

Summary
Objectives and Design
In this study, we examined the role of MALAT1, a highly conserved nuclear long non‐coding RNA molecule, in chronic diabetic complications affecting the heart and kidneys using both in vitro and in vivo models: human endothelial cell culture and a Malat1 knockout mice model.
Results
Findings from our in vitro experiments demonstrated that MALAT1 was predominantly localized to nuclear speckles in endothelial cells and MALAT1 expression was significantly increased following incubation with high glucose in association with increased expression of inflammatory cytokines. As for our in vivo experiments, we used Malat1 knockout mice and wild‐type controls with or without streptozotocin‐induced diabetes over 2 months of follow‐up, where all of our diabetic animals showed hyperglycaemia and polyuria. Examination of cardiac and renal tissues demonstrated altered MALAT1 RNA expression in wild‐type diabetic animals. Such changes were associated with augmented production of downstream inflammatory molecules at the mRNA and protein levels. Diabetes‐induced elevations of inflammatory markers were significantly decreased in Malat1 knockout diabetic animals. In addition to transcript and protein analyses, we examined functional changes in the heart and kidneys. Organ functions were affected in the wild‐type diabetic mice but were rescued in Malat1 knockout mice.
Conclusions
Taken together, findings from this study will provide direct evidence and insight into the importance of MALAT1 in the pathogenesis of chronic diabetic complications involving the heart and kidneys.
Keywords: cellular research, diabetic complications, long non‐coding RNAs, mouse model
1. INTRODUCTION
Affecting around 400 million people worldwide, diabetes mellitus (DM) is one of the fastest growing health issues in the world.1 DM is also a tremendous burden on the healthcare system where diabetes and its complications accounted for at least 376 billion US dollars of total global health expenditure in 2010.2 Chronic complications, due to diabetes, lead to functional and structural alterations in the organs through multiple abnormalities arising as a result of chronic hyperglycaemia.3 Hyperglycaemia leads to signalling alterations, along with DNA damage due to oxidative stress, in endothelial cells in susceptible tissues.3, 4 As a consequence of hyperglycaemia, a large number of transcriptional alterations in gene expression transpire to compensate for the damage and biochemical changes that follow.4, 5 Many of these changes are attributed to oxidative stress, which is known to cause changes in cell cycle regulation and survival.6, 7 Further, the elevated presence of oxygen radicals can be detected by receptors of the innate immune system.8 When these receptors are activated, downstream signalling cascades are evoked to further stimulate the production of inflammatory cytokines and extracellular matrix proteins that promote a pro‐inflammatory environment, thereby resulting in inflammatory‐mediated damage to the affected tissues.8, 9 At the transcriptional level, regulating the production of downstream transcriptional factors is a major mechanism in the genesis of downstream effects.10, 11 We have previously shown that alterations in epigenetic modifications such as histone acetylation and specific microRNAs (miRs) can affect this transcriptional regulation and mediate glucose‐induced abnormalities.12, 13 Nevertheless, the transcriptional process can also be controlled by long noncoding RNAs (lncRNAs).14
LncRNAs are >200 nucleotides in length and generally do not code for proteins.14, 15 Despite possessing little or no protein‐coding capabilities, lncRNAs are still capable of regulating protein and non–protein‐coding genes through both cis‐ and trans‐acting effects.15 LncRNA MALAT1 (metastasis‐associated lung adenocarcinoma transcript 1, also known as NEAT2) was first identified in early‐stage non–small‐cell lung carcinoma cells.16 MALAT1 encodes an 8.7‐kb exonic transcript that has high levels of transcription in many tissues including the heart, kidney and brain.17, 18 MALAT1 is highly conserved among mammals; however, particular conserved regions of MALAT1 have been found in other species such as frogs and fish.19, 20 MALAT1 is primarily retained in the nucleus, where it associates with other molecules in nuclear speckles and has been shown to regulate gene transcription.21, 22
The definitive roles of lncRNAs in DM remain largely elusive; however, studies have indicated that lncRNAs may be implicated in β‐cell function,23, 24 glucose homeostasis,25 end‐stage kidney disease 26 and endothelial cell dysfunction.27 Earlier work completed by our laboratory showed for the first time that MALAT1 expression is elevated in large vessel endothelial cells (human umbilical vein endothelial cells; HUVECs) following high glucose incubation in vitro.28 Furthermore, the increase in MALAT1 transcript expression correlated with increases in multiple inflammatory proteins (IL‐6 and TNF‐α) and siRNA‐mediated MALAT1 knockdown subsequently prevented the upregulation of glucose‐induced inflammatory cytokines.28 To further confirm our previous findings, a recent study has shown that TNF‐α expression in splenocytes was lower in mice lacking MALAT1 compared to wild‐type (WT) mice.29 Although MALAT1 is thought to be associated with the generation of pro‐inflammatory cytokines,27, 28, 30 use of a novel Malat1 knockout (KO) mice model will provide the opportunity to demonstrate a direct causal relationship between MALAT1 and the inflammatory network.
Mouse knockout models of Malat1 have been generated previously that remove either portions or the entire gene segment of Malat1.21 These knockouts are not only nonlethal, but also show no observable phenotype.21, 22 To date, very few studies have implemented the Malat1 KO mouse model to examine the role of MALAT1 in diabetic complications. In fact, recent research that investigated MALAT1 in diabetic cardiomyopathy used a diabetic rat model in which intracoronary injections of small hairpin RNA (shRNA) targeting Malat1 were administered.30, 31 Knockdown of Malat1 resulted in improved left ventricular systolic function, reduced diabetes‐induced myocardial inflammation and decreased apoptosis of cardiomyocytes.30, 31 Furthermore, in the context of diabetic nephropathy, ex vivo approaches with MALAT1 siRNA delivery demonstrated reduced high glucose‐induced injury in cultured mice podocytes.32 Despite the revelations from these reports, whether similar patterns exist in the diabetic heart and kidneys after Malat1 KO still remains unclear.
In this study, we investigated the regulatory role of MALAT1 on the expression of inflammatory cytokines and subsequent structural and functional effects in the diabetic heart and kidneys using a Malat1 knockout mice model with chronically induced diabetes as well as in a cell culture system using microvascular endothelial cells.
2. MATERIALS AND METHODS
2.1. Animal experiments
Mice with a homozygous deletion in the Malat1 gene (Malat1 KO) were obtained through collaboration with Dr. Spector (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, USA). These mice, generated on a C57/BL6 background, have a approximately 3‐kb deletion in the 5′ end and promoter of the Malat1 gene.21 This homozygous deletion is a global knockout eliminating MALAT1 transcript expression in all tissues. Diabetes was induced in 10‐week‐old male mice using daily intraperitoneal injections of 50 mg/kg streptozotocin (STZ) dissolved in citrate buffer (0.1 mol/L, pH 4.5) for 5 days as described previously.33 Age‐ and gender‐matched littermate control mice were administered injections of the same volume of citrate buffer alone. Male mice that were hyperglycaemic were evaluated based on having a blood glucose level >15 mmol/L. All animals were housed with access to standard diet (Harlan 2018 rodent diet) and water ad libitum. Animals were monitored up until the time of sacrifice at 1, 2, 4 or 8 weeks postdiabetes. Blood glucose levels, urine glucose and ketones, and body weight were recorded and monitored.
For specific experiments, WT mice at 10 weeks of age were fed for a week with either a standard rodent diet containing 19% protein, 5% fat and 5% crude fibre or a similar feed enriched with 30% galactose (Test Diet, IN, USA) to promote a hyperhexosemic nonketotic model of diabetic complications.
Following sacrifice, left ventricular and renal cortical tissues were snap‐frozen for RNA and protein analyses. All procedures were approved by the University's Animal Care Committee.33
2.2. Cell culture
As the microvascular endothelium is a main target for glucose‐induced cell damage, we used human microvascular endothelial cells (retinal origin; HRECs) for our in vitro experiments. HRECs were purchased from Olaf Pharmaceuticals (Worcester, MA, USA) and grown at 37°C, 5% CO2, in EBM‐2 (Lonza) supplemented with 10% foetal bovine serum, growth factors and antibiotics (EBM‐2 Single Quots). Cells were plated at a density of 2500 cells/cm2. Once the cells had reached a confluency of 80%, they were serum starved for 24 hours and were then incubated with either 5 mmol/L (NG, mimicking euglycemia) or 25 mmol/L (HG, mimicking hyperglycaemia) d‐glucose. l‐glucose (25 mmol/L) was used as osmotic control (Osm). Following glucose addition, cells were incubated for 48 hours and were later trypsinized prior to RNA collection.34 In addition to our in vitro work with HRECs, we cultured human cardiac microvascular endothelial cells (HCMECs; purchased from Olaf Pharmaceuticals) in high glucose to determine the MALAT1 RNA expression levels at multiple time points (Figure S1).
2.3. Cardiac imaging
Echocardiographic and Doppler studies were performed using the Vevo™ 2100 imaging system (Visualsonic, Toronto, ON, Canada). All examinations were performed 1 week prior to the time of sacrifice between 1 pm and 5 pm. Each mouse was anesthetized in a sealed chamber with inhalant isoflurane at 3% in 100% oxygen. Once anesthetized, the mouse was transferred and secured on its dorsal side to a heated imaging platform and isoflurane was maintained at 1% for the duration of the experiment. The hair on the mouse chest was carefully removed to reduce signal attenuation, and an electrode gel was applied to the chest before placing the transducer to the left of the sternum. At this position, images were obtained of the left ventricle along its long axis. The blood flow velocity was measured using Doppler ultrasonography to directly measure the flow motion (Pulse Wave Doppler). M‐mode tracings of the heart were acquired by rotating the transducer to be perpendicular to the left ventricle and aligned along the short axis of the heart. Measurements were averaged from 3 consecutive cardiac cycles.12, 35
2.4. RNA isolation, cDNA synthesis and qRT‐PCR
RNA extraction was carried out from tissues using the PureLink™ RNA Mini Kit (Ambion Life Technologies, Carlsbad, CA, USA) following the manufacturer's protocol. The tissues were homogenized in lysis buffer using a mechanical homogenizer or a mortar and passaged through a 20‐gauge needle. After washing, RNA was eluted from the membrane by molecular grade water. RNA purity and concentration were assessed by NanoDrop. RNA was then immediately reverse‐transcribed to cDNA or kept at −70°C. RNA extractions of HRECs were carried out using TRIzol™ Reagent (Invitrogen INC, Burlington, ON, Canada) following the manufacturer's instructions.
One μicrogram of total extracted RNA was reverse‐transcribed to cDNA using High Capacity CDNA Reverse Transcription Kit (Applied Biosystems Inc., Foster City, CA, USA). qRT‐PCR was performed in duplicate using SYBR Green Master Mix (Clontech, Mountain View, CA, USA) and LightCycler™ 96 (RocheDiagnotstics Canada, Laval, QC, Canada). Gene‐specific primer sets were designed using the basic local alignment program (BLAST) from NCBI, or predesigned sets were ordered from Qiagen (Toronto, ON, Canada) (Table S1). Target genes were analysed using standard curves to determine relative levels of gene expression. The levels of specific RNAs were analysed using the Roche LightCycler™ 96 System. Individual RNA samples were normalized to the level of β‐actin.12
2.5. RNA in situ hybridization
RNA fluorescent in situ hybridization (FISH) was carried out using specific probes and reagents designed by Stellaris™ RNA FISH (Biosearch Technologies, Petalume, CA, USA). To detect MALAT1 RNA, HRECs were grown on coverslips and then fixed in 2% formaldehyde. Cells were made permeable in 70% (vol./vol.) ethanol diluted in diethylpyrocarbonate (DEPC)‐treated water overnight at 4°C. Hybridization was carried out using Stellaris™ RNA FISH probes at a concentration of 125 nmol/L, which was incubated with the cells in a moist chamber at room temperature for 4‐6 hours. For nuclear visualization, nuclei were counterstained with Hoechst stain (5 ng/mL). Coverslips were then mounted on the slides to be visualized by immunofluorescence.36
2.6. ELISA
Proteins were isolated from mice tissues using radioimmunoprecipitation assay buffer (Sigma‐Aldrich, St. Louis, MO, USA). Protein concentrations were measured using a BCA protein assay kit (Pierce, Rockford, IL, USA). Protein concentrations were kept constant across protein lysates used for ELISA. For detection of proteins in tissue homogenate, antigen‐specific precoated plates were obtained from R&D systems (Minneapolis, MN, USA) and the protocol was followed to the manufacturer's instructions. Plates were measured at 450 and 570 nm (to correct for plate defects) using a plate reader. For detection of albumin and creatinine in mouse urine, Albuwell™ and Creatinine Companion kits were used from Exocell (Philadelphia, PA, USA).28, 37
2.7. Statistical analysis
All experimental data are expressed as mean ± SEM. Student's t test was used when comparing 2 conditions, whereas ANOVA, followed by Tukey's post hoc test, was employed when comparing multiple conditions. A P value of <5% (P < .05) was considered statistically significant.
3. RESULTS
3.1. High glucose leads to increased endothelial production of MALAT1, which is associated with increased pro‐inflammatory transcripts
Endothelial cells are primary targets of glucose‐induced alterations due to their location and function as the proximal barrier to blood. Considering these characteristics, we examined microvascular endothelial cells cultured under normal (5 mmol/L; NG) or high glucose conditions (25 mmol/L; HG) to evaluate MALAT1 expression. Our initial experiments showed that MALAT1 expression peaks after 48 hours of HG incubation; hence, we used this time point for all of our in vitro experiments. We found that at 48 hours, both MALAT1 and RNA transcripts coding for pro‐inflammatory cytokines (TNF‐α, IL‐6 and IL‐1β) were significantly elevated (Figure 1A‐D) following HG incubation. There were no significant changes in MALAT1 expression with 25 mmol/L l‐glucose (Osm; osmotic control). In situ hybridization confirmed the presence of MALAT1 in the nucleus of endothelial cells, and no glucose‐induced alterations of microscopic distribution were observable (Figure 1E). Furthermore, extending our findings into HCMECs, MALAT1 was significantly elevated at 12 hours following HG incubation—suggesting a cell‐specific phenotype for MALAT1 (Figure S1).
Figure 1.
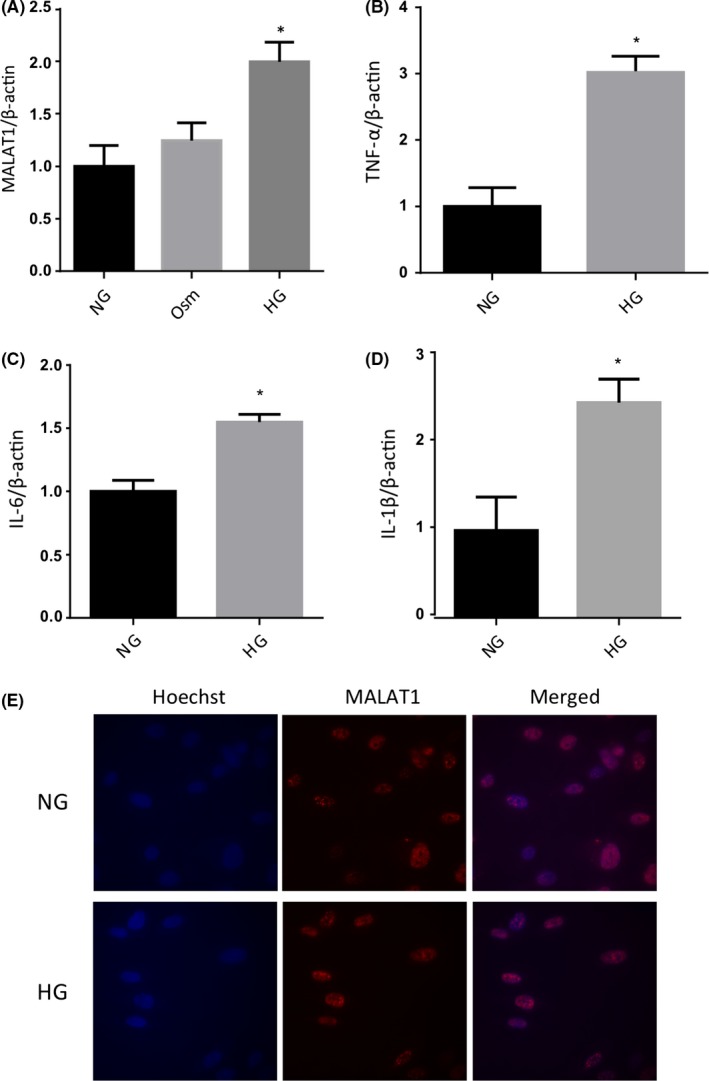
qRT‐PCR and FISH analyses in human retinal microvascular endothelial cells. Real‐time reverse transcription PCR analysis showing 25 mmol/L glucose (HG) induced upregulation of (A) MALAT1 transcript and (B‐D) pro‐inflammatory cytokine (TNF‐α, IL‐6 and IL‐1β) transcripts in endothelial cells compared to 5 m mmol/L glucose (NG) at 48 hours. No changes were seen following incubation with 25 m mmol/L l‐glucose (osmotic control, Osm). E, Fluorescent in situ hybridization, using MALAT1‐specific probes (red), confirmed MALAT1's localization to nuclear speckles without any demonstrable glucose‐induced changes in the subcellular distribution [data expressed as mean ± SEM; normalized to NG, *=P < .05 compared to NG; experiment performed in triplicate from 3 independent experiments; Hoechst (blue) counterstain; original magnification, 60×]
3.2. Animal monitoring
As the primary aim of this study was to delineate the role for MALAT1 in the regulation of inflammatory cytokines in tissues affected by hyperglycaemia, we used both WT C57/BL6 mice and Malat1 KO animals with STZ‐induced diabetes and nondiabetic controls. Both diabetic WT and KO animals developed hyperglycaemia and had reduced body weight gain compared to the nondiabetic WT and KO mice (Figure 2A,B). There were no observable differences between WT and KO control animals.
Figure 2.
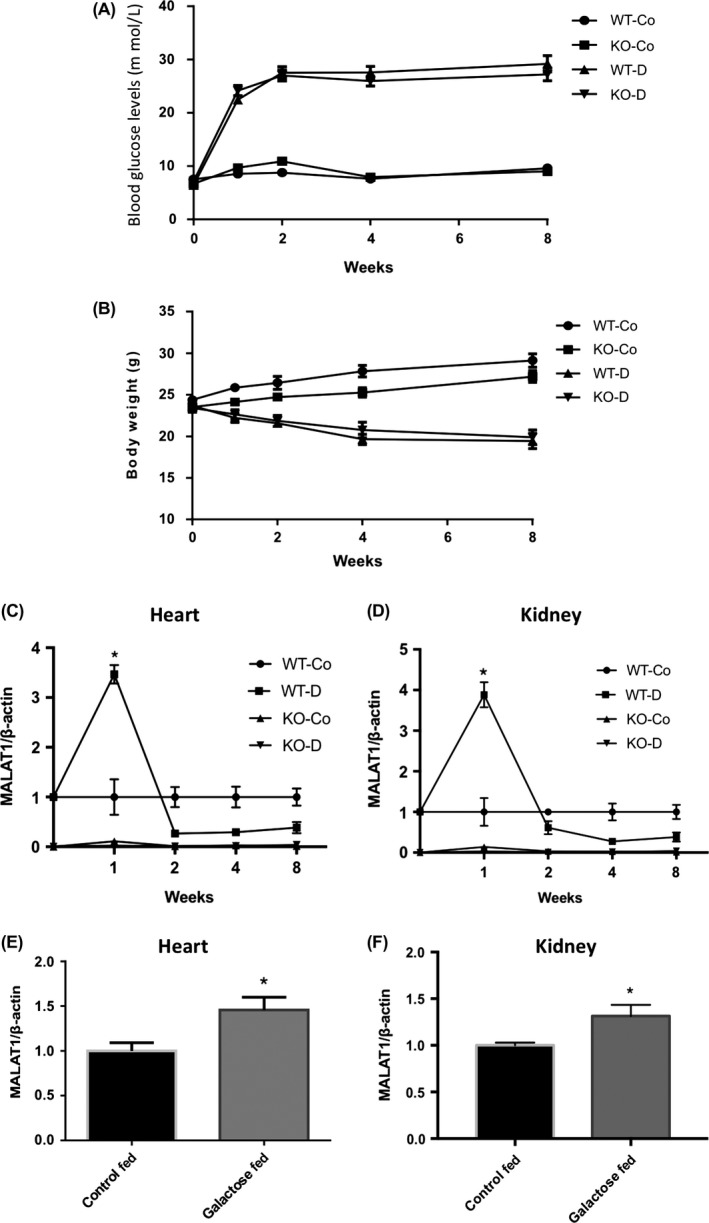
Animal monitoring data and MALAT1 expression in mice heart and kidney tissues. A, Blood glucose levels (mmol/L) and (B) body weights (g) of mice during the follow‐up period. Both wild‐type diabetic (WT‐D) and MALAT1−/− diabetic (KO‐D) mice were hyperglycaemic and had reduced body weight gain at all time points compared to wild‐type nondiabetic controls (WT‐Co) and MALAT1−/− nondiabetic controls (KO‐Co) animals. Loss of MALAT1 had no significant effect on these parameters [n = 8‐13; data expressed as mean ± SEM]. Real‐time reverse transcription PCR analysis of MALAT1 transcripts in the (C) hearts and (D) kidneys from WT control (WT‐Co), WT diabetic (WT‐D), MALAT1−/− control (KO‐Co) and MALAT1−/− diabetic (KO‐D) mice at several time points showed an early increase in MALAT1 expression in the WT‐D animals. Increased MALAT1 RNA expressions were also seen in the (E) hearts and (F) kidneys of diabetic animals following 1 week of galactose feeding (“Galactose Fed”) (*P < 0.05 compared to WT‐Co or Control Fed; n = 8 or more/group; data expressed as mean ± SEM; normalized to β‐actin; and expressed as a fold change in WT‐Co or Control Fed)
3.3. Diabetes‐induced cardiac and kidney inflammatory gene expressions are regulated by MALAT1
With the heart and kidneys being target sites for tissue damage in diabetes, we first wanted to establish the effects of MALAT1 on animal hearts and kidneys affected by diabetes. Control and diabetic mice were sacrificed at 5 time points over 2 months following the onset of diabetes (0, 1, 2, 4 and 8 weeks). We found that MALAT1 expression levels in heart and kidney tissues of WT diabetic animals were initially elevated more than threefold at 1 week when compared to WT nondiabetic animals. However, expression levels of MALAT1 in the WT diabetic animals decreased after 1 week and remained at this level for the remaining time points. Malat1 KO animals sacrificed at the same time points had no detectable levels of MALAT1 (Figure 2C,D).
To further examine whether this early increase in MALAT1 might be a result of the STZ, we implemented a metabolic model of hyperhexosemia using a custom diet high in galactose, where mice fed this custom diet were compared to mice on a standard diet—we have used this approach in the past.38, 39 We found that 1 week of galactose feeding led to significantly elevated levels of MALAT1 in heart and kidney tissues compared to age‐ and gender‐matched mice that were fed a standard diet (Figure 2E,F).
3.4. MALAT1 regulates increased levels of inflammatory cytokines in the heart during diabetes
We examined pro‐inflammatory cytokines (IL‐1β, IL‐6, TNF‐α and IFN‐λ) in mice cardiac tissues at 1 and 2 months postonset of diabetes (Figure 3). WT diabetic animals had significantly elevated levels of pro‐inflammatory transcripts at both time points compared to controls. Interestingly, we found that diabetic KO animals had significantly lower levels of these transcripts. These levels were similar to the baseline levels found in control animals. No significant differences were found between WT control and KO control animals. We further expanded the study and examined protein levels of IL‐6 and INF‐γ in the hearts of WT and Malat1 KO mice. Diabetes caused upregulation of IL‐6 and IFN‐λ protein levels in WT diabetic animals; however, such changes were absent in diabetic Malat1 KO animals (Figure 4A,B).
Figure 3.
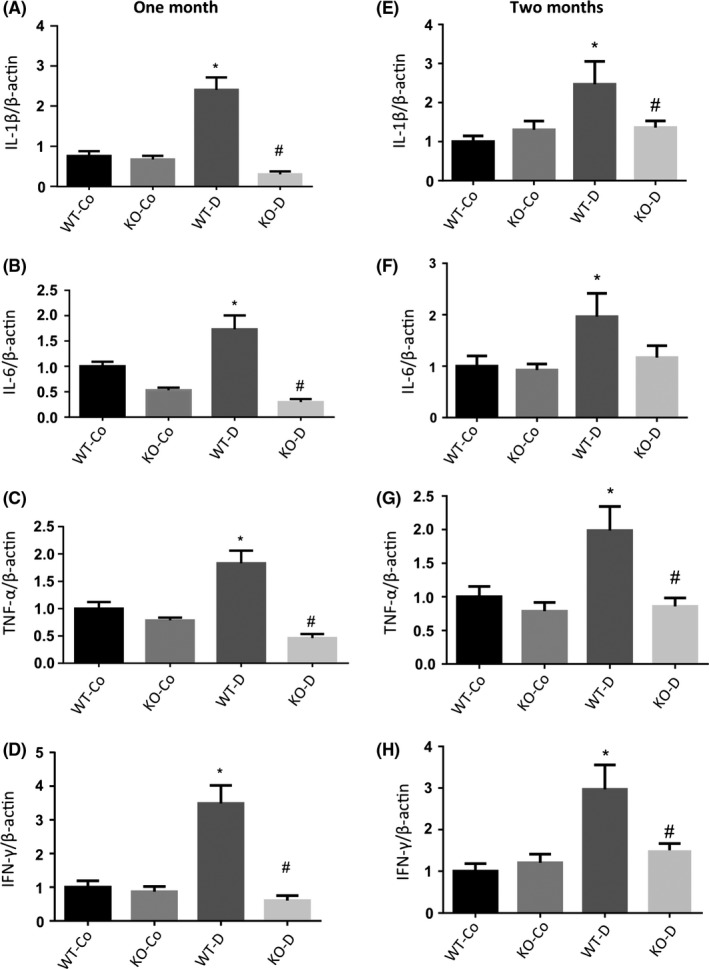
qRT‐PCR analysis of inflammatory markers in mice hearts at 1 and 2 months. Real‐time reverse transcription PCR analysis of the hearts from WT control (WT‐Co), WT diabetic (WT‐D), MALAT1−/− control (KO‐Co) and MALAT1−/− diabetic (KO‐D) mice at 1 (left panel) and 2 months (right panel) showed upregulations of (A,E) IL‐1β; (B,F) IL‐6; (C,G) TNF‐α and (D,H) INF‐γ in WT diabetic animals. Loss of MALAT1 prevented such increases in the diabetic KO group (KO‐D). No changes in normal glucose expression were observed in the KO‐Co animals (*P < .05 compared to WT‐Co, #P < .05 compared to WT‐D; n = 6/group; data expressed as mean ± SEM; normalized to β‐actin; and expressed as a fold change in WT‐Co)
Figure 4.
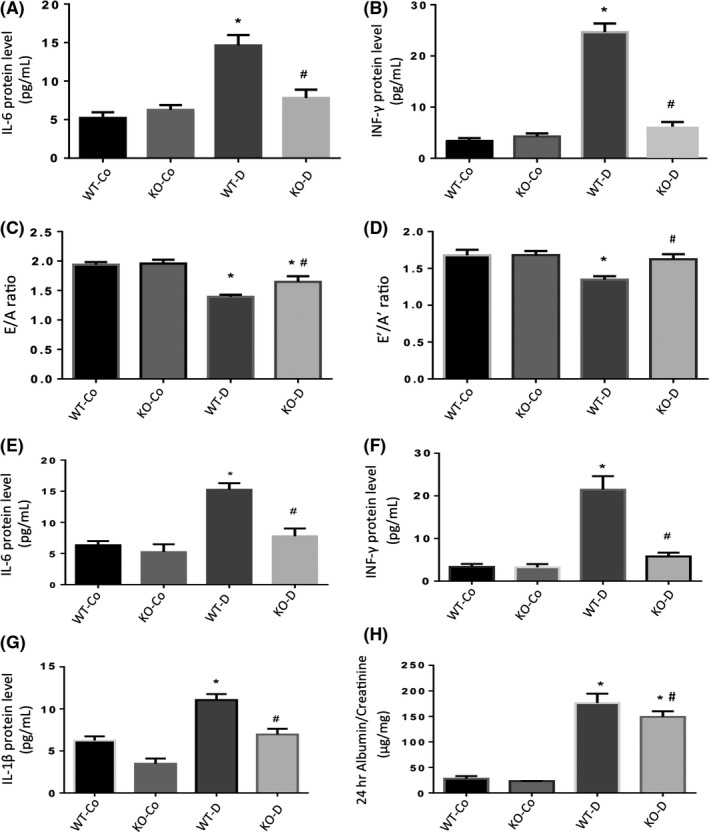
Protein levels, echocardiographic and albumin: creatinine analyses in mice hearts and kidneys at 2 months. Protein expressions of (A) IL‐6 and (B) INF‐γ (upper panel, measured by ELISA) were significantly increased in the hearts of WT‐D mice following 2 months of diabetes compared to WT‐Co mice. Such diabetes‐induced upregulations were prevented in the KO‐D animals. Echocardiographic analyses of mice showed a significant decrease in the (C) E/A ratio and (D) E′/A′ ratio in WT‐D animals after 2 months of diabetes. Such diabetes‐induced changes were corrected in the KO‐D mice. Protein expressions of (E) IL‐6, (F) INF‐γ and (G) IL‐1β (measured by ELISA) were significantly increased in the kidneys of WT‐D mice compared to WT‐Co mice following 2 months of diabetes. Such diabetes‐induced upregulations were prevented in the KO‐D animals. (H) Albumin: creatinine ratios (ACr) were elevated in the WT‐D animals following 2 months of diabetes compared to WT‐Co animals. Loss of MALAT1 partially reduced such levels in the KO‐D animals. KO‐Co and WT‐Co animals had no differences in any of these parameters (*P < .05 compared to WT‐Co, #P < .05 compared to WT‐D; n ≥ 8/group; data expressed as mean ± SEM)
3.5. Loss of MALAT1 reduced cardiac dysfunction postdiabetes
We also examined whether such RNA and protein changes affected cardiac function. Hence, we analysed echocardiographic data from 2‐month diabetic animals at the time of sacrifice. Both nondiabetic Malat1 KO and WT control animals showed normal E/A ratios. In contrast, WT diabetic animals had significantly decreased E/A ratios, indicating diastolic dysfunction. Diabetic Malat1 KO animals had a partial protective effect on diabetes‐induced cardiac dysfunction (Figure 4C,D). No significant differences in heart rates were observed between all groups (Table S2).
3.6. Loss of MALAT1 protects against increased levels of inflammatory cytokines in the kidneys during diabetes
Similarly, we examined renal tissues for changes in cytokines at the mRNA and protein levels after 1 and 2 months of follow‐up. We found that these cytokine transcripts (IL‐1β, IL‐6, TNF‐α and IFN‐λ) were elevated in renal tissues from WT diabetic animals. However, such elevations in RNA and protein levels were prevented in diabetic KO animals at both time points (Figures 4E‐G and 5). To assess renal function, we used the 24‐hour albumin: creatinine (ACr) ratio. The WT diabetic animals showed significantly increased ACr ratio compared to the WT control animals. Diabetic animals with MALAT1 KO showed a partial reduction in ACr ratio (Figure 4H).
Figure 5.
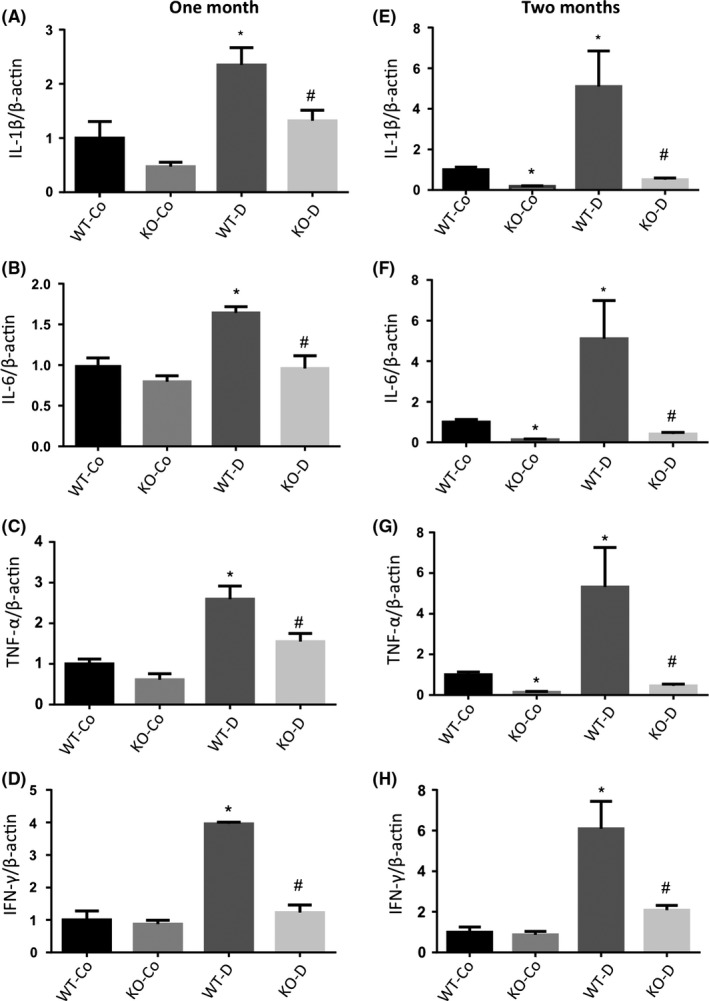
qRT‐PCR analysis of inflammatory markers in mice kidneys at 1 and 2 months. Real‐time reverse transcription PCR analysis of the kidneys from WT control (WT‐Co), WT diabetic (WT‐D), MALAT1−/− control (KO‐Co) and MALAT1−/− diabetic (KO‐D) mice at 1 (left panel) and 2 months (right panel) showed upregulations of (A,E) IL‐1β; (B,F) IL‐6; (C,G) TNF‐α and (D,H) INF‐γ in WT diabetic animals. Loss of MALAT1 prevented such increases in the diabetic KO group (KO‐D). No changes in expressions were observed in the KO‐Co and WT‐Co animals (*P < .05 compared to WT‐Co, #P < .05 compared to WT‐D; n = 6/group; data expressed as mean ± SEM; normalized to β‐actin; and expressed as a fold change in WT‐Co)
4. DISCUSSION
Our current study demonstrated that the lncRNA MALAT1 regulates inflammatory cytokine production in diabetes. We have demonstrated such regulation in endothelial cells, the primary target of glucose‐induced vascular damage, as well as the kidneys and heart in diabetes. Furthermore, we have shown that such changes and subsequent organ dysfunction in diabetes are prevented in Malat1 KO mice.
It has been previously established that secondary to sustained hyperglycaemia in diabetes, persistent low‐grade inflammatory processes resulting from increased cytokine expression leads to cellular insult, tissue injury and functional impairments.9, 40, 41 This study delineates a novel mechanism mediating such processes at the transcriptional level. Our study further implies that the nuclear‐retained lncRNA MALAT1 is a regulator of these cytokines during hyperglycaemic conditions.
Extending the findings from our previous MALAT1 study, here, we targeted the heart and kidneys because both organs are substantially affected by diabetes and highly express MALAT1.17, 18, 28, 42 We examined the mechanisms at various levels of complexities, using microvascular endothelial cells, and then translated our in vitro work to an animal model.
Moreover, lncRNAs work by diverse molecular mechanisms to regulate global gene expression.43, 44, 45 To demonstrate MALAT1′s regulatory capabilities directly, West and colleagues adapted a CHART (Capture Hybridization Analysis of RNA Targets)–mass spectrometry approach, in which they identified that MALAT1 is capable of binding to hundreds of active genes.46 Based on their findings, West et al46 hypothesize that MALAT1 may have roles in interacting with nuclear bodies and providing structural support to various proteins at highly transcribed loci. Further, histone methylation and specific transcription factors may also regulate the expression of lncRNAs.47
In keeping with previous studies,48, 49 we found that MALAT1 is localized to nuclear speckles in endothelial cells. Interestingly, pre‐mRNA splicing factors are also localized in these nuclear domains, which may allude to other implications of MALAT1 in nuclear organization.50 Although the exact mechanism of glucose‐induced regulation of MALAT1 remains to be deciphered, it may be regulated epigenetically through its association with Polycomb repressive complex 2.51 Intriguingly, we have previously shown glucose‐induced alterations of miRs and another lncRNA through such pathways.43, 52
It is of further interest to note that other substrates (eg fatty acids) may also change MALAT1 expression.53 Relationships of such alterations, with respect to chronic diabetic complications, need additional exploration and may uncover novel links between MALAT1 function and pathological processes. Moreover, this study has shown that although MALAT1 upregulation was seen after 1 week of high glucose incubation, the initial increase in MALAT1 showed long‐lasting effects. Most probably, this can be explained by the “metabolic memory” phenomenon. In keeping with this theory, El‐Osta et al54 have previously shown that transient hyperglycaemia can lead to long‐term histone methylation alterations and transcription factor activation. However, we do recognize that additional experiments are needed to confirm this notion.
Our findings, in general, show that MALAT1 is an important contributor to increased levels of inflammatory cytokines in diabetic mice at 1 and 2 months. As observed from our diabetic KO group, globally eliminating Malat1 had a significant impact on inflammatory cytokines at 1 and 2 months—suggesting that MALAT1 may have a role in inflammatory regulation during diabetes. Furthermore, Hu et al32 have recently suggested that early high glucose‐induced increases of MALAT1 may be capable of activating and sustaining splicing patterns for certain pre‐mRNAs, as well as potentially activating a degradation mechanism that “overcorrects” the MALAT1 expression at later time points. Interestingly, a similar phenomenon was observed in our cardiac endothelial cells exposed to HG (Figure S1) and diabetic animal kidney and heart tissues (Figure 2C,D), in which earlier time points showed an early upregulation of MALAT1 and a subsequent reduction in expression at later time points. Nevertheless, it is always possible and almost certain that other molecules may further contribute to the regulation of MALAT1 in hyperglycaemic environments.
The exact mechanisms of MALAT1 mediating the regulation of inflammatory cytokines also remain to be explored. However, we have previously demonstrated that such changes may occur through one regulatory pathway involving serum amyloid antigen protein 3 (SAA3).28
Unlike its typically high levels of expression in various cancers,19, 55, 56 we report a transient increase in MALAT1 expression at 1‐week postonset of diabetes, followed by a reduction in MALAT1 expression in tissues from diabetic mice. In contrast, in the retinas of diabetic db/db mice, other investigators have shown increased MALAT1 expression throughout the follow‐up period.27 Interestingly, in our WT diabetic mice, this initial upregulation of MALAT1 was capable of sustaining the inflammatory response and lack of MALAT1 was sufficient to eliminate such changes in the KO mice. The changes in MALAT1 expression, following the increase at 1‐week postonset of diabetes, may indicate the induction of an epigenetic regulatory change. A mechanism of this nature could be initiated by MALAT1's ability to regulate epigenetic histone modifications in the chromatin early on, which may ultimately contribute to sustained inflammation, even in the presence of reduced MALAT1 RNA levels.44 Insights into how MALAT1 modulates the epigenetic landscape in diabetic complications warrant further investigation.
The E/A and E′/A′ ratios are measurements of left ventricular diastolic function and were significantly decreased in WT diabetic animals. Such changes are indicative of cardiac/diastolic dysfunction.57, 58 However, this decrease was partially or fully (in the case of E′/A′ ratio) eliminated in Malat1 knockout diabetic animals—supporting our findings that the loss of MALAT1 may have a protective effect. However, our analysis of renal function using collected urine to examine the ACr ratio showed only partial functional gain. Such findings may suggest that the role of MALAT1 in the pathogenesis of diabetic complications may vary between tissues. A pathogenetic role of MALAT1 is further supported by the demonstration of increased MALAT1 expression and pathological sequelae in the retina of diabetic animals.27
Hence in future studies, conducting cell‐specific experiments for these tissues may help to elucidate the function of MALAT1 in these organs and the cells that comprise them. In this study, we report that the loss of MALAT1 eliminates the increased production of inflammatory cytokines that is typical of diabetes in both cardiac and renal tissues. We also demonstrated in vivo that the loss of MALAT1 is protective against cardiac dysfunction, which is observed in diabetes.
Nevertheless, this is the first study to use a chronically diabetic KO animal model to examine MALAT1 in mediating cardiac and renal damage. In accordance with past findings,30, 31 our study demonstrates that MALAT1 plays a significant role in mediating diabetes‐induced tissue damage through regulation of the inflammatory process. Using a well‐established model of type 1 diabetes, our research provides direct evidence that MALAT1 plays a pathogenetic role in chronic diabetic complications affecting the heart and kidneys. Establishing this link between lncRNAs and diabetes opens the door for novel RNA‐based therapeutic targets. However, it becomes important to recognize that there are other epigenetic mechanisms (eg histone methylation and acetylation, and the activity of miRs) in play that may modulate such processes.44 Furthermore, in the current context, whether epigenetic mechanisms observed here can lead to permanent alterations require additional investigation.59 Well‐designed long‐term studies are needed to address such questions and improve our understanding of lncRNAs and their functions.
CONFLICT OF INTEREST
The authors have declared no conflict of interests.
AUTHOR CONTRIBUTIONS
ADG and SC contributed to experimental conception and design. ADG, BF and SB performed the experiments. ADG, SB, BF and SC contributed reagents/materials/analysis tools. ADG, SB and SC performed writing of manuscript.
DISCLOSURE
The authors do not have any conflicts of interest to disclose.
Supporting information
ACKNOWLEDGEMENTS
We gratefully acknowledge Dr. David Spector and his team from the Cold Spring Harbor Laboratory for the generation and generous contribution of the Malat1 KO mice. This research was supported by the Heart and Stroke Foundation of Canada.
Gordon AD, Biswas S, Feng B, Chakrabarti S. MALAT1: A regulator of inflammatory cytokines in diabetic complications. Endocrinol Diab Metab. 2018;1:e10 10.1002/edm2.10
Funding information
This research was supported by grants from the Heart and Stroke Foundation of Canada.
REFERENCES
- 1. Heron M. Deaths: leading causes for 2013. Natl Vital Stat Rep. 2016;65:1‐95. [PubMed] [Google Scholar]
- 2. Zhang P, Zhang X, Brown J, et al. Global healthcare expenditure on diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87:293‐301. [DOI] [PubMed] [Google Scholar]
- 3. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813‐820. [DOI] [PubMed] [Google Scholar]
- 4. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058‐1070. 10.1161/circresaha.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152:1237‐1251 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rains JL, Jain SK. Oxidative stress, insulin signaling, and diabetes. Free Radic Biol Med. 2011;50:567‐575. 10.1016/j.freeradbiomed.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kayama Y, Raaz U, Jagger A, et al. Diabetic cardiovascular disease induced by oxidative stress. Int J Mol Sci. 2015;10:25234‐25263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Graves DT, Kayal RA. Diabetic complications and dysregulated innate immunity. Front Biosci. 2008;13:1227‐1239. 10.2741/2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wei M, Li Z, Xiao L, Yang Z. Effects of ROS‐relative NF‐κB signaling on high glucose‐induced TLR4 and MCP‐1 expression in podocyte injury. Mol Immunol. 2015;68:261‐271. [DOI] [PubMed] [Google Scholar]
- 10. Yerneni KK, Bai W, Khan BV, Medford RM, Natarajan R. Hyperglycemia‐induced activation of nuclear transcription factor kappaB in vascular smooth muscle cells. Diabetes. 1999;48:855‐864. [DOI] [PubMed] [Google Scholar]
- 11. Iwasaki Y, Kambayashi M, Asai M, Yoshida M, Nigawara T, Hashimoto K. High glucose alone, as well as in combination with proinflammatory cytokines, stimulates nuclear factor kappa‐B‐mediated transcription in hepatocytes in vitro. J Diabetes Complications. 2007;21:56‐62. [DOI] [PubMed] [Google Scholar]
- 12. Feng B, Cao Y, Chen S, Chu X, Chu Y, Chakrabarti S. miR‐200b Mediates endothelial‐to‐mesenchymal transition in diabetic cardiomyopathy. Diabetes. 2016;3:768‐779. [DOI] [PubMed] [Google Scholar]
- 13. Mortuza R, Feng B, Chakrabarti S. SIRT1 reduction causes renal and retinal injury in diabetes through endothelin 1 and transforming growth factor β1. J Cell Mol Med. 2015;8:1857‐1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Spurlock CF, Crooke PS, Aune TM. Biogenesis and transcriptional regulation of long noncoding RNAs in the human immune system. J Immunol. 2016;197:4509‐4517. 10.4049/jimmunol.1600970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roberts TC, Morris KV, Weinberg MS. Perspectives on the mechanism of transcriptional regulation by long non‐coding RNAs. Epigenetics. 2014;9:13‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ji P, Diederichs S, Wang W, et al. MALAT‐1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early‐stage non‐small cell lung cancer. Oncogene. 2003;22:8031‐8041. [DOI] [PubMed] [Google Scholar]
- 17. Ma X‐Y, Wang J‐H, Wang J‐L, Ma CX, Wang X‐C, Liu F‐S. Malat1 as an evolutionarily conserved lncRNA, plays a positive role in regulating proliferation and maintaining undifferentiated status of early‐stage hematopoietic cells. BMC Genomics. 2015;16:676 10.1186/s12864-015-1881-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guo F, Guo L, Li Y, Zhou Q, Li Z. MALAT1 is an oncogenic long non‐coding RNA associated with tumor invasion in non‐small cell lung cancer regulated by DNA methylation. Int J Clin Exp Pathol. 2015;8:15903‐15910. [PMC free article] [PubMed] [Google Scholar]
- 19. Li T, Mo X, Fu L, Xiao B, Guo J. Molecular mechanisms of long noncoding RNAs on gastric cancer. Oncotarget. 2016;7:8601‐8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011;147:1537‐1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang B, Arun G, Mao YS, et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis‐regulatory role in the adult. Cell Rep. 2012;2:111‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eißmann M, Gutschner T, Hämmerle M, et al. Loss of the abundant nuclear non‐coding RNA MALAT1 is compatible with life and development. RNA Biol. 2012;9:1076‐1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morán I, Akerman I, Van De Bunt M, et al. Human β cell transcriptome analysis uncovers lncRNAs that are tissue‐specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab. 2012;16:435‐448. 10.1016/j.cmet.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. You L, Wang N, Yin D, et al. Downregulation of long noncoding RNA Meg3 affects insulin synthesis and secretion in mouse pancreatic beta cells. J Cell Physiol. 2016;231:852‐862. 10.1002/jcp.25175. [DOI] [PubMed] [Google Scholar]
- 25. Petry CJ, Evans ML, Wingate DL, et al. Raised late pregnancy glucose concentrations in mice carrying pups with targeted disruption of H19Δ13. Diabetes. 2010;59:282‐286. 10.2337/db09-0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Millis MP, Bowen D, Kingsley C, Watanabe RM, Wolford JK. Variants in the plasmacytoma variant translocation gene (PVT1) are associated with end‐stage renal disease attributed to type 1 diabetes. Diabetes. 2007;56:3027‐3032. 10.2337/db07-0675. [DOI] [PubMed] [Google Scholar]
- 27. Liu J‐Y, Yao J, Li X‐M, et al. Pathogenic role of lncRNA‐MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis. 2014;5:e1506 10.1038/cddis.2014.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Puthanveetil P, Chen S, Feng B, Gautam A, Chakrabarti S. Long non‐coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J Cell Mol Med. 2015;19:1418‐1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gast M, Schroen B, Voigt A, et al. Long noncoding RNA MALAT1‐derived mascRNA is involved in cardiovascular innate immunity. J Mol Cell Biol. 2016;8:178‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang M, Gu H, Chen J, Zhou X. Involvement of long noncoding RNA MALAT1 in the pathogenesis of diabetic cardiomyopathy. Int J Cardiol. 2016;202:753‐755 10.1016/j.ijcard.2015.10.019. [DOI] [PubMed] [Google Scholar]
- 31. Zhang M, Gu H, Xu W, Zhou X. Down‐regulation of lncRNA MALAT1 reduces cardiomyocyte apoptosis and improves left ventricular function in diabetic rats. Int J Cardiol. 2016;203:214‐216 10.1016/j.ijcard.2015.10.136. [DOI] [PubMed] [Google Scholar]
- 32. Hu M, Wang R, Li X, et al. LncRNA MALAT1 is dysregulated in diabetic nephropathy and involved in high glucose‐induced podocyte injury via its interplay with β‐catenin. J Cell Mol Med. 2017;21:2732‐2747. 10.1111/jcmm.13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Feng B, Chen S, George B, Feng Q, Chakrabarti S. miR133a regulates cardiomyocyte hypertrophy in diabetes. Diabetes Metab Res Rev. 2010;26:40‐49. [DOI] [PubMed] [Google Scholar]
- 34. Chen S, Khan ZA, Cukiernik M, Chakrabarti S. Differential activation of NF‐kappa B and AP‐1 in increased fibronectin synthesis in target organs of diabetic complications. Am J Physiol Endocrinol Metab. 2003;284:E1089‐E1097. [DOI] [PubMed] [Google Scholar]
- 35. Pistner A, Belmonte S, Coulthard T, Blaxall B. Murine echocardiography and ultrasound imaging. J Vis Exp. 2010;42:pii: 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Coassin SR, Orjalo AV, Semaan SJ, Johansson HE. Simultaneous detection of nuclear and cytoplasmic RNA variants utilizing Stellaris® RNA fluorescence in situ hybridization in adherent cells. Methods Mol Biol. 2014;1211:189‐199. [DOI] [PubMed] [Google Scholar]
- 37. Chen S, Feng B, Thomas AA, Chakrabarti S. MiR‐146a regulates glucose induced upregulation of inflammatory cytokines extracellular matrix proteins in the retina and kidney in diabetes. PLoS One. 2017;12:e0173918 10.1371/journal.pone.0173918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xu B, Chiu J, Feng B, Chen S, Chakrabarti S. PARP activation and the alteration of vasoactive factors and extracellular matrix protein in retina and kidney in diabetes. Diabetes Metab Res Rev. 2008;24:404‐412. 10.1002/dmrr.842. [DOI] [PubMed] [Google Scholar]
- 39. Xin X, Chen S, Khan ZA, Chakrabarti S. Akt activation and augmented fibronectin production in hyperhexosemia. Am J Physiol Endocrinol Metab. 2007;293:E1036‐E1044. 10.1152/ajpendo.00271.2007. [DOI] [PubMed] [Google Scholar]
- 40. Van Greevenbroek MMJ, Schalkwijk CG, Stehouwer CD. Obesity‐associated low‐grade inflammation in type 2 diabetes mellitus: causes and consequences. Neth J Med. 2013;71:174‐187. [PubMed] [Google Scholar]
- 41. Minihane AM, Vinoy S, Russell WR, et al. Low‐grade inflammation, diet composition and health: current research evidence and its translation. Br J Nutr. 2015;114:999‐1012. 10.1017/S0007114515002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lorenzen JM, Thum T. Long noncoding RNAs in kidney and cardiovascular diseases. Nat Rev Nephrol. 2016;12:360‐373. 10.1038/nrneph.2016.51. [DOI] [PubMed] [Google Scholar]
- 43. Thomas AA, Feng B, Chakrabarti S. ANRIL: a regulator of VEGF in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2017;58:470‐480. 10.1167/iovs.16-20569. [DOI] [PubMed] [Google Scholar]
- 44. Reddy MA, Zhang E, Natarajan R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia. 2014;58:443‐455. 10.1007/s00125-014-3462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145‐166. 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. West JA, Davis CP, Sunwoo H, et al. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol Cell. 2014;55:791‐802. 10.1016/j.molcel.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dey BK, Mueller AC, Dutta A. Long non‐coding RNAs as emerging regulators of differentiation, development, and disease. Transcription. 2014;5:e944014 10.4161/21541272.2014.944014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hutchinson JN, Ensminger AW, Clemson CM, Lynch CR, Lawrence JB, Chess A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics. 2007;8:39 10.1186/1471-2164-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tripathi V, Shen Z, Chakraborty A, et al. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B‐MYB. PLoS Genet. 2013;9:e1003368 10.1371/journal.pgen.1003368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lamond AI, Spector DL. Nuclear speckles: a model for nuclear organelles. Nat Rev Mol Cell Biol. 2003;4:605‐612. 10.1038/nrm1172. [DOI] [PubMed] [Google Scholar]
- 51. Davidovich C, Zheng L, Goodrich KJ, Cech TR. Promiscuous RNA binding by Polycomb repressive complex 2. Nat Struct Mol Biol. 2013;20:1250‐1257. 10.1038/nsmb.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ruiz MA, Feng B, Chakrabarti S. Polycomb repressive complex 2 regulates MiR‐200b in retinal endothelial cells: potential relevance in diabetic retinopathy. PLoS One. 2015;10:e0123987 10.1371/journal.pone.0123987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yan C, Chen J, Chen N. Long noncoding RNA MALAT1 promotes hepatic steatosis and insulin resistance by increasing nuclear SREBP‐1c protein stability. Sci Rep. 2016;6:22640 10.1038/srep22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. El‐Osta A, Brasacchio D, Yao D, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008;205:2409‐2417. 10.1084/jem.20081188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tani H, Nakamura Y, Ijiri K, Akimitsu N. Stability of MALAT‐1, a nuclear long non‐coding RNA in mammalian cells, varies in various cancer cells. Drug Discov Ther. 2010;4:235‐239. [PubMed] [Google Scholar]
- 56. Luo JH, Ren B, Keryanov S, et al. Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas. Hepatology. 2006;44:1012‐1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Semeniuk LM, Kryski AJ, Severson DL. Echocardiographic assessment of cardiac function in diabetic db/db and transgenic db/db‐hGLUT4 mice. Am J Physiol Heart Circ Physiol. 2002;283:H976‐H982. 10.1152/ajpheart.00088.2002. [DOI] [PubMed] [Google Scholar]
- 58. Yuan X, Xiao Y‐C, Zhang G‐P, et al. Chloroquine improves left ventricle diastolic function in streptozotocin‐induced diabetic mice. Drug Des Devel Ther. 2016;10:2729‐2737. 10.2147/DDDT.S111253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ciabrelli F, Comoglio F, Fellous S, et al. Stable polycomb‐dependent transgenerational inheritance of chromatin states in Drosophila . Nat Genet. 2017;49:876‐886. 10.1038/ng.3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials