

Abstract
Motivation
Precise assessment of ligand bioactivities (including IC50, EC50, Ki, Kd, etc.) is essential for virtual screening and lead compound identification. However, not all ligands have experimentally determined activities. In particular, many G protein-coupled receptors (GPCRs), which are the largest integral membrane protein family and represent targets of nearly 40% drugs on the market, lack published experimental data about ligand interactions. Computational methods with the ability to accurately predict the bioactivity of ligands can help efficiently address this problem.
Results
We proposed a new method, WDL-RF, using weighted deep learning and random forest, to model the bioactivity of GPCR-associated ligand molecules. The pipeline of our algorithm consists of two consecutive stages: (i) molecular fingerprint generation through a new weighted deep learning method, and (ii) bioactivity calculations with a random forest model; where one uniqueness of the approach is that the model allows end-to-end learning of prediction pipelines with input ligands being of arbitrary size. The method was tested on a set of twenty-six non-redundant GPCRs that have a high number of active ligands, each with 200–4000 ligand associations. The results from our benchmark show that WDL-RF can generate bioactivity predictions with an average root-mean square error 1.33 and correlation coefficient (r2) 0.80 compared to the experimental measurements, which are significantly more accurate than the control predictors with different molecular fingerprints and descriptors. In particular, data-driven molecular fingerprint features, as extracted from the weighted deep learning models, can help solve deficiencies stemming from the use of traditional hand-crafted features and significantly increase the efficiency of short molecular fingerprints in virtual screening.
Availability and implementation
The WDL-RF web server, as well as source codes and datasets of WDL-RF, is freely available at https://zhanglab.ccmb.med.umich.edu/WDL-RF/ for academic purposes.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
G protein-coupled receptors (GPCRs) are an important superfamily of transmembrane proteins involved in various signal transduction pathways. They play critical roles in many physiological processes by binding with G proteins or arrestins to regulate downstream activities (Miller and Lefkowitz, 2001). GPCRs are closely tied to many human diseases, such as cancer and diabetes, and are the targets of approximately 40% of modern medical drugs (Overington et al., 2006). Since many GPCRs are unstable or challenging to crystallize (Tautermann, 2014), obtaining their three-dimensional (3D) structures has remained challenging. At present, only a very small portion of human GPCRs have 3D structures available in the PDB (Berman et al., 2000; Zhang et al., 2015) (see also https://zhanglab.ccmb.med.umich.edu/GPCR-EXP/). As a result, this lack of GPCR structural information has proven to be a major barrier in a multitude of virtual screening and rational drug design studies, particularly those targeting GPCRs (Becker et al., 2004; Wootten et al., 2013).
In general, drug discovery campaigns often start with the screening of thousands to millions of chemical compounds against a therapeutic target by biological high-throughput assays, where bioactivities are typically measured by IC50, EC50, Ki and Kd values. Subsequently, hits are chosen based on their activity and modified to become stronger binders or more selective for their target (Unterthiner et al., 2014). However, biological high-throughput assays for screening compounds are usually time consuming and labor intensive. Even worse, there is only a very small population of ‘available compounds’, and not all GPCR targets are suitable for direct high-throughput screening assays to obtain their bioactivities interacting with compounds (Blum and Reymond, 2009). Thus, the use of computationally based virtual screening has been implemented as a complement to experimental efforts.
Virtual screening can be divided into structure-based and ligand-based techniques (Cereto-Massagué et al., 2015). The structure-based techniques perform compound screening by simulating physical interaction between known compounds and a biomolecular target, but they are only applicable if the 3D structure of the target protein is available (Cereto-Massagué et al., 2015). On the other hand, ligand-based techniques predict the activity of a compound on a biomolecular target through known experimental data, where machine learning-based methods have found significant usefulness and have been widely used in drug design (Cereto-Massagué et al., 2015; Shang et al., 2017).
Many of the machine learning-based virtual screening methods implement the high-throughput screening experiments using approaches such as Bayesian statistical methods, nearest neighbor methods, support vector machines and artificial neural network (Unterthiner et al., 2014). In recent years, deep learning methods have been particularly successful in several studies employing ligand-based virtual screening and the generation of molecular fingerprints. In 2012, for instance, Merck organized a Kaggle challenge, where participants developed machine learning models to predict the bioactivities of ligands interacting with drug targets, whereupon approaches using deep learning performed the best. In 2014, researchers from Johannes Kepler University of Austria and Johnson & Johnson Pharmaceutical Research & Development developed a deep learning-based virtual screening model and successfully applied it to the benchmark containing more than 1200 targets and 1.3 M compounds (Unterthiner et al., 2014). In 2015, Adams and coworkers designed a molecular fingerprint generation method based on a convolutional neural network that operates directly on graphs and applied it to the prediction of drug activity, molecular solubility and other properties (Duvenaud et al., 2015). Most recently, Shang et al. proposed HybridSim-VS, which combines 2D fingerprint and 3D shape-based methods for virtual screening, demonstrating advantage of the combined method over the individual approaches (Cereto-Massagué et al., 2015; Shang et al., 2017); the web server of HybridSim-VS has access to more than 17 M compounds.
The common strategy of machine learning-based virtual screening is to first use off-the-shelf software to compute the hand-crafted features with fixed length, such as molecular fingerprints and molecular descriptors, and then to call standard machine learning methods to construct prediction models. The shortcoming of this strategy is that the hand-crafted features to describe a compound are invariable and independent of its targets. More specifically, hand-crafted features are data-independent, indicating that the semantic gap between the features and the bioactivities cannot be solved (Li et al., 2016). In addition, the extraction of hand-crafted features usually requires researchers to have an intimate understanding of their generation, limiting their popularization with the typical end user.
There are several types of molecular fingerprints, depending on the method by which it is generated (Cereto-Massagué et al., 2015). The main approaches consist of substructure keys-based fingerprints, topological or path-based fingerprints and circular fingerprints (Cereto-Massagué et al., 2015). In ligand-based virtual screening, molecular fingerprints with good performance are usually large in length. For example, Unterthiner et al. used an extended-connectivity circular fingerprints (ECFP) vector of size 43 000, after having removed rarely occurring features (Unterthiner et al., 2014). Since the number of compounds to be used in virtual screening is usually very large, the construction of effective virtual screening models can be extremely time consuming and therefore difficult to attain in real-life applications. Therefore, it is of utmost importance to develop molecular fingerprints that are short and effective for use in the virtual screening of drugs.
In commercial drug design, virtual screening results are acceptable only if the prediction accuracy is high. Motivated by the success of applying deep learning to virtual screening (Duvenaud et al., 2015; Unterthiner et al., 2014), we proposed to develop a deep learning algorithm designed to predict the bioactivities of ligands that potentially interact with GPCRs. One difficulty with this task is that the input to the model, a ligand, can be of arbitrary size. Currently, deep learning pipelines can only handle inputs of a fixed size. Our two-stage algorithm, WDL-RF, which combines a weighted deep learning (WDL) and a random forest (RF) model, allows end-to-end learning of prediction pipelines whose inputs are of arbitrary size. Additionally, the molecular fingerprint generation stage is comprised of a new weighted deep learning method, while the bioactivity prediction stage utilizes a random forest model. The results indicate that our algorithm, WDL-RF, achieves the best performance in the prediction of ligand bioactivities in twenty-six human GPCR datasets, suggesting that our algorithm has a potential application in drug development. Moreover, the data-driven molecular fingerprint features, which are generated using weighted deep learning, solves the deficiencies of traditional hand-crafted features and makes up for the insufficiencies stemming from the usage of short molecular fingerprints in drug design.
There has been an unfortunate dearth of open-source code of virtual screening software, as most have been developed into commercial products. In this study, we provided three demo programs and shared the source codes and data on our webserver for the benefit of academic community. Since our approach is built on a general method of ligand-based virtual screening, it is straightforward for users to develop virtual screening models with these codes, for the targets of their own interest. All the codes and database of WDL-RF, together with an on-line server, are freely available at https://zhanglab.ccmb.med.umich.edu/WDL-RF/.
2 Datasets and methods
2.1 Datasets
We first downloaded the 7tmrlist file, which contains 3052 G protein-coupled receptors (GPCRs), from UniProt database (http://www.uniprot.org/docs/7tmrlist) (Consortium, 2008). Then, a total of 825 human GPCR proteins were acquired after parsing the 7tmrlist file. Next, we downloaded the ‘all interaction data file’ from GLASS database (http://zhanglab.ccmb.med.umich.edu/GLASS/), which includes 519 051 unique GPCR-ligand interaction entries (Chan et al., 2015). Subsequently, the 825 human GPCRs were sorted by the number of interacting ligands they each had. Twenty-six representative GPCRs, which have at least 200 ligands, were selected as the experimental targets. These GPCRs cover four GPCR families (A, B, C and F) and 13 subfamilies (see Table 1). Many other subfamilies that have none or too few known ligands are not included because no trustworthy models could be trained due to the lack of sufficient samples; these include, for example, the subfamily ‘Sensory receptors’ in Family A, ‘Adhesion receptors’ in Family B, ‘Sensory receptors’ and ‘Orphan receptors’ in Family C and others (Chan et al., 2015; Isberg et al., 2014).
Table 1.
Descriptions of datasets used in this study
| UniProt ID | Gene name | Protein name | Family | Subfamily | # of ligands | # of controls | Clinical significance |
|---|---|---|---|---|---|---|---|
| P08908 | HTR1A | 5-Hydroxytryptamine receptor 1A | A | Aminergic receptors | 2294 | 400 | Blood pressure, heart rate, antidepressant, anxiolytic, schizophrenia and Parkinson (Ito et al., 1999) |
| P50406 | HTR6 | 5-Hydroxytryptamine receptor 6 | A | Aminergic receptors | 1421 | 300 | Motor control, emotionality, cognition and memory (Woolley et al., 2004) |
| P08912 | CHRM5 | Muscarinic acetylcholine receptor M5 | A | Aminergic receptors | 369 | 71 | Nervous system activity (Anney et al., 2007) |
| P35348 | ADRA1A | Alpha-1A adrenergic receptor | A | Aminergic receptors | 1027 | 200 | Fight-or-flight response |
| P21917 | DRD4 | D(4) dopamine receptor | A | Aminergic receptors | 1679 | 300 | Neurological and psychiatric conditions (Zhang et al., 2007) |
| Q9Y5N1 | HRH3 | Histamine H3 receptor | A | Aminergic receptors | 2092 | 400 | Cognitive disorders (Esbenshade et al., 2008) |
| P30968 | GNRHR | Gonadotropin-releasing hormone receptor | A | Peptide receptors | 1124 | 200 | Hypogonadotropic hypogonadism (Layman et al., 1998) |
| P24530 | EDNRB | Endothelin receptor type B | A | Peptide receptors | 1019 | 200 | Hirschsprung disease type 2 (Tanaka et al., 1998) |
| Q99705 | MCHR1 | Melanin-concentrating hormone receptors 1 | A | Peptide receptors | 2052 | 400 | Appetite, anxiety and depression (Rivera et al., 2008) |
| P35372 | OPRM1 | Mu-type opioid receptor | A | Peptide receptors | 3828 | 700 | Morphine-induced analgesia and itching (Liu et al., 2011) |
| P46663 | BDKRB1 | B1 bradykinin receptor | A | Peptide receptors | 452 | 90 | Inflammatory responses (Souza et al., 2004) |
| P35346 | SSTR5 | Somatostatin receptor type 5 | A | Peptide receptors | 689 | 130 | Inhibit the release of many hormones and other secretory proteins (Tulipano et al., 2001) |
| P21452 | TACR2 | Substance-K receptor | A | Peptide receptors | 696 | 155 | Anxiolytic and antidepressant (Hanley and Jackson, 1987) |
| P30542 | ADORA1 | Adenosine receptor A1 | A | Nucleotide receptors | 3016 | 600 | Tachyarrhythmias, neonatal medicine (Phillis, 1991) |
| Q99500 | S1PR3 | Sphingosine 1-phosphate receptor 3 | A | Lipid receptors | 317 | 63 | Regulation of angiogenesis and vascular endothelial cell function (Barthomeuf et al., 2006) |
| Q9Y5Y4 | PTGDR2 | Prostaglandin D2 receptor 2 | A | Lipid receptors | 641 | 130 | Allergy and inflammation (Nantel et al., 2004) |
| P34995 | PTGER1 | Prostaglandin E2 receptor EP1 subtype | A | Lipid receptors | 236 | 45 | Hyperalgesia (Kawahara et al., 2001) |
| P51677 | CCR3 | C-C chemokine receptor type 3 | A | Protein receptors | 781 | 160 | Binds and responds to a variety of chemokines (Choe et al., 1996) |
| P48039 | MTNR1A | Melatonin receptor type 1A | A | Melatonin receptors | 684 | 135 | Circadian rhythm(Slaugenhaupt et al., 1995) |
| Q8TDU6 | GPBAR1 | G-protein coupled bile acid receptor 1 | A | Steroid receptors | 1153 | 230 | Suppression of macrophage functions and regulation of energy homeostasis by bile acids(Wang et al., 2011) |
| Q8TDS4 | HCAR2 | Hydroxycarboxylic acid receptor 2 | A | Alicarboxylic acid receptors | 271 | 55 | Dyslipidemia(Hu et al., 2015) |
| Q9HC97 | GPR35 | G-protein coupled receptor 35 | A | Orphan receptors | 1589 | 320 | Brachydactyly mental retardation syndrome(Shrimpton et al., 2004) |
| P47871 | GCGR | Glucagon receptor | B | Peptide receptors | 1129 | 220 | Diabetes mellitus type 2 (Hager et al., 1995) |
| P41180 | CASR | Extracellular calcium-sensing receptor | C | Ion receptors | 940 | 190 | Alzheimer's disease, asthma(Kim et al., 2014) |
| Q14416 | GRM2 | Metabotropic glutamate receptor 2 | C | Amino acid receptors | 1810 | 360 | Hallucinogenesis |
| Q99835 | SMO | Smoothened homolog | F | Protein receptors | 1523 | 300 | Developmental disorders |
All ligands of the GPCRs were reacquired from the CHEMBL database (Gaulton et al., 2012) with the match term of ‘Assay type = B and Standard units = nM and Confidence score 5’, where ‘B’ means ‘binding’ assayed by in vitro experiments, except that all ligands of five GPCRs, i.e. Q8TDU6, Q9HC97, P41180, Q14416 and Q99835 were directly collected from GLASS database (Chan et al., 2015) with the match term of ‘Standard units = nM’ due to the satisfied number of samples. The canonical SMILES strings and the target-associated bioactivities of these ligands were saved as experimental datasets. Since the value of the raw bioactivities of ligands varies over a large range, we used the p-bioactivity throughout this study; this is defined as , where is the raw bioactivity and can be measured using IC50, EC50, Ki, Kd, etc. (Cortes-Ciriano, 2016). In our experimental datasets, the range of p-bioactivity is from -10 to 4, where the smaller the value is, the lower the activity of the ligand will be. If a ligand has multiple p-bioactivity values, the mean is adopted.
For each GPCR dataset, we added some control ligands to obtain a more robust regression model for predicting the bioactivities of ligands. The control ligands that do not interact with the target GPCR were randomly chosen from the remaining subfamily-irrelevant GPCR datasets, which is about 20% of that of the original ligands. For the control ligands, the p-bioactivity is set to -10, which is the upper bound of that of acting ligands. Table 1 gives the detailed descriptions of the nineteen GPCR datasets used in the present study.
2.2 Algorithm
WDL-RF operates by first generating molecular fingerprints from the canonical smile string as the sole input through a novel weighted deep learning (WDL), followed by bioactivity prediction using a random forest (RF) regression model.
2.2.1 Molecular fingerprint generation by weighted deep learning
Figure 1 shows the feedforward structure of the WDL algorithm, which is comprised of three parts of molecular fingerprint generation (I), weighted molecular fingerprint generation (II) and bioactivity output (III). The molecular fingerprint generation consists of multiple module units, each of which contains four layers, i.e. sum pooling, convolution, convolution and sum pooling. The weighted molecular fingerprint generation involves one layer, which is weighted by the molecular fingerprints from each module unit. The bioactivity output is made up of two fully connected layers.
Fig. 1.
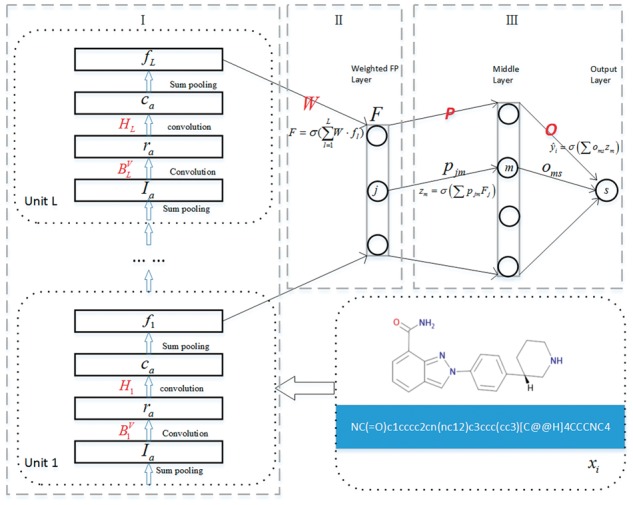
Feedforward structure of the proposed weighted deep learning algorithm WDL. The algorithm consists of three parts: molecular fingerprint generation (I), weighted molecular fingerprint generation (II) and bioactivity output (III). The characters highlighted in red represent the model parameters that need to be updated
Given the ligand molecular dataset , where denotes the ith ligand molecule which takes as input the canonical SMILES string encoding of each molecule, and represents its p-bioactivity. For the ligand molecule that contains atoms, we obtain the attribute vector of each atom using RDkit. The initial atom attributes concatenate a one-hot encoding of the atom’s element, its degree, the number of attached hydrogen atoms, and the implicit valence, and an aromaticity indicator (Duvenaud et al., 2015).
Assume that the first part (I) of WDL contains L module units. In the lth module unit, denotes the attribute vector of atom , and the attribute information of atom and its neighboring atoms were taken into consideration by
| (1) |
where is the number of neighboring atoms.
The bond information is a concatenation of whether the bond type was single, double, triple, or aromatic, whether the bond was conjugated, and whether the bond was part of a ring (Duvenaud et al., 2015). The bond information of atom was involved by the first convolution operation,
| (2) |
where ; V denotes the number of chemical bonds that atom links, ; the weight matrix is to imply the linked chemical bond information; denotes the Rectified linear unit (ReLU) activation function,
| (3) |
Then, we implement the second convolution operation by the weight matrix ,
| (4) |
where ; denotes the softmax normalization which is used to reduce the influence of extreme values or outliers in the data without removing them,
| (5) |
Then, the molecular fingerprint generated by each module unit is adjusted by the sum pooling operation,
| (6) |
Szegedy et al. demonstrated that a combination of all layers with their outputs into a single output vector forming the input of the next stage has a beneficial effect in their deep convolutional neural network architecture, GoogLeNet (Szegedy et al., 2015). In this paper, the weighted molecular fingerprint is combined by weighting the molecular fingerprints obtained by each module unit,
| (7) |
where is the number of module units and ; denotes the weight matrix; and represents the ReLU activation function.
After obtaining the weighted molecular fingerprint, , the predicted bioactivity value of the ligand molecule is calculated by two fully connected layers. Let be the connection weight between the jth neuron of the weighted molecular fingerprint layer and the mth neuron of the middle layer, then
| (8) |
Let be the connection weight between the mth neuron of the middle layer and the neuron() of the output layer, then
| (9) |
where means the ReLU activation function.
After gaining the predicted p-bioactivity value , the optimization problem we address is
| (10) |
where n is the number of ligands in the training dataset; and respectively denote the real and predicted p-bioactivity of the ligand ; represents all the weight parameters that need to be solved. The first term is the regularized quadratic cost function, which penalizes the deviation of estimated entries from the observations. The second term is the regularization term to control the model complexity and avoid overfitting, where is the regularization parameter for balancing the loss function term and the regularization constraint term. Given A dimensions for the attribute vector at each module unit, a fingerprint length B, and M neurons in the middle layer, the weight parameters consist of . Thus, the total number of parameters optimized in all layers of the weighted deep learning is ‘A × A×L + A × B×L + B × B + B × M + M’.
The Adam algorithm is used to update all the weight parameters, , which is an algorithm for first-order gradient-based optimization of stochastic objective functions, based on adaptive estimates of lower-order moments (Kingma and Ba, 2014). Let be the objective function, i.e. Eq. (10), and with we denote the gradient, i.e. the vector of partial derivatives of with respect to evaluated at timestep t. The algorithm updates exponential moving averages of the gradient () and the squared gradient (), where the hyper-parameters and () control the exponential decay rates of these moving averages,
| (11) |
| (12) |
where indicates the elementwise square .
The moving averages themselves are estimates of the 1st moment (the mean) and the 2nd raw moment (the uncentered variance) of the gradient. However, these moving averages are initialized as (vectors of) 0s, leading to moment estimates that are biased towards zero, especially during the initial time steps when the decay rates are small (i.e. the and are close to 1) (Kingma and Ba, 2014).The good news is that this initialization bias can be easily counteracted, resulting in bias-corrected estimates and (Kingma and Ba, 2014),
| (13) |
| (14) |
where and are and to the power of , respectively.
Finally, the weight parameters are updated by
| (15) |
where is the step size. In this paper, good default settings of hyperparameters are and .
In the optimization process by the Adam algorithm, we adopt the evaluation at random subsamples (minibatches) of data points where 100 samples are randomly selected in each round of iteration, and the maximum number of iterations is set to 250. While training, the popular regularization technique dropout (Srivastava et al., 2014) is implemented by only keeping a neuron active with some probability or setting it to zero otherwise, to avoid overfitting.
2.2.2 Predicting bioactivities of ligands by random forest regression models
Random forest, which was first proposed by Breiman (2001), is an ensemble of M decision trees. The Random forest model produces M outputs where is the prediction value for a ligand by the mth tree. Outputs of all trees are assembled to produce one final prediction . For regression problems, is the average value of the individual tree predictions.
Given data on a set of n ligands for training, where is a vector of fingerprints and is the p-bioactivity value of ligands, the training procedure is as follows:
From the training data of n ligands, draw a bootstrap sample dataset to produce n training examples by randomly sampling with replacement from the training dataset;
For each bootstrap sample dataset, generate a tree with the following scheme: at each node, choose the best split among a randomly selected subset of features. The tree is grown to the maximum size (i.e. till no more splits are possible) and not pruned back;
Repeat the above steps until M such trees are grown.
The prediction performance of the random forest regression model is assessed by the so-called Out-Of-Bags (OOB) samples. On average, each tree is grown using about of the training ligands, leaving as OOB, indicating that 2/3 of the elements are in the training dataset and the remaining 1/3 of them are used for the test. The training and testing date sets are selected by random sampling. The random forest regression models are implemented in Python 2.7. The pseudocode of our proposed algorithm WDL-RF is shown in Table 2.
Table 2.
The pseudocode of WDL-RF
|
Algorithm WDL-RF |
| Inputs: the training dataset |
|
Outputs: model performance |
| Process: |
| Stage1: |
| 1: Initialization: |
| 2: Repeat: |
| 3: Randomly sample a mini-batch of subset S from D |
| 4: for |
| 5: for each atom in molecular |
| 6: |
| 7: for l = 1 to L |
| 8: for each atom in molecular |
| 9: Compute by Eq.(1) |
| 10: Compute by Eq.(2) |
| 11: Compute by Eq.(4) |
| 12: Obtain the molecular fingerprint of each module unit by Eq.(6) |
| 13: Obtain the weighted molecular fingerprintby Eq.(7) |
| 14: Predict p-bioactivity of ligands by Eq.(9) |
| 15: Compute the loss function by Eq.(10) |
| 16: Updateby Eq.(15) |
| 17: Until stop criterion reached |
| 18: Return: |
| Stage2: |
| 19: Construct random forest regression prediction models: P = Predictor (F, Y) |
2.3 Evaluation criterion
Root mean square error (RMSE) is a commonly used metric for evaluating regression models,
| (16) |
where and are the true and the predicted activity values, respectively, and n is the number of ligands. The smaller the RMSE value is, the better the model performance will be.
Correlation coefficient (r2) was used in evaluating the performance of the predictions in the Kaggle challenge on drug activity prediction organized by Merck in 2012,
| (17) |
where is the known activity, is the mean of the known activity, is the predicted activity, is the mean of the predicted activity, and n is the number of molecules in the dataset. The larger the r2 value is, the better the model performance will be.
To eliminate the effect of random selection, three sets of control ligands were created for each GPCR dataset, and the regression model for predicting bioactivities of ligands was built separately. The mean of the performance among the three models was calculated and designated as the final result. Moreover, the Wilcoxon signed-ranked test was implemented to compare the differences in the performances of the compared methods and to determine whether it was statistically significant.
3 Results and discussion
3.1 Comparison with short molecular fingerprints
We benchmarked WDL-RF with ten types of short molecular fingerprints, which include the neural graph fingerprint, the MACCS fingerprint, four kinds of ECFP fingerprints and four kinds of FCFP fingerprints. The neural graph fingerprint (NGFP) is a short one with continuous values, which is based on the molecular graph of compounds and trained by the convolution neural network algorithm (Duvenaud et al., 2015). The MACCS fingerprint is widely used, with 166 bits, and covers most of the interesting chemical features for drug discovery and virtual screening (Durant et al., 2002). The Extended-Connectivity Fingerprints (ECFPs) are the most-widely used molecular fingerprints, based on the Morgan algorithm (Morgan, 1965), and specifically designed for the use in structure-activity modeling (Rogers et al., 2010). In this study, we employ four types of ECFPs including ECFP2, ECFP4, ECFP8 and ECFP10, where the digits indicate the diameter of the fingerprints. The Functional-Class Fingerprints (FCFPs) are a variation of ECFP, which generates molecular fingerprints by determining whether an atom is a hydrogen bond acceptor, a hydrogen bond donor, a cation, an anion, an aromatic, or a halogen etc (Cereto-Massagué et al., 2015). Here, we adopt four types of FCFPs, i.e. FCFP2, FCFP4, FCFP8 and FCFP10, where the digits indicate the diameter. The NGFP fingerprint was generated with a custom script, while the remaining nine molecular fingerprints were produced with RDKit.
The neural graph fingerprint (NGFP) is the only data-driven feature in all baseline molecular fingerprints, and its default dimension is 50. To reach a fair comparison, the dimension of each type of molecular fingerprint is set to 50 except for the MACCS fingerprint (Durant et al., 2002) with the fixed dimension of 166, and the same random forest regression model is adopted for each type of molecular fingerprint.
In the experiments, the random forest regression method is used for each type of molecular fingerprint to construct the models for predicting the bioactivities of ligands. The parameters of random forest, n_estimates and max_features, are respectively set to 100 and sqrt(m), where m is the dimension of the molecular fingerprint. In WDL-RF, the number of module units (L) is set to 4, and the regularization parameter (i.e. in Eq. 10) is set to . As shown from Table 3, WDL-RF is optimal on almost all GPCR datasets and evaluation criteria.
Table 3.
Comparison of various short molecular fingerprints
| ECa | GPCR | MFPb |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MACCS | ECFP2 | ECFP4 | ECFP8 | ECFP10 | FCFP2 | FCFP4 | FCFP8 | FCFP10 | NGFP | WDL | ||
| RMSE (↓) | P08908 | 1.85* | 1.88* | 1.97* | 2.30* | 2.41* | 1.77* | 1.98* | 2.22* | 2.25* | 1.56 | 1.50 |
| P50406 | 1.81* | 1.90* | 2.10* | 2.54* | 2.61* | 1.83* | 2.04* | 2.39* | 2.55* | 1.60* | 1.45 | |
| P08912 | 2.17* | 2.15 | 2.33* | 2.92* | 3.13* | 2.11 | 2.16 | 2.41* | 2.84* | 2.30* | 2.14 | |
| P35348 | 2.20* | 2.25* | 2.58* | 2.69* | 3.01* | 2.28* | 2.06* | 2.94* | 2.77* | 2.03* | 1.72 | |
| P21917 | 2.08* | 2.25* | 2.41* | 2.91* | 3.27* | 2.21* | 2.47* | 2.92* | 3.02* | 1.99* | 1.76 | |
| Q9Y5N1 | 2.17* | 2.46* | 2.66* | 3.23* | 3.45* | 2.17* | 2.30* | 3.03* | 3.08* | 1.62* | 1.42 | |
| P30968 | 1.33* | 1.53* | 1.57* | 2.10* | 2.25* | 1.49* | 1.59* | 1.89* | 2.02* | 1.23 | 1.21 | |
| P24530 | 1.32* | 1.58* | 1.73* | 2.01* | 2.16* | 1.49* | 1.67* | 1.80* | 1.92* | 1.11* | 1.04 | |
| Q99705 | 1.82* | 1.92* | 2.10* | 2.48* | 2.58* | 1.80* | 1.90* | 2.28* | 2.44* | 1.41* | 1.33 | |
| P35372 | 1.74* | 1.87* | 2.13* | 2.47* | 2.52* | 1.80* | 1.93* | 2.24* | 2.45* | 1.53 | 1.51 | |
| P46663 | 2.19* | 2.29* | 2.67* | 3.42* | 3.39* | 2.49* | 2.46* | 2.93* | 3.25* | 2.33* | 1.77 | |
| P35346 | 1.53* | 1.70* | 2.00* | 2.21* | 2.21* | 1.37* | 1.57* | 2.28* | 2.01* | 1.18* | 1.13 | |
| P21452 | 2.36* | 2.50* | 2.42* | 2.75* | 3.14* | 2.46* | 2.27* | 2.67* | 2.85* | 2.07* | 1.86 | |
| P30542 | 1.14* | 1.45* | 1.79* | 2.07* | 2.21* | 1.30* | 1.49* | 1.86* | 1.96* | 1.00 | 0.96 | |
| Q99500 | 1.29* | 1.78* | 1.88* | 2.23* | 2.44* | 1.67* | 1.92* | 2.02* | 2.06* | 1.33* | 1.02 | |
| Q9Y5Y4 | 1.41* | 1.70* | 1.74* | 2.41* | 2.54* | 1.48* | 1.78* | 2.08* | 2.35* | 1.46* | 1.28 | |
| P34995 | 2.00* | 2.12* | 2.11* | 2.76* | 3.28* | 3.10* | 2.45* | 2.59* | 2.91* | 2.06* | 1.70 | |
| P51677 | 1.57* | 1.64* | 2.00* | 2.29* | 2.52* | 1.90* | 1.75* | 2.11* | 2.51* | 1.41* | 1.15 | |
| P48039 | 1.65 | 1.72 | 2.06* | 2.38* | 2.53* | 1.60 | 1.79 | 2.25* | 2.32* | 1.84* | 1.69 | |
| Q8TDU6 | 0.99* | 1.37* | 1.25* | 1.81* | 2.00* | 1.41* | 1.13* | 1.37* | 1.88* | 0.87 | 0.87 | |
| Q8TDS4 | 1.37 | 1.45 | 2.61* | 3.15* | 3.00* | 2.03* | 2.12* | 2.93* | 2.94* | 1.59 | 1.52 | |
| Q9HC97 | 1.22* | 1.31* | 1.36* | 1.47* | 1.65* | 1.41* | 1.46* | 1.22* | 1.40* | 1.39* | 0.85 | |
| P47871 | 1.17* | 1.56* | 1.71* | 2.47* | 2.51* | 1.49* | 1.54* | 2.08* | 2.25* | 1.06 | 1.05 | |
| P41180 | 1.08* | 1.28* | 1.34* | 2.07* | 2.16* | 1.31* | 1.10* | 1.44* | 1.90* | 1.16* | 0.76 | |
| Q14416 | 1.08* | 1.23* | 1.42* | 1.81* | 1.91* | 1.12* | 1.42* | 1.88* | 1.70* | 1.05* | 0.81 | |
| Q99835 | 1.25* | 1.48* | 1.44* | 2.31* | 2.12* | 1.43* | 1.08 | 1.61* | 1.73* | 1.11 | 1.01 | |
| r2 (↑) | P08908 | 0.58* | 0.56* | 0.53* | 0.29* | 0.22* | 0.61* | 0.51* | 0.37* | 0.33* | 0.67 | 0.70 |
| P50406 | 0.68* | 0.68* | 0.60* | 0.37* | 0.33* | 0.70* | 0.64* | 0.48* | 0.38* | 0.75 | 0.79 | |
| P08912 | 0.46 | 0.49 | 0.40* | 0.18* | 0.14* | 0.53 | 0.53 | 0.38* | 0.20* | 0.47 | 0.47 | |
| P35348 | 0.61* | 0.54* | 0.43* | 0.37* | 0.29* | 0.57* | 0.59* | 0.29* | 0.38* | 0.65* | 0.70 | |
| P21917 | 0.54* | 0.44* | 0.40* | 0.21* | 0.12* | 0.51* | 0.42* | 0.25* | 0.18* | 0.59* | 0.66 | |
| Q9Y5N1 | 0.66* | 0.55* | 0.47* | 0.28* | 0.20* | 0.64* | 0.60* | 0.36* | 0.35* | 0.80 | 0.83 | |
| P30968 | 0.85 | 0.82 | 0.80* | 0.60* | 0.52* | 0.82 | 0.80* | 0.72* | 0.64* | 0.85 | 0.86 | |
| P24530 | 0.80* | 0.71* | 0.62* | 0.44* | 0.31* | 0.73* | 0.69* | 0.59* | 0.48* | 0.82 | 0.85 | |
| Q99705 | 0.69* | 0.69* | 0.64* | 0.40* | 0.32* | 0.69* | 0.71* | 0.53* | 0.42* | 0.80 | 0.82 | |
| P35372 | 0.67* | 0.62* | 0.50* | 0.29* | 0.26* | 0.64* | 0.59* | 0.43* | 0.30* | 0.73 | 0.74 | |
| P46663 | 0.71* | 0.66* | 0.53* | 0.27* | 0.17* | 0.60* | 0.62* | 0.45* | 0.34* | 0.66* | 0.78 | |
| P35346 | 0.74* | 0.69* | 0.58* | 0.50* | 0.48* | 0.80* | 0.73* | 0.45* | 0.56* | 0.85 | 0.86 | |
| P21452 | 0.53 | 0.44* | 0.59* | 0.31* | 0.16* | 0.48* | 0.55* | 0.41* | 0.36* | 0.66 | 0.70 | |
| P30542 | 0.83 | 0.75* | 0.58* | 0.35* | 0.23* | 0.75* | 0.71* | 0.50* | 0.43* | 0.84 | 0.86 | |
| Q99500 | 0.80* | 0.52* | 0.51* | 0.45* | 0.35* | 0.60* | 0.48* | 0.47* | 0.38* | 0.82* | 0.87 | |
| Q9Y5Y4 | 0.84 | 0.74* | 0.75* | 0.52* | 0.44* | 0.81* | 0.73* | 0.60* | 0.52* | 0.82 | 0.86 | |
| P34995 | 0.56* | 0.52* | 0.45* | 0.26* | 0.50* | 0.54* | 0.35* | 0.27* | 0.17* | 0.54* | 0.64 | |
| P51677 | 0.76* | 0.72* | 0.58* | 0.50* | 0.43* | 0.65* | 0.69* | 0.56* | 0.40* | 0.80* | 0.86 | |
| P48039 | 0.75 | 0.75 | 0.56* | 0.53* | 0.44* | 0.74 | 0.67 | 0.52* | 0.51* | 0.74 | 0.72 | |
| Q8TDU6 | 0.88 | 0.77* | 0.82* | 0.60* | 0.51* | 0.76* | 0.85 | 0.77* | 0.58* | 0.91 | 0.92 | |
| Q8TDS4 | 0.86 | 0.84 | 0.44* | 0.22* | 0.32* | 0.65* | 0.67 | 0.25* | 0.21* | 0.76 | 0.77 | |
| Q9HC97 | 0.77* | 0.72* | 0.70* | 0.64* | 0.56* | 0.70* | 0.67* | 0.75* | 0.68* | 0.72* | 0.89 | |
| P47871 | 0.87 | 0.78* | 0.73* | 0.47* | 0.43* | 0.80* | 0.77* | 0.58* | 0.50* | 0.89 | 0.90 | |
| P41180 | 0.86 | 0.81* | 0.77* | 0.51* | 0.47* | 0.81* | 0.86 | 0.76* | 0.59* | 0.85* | 0.93 | |
| Q14416 | 0.86 | 0.82* | 0.78* | 0.62* | 0.60* | 0.86 | 0.80* | 0.66* | 0.69* | 0.88 | 0.92 | |
| Q99835 | 0.85 | 0.79* | 0.80* | 0.51* | 0.58* | 0.81* | 0.89 | 0.76* | 0.71* | 0.89 | 0.90 | |
Evaluation Criterion: ↑ (↓) indicates the larger (smaller), the better the model performance; the best results on each evaluation criterion are highlighted in boldface.
Molecular Fingerprints: * indicates the performance of the compared short molecular fingerprint is significantly worse than that of WDL based on Wilcoxon signed-ranked test.
Data-driven features have already replaced hand-crafted features in speech recognition, machine vision and natural-language processing (Duvenaud et al., 2015). In this paper, the proposed molecular fingerprints (WDL) and the neural graph fingerprint (NGFP) (Duvenaud et al., 2015) both contain data-driven features, which fill in the semantic gap between the hand-crafted features of molecular fingerprints and the bioactivities of ligands interacting with different drug targets. The data shows that their performance is significantly better than the nine types of molecular fingerprints which employ hand-crafted features (Table 3).
It is also observed that, in most datasets, for each kind of ECFP or FCFP fingerprints, when its diameter becomes larger, its performance instead decreases significantly. The reason is that, when the diameter gets bigger, the number of possible substructures will grow exponentially, but what we care about are short molecular fingerprints with low and fixed number of dimensions. Thus, when the diameter becomes larger, the missing substructure information will increase dramatically, resulting in a significant decrease in the performance.
3.2 Comparison with various molecular descriptors
Molecular descriptors can also be used as the features to build models for predicting the bioactivities of ligands. Ten types of molecular descriptors commonly used in virtual screening were compared with our method. They include Charge descriptors, Connectivity descriptors, Constitutional descriptors, E-state descriptors, Geary autocorrelation descriptors, MOE-type descriptors, Moran autocorrelation descriptors, Moreau-Broto autocorrelation descriptors, Topology descriptors and Basak descriptors (Jie et al., 2015), all of which were generated through the ChemoPy program (Cao et al., 2013). Their dimensions are 25, 44, 30, 245, 32, 60, 32, 32, 35 and 21, respectively.
For each type of molecular descriptor, the strategy of building random forest regression models is the same as that of our WDL-RF algorithm. The experimental results are indicated in Table 4, which shows that WDL-RF achieves the best performance on almost all GPCR datasets and evaluation criterions (Table 4).
Table 4.
Comparison of various molecular descriptors
| ECa | GPCR | MDb |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Charge | Connect | Constitut | E-state | Geary | MOE | Moran | MB | Topology | Basak | WDL | ||
| RMSE (↓) | P08908 | 1.90* | 1.97* | 2.10* | 1.79* | 2.20* | 1.86* | 2.19* | 1.99* | 2.09* | 2.28* | 1.50 |
| P50406 | 1.88* | 2.04* | 1.93* | 1.74* | 2.27* | 1.78* | 2.33* | 2.14* | 2.09* | 2.40* | 1.45 | |
| P08912 | 2.15 | 2.19* | 2.00* | 1.87* | 2.30* | 1.85 | 2.48* | 2.11 | 2.25* | 2.32* | 2.14 | |
| P35348 | 2.36* | 2.36* | 2.28* | 2.16* | 2.49* | 2.10* | 2.47* | 2.31* | 2.37* | 2.78* | 1.72 | |
| P21917 | 2.13* | 2.13* | 2.16* | 1.95* | 2.43* | 1.92* | 2.40* | 2.17* | 2.09* | 2.22* | 1.76 | |
| Q9Y5N1 | 1.99* | 2.18* | 2.02* | 1.74* | 2.58* | 2.04* | 2.51* | 2.31* | 2.13* | 2.58* | 1.42 | |
| P30968 | 1.67* | 1.69* | 1.68* | 1.37* | 1.88* | 1.49* | 1.86* | 1.79* | 1.86* | 1.92* | 1.21 | |
| P24530 | 1.23* | 1.54* | 1.45* | 1.20* | 1.70* | 1.38* | 1.62* | 1.52* | 1.58* | 1.95* | 1.04 | |
| Q99705 | 1.95* | 2.08* | 1.95* | 1.55* | 2.16* | 1.62* | 2.36* | 2.13* | 2.05* | 2.58* | 1.33 | |
| P35372 | 1.96* | 2.05* | 1.98* | 1.79* | 2.28* | 1.87* | 2.19* | 2.18* | 2.12* | 2.32* | 1.51 | |
| P46663 | 2.10* | 2.33* | 2.24* | 1.70 | 2.44* | 1.86* | 2.28* | 2.54* | 2.28* | 2.88* | 1.77 | |
| P35346 | 1.64* | 1.64* | 1.59* | 1.35* | 2.08* | 1.44* | 1.92* | 1.98* | 1.77* | 1.76* | 1.13 | |
| P21452 | 2.02* | 2.17* | 2.11* | 1.96* | 2.08* | 1.93 | 2.22* | 2.22* | 2.28 | 2.25 | 1.86 | |
| P30542 | 1.31* | 1.66* | 1.44* | 1.21* | 2.01* | 1.24* | 2.00* | 1.85* | 1.86* | 1.87* | 0.96 | |
| Q99500 | 1.46* | 1.51* | 1.38* | 1.37* | 1.48* | 1.39* | 1.54* | 1.36* | 1.54* | 1.55* | 1.02 | |
| Q9Y5Y4 | 1.53* | 2.02* | 1.64* | 1.46* | 2.02* | 1.71* | 2.03* | 2.18* | 1.94* | 2.42* | 1.28 | |
| P34995 | 1.73 | 2.18* | 1.86* | 1.72 | 1.69* | 1.66 | 1.73 | 1.90* | 2.02* | 2.28* | 1.70 | |
| P51677 | 1.66* | 1.82* | 1.74* | 1.56* | 2.03* | 1.54* | 2.04* | 2.05* | 2.04* | 2.05* | 1.15 | |
| P48039 | 1.67 | 1.71 | 1.71 | 1.56 | 2.15* | 1.63 | 2.09* | 1.79 | 1.67 | 2.07* | 1.69 | |
| Q8TDU6 | 1.36* | 1.36* | 1.63* | 1.24* | 1.59* | 1.24* | 1.64* | 1.57* | 1.85* | 1.76* | 0.87 | |
| Q8TDS4 | 1.27 | 2.21* | 1.76* | 1.57 | 2.29* | 1.57 | 2.05* | 2.38* | 2.12* | 2.64* | 1.52 | |
| Q9HC97 | 1.07* | 1.29* | 1.07* | 1.07* | 1.36* | 1.01* | 1.24* | 1.21* | 1.22* | 1.17* | 0.85 | |
| P47871 | 1.25* | 1.56* | 1.37* | 1.08 | 1.52* | 1.17* | 1.50* | 1.96* | 1.59 | 1.94 | 1.05 | |
| P41180 | 1.31* | 1.51* | 1.33* | 1.50* | 2.00* | 1.45* | 1.97* | 1.68* | 1.84* | 1.86* | 0.76 | |
| Q14416 | 1.22* | 1.50* | 1.40* | 1.13* | 1.59* | 1.11* | 1.55* | 1.54* | 1.34* | 1.62* | 0.81 | |
| Q99835 | 1.18* | 1.28* | 1.25* | 1.06 | 1.44* | 1.15* | 1.59* | 1.16* | 1.39* | 1.56* | 1.01 | |
| r2 (↑) | P08908 | 0.53* | 0.46* | 0.41* | 0.60* | 0.35* | 0.56* | 0.34* | 0.45* | 0.41* | 0.29* | 0.70 |
| P50406 | 0.66* | 0.58* | 0.63* | 0.71* | 0.50* | 0.69* | 0.46* | 0.55* | 0.56* | 0.43* | 0.79 | |
| P08912 | 0.48 | 0.45 | 0.56* | 0.65* | 0.41* | 0.67* | 0.31* | 0.49 | 0.43* | 0.39* | 0.47 | |
| P35348 | 0.45* | 0.46* | 0.52* | 0.59* | 0.40* | 0.61* | 0.42* | 0.47* | 0.45* | 0.24* | 0.70 | |
| P21917 | 0.49* | 0.50* | 0.48* | 0.60* | 0.35* | 0.61* | 0.36* | 0.47* | 0.52* | 0.47* | 0.66 | |
| Q9Y5N1 | 0.68* | 0.65* | 0.70* | 0.79 | 0.47* | 0.69* | 0.50* | 0.58* | 0.56* | 0.55* | 0.83 | |
| P30968 | 0.74* | 0.72* | 0.73* | 0.84 | 0.68* | 0.81 | 0.70* | 0.68* | 0.66* | 0.63* | 0.86 | |
| P24530 | 0.81 | 0.68* | 0.73* | 0.84 | 0.63* | 0.76* | 0.64* | 0.70* | 0.68* | 0.47* | 0.85 | |
| Q99705 | 0.61* | 0.56* | 0.60* | 0.78 | 0.54* | 0.75* | 0.43* | 0.53* | 0.55* | 0.30* | 0.82 | |
| P35372 | 0.56* | 0.52* | 0.55* | 0.65* | 0.41* | 0.62* | 0.46* | 0.45* | 0.49* | 0.38* | 0.74 | |
| P46663 | 0.69* | 0.62* | 0.64* | 0.82 | 0.57* | 0.80 | 0.67* | 0.53* | 0.63* | 0.37* | 0.78 | |
| P35346 | 0.71* | 0.70* | 0.71* | 0.83 | 0.52* | 0.79* | 0.60* | 0.57* | 0.64* | 0.66* | 0.86 | |
| P21452 | 0.59* | 0.53* | 0.57* | 0.63* | 0.61* | 0.63* | 0.54* | 0.48* | 0.46* | 0.47* | 0.70 | |
| P30542 | 0.74* | 0.58* | 0.68* | 0.79* | 0.37* | 0.78* | 0.39* | 0.47* | 0.47* | 0.45* | 0.86 | |
| Q99500 | 0.66* | 0.66* | 0.73* | 0.73* | 0.67* | 0.71* | 0.62* | 0.70* | 0.65* | 0.61* | 0.87 | |
| Q9Y5Y4 | 0.80* | 0.62* | 0.76* | 0.83 | 0.62* | 0.72* | 0.62* | 0.53* | 0.65* | 0.42* | 0.86 | |
| P34995 | 0.56* | 0.35* | 0.52* | 0.61 | 0.59* | 0.66 | 0.60* | 0.49* | 0.44* | 0.26* | 0.64 | |
| P51677 | 0.72* | 0.66* | 0.69* | 0.78* | 0.61* | 0.79* | 0.60* | 0.53* | 0.56* | 0.47* | 0.86 | |
| P48039 | 0.72 | 0.73 | 0.73 | 0.80 | 0.53* | 0.77 | 0.54* | 0.69 | 0.73 | 0.54* | 0.72 | |
| Q8TDU6 | 0.83 | 0.84 | 0.73* | 0.86 | 0.71* | 0.87 | 0.70* | 0.78* | 0.62* | 0.66* | 0.92 | |
| Q8TDS4 | 0.86 | 0.50* | 0.71 | 0.77 | 0.52* | 0.77 | 0.60* | 0.42* | 0.54* | 0.28* | 0.77 | |
| Q9HC97 | 0.84 | 0.76* | 0.84 | 0.85 | 0.77* | 0.83 | 0.82 | 0.79* | 0.79* | 0.80* | 0.89 | |
| P47871 | 0.85* | 0.81* | 0.84* | 0.90 | 0.79* | 0.88 | 0.79* | 0.64* | 0.75* | 0.64* | 0.90 | |
| P41180 | 0.85 | 0.76* | 0.87 | 0.83* | 0.61* | 0.84 | 0.63* | 0.73* | 0.65* | 0.61* | 0.93 | |
| Q14416 | 0.85 | 0.76* | 0.80* | 0.88 | 0.80* | 0.89 | 0.80* | 0.76* | 0.82 | 0.72* | 0.92 | |
| Q99835 | 0.87 | 0.85 | 0.86 | 0.92 | 0.86 | 0.89 | 0.83 | 0.88 | 0.83 | 0.81* | 0.90 | |
Evaluation Criterion: ↑ (↓) indicates the larger (smaller), the better the model performance; the best results on each evaluation criterion are highlighted in boldface.
Molecular Descriptors: Charge: Charge descriptors; Connect: Connectivity descriptors; Constitut: Constitutional descriptors; Estate: E-state descriptors; Geary: Geary autocorrelation descriptors; MOE: MOE-type descriptors; Moran: Moran autocorrelation descriptors; MB: Moreau-Broto autocorrelation descriptors; Topology: Topology descriptors; Basak: Basak descriptors. * indicates the performance of the compared molecular descriptor is significantly worse than that of WDL based on Wilcoxon signed-ranked test.
We found that the performance of random forest prediction model based on our data-driven feature, i.e. the weighted deep learning molecular fingerprint (WDL), is significantly better than that based on hand-crafted molecular descriptors (Table 4), further illustrating the contributions of data-driven features. Moreover, comparable performance is achieved by all nine kinds of hand-crafted molecular fingerprints (Table 3) and all ten types of hand-crafted molecular descriptors (Table 4). For the molecular descriptors, it is usually easier to get better performance by the usage of longer one. For example, the E-state molecular descriptor (245 dimensions) or the MOE-type molecular descriptor (60 dimensions) with the higher feature dimensions achieves the second best predictive performance (Table 4).
3.3 Performance of molecular fingerprints from different module units
The information extracted from different module units is distinct. Figure 2 summarizes the comparison results of model performance based on the molecular fingerprint generated by different module units, where each model was built by random forest with the same parameter configuration. The number of module units (L) of our WDL-RF algorithm is set to 4, in which the 1st to 4th layers denote respectively the models with the molecular fingerprint generated by the first to the fourth module unit, while WDL represents the default molecular fingerprint, i.e. the weighted molecular fingerprint of all module units. It was observed that, for most targets, the model performance increases as the number of the module units increases; this is probably due to the fact that the molecular fingerprint from higher module units can achieve a higher-level of semantic information which helps to improve the predictive performance of models. However, when we increase the number of module units beyond 4, the results show that the performance slightly decreases (see data listed in Supplementary Table S1), indicating that L = 4 represents an approximately optimal setting for our modeling.
Fig. 2.

Performance of molecular fingerprints from different module units. The 1st to 4th respectively denote the molecular fingerprint generated by the first to the fourth module unit of WDL-RF. The WDL is the default molecular fingerprint used in this study, i.e. the weighted molecular fingerprint of all module units. The x-axis denotes the different GPCR datasets, i.e. P1: P08908; P2: P50406; P3: P08912; P4: P35348; P5: P21917; P6: Q9Y5N1; P7: P30968; P8: P24530; P9: Q99705; P10: P35372; P11: P46663; P12: P35346; P13: P21452; P14: P30542; P15: Q99500; P16: Q9Y5Y4; P17: P34995; P18: P51677; P19: P48039; P20: Q8TDU6; P21: Q8TDS4; P22: Q9HC97; P23: P47871; P24: P41180; P25: Q14416; P26: Q99835
Overall, the performance of our weighted molecular fingerprint, WDL, is better than that of each kind of molecular fingerprint from different module units on almost all GPCR datasets and evaluation criteria. The reason is probably that molecular fingerprints from different module units contain different information, and the weighted molecular fingerprint combines the different information and therefore improves the predictive performance of models.
3.4 Effect of parameters
There are several parameters in WDL-RF which have been determined in our training datasets to be of critical importance. Here, we examine how the performance of predicting ligand bioactivities is affected by these parameters in our testing dataset.
The first key parameter of WDL-RF is the regression model, where Neural Network (NN), Support Vector Regression (SVR) and Random Forest (RF) were taken into consideration. The input of each regression model is the default weighted deep learning (WDL) molecular fingerprint. The optimal parameters of NN and SVR models are obtained through a standard grid search method. Figure 3A presents the dependence of performance of WDL-RF on different regression models. Here, a lower RMSE or higher r2 value indicates better model performance. The results show that the random forest regression model is slightly better than the other two regression models, i.e. NN and SVR, in most GPCR datasets and evaluations (Fig. 3A). Thus, the random forest regression model was adopted into our WDL-RF algorithm mainly due to its exceptional, robust performance on different parameter values.
Fig. 3.

Dependence of WDL-RF performance on (A) regression models, (B) the parameter n_estimates and (C) max_features of random forest. The x-axis denotes the different GPCR datasets, i.e. P1: P08908; P2: P50406; P3: P08912; P4: P35348; P5: P21917; P6: Q9Y5N1; P7: P30968; P8: P24530; P9: Q99705; P10: P35372; P11: P46663; P12: P35346; P13: P21452; P14: P30542; P15: Q99500; P16: Q9Y5Y4; P17: P34995; P18: P51677; P19: P48039; P20: Q8TDU6; P21: Q8TDS4; P22: Q9HC97; P23: P47871; P24: P41180; P25: Q14416; P26: Q99835
The influence of the parameters n_estimates and max_features of random forest on the performance were examined in Figure 3B and C, respectively, where n_estimates denotes the size of the decision trees growth in random forest, and max_features represents the number of the randomly selected subset of features. The numerical parameters for n_estimates were generated first, for which four alternative values (50, 80, 100 and 120) are presented in Figure 3B. Three alternative options are compared for the max_features parameter, which include all(m), sqrt(m) and log2(m), where m is the dimension of the molecular fingerprint, and all(m) means all dimensions. Thus, there are in total seven options for the two parameters that were optimized in random forest. The results in Figure 3 indicate that the parameter n_estimates has little effect on model performance, the default value of which was set to 100 in WDL-RF.
A similar situation occurred for the parameter max_features (Fig. 3C), so the default configuration for max_features was set to sqrt(m) in WDL-RF.
3.5 Code usage
We have developed three demo programs for different applications in ligand-based virtual screening, with the source codes and datasets released through https://zhanglab.ccmb.med.umich.edu/WDL-RF/. The code for WDL-RF was written in Python 2.7, which can easily be implemented across multiple platforms, including Windows 10 and Linux. The pipelines have three major functions.
(1) demo_new: This provides a general framework on ligand-based virtual screening, and it is easy for users to develop their own virtual screening tools for drug targets of their choice on the basis of our code. Input: Compounds in the format of canonical SMILES and their bioactivity values. Output: Model performance (RMSE, r2). The procedure is as follows: To input compounds in the format of canonical SMILES and their bioactivity values →To train the weighted deep learning model →To get the weighted molecular fingerprints →To construct random forest regression models → To obtain the model performance.
(2) demo_activity: This offers the ligand-based virtual screening models of nineteen important human GPCR drug targets, and users can predict the bioactivities of new compounds acting with these targets, which is important in implementations of drug design against these drug targets, the prediction of side effects of multi-target drugs, and the risk assessment of drug development. Input: Compounds in the format of canonical SMILES. Output: Bioactivity values interacting with these GPCR drug targets. The steps are as follows: To input compounds in the format of canonical SMILES →To get the weighted molecular fingerprints by our trained weighted deep learning models →To obtain the bioactivity values based on our trained random forest models.
(3) demo_fp: Users can obtain multiple types of short molecular fingerprints for a compound, which can be used in compound similarity search, pharmacophore search and bioactivity prediction. Input: Compounds in the format of canonical SMILES. Output: Molecular fingerprints. The steps are as follows: To input compounds in the format of canonical SMILES →To obtain molecular fingerprints based on our trained weighted deep learning models. For all 26 GPCR drug targets, five types of short molecular fingerprints are produced for a compound. Therefore, a total 130 (=26 × 5) kinds of different molecular fingerprints will be generated for each compound, where ‘26’ means twenty-six GPCR drug targets used in this paper and ‘5’ is the number of fingerprint types.
It should be mentioned that the training of WDL-RF has taken the standard SMILES strings as input, which does not distinguish the structural difference of ‘stereoisomer’ ligands that have the same molecular formula and bonding sequence but differ in the 3D orientations of their atoms in space. Therefore, the current version of WDL-RF cannot deal with the stereoisomerism problem. However, since the pipeline is built on a multi-fold training platform, in which the bioactivity and structure information of stereoisomers can be conveniently integrated, it has the potential to include the specificity of stereoisomers in the model construction. The work on this issue is currently in progress.
4 Conclusions
Accurate determination of ligand bioactivities is essential for virtual screening and lead compound identification. Inspired by the success of deep learning on virtual screening, a novel method, WDL-RF, was developed to predict the bioactivities of GPCR-associated ligand molecules.
The algorithm of WDL-RF is comprised of two steps: (i) molecular fingerprint generation through a new weighted deep learning and, (ii) bioactivity prediction by a random forest model; this allows end-to-end learning of prediction pipelines whose input ligand can be of arbitrary size. Large-scale benchmark tests show that WDL-RF can generate high-accuracy bioactivity predictions with an average root-mean square error (RMSE) of 1.33 and a correlation coefficient (r2) of 0.80, which are significantly better than that from several control predictors benchmarked. Moreover, the data-driven molecular fingerprint features, extracted from our weighted deep learning, can help solve the deficiency of traditional hand-crafted features and make up for the insufficiency of short molecular fingerprints in drug design.
The source codes and databases of WDL-RF have been made freely available through our dedicated web server, where users can use the package to predict bioactivities of compounds against a GPCR target or generate molecular fingerprints for new compounds acting with these known GPCR drug targets, as well as to develop their own virtual screening models for their drug targets of choice on the basis of the developed general learning framework.
Overall, deep learning is slowly coming to fruition in various quantitative biomedical investigations. This study demonstrated a novel application of the deep learning approach to ligand bioactivity prediction, in addition to that of other domains of virtual screening experiments, including materials design and organic photovoltaic efficiency (Duvenaud et al., 2015).
Funding
This work was supported in part by the National Science Foundation of China (81771478, 61571233), the key University Science Research Project of Jiangsu Province (17KJA510003) and the National Science Foundation (DBI1564756).
Conflict of Interest: none declared.
Supplementary Material
References
- Anney R.J. et al. (2007) Variation in the gene coding for the M5 Muscarinic receptor (CHRM5) influences cigarette dose but is not associated with dependence to drugs of addiction: evidence from a prospective population based cohort study of young adults. BMC Genet., 8, 46.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthomeuf C. et al. (2006) Inhibition of sphingosine-1-phosphate- and vascular endothelial growth factor-induced endothelial cell chemotaxis by red grape skin polyphenols correlates with a decrease in early platelet-activating factor synthesis. Free Radic. Biol. Med., 40, 581–590. [DOI] [PubMed] [Google Scholar]
- Becker O.M. et al. (2004) G protein-coupled receptors: in silico drug discovery in 3D. Proc. Natl. Acad. Sci. USA, 101, 11304–11309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M. et al. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum L.C., Reymond J.L. (2009) 970 Million druglike small molecules for virtual screening in the chemical universe database GDB-13. J. Am. Chem. Soc., 131, 8732.. [DOI] [PubMed] [Google Scholar]
- Breiman L. (2001) Random forest. Mach. Learn., 45, 5–32. [Google Scholar]
- Cao D.S. et al. (2013) ChemoPy: freely available python package for computational biology and chemoinformatics. Bioinformatics, 29, 1092.. [DOI] [PubMed] [Google Scholar]
- Cereto-Massagué A. et al. (2015) Molecular fingerprint similarity search in virtual screening. Methods, 71, 58–63. [DOI] [PubMed] [Google Scholar]
- Chan W.K.B. et al. (2015) GLASS: a comprehensive database for experimentally validated GPCR-ligand associations. Bioinformatics, 31, 3035–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe H. et al. (1996) The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell, 85, 1135–1148. [DOI] [PubMed] [Google Scholar]
- Consortium T.U. (2008) The Universal Protein Resource. Nucleic Acids Res., 35, 193–197. [Google Scholar]
- Cortes-Ciriano I. (2016) Benchmarking the predictive power of ligand efficiency indices in QSAR. J. Chem. Inf. Model., 56, 1576.. [DOI] [PubMed] [Google Scholar]
- Durant J.L. et al. (2002) Reoptimization of MDL keys for use in drug discovery. Cheminform, 42, 1273–1280. [DOI] [PubMed] [Google Scholar]
- Duvenaud D. et al. (2015) Convolutional networks on graphs for learning molecular fingerprints. In: Advances in Neural Information Processing Systems, pp. 2215–2232.
- Esbenshade T.A. et al. (2008) The histamine H3 receptor: an attractive target for the treatment of cognitive disorders. Br. J. Pharmacol., 154, 1166.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton A. et al. (2012) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res., 40, D1100–D1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager J. et al. (1995) A missense mutation in the glucagon receptor gene is associated with non-insulin-dependent diabetes mellitus. Nat. Genet., 9, 299–304. [DOI] [PubMed] [Google Scholar]
- Hanley M.R., Jackson T. (1987) Substance K receptor: return of the magnificent seven. Nature, 329, 766.. [DOI] [PubMed] [Google Scholar]
- Hu M. et al. (2015) Pharmacogenetics of cutaneous flushing response to niacin/laropiprant combination in Hong Kong Chinese patients with dyslipidemia. Pharmacogenomics, 16, 1387–1397. [DOI] [PubMed] [Google Scholar]
- Isberg V. et al. (2014) GPCRDB: an information system for G protein-coupled receptors. Nucleic Acids Res., 42, D422.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H. et al. (1999) Localization of 5-HT1A receptors in the living human brain using [carbonyl-11C]WAY-100635: pET with anatomic standardization technique. J. Nuclear Med., 40, 102–109. [PubMed] [Google Scholar]
- Jie D. et al. (2015) ChemDes: an integrated web-based platform for molecular descriptor and fingerprint computation. J. Cheminf., 7, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara H. et al. (2001) A prostaglandin E2 receptor subtype EP1 receptor antagonist (ONO-8711) reduces hyperalgesia, allodynia, and c-fos gene expression in rats with chronic nerve constriction. Anesthesia Analgesia, 93, 1012.. [DOI] [PubMed] [Google Scholar]
- Kim J.Y. et al. (2014) Calcium-sensing receptor (CaSR) as a novel target for ischemic neuroprotection. Ann. Clin. Transl. Neurol., 1, 851–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingma D.P., Ba J. (2014) Adam: A Method for Stochastic Optimization, arXiv preprint arXiv: 1412.6980.
- Layman L.C. et al. (1998) Mutations in gonadotropin-releasing hormone receptor gene cause hypogonadotropic hypogonadism. Nat. Genet., 18, 14–15. [DOI] [PubMed] [Google Scholar]
- Li W.J. et al. (2016) Feature learning based deep supervised hashing with pairwise labels. Int. Jt. Conf. Artif. Intell., p 1711–1717. [Google Scholar]
- Liu X.Y. et al. (2011) Unidirectional cross-activation of GRPR by MOR1D uncouples itch and analgesia induced by opioids. Cell, 147, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller W.E., Lefkowitz R.J. (2001) Expanding roles for β-arrestins as scaffolds and adapters in GPCR signaling and trafficking. Curr. Opin. Cell Biol., 13, 139–145. [DOI] [PubMed] [Google Scholar]
- Morgan H.L. (1965) The generation of a unique machine description for chemical structures—a technique developed at chemical abstracts service. J. Chem. Document., 5, 107–113. [Google Scholar]
- Nantel F. et al. (2004) Expression of prostaglandin D synthase and the prostaglandin D2 receptors DP and CRTH2 in human nasal mucosa. Prostaglandins Other Lipid Mediators, 73, 87. [DOI] [PubMed] [Google Scholar]
- Overington J.P. et al. (2006) How many drug targets are there? Nat. Rev. Drug Discov., 5, 993–996. Nature Reviews Drug Discovery, 5, 993-996. [DOI] [PubMed] [Google Scholar]
- Phillis J.W. (1991) Adenosine and Adenine Nucleotides as Regulators of Cellular Function. CRC Press, Boca Raton, Florida.
- Rivera G. et al. (2008) Melanin-concentrating hormone receptor 1 antagonists: a new perspective for the pharmacologic treatment of obesity. Curr. Med. Chem., 15, 1025.. [DOI] [PubMed] [Google Scholar]
- Rogers D., Hahn M.. et al. (2010) Extended-connectivity fingerprints. J. Chem. Inf. Model., 50, 742. [DOI] [PubMed] [Google Scholar]
- Shang J. et al. (2017) HybridSim-VS: a web server for large-scale ligand-based virtual screening using hybrid similarity recognition techniques, Bioinformatics, 33, 3480–3481. [DOI] [PubMed] [Google Scholar]
- Shrimpton A.E. et al. (2004) Molecular delineation of deletions on 2q37.3 in three cases with an Albright hereditary osteodystrophy-like phenotype. Clin. Genet., 66, 537. [DOI] [PubMed] [Google Scholar]
- Slaugenhaupt S.A. et al. (1995) Mapping of the gene for the Mel 1a-melatonin receptor to human chromosome 4 (MTNR1A) and mouse chromosome 8 (Mtnr1a). Genomics, 27, 355–357. [DOI] [PubMed] [Google Scholar]
- Souza D.G. et al. (2004) Role of bradykinin B2 and B1 receptors in the local, remote, and systemic inflammatory responses that follow intestinal ischemia and reperfusion injury. J. Immunol., 172, 2542.. [DOI] [PubMed] [Google Scholar]
- Srivastava N. et al. (2014) Dropout: a simple way to prevent neural networks from overfitting. J. Mach. Learn. Res., 15, 1929–1958. [Google Scholar]
- Szegedy C. et al. (2015) Going deeper with convolutions. In: IEEE Conference on Computer Vision and Pattern Recognition pp. 1–9.
- Tanaka H. et al. (1998) Novel mutations of the endothelin B receptor gene in patients with Hirschsprung’s disease and their characterization. J. Biol. Chem., 273, 11378–11383. [DOI] [PubMed] [Google Scholar]
- Tautermann C.S. (2014) GPCR structures in drug design, emerging opportunities with new structures. Bioorg. Med. Chem. Lett., 24, 4073–4079. [DOI] [PubMed] [Google Scholar]
- Tulipano G. et al. (2001) Differential inhibition of growth hormone secretion by analogs selective for somatostatin receptor subtypes 2 and 5 in human growth-hormone-secreting adenoma cells in vitro. Neuroendocrinology, 73, 344–351. [DOI] [PubMed] [Google Scholar]
- Unterthiner T. et al. (2014) Deep learning as an opportunity in virtual screening. In: Advances in Neural Information Processing Systems, pp. 1–9.
- Wang Y.D. et al. (2011) The G‐Protein‐coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light‐chain enhancer of activated B cells (NF‐κB) in mice. Hepatology, 54, 1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley M.L. et al. (2004) 5-ht6 receptors. Curr. Drug Targets CNS Neurol. Disorders, 3, 59.. [DOI] [PubMed] [Google Scholar]
- Wootten D. et al. (2013) Emerging paradigms in GPCR allostery: implications for drug discovery. Nat. Rev. Drug Discov., 12, 630–644. [DOI] [PubMed] [Google Scholar]
- Zhang A. et al. (2007) Recent progress in development of dopamine receptor subtype-selective agents: potential therapeutics for neurological and psychiatric disorders. Chem. Rev., 38, 274–302. [DOI] [PubMed] [Google Scholar]
- Zhang J. et al. (2015) GPCR-I-TASSER: a hybrid approach to G protein-coupled receptor structure modeling and the application to the human genome. Structure, 23, 1538–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.