limited to childhood. Further studies of the associations between testicular histology and molecular genetic factors and endocrine function in PWS are therefore necessary.
In conclusion, testicular histology of PWS men in childhood varies from normal to SCOS. Regardless of the testicular histology in childhood, hypogonadism in PWS adults arises as a consequence of primary testicular dysfunction with highly elevated FSH and insufficient testosterone levels.
CONFLICT OF INTEREST
Nothing to declare.
AUTHORS CONTRIBUTION
Satoko Matsuyama collected data, wrote the manuscript and analyzed the data. Futoshi Matsui designed the research, wrote the manuscript and performed the operations. Keiko Matsuoka and Makoto Takeuchi performed pathology review. Shinobu Ida collected endocrinological data. Fumi Matsumoto performed the operations and review the manuscript. Masashi Iijima and Atsushi Mizokami review the manuscript.
ETHICS STATEMENT
Not required.
ACKNOWLEDGEMENTS
The authors are very grateful to Dr. K Shimada and Dr. K Yazawa (Departments of Urology, Osaka Women's and Children's Hospital) for providing surgical data.
Matsuyama S, Matsui F, Matsuoka K, et al. Gonadal function and testicular histology in males with Prader‐Willi syndrome. Endocrinol Diab Metab. 2019;2:e49 10.1002/edm2.49
DATA ACCESSIBILITY
All data are included within the manuscript.
REFERENCES
- 1. Cassidy SB. Prader‐Willi syndrome. J Med Genet. 1997;34:917‐923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goldstone AP, Holland AJ, Hauffa BP, Hokken‐Koelega AC, Tauber M. Recommendations for the diagnosis and management of Prader‐Willi syndrome. J Clin Endocrinol Metab. 2008;93:4183‐4197. [DOI] [PubMed] [Google Scholar]
- 3. Swaab DF. Prader‐Willi syndrome and the hypothalamus. Acta Paediatr Suppl. 1997;423:50‐54. [DOI] [PubMed] [Google Scholar]
- 4. Muscatelli F, Abrous DN, Massacrier A, et al. Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader‐Willi syndrome. Hum Mol Genet. 2000;9:3101‐3110. [DOI] [PubMed] [Google Scholar]
- 5. Hoybye C, Hilding A, Jacobsson H, Thoren M. Metabolic profile and body composition in adults with Prader‐Willi syndrome and severe obesity. J Clin Endocrinol Metab. 2002;87:3590‐3597. [DOI] [PubMed] [Google Scholar]
- 6. Siemensma EP, de Lind van Wijngaarden RF, Hokken‐Koelega AC, et al. Testicular failure in boys with Prader‐Willi syndrome: longitudinal studies of reproductive hormones. J Clin Endocrinol Metab. 2012;97:E452–E459. [DOI] [PubMed] [Google Scholar]
- 7. Radicioni AF, Di Giorgio G, Grugni G, et al. Multiple forms of hypogonadism of central, peripheral or combined origin in males with Prader‐Willi syndrome. Clin Endocrinol (Oxf). 2012;76:72‐77. [DOI] [PubMed] [Google Scholar]
- 8. Hirsch HJ, Eldar‐Geva T, Gross‐Tsur V, Benarroch F, Roger M, Lahlou N. Normal insulin‐like peptide‐3 levels despite low testosterone in adult males with Prader‐Willi syndrome: variations in Leydig cell function from infancy through adulthood. J Clin Endocrinol Metab. 2013;98:E135–E143. [DOI] [PubMed] [Google Scholar]
- 9. Gross‐Tsur V, Hirsch HJ, Benarroch F, Eldar‐Geva T. The FSH‐inhibin axis in prader‐willi syndrome: heterogeneity of gonadal dysfunction. Reprod Biol Endocrinol. 2012;10:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bakker NE, Wolffenbuttel KP, Looijenga LH, Hokken‐Koelega AC. Testes in infants with Prader‐Willi syndrome: human chorionic gonadotropin treatment, surgery and histology. J Urol. 2015;193:291‐298. [DOI] [PubMed] [Google Scholar]
- 11. Vogels A, Moerman P, Frijns JP, Bogaert GA. Testicular histology in boys with Prader‐Willi syndrome: fertile or infertile? J Urol. 2008;180:1800‐1804. [DOI] [PubMed] [Google Scholar]
- 12. Marshall WA, Tanner JM. Variations in the pattern of pubertal changes in boys. Arch Dis Child. 1970;45:13‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Karino K, Moriyama H, Furuse H, Shibata H, Masuda J. Improvement of LH assay system by full‐automatic EIA analyzer. Horumon to Rinsho. 2000;48:1157‐1161. [Google Scholar]
- 14. Saito H, Okuyama D, Kozuka K, et al. Study on the assay of serum LH, FSH, PRL, hCG, β‐hCG by automatic enzyme immunoassay system (AIA‐1200). Horumon to Rinsho. 1992;40:651‐657. [Google Scholar]
- 15. Yamaguchi H, Aono Y, Fukumoto S, Nakahara K, Uchimura H. Study on the assay of serum testosterone by automatic enzyme immunoassay system (AIA21) [Translated from Japanese.] JJCLA. 2003;28:428. [Google Scholar]
- 16. Aono T, Kumamoto E, Sasaki R, et al. Multi centric clinical studies on immunoradiometric assays (SPAC‐S LH, SPAC‐S FSH) for measurement of serum LH and FSH using the pituitary gonadotropin standards. Horumon to Rinsho. 1988;36:1087‐1097. [Google Scholar]
- 17. Date on files at Roche Diagnotics. https://www.crlcorp.com/getDocument.cfm?documentID=44. Accessed October 17, 2016.
- 18. Sato N, Katsumata N, Horikawa R, Tanaka T. The usefulness of GnRH test and HCG test for differential diagnosis between delayed puberty and hypogonadotropic hypogonadism in prepubertal boys. Jpn J Reprod Endocrinol. 2003;8:49‐53. [Google Scholar]
- 19. Tanaka T, Hibi I, Tanae A. Predictability of sexual maturation by human chorionic gonadotropin (hCG) test and gonadotropin‐releasing hormone (GnRH) test in prepubertal boys with idiopathic growth hormone deficiency (GHD): diagnosis of combined gonadotropin deficiency (GnD) before puberty. Clin Pediatr Endocrinol. 1992;1:21‐25. [Google Scholar]
- 20. Nistal M, Paniagua R, Diez‐Pardo JA. Histologic classification of undescended testes. Hum Pathol. 1980;11:666‐674. [DOI] [PubMed] [Google Scholar]
- 21. Nistal M, Riestra ML, Paniagua R. Correlation between testicular biopsies (prepubertal and postpubertal) and spermiogram in cryptorchid men. Hum Pathol. 2000;31:1022‐1030. [DOI] [PubMed] [Google Scholar]
- 22. Hirsch HJ, Eldar‐Geva T, Bennaroch F, Pollak Y, Gross‐Tsur V. Sexual dichotomy of gonadal function in Prader‐Willi syndrome from early infancy through the fourth decade. Hum Reprod. 2015;30:2587‐2596. [DOI] [PubMed] [Google Scholar]
- 23. Eiholzer U, l'Allemand D, Rousson V, et al. Hypothalamic and gonadal components of hypogonadism in boys with Prader‐Labhart‐ Willi syndrome. J Clin Endocrinol Metab. 2006;91:892‐898. [DOI] [PubMed] [Google Scholar]
- 24. Fillion M, Deal CL, Van Vliet G. Normal minipuberty of infancy in boys with Prader‐Willi syndrome. J Pediatr. 2006;149:874‐876. [DOI] [PubMed] [Google Scholar]
- 25. Sato N, Katsumata N, Kagami M, et al. Clinical assessment and mutation analysis of Kallmann syndrome 1 (KAL1) and fibroblast growth factor receptor 1 (FGFR1, or KAL2) in five families and 18 sporadic patients. J Clin Endocrinol Metab. 2004;89:1079‐1088. [DOI] [PubMed] [Google Scholar]
- 26. Crino A, Schiaffini R, Ciampalini P, et al. Hypogonadism and pubertal development in Prader‐Willi syndrome. Eur J Pediatr. 2003;162:327‐333. [DOI] [PubMed] [Google Scholar]
- 27. Crino A, Di Giorgio G, Schiaffini R, et al. Central precocious puberty and growth hormone deficiency in a boy with Prader‐Willi syndrome. Eur J Pediatr. 2008;167:1455‐1458. [DOI] [PubMed] [Google Scholar]
- 28. Ludwig NG, Radaeli RF, Silva MM, et al. A boy with Prader‐Willi syndrome: unmasking precocious puberty during growth hormone replacement therapy. Arch Endocrinol Metab. 2016;60:596‐600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Abreu AP, Macedo DB, Brito VN, Kaiser UB, Latronico AC. A new pathway in the control of the initiation of puberty: the MKRN3 gene. J Mol Endocrinol. 2015;54:R131–R139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abreu AP, Dauber A, Macedo DB, et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N Engl J Med. 2013;368:2467‐2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aksglaede L, Juul A. Testicular function and fertility in men with Klinefelter syndrome: a review. Eur J Endocrinol. 2013;168:R67–R76. [DOI] [PubMed] [Google Scholar]
- 32. Farber C, Gross S, Neesen J, Buiting K, Horsthemke B. Identification of a testis‐specific gene (C15orf2) in the Prader‐Willi syndrome region on chromosome 15. Genomics. 2000;65:174‐183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are included within the manuscript.