

Abstract
Hutchinson-Gilford progeria syndrome (HGPS) is a fatal disease characterized by premature aging in which young children fail to thrive and adolescents die from myocardial infarction or stroke. The pathogenesis of HGPS is studied intensively because the mechanisms of premature aging may lead to a better understanding of normal aging. In this issue of the JCI, Osmanagic-Myers and colleagues identify the cellular mechanisms that lead to vascular abnormalities and death in children with HGPS.
Hutchinson-Gilford progeria syndrome
Hutchinson-Gilford progeria syndrome (HGPS) is a rare disease with an incidence of approximately 1 out of 10 million and a world-wide prevalence of less than 500. The clinical features of HGPS are characterized by accelerated aging; young children develop skin changes such as alopecia and dry skin, musculoskeletal abnormalities including skeletal dysplasia and joint contractures, and endocrine findings like lipodystrophy and insulin resistance (1, 2). Cardiovascular abnormalities include left ventricular hypertrophy and decreased vascular function. Adolescents with HGPS develop atherosclerosis, leading to strokes and myocardial infarction. The average age of death in HGPS is 15 years old.
HGPS is an autosomal dominant disease caused by a mutation in the LMNA gene encoding the lamin A protein (3, 4). A single base-pair mutation in LMNA generates a cryptic splice site, creating an abnormal mRNA encoding a truncated protein. This abnormal protein, called progerin, fails to be processed normally; instead of a farnesyl group temporarily attached to the protein and then cleaved off, a farnesyl group is permanently affixed to progerin.
Progerin is a dominant gain-of-function protein that causes a host of abnormal cellular reactions (5). Normal lamin proteins play many roles in the nucleus of the cell, maintaining the structure of the nuclear lamina, transducing signals from the periphery into the nucleus, and serving as a scaffold for transcription factors (6). However, progerin disrupts the morphology of the nucleus, alters epigenetic regulation of gene transcription, dysregulates transcription, inhibits DNA repair, shortens telomeres, and accelerates cellular senescence.
The major cause of premature death of subjects with HGPS is myocardial infarction, but the pathogenesis remained obscure until recently. Postmortem studies revealed atherosclerosis in the coronary arteries of children with HGPS, but the vascular lesions were characterized by dense fibrosis rather than atheromatous cores seen in typical coronary artery disease of normally aging individuals (7). Furthermore, the vasculopathy of HGPS is characterized by loss of smooth muscle cells in the medial layer of the arterial wall (8–11). This loss of vascular smooth muscle is also seen in a mouse model for progeria (12). However, this mouse model of HGPS expresses progerin in all cells, obscuring the contribution of individual cell types and specific pathways to accelerated atherosclerosis in HGPS.
Progerin in endothelial cells drives tissue fibrosis
What is the key vascular cell that leads to severe atherosclerosis in HGPS? In the current issue of the JCI, Osmanagic-Myers and colleagues show that endothelial cells play a major role in the pathogenesis of HGPS (13). The research team created a unique mouse overexpressing progerin only in endothelial cells. These mice have a shortened life span, confirming that vascular pathology is a major cause of premature death in HGPS.
Next, these transgenic mice have left ventricular hypertrophy and diastolic dysfunction, which are seen in patients with HGPS. Furthermore, the transgenic mice have cardiac fibrosis in an interstitial and perivascular pattern, a pattern that also occurs in some humans with HGPS. Thus, progerin enhances cardiac fibrosis. The question remains — how is this achieved?
Exploring the endothelial pathway underlying cardiac fibrosis, the investigators discovered that progerin impairs signal transduction from the cytoskeleton into the nucleus. In particular, abnormal mechanosensing limited the activity of the transcription factor MRTF-A, decreasing endothelial nitric oxide synthase (eNOS) expression and NO production (Figure 1). NO is a messenger molecule well known to inhibit fibrosis by limiting fibroblast proliferation and activation (14, 15). Thus, progerin in endothelial cells permitted exuberant tissue fibrosis by limiting endothelial synthesis of NO.
Figure 1. In healthy endothelial cells, shear stress activates the mechanoresponsive myocardin-related transcription factor MRTF-A, increasing eNOS expression and NO production, which in turn inhibits tissue fibrosis.
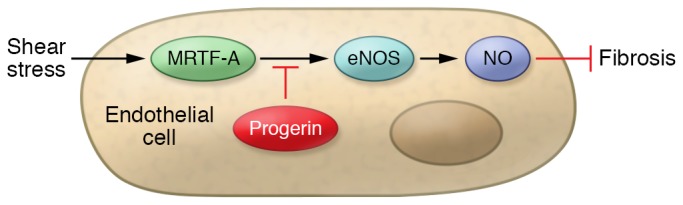
However, in endothelial cells of subjects with Hutchinson-Gilford progeria syndrome, the mutant lamin called progerin interferes with normal mechanosensing, inhibits eNOS expression and NO synthesis, and permits accelerated fibrosis.
Progerin in vascular smooth muscle cells drives vascular fibrosis
Endothelial cells are not the only critical effector cells of HGPS in the vessel wall. Another team recently identified vascular smooth muscle as a major target of progerin (16). Hamczyk and colleagues created a mouse overexpressing progerin only in vascular smooth muscle cells. These transgenic mice die sooner than control mice and display the vascular phenotype characteristic of humans with HGPS: vascular smooth muscle loss in the medial layer of arteries and fibrosis in the adventitial layer. These studies show that vascular smooth muscle cells are particularly sensitive to progerin — although the underlying mechanisms are unknown. Thus, progerin in vascular smooth muscle cells leads to vascular fibrosis.
Progerin during aging in the normal vasculature
Tremendous advances have been made in research uncovering the pathophysiology of HGPS — but a cure remains elusive (17). The current new studies reveal novel insights into accelerated atherosclerosis in HGPS. Progerin in the vascular wall drives the cardiovascular phenotype of HGPS: endothelial progerin enhances cardiac fibrosis, whereas vascular smooth muscle progerin causes vascular fibrosis. These studies suggest new approaches to treat HGPS, such as drugs targeting progerin pathways in endothelial cells and vascular smooth muscle cells. The current study in the JCI suggests that therapies that target NO pathways might improve cardiac and vascular function in HGPS. In addition, therapies targeted at vascular smooth muscle cells might limit the severe atherosclerosis that can cause premature death in HGPS.
These studies also reveal new pathways for vascular disease during normal aging. Progerin biology is relevant for normal aging, since progerin is found in the vessel wall of humans as they age normally — although at levels much lower than in subjects with HGPS (7, 18). Pathways activated by progerin, such as shortened telomeres or senescence, may play a role in vascular disease during physiological aging. Novel therapies that target progerin pathways in the vasculature may not only lead to cures for the accelerated aging of HGPS, but to new approaches to slow atherosclerosis during the process of natural aging.
Version 1. 12/18/2018
Electronic publication
Version 2. 02/01/2019
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2019;129(2):492–493. https://doi.org/10.1172/JCI125616.
See the related article at Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse.
References
- 1. Gordon LB, Brown WT, Collins FS. Hutchinson-Gilford progeria syndrome. In: Adam MP, et al. eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. [PubMed] [Google Scholar]
- 2.Merideth MA, et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008;358(6):592–604. doi: 10.1056/NEJMoa0706898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Sandre-Giovannoli A, et al. Lamin A truncation in Hutchinson-Gilford progeria. Science. 2003;300(5628):2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 4.Eriksson M, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423(6937):293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalo S, Kreienkamp R, Askjaer P. Hutchinson-Gilford progeria syndrome: A premature aging disease caused by LMNA gene mutations. Ageing Res Rev. 2017;33:18–29. doi: 10.1016/j.arr.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gruenbaum Y, Foisner R. Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu Rev Biochem. 2015;84:131–164. doi: 10.1146/annurev-biochem-060614-034115. [DOI] [PubMed] [Google Scholar]
- 7.Olive M, et al. Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol. 2010;30(11):2301–2309. doi: 10.1161/ATVBAHA.110.209460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker PB, Baba N, Boesel CP. Cardiovascular abnormalities in progeria. Case report and review of the literature. Arch Pathol Lab Med. 1981;105(7):384–386. [PubMed] [Google Scholar]
- 9.Stehbens WE, Wakefield SJ, Gilbert-Barness E, Olson RE, Ackerman J. Histological and ultrastructural features of atherosclerosis in progeria. Cardiovasc Pathol. 1999;8(1):29–39. doi: 10.1016/S1054-8807(98)00023-4. [DOI] [PubMed] [Google Scholar]
- 10.Stehbens WE, Delahunt B, Shozawa T, Gilbert-Barness E. Smooth muscle cell depletion and collagen types in progeric arteries. Cardiovasc Pathol. 2001;10(3):133–136. doi: 10.1016/S1054-8807(01)00069-2. [DOI] [PubMed] [Google Scholar]
- 11.Ackerman J, Gilbert-Barness E. Hutchinson-Gilford progeria syndrome: a pathologic study. Pediatr Pathol Mol Med. 2002;21(1):1–13. doi: 10.1080/pdp.21.1.1.13. [DOI] [PubMed] [Google Scholar]
- 12.Varga R, et al. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2006;103(9):3250–3255. doi: 10.1073/pnas.0600012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Osmanagic-Myers S, et al. Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse. J Clin Invest. 2019;129(2):531–545. doi: 10.1172/JCI121297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hou J, Kato H, Cohen RA, Chobanian AV, Brecher P. Angiotensin II-induced cardiac fibrosis in the rat is increased by chronic inhibition of nitric oxide synthase. J Clin Invest. 1995;96(5):2469–2477. doi: 10.1172/JCI118305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelley TJ, Drumm ML. Inducible nitric oxide synthase expression is reduced in cystic fibrosis murine and human airway epithelial cells. J Clin Invest. 1998;102(6):1200–1207. doi: 10.1172/JCI2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamczyk MR, et al. Vascular smooth muscle-specific progerin expression accelerates atherosclerosis and death in a mouse model of Hutchinson-Gilford progeria syndrome. Circulation. 2018;138(3):266–282. doi: 10.1161/CIRCULATIONAHA.117.030856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collins FS. Seeking a cure for one of the rarest diseases: Progeria. Circulation. 2016;134(2):126–129. doi: 10.1161/CIRCULATIONAHA.116.022965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312(5776):1059–1063. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]