

Abstract
The adenomatous polyposis coli (APC) gene plays a pivotal role in the pathogenesis of colorectal carcinoma (CRC) but remains a challenge for drug development. Long noncoding RNAs (lncRNAs) are invaluable in identifying cancer pathologies and providing therapeutic options for patients with cancer. Here, we identified a lncRNA (lncRNA-APC1) activated by APC through lncRNA microarray screening and examined its expression in a large cohort of CRC tissues. A decrease in lncRNA-APC1 expression was positively associated with lymph node and/or distant metastasis, a more advanced clinical stage, as well as a poor prognosis for patients with CRC. Additionally, APC could enhance lncRNA-APC1 expression by suppressing the enrichment of PPARα on the lncRNA-APC1 promoter. Furthermore, enforced lncRNA-APC1 expression was sufficient to inhibit CRC cell growth, metastasis, and tumor angiogenesis by suppressing exosome production through the direct binding of Rab5b mRNA and a reduction of its stability. Importantly, exosomes derived from lncRNA-APC1–silenced CRC cells promoted angiogenesis by activating the MAPK pathway in endothelial cells, and, moreover, exosomal Wnt1 largely enhanced CRC cell proliferation and migration through noncanonicial Wnt signaling. Collectively, lncRNA-APC1 is a critical lncRNA regulated by APC in the pathogenesis of CRC. Our findings suggest that an APC-regulated lncRNA-APC1 program is an exploitable therapeutic approach for the treatment of patients with CRC.
Keywords: Gastroenterology, Oncology
Keywords: Cell migration/adhesion, Colorectal cancer
Introduction
As one of the most common human malignancies, colorectal carcinoma (CRC) is a leading cause of cancer-related deaths worldwide. It is well established that the pathogenesis of CRC follows the adenoma-carcinoma sequence and involves multistep tumorigenesis through the progressive accumulation of abnormalities in both tumor suppressor genes and oncogenes (1, 2). Mutations in the gene adenomatous polyposis coli (APC) play a pivotal role in tumorigenesis and progression of CRC (3). To date, targeting certain oncogenes and their related pathways represents the best option for cancer treatment and improving the survival of patients at advanced stages of the disease. However, because it is a large scaffold protein with multiple functions, APC remains a challenge to target for translation into drug development.
Long noncoding RNAs (lncRNAs) are a large class of transcripts longer than 200 bases, with no protein-coding potential (4). Current research indicates that lncRNAs are exquisitely regulated and that they can control gene expression to regulate various aspects of biological and/or pathological processes (5, 6). It has been shown that lncRNAs modulate several important cancer phenotypes, including cellular proliferation, apoptosis, immortality, motility, as well as angiogenesis (7, 8). As a result, it is now widely understood that lncRNAs are invaluable in their ability to identify cancer pathologies as well as to provide other prognostic value, or even inform therapeutic options for cancer patients. Despite this knowledge, the functions of certain lncRNAs involved in mediating the anticancer role of APC in CRC and any abnormalities they might possess have yet to be elucidated.
Here, we used lncRNA microarray screening to identify a lncRNA (TCONS_00027227) activated by APC through PPARα, which we named lncRNA-APC1. Examination and function analysis of CRC tissues collected from a large patient cohort showed that lncRNA-APC1 plays a crucial tumor-suppressive role in the pathogenesis of CRC. Further mechanistic studies revealed that lncRNA-APC1 exerts its effects through the direct binding of Rab5b mRNA, thereby reducing its stability and ultimately leading to decreased exosome production. This action inhibits the overactivation of the MAPK pathway in endothelial cells and the subsequent suppression of angiogenesis. Importantly, we reveal for the first time to our knowledge an oncogenic role of CRC-derived exosomal Wnt1, which acts in an autocrine manner through noncanonical Wnt signaling. Collectively, our data uncovered an APC signaling mechanism, APC/PPARα/lncRNA-APC1/Rab5b, in the pathogenic process of CRC and revealed the potential for several prognostic and/or therapeutic targets for human CRC.
Results
lncRNA-APC1 is upregulated by APC in CRCs.
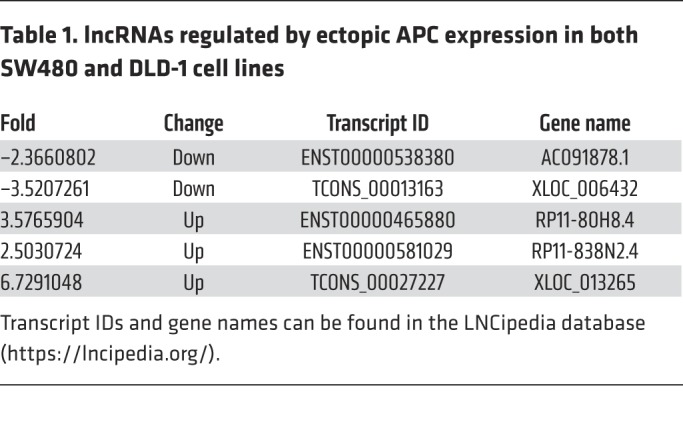
Inactivated mutations in the APC gene are the initiating mutation driving CRC tumorigenesis and/or progression (3). In this study, we sought to investigate the abnormal dynamics and underlying roles of certain lncRNAs that are involved in this process and applied a lncRNA microarray technique to select and identify which lncRNAs were regulated by APC in CRC cells. We first reinduced WT APC full-length coding sequence (CDS) into the SW480 and DLD-1 human CRC cell lines (Figure 1, A and B), both of which express an endogenous truncated APC protein (mutated at aa 1338 and 1427, respectively) that constitutively activates β-catenin/T cell factor 4–mediated (β-catenin/TCF4–mediated) transcription. The 2 cell lines were examined in 2 independently repeated microarray tests. We found that 3 lncRNAs were upregulated and 2 lncRNAs were downregulated by more than 2-fold and that these events were induced after ectopic overexpression of WT APC in both lines (Figure 1C and Table 1). Among these, TCONS_00027227, which we named lncRNA-APC1, is encoded by a gene at chromosome 19p12 and was consistently upregulated by more than 17-fold, as confirmed by quantitative reverse transcription PCR (qRT-PCR) (Figure 1D).
Figure 1. Upregulation of lncRNA-APC1 by APC.

Expression of APC in the indicated cell lines transfected with control or WT APC vector, as measured by qRT-PCR (A) and Western blotting (B). (C) Number of altered lncRNAs in the indicated cells examined in 2 independently repeated lncRNA microarray tests. (D) qRT-PCR verification of lncRNAs potentially regulated by APC. (E) Expression of lncRNA-APC1 was detected by FISH. Scale bars: 20 μm. (F) Relative expression of lncRNA-APC1 in paired CRC primary tumor tissues and nontumor colonic tissues (n = 30). (G) Kaplan-Meier survival analysis of patients with CRC (n = 110) according to lncRNA-APC1 expression (cutoff value is the median). Experiments in F and G were repeated twice with similar results. Data in A, E, and F represent the mean ± SD of 3 separate experiments. **P < 0.01, ***P < 0.001, and ****P < 0.0001, by independent Student’s t test (A and F) or log-rank test (G). NC, negative control.
Table 1. lncRNAs regulated by ectopic APC expression in both SW480 and DLD-1 cell lines.
Using the 5′ and 3′ rapid amplification of cDNA ends (RACE) assay, we discovered that lncRNA-APC1 was a 1580-nt intergene transcript and poly(A) positive. The sequence of full-length lncRNA-APC1 is presented in Supplemental Figure 1, A and C (supplemental material available online with this article; https://doi.org/10.1172/JCI122478DS1). Northern blot analysis confirmed the size of lncRNA-APC1 in the CRC cell lines (Supplemental Figure 1B). Further analysis of the sequences using the NCBI’s National Center for Biotechnology Information ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/) failed to predict a protein of more than 55 aa. Additionally, we calculated its coding potential using the Coding Potential Calculator (CPC) (http://cpc.cbi.pku.edu.cn/) and the Coding Potential Assessment Tool (CPAT) (http://cpc.cbi.pku.edu.cn/). The CPC (using ORF_ FRAME FINDER) predicted a lncRNA-APC1 score of 36.13, and the CPAT predicted a coding probability of 0.008, further supporting the notion that lncRNA-APC1 has no protein-coding potential. Moreover, FISH analysis showed that lncRNA-APC1 was primarily located in the cytoplasm (Figure 1E).
Subsequent qRT-PCR analysis in our study revealed that expression of lncRNA-APC1 was significantly lower in CRC tissues than that in the 30 corresponding samples of nontumor colorectal tissues (Figure 1F). Furthermore, we measured the expression levels of lncRNA-APC1 in CRC tissues from 110 patients, and our correlation analysis revealed that low expression levels of lncRNA-APC1 were positively correlated with lymph node and/or distant metastasis of CRC as well as with a more advanced clinical stage (P < 0.05, Table 2). Kaplan-Meier analysis showed that CRC patients with low levels of lncRNA-APC1 expression had shorter survival (41.4 months, 95% CI: 36.2–46.7) when compared with survival of patients with normal expression levels of lncRNA-APC1 (64.3 months, 95% CI: 60.1–68.7; P < 0.001, log-rank test) (Figure 1G and Table 3). These results indicated that a decrease in lncRNA-APC1, which is downstream of APC, could play an important oncogenic role in regulating CRC progression.
Table 2. Relationship between lncRNA-APC1 expression levels and clinicopathological parameters of CRC.
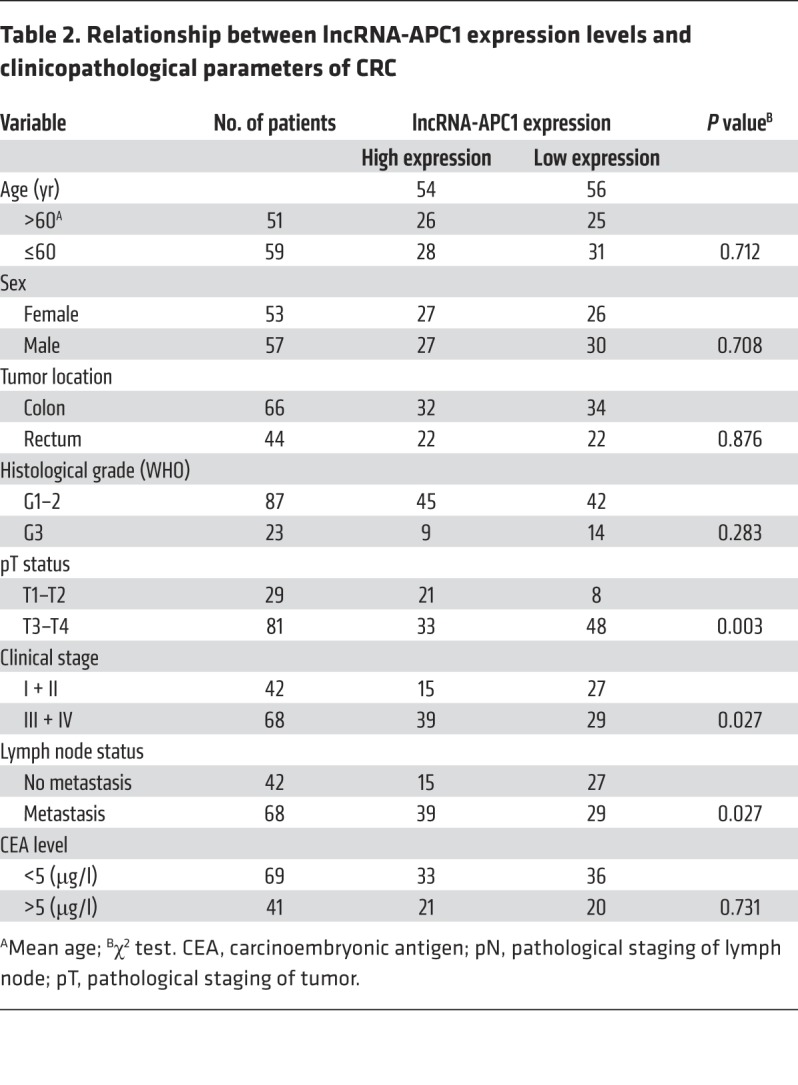
Table 3. Univariate and multivariate Cox regression analysis of different prognostic variables for patients with CRC.

lncRNA-APC1 is regulated by APC through PPARα in CRC cells.
Previous studies have found that truncated APC in CRC cells contributes to tumor cell migration via interaction with the Rac-specific guanine nucleotide exchange factor Asef (9, 10). Hence, we further examined whether mutant APC affects lncRNA-APC1 expression. Two common mutant APC plasmids (gifts of Bert Vogelstein, Johns Hopkins University, Baltimore, Maryland, USA), shown in Supplemental Figure 2A, were transfected into the CRC cell line HCT116 (WT APC) (11). qRT-PCR and immunoblot analysis showed that the 2 APC plasmids produced comparable amounts of APC mRNA and protein (Supplemental Figure 2, B and C). Surprisingly, we found that mutant APC331Δ had no effect on lncRNA-APC1 expression and that APC1309Δ-mutant overexpression slightly suppressed lncRNA-APC1 expression (Supplemental Figure 2D). In addition, we transfected siRNAs specific for APC into SW480 and DLD-1 cells and observed similar results (Supplemental Figure 2, E and F). These results suggest that WT but not truncated APC is mainly responsible for regulating lncRNA-APC1 expression.
It is well accepted that regulating β-catenin stabilization is the most prominent function of APC (12). Therefore, we tested whether β-catenin could regulate lncRNA-APC1 expression in CRC. Our results showed that inhibition of β-catenin by a specific siRNA resulted in only slightly elevated expression of lncRNA-APC1 in the CRC cell lines HCT116 (WT APC, β-catenin mutant) and DLD-1 (Supplemental Figure 3, A and B). We consistently found that neither WT nor mutant β-catenin (45th aa deletion mutation in HCT116 or S33Y) overexpression had a significant effect on lncRNA-APC1 (Supplemental Figure 3, C and D) (11). These results suggest that β-catenin may not be required for the regulation of lncRNA-APC1 expression by APC.
To further explore the potential mechanisms by which APC regulates lncRNA-APC1 expression, we first analyzed the promoter of lncRNA-APC1. Results from the luciferase reporter assay showed that enforced expression of APC could significantly enhance the transcriptional activity of the lncRNA-APC1 promoter (Figure 2A). To identify which domains might be responsible for the induction of transcriptional activity mediated by APC, we constructed 3 more reporter genes, as indicated in Figure 2B. The data from those assays showed that –160 to –374 bp of the promoter had the greatest effect on reporter activity (Figure 2C). Further analysis of the transcription factor binding motif by MatIspector software revealed an enrichment for binding motifs of the nuclear receptor peroxisome proliferator–activated receptor α (PPARα), located –60 to –37 bp (5′-AAAAGAACTGTGACATACCACAG-3′) upstream of the lncRNA-APC1 transcription start site (TSS). It has been reported that the PPAR family includes 3 members, α, Δ, and γ, which encode proteins sharing a highly conserved structure and molecular mode of action. PPARs play central roles in the regulation of glucose and lipid homeostasis and have been shown to be of critical importance in CRC pathogenesis (13, 14). To test whether PPARα regulates lncRNA-APC1 expression, we first transfected siRNAs specific for PPARα into CRC cells (Figure 2D). We found that lncRNA-APC1 expression was significantly enhanced by PPARα silencing (Figure 2E) and, moreover, that the PPARα binding motif deletion mutation significantly increased lncRNA-APC1 promoter–driven luciferase reporter activity (Figure 2F). The data from the dual luciferase reporter assays consistently revealed that enforced expression of PPARα could abrogate the transcriptional activity of the lncRNA-APC1 promoter induced by APC (Figure 2, G and H). Further ChIP analysis confirmed the enrichment of PPARα binding sites on the promoter of lncRNA-APC1 and that ectopic overexpression of APC substantially suppressed the enrichment of PPARα on the promoter of lncRNA-APC1 (Figure 2I). However, we did not observe the same altered levels of PPARα after the enforced expression of APC (data not shown). Together, these findings revealed that APC enhances the expression of lncRNA-APC1 by inhibiting the binding of PPARα to the promoter of lncRNA-APC1.
Figure 2. Upregulation of lncRNA-APC1 by APC is partly dependent on the binding of PPARα to the lncRNA-APC1 promoter.

(A) Luciferase activity of pGL3-lncRNA-APC1 promoter luciferase in DLD-1 cells. (B and C) Luciferase activity of the indicated pGL3-lncRNA-APC1 promoter luciferase vectors. (D) PPARα was efficiently knocked down by siRNA, as detected by Western blotting. (E) qRT-PCR analysis of lncRNA-APC1 expression induced by PPARα knockdown. (F) Luciferase activity in the indicated cells cotransfected with WT or PPARα binding motif deletion (Mut-del) pGL3-lncRNA-APC1 promoter luciferase. (G) Ectopic expression of PPARα was substantially increased in DLD-1 cells, as shown by Western blotting. (H) Luciferase activity of the lncRNA-APC1 promoter cotransfected with APC and/or the PPARα construct. (I) ChIP analysis to detect the enrichment of PPARα on the promoter of lncRNA-APC1. All luciferase data were normalized to Renilla luciferase activity. Data represent the mean ± SD of 3 separate experiments. All experiments were repeated at least 3 times. *P < 0.05, **P < 0.01, and ***P < 0.001, and ****P < 0.0001, by independent Student’s t test (A, F, and I) or 1-way ANOVA (C, E, and H).
lncRNA-APC1 suppresses the proliferative and invasive capacities of CRC cells and inhibits angiogenesis.
It is also well established that APC mutations lead to CRC initiation and/or progression by influencing multiple cellular processes, including cell apoptosis, adhesion, and migration, in tumor cells (15). To investigate whether lncRNA-APC1 plays an important role in APC-mediated biological functions in CRC, we first stably overexpressed lncRNA-APC1 in DLD-1 and SW480 cells and examined the effect of lncRNA-APC1 on cellular biological functions (Supplemental Figure 4A). The data from these in vitro studies showed that ectopic overexpression of lncRNA-APC1 largely inhibited CRC cell proliferation and migration (Figure 3, A and B, and Supplemental Figure 4B). Furthermore, in vivo assays demonstrated that overexpression of lncRNA-APC1 dramatically inhibited the oncogenic and metastatic potential of CRC cells in nude mice (Figure 3, C–E). Next, to test the contribution of lncRNA-APC1 to APC functions in the pathogenesis of CRC, we stably overexpressed WT APC in the DLD-1 cell line and then stably silenced lncRNA-APC1 expression with specific shRNAs (Figure 4, A and B). As anticipated, we observed that overexpression of APC significantly inhibited the proliferation and migration of CRC cells in vivo and that silencing of lncRNA-APC1 in APC-overexpressed CRC cells could largely abrogate the APC-inhibited cell proliferation and migration (Figure 4, C and D).
Figure 3. Suppression of CRC tumorigenesis and metastasis by lncRNA-APC1.

(A) Proliferation rate of empty vector– or lncRNA-APC1–transfected cells as determined by CCK8 assay (P < 0.05). (B) Representative images of decreased foci formation. (C) Images of xenograft tumors formed in nude mice. (D) Representative images of H&E-stained sections from metastatic nodules in the lung (original magnification, ×100). (E) Representative images of hepatic and splenic tissue in a nude mouse metastasis model. Black arrow indicates CRC cells; green arrow indicates liver tissue; blue arrow indicates splenic tissue (original magnification, ×100). Data represent the mean ± SD of 3 independent experiments. Scale bars: 50 μm. **P < 0.01, ***P < 0.001, and ****P < 0.0001, by independent Student’s t test.
Figure 4. APC function is partly dependent on lncRNA-APC1.

(A) APC expression in the DLD-1 stable cell line. (B) Relative expression of lncRNA-APC1 in DLD-1 cells transfected with shRNAs specific for silencing lncRNA-APC1. (C) Images of xenograft tumors formed in nude mice. (D) Representative images of H&E-stained sections from metastatic nodules in the lung (original magnification, ×100). Scale bars: 50 μm. Data represent the mean ± SD of 3 independent experiments. *P < 0.05, **P < 0.01, and ****P < 0.0001, by 1-way ANOVA.
To deepen our understanding of this process, we performed analysis of tumor tissues in a subcutaneous xenograft model and observed that ectopic overexpression of lncRNA-APC1 led to marked tumor tissue necrosis (almost more than 70% of the tumor tissue) (Figure 5A). Although overexpression of lncRNA-APC1 had little effect on CRC cell apoptosis (Supplemental Figure 4C), it induced moderate cell-cycle arrest at the G1 phase (Figure 5, B and C). Meanwhile, we found that neither β-catenin nor c-Myc (2 well-known downstream targets of APC) expression levels were influenced by the enforced depletion or overexpression of lncRNA-APC1 (Supplemental Figure 4D). Moreover, T cell factor/lymphoid enhancer factor (TCF/LEF) reporter gene and c-Myc reporter gene activity assays showed that lncRNA-APC1 had no effect on β-catenin or c-Myc transcriptional activity (Supplemental Figure 4E). These results suggest that lncRNA-APC1 exerts its effects independently of β-catenin or c-Myc. We know that angiogenesis is required to provide the nutrients and oxygen for the survival of tumor cells and that it is also essential for their metastasis. We were also able to identify a drastically decreased microvascular density (MVD) in the lncRNA-APC1–overexpressed tumor tissues in our mouse model (Figure 5D), and our in vitro assays consistently and clearly showed significantly decreased tube formation and migration of HUVECs using exosomes derived from lncRNA-APC1–overexpressed CRC cells (Figure 5, E and F).
Figure 5. Suppression of CRC tumor growth by lncRNA-APC1 acts through the inhibition of tumor angiogenesis.

(A) Representative images of H&E-stained sections from xenograft tumors formed in nude mice. Original magnification, ×40 (top); ×400 (bottom). (B) Expression of cell-cycle checkpoint markers as revealed by Western blotting. (C) Flow cytometric analysis of the cell cycle. (D) MVD of the indicated xenograft tumors detected by CD34 staining. Capillary tube formation assay (E) and Transwell invasion assay (F) of HUVECs treated with the indicated exosomes derived from transfected HCT116 cells. Scale bars: 100 μm (D and F). Original magnification, ×40 (E). Data represent the mean ± SD of 3 independent experiments (E and F). ****P < 0.0001, by independent Student’s t test.
lncRNA-APC1 inhibits angiogenesis by reducing exosome production in CRC cells.
We then asked whether lncRNA-APC1 might affect the expression of certain important angiogenesis-associated factors in CRC, including VEGFA, EGF, PDECFG, ANG1, ANG2, and TGF-β1. However, in our study, we did not observe any significant changes in the expression levels in the CRC cells before or after ectopic overexpression of lncRNA-APC1 (Figure 6A). Recently, it has been suggested that exosomes can act as paracrine or autocrine factors to affect important biological functions mediating cell-to-cell interactions (16–18), and, further, growing evidence suggests that exosomes released from cancer cells can contribute to tumor angiogenesis and metastasis (19–21). Notably, Irina Nazarenko et al. reported that tumor-derived exosomes could efficiently induce angiogenesis without an initial requirement for known angiogenic factors (22). Therefore, in the next part of our study, we isolated exosomes from CRC cell culture medium and confirmed their identity by Western blot analysis (Figure 6B) and electron microscopy (Figure 6C). We quantified the size distribution of exosomes using NanoSight analysis (Figure 6D). As shown in Figure 6C, the shape and size distribution of the exosomes in the control and lncRNA-APC1–overexpressed groups were not significantly different. Surprisingly, the concentrations of the exosome lysates were drastically decreased in the lncRNA-APC1–overexpressed CRC cells as compared with those from the same amount of control cells (Figure 6E), and we confirmed this by quantification using NanoSight (Figure 6F). Furthermore, we found that induced overexpression of APC clearly inhibited the production of exosomes in CRC cells, which could be reversed in part by silencing lncRNA-APC1 (Figure 6G).
Figure 6. lncRNA-APC1 suppresses exosome production.

(A) Relative expression of angiogenesis-associated factors in cells stably overexpressing lncRNA-APC1 or in control cells. (B) Western blot analysis of exosome markers. (C) Representative images of exosomes by electron microscopic detection. Scale bars: 200 nm. (D) Size distribution of exosomes analyzed by NanoSight. Relative fold changes in protein concentrations of exosome lysates as determined by (E) bicinchoninic acid (BCA) assay or (F) NanoSight analysis. (G) Relative fold changes in protein concentrations of exosome lysates as revealed by BCA assay. All experiments were repeated at least 3 times. Data represent the mean ± SD of 3 independent experiments. ***P < 0.001 and ****P < 0.0001, by independent Student’s t test (E and F) or 1-way ANOVA (G).
We further investigated whether lncRNA-APC1 could inhibit tumor angiogenesis through exosomes. We observed that exosomes derived from lncRNA-APC1–overexpressed DLD-1 and SW480 cells showed a substantial decrease in the migration and tube formation of HUVECs when compared with those from control cells (Figure 7, A and B), while exosomes derived from lncRNA-APC1–depleted HCT116 cells largely enhanced the migration and tube formation of HUVECs (Figure 7, C and D). In addition, our immunofluorescence assay demonstrated that exosomes were directly taken up by HUVECs in vitro (Supplemental Figure 5A) and by endothelial cells in vivo (Supplemental Figure 5B). Collectively, these results suggest that lncRNA-APC1 exerts a strong antiangiogenic effect on CRC cells by suppressing exosome production.
Figure 7. lncRNA-APC1 inhibits tumor angiogenesis through exosomes.

(A) Transwell invasion assay of HUVECs treated with the indicated exosomes (exo). Scale bar: 50 μm. (B) Capillary tube formation assay of HUVECs treated with the indicated exosomes. (C) Transwell invasion assay of HUVECs treated with the indicated exosomes derived from transfected HCT116 cells. Scale bars 100 μm. (D) Capillary tube formation assay of HUVECs treated with the indicated exosomes. Data represent the mean ± SD of 3 independent experiments. **P < 0.01 and ***P < 0.001, by independent Student’s t test.
lncRNA-APC1 reduces the production of CRC exosomes through Rab5b.
Since Ras-related Rab proteins control exosome biogenesis and release (23), we first analyzed the expression levels of certain Rab genes (i.e., Rab1a, Rab5a, Rab5b, Rab7, Rab27a, and Rab27b) between the lncRNA-APC1–overexpressed and control CRC cells. Notably, the mRNA levels of both Rab5b, which regulates the motility and fusion of early endosomes, and Rab27b, which plays a vital role in exosome release (24, 25), were significantly decreased with ectopic overexpression of lncRNA-APC1 (Figure 8A). However, since the expression of Rab27b protein was barely detectable in either of our SW480 or DLD-1 cell lines, we focused on Rab5b in our subsequent experiments (Figure 8B). As anticipated, we observed that lncRNA-APC1 silencing largely prevented the decrease in mRNA and protein levels of Rab5b in APC-overexpressed CRC cells (Figure 8, C and D). Additionally, neither truncated APC nor β-catenin significantly regulated Rab5b expression (Supplemental Figure 6, A–C). To further determine whether Rab5b is involved in APC- and lncRNA-APC1–mediated cellular functions, we constructed Rab5b–stably silenced CRC cells (Figure 8E). We found that silencing of Rab5b in DLD-1 cells resulted in an almost 70% decrease in exosome secretion, as determined by NanoSight analysis (Figure 8F). Further in vivo assays showed that knockdown of Rab5b could significantly suppress tumor growth and distant colonization of CRC cells (Figure 8G and Supplemental Figure 6D). Furthermore, we assessed the expression levels of Rab5b protein by IHC in a large collection of 229 primary CRC tissue samples. Survival analysis showed that CRC patients with high expression of Rab5b protein had a poorer prognosis than did patients with low expression of Rab5b (Supplemental Figure 6, E and F). These data suggested a critical role of Rab5b in exosome production during CRC cell progression. Subsequently, we tested the contribution of Rab5b to the function of lncRNA-APC1. Our results showed that knockdown of lncRNA-APC1 significantly enhanced the in vitro proliferation (Figure 9, A and B) and migration (Figure 9C) of CRC cells, an effect that could be markedly prevented by Rab5b silencing.
Figure 8. lncRNA-APC1 reduces the production of CRC exosomes through Rab5b.

(A) Relative expression of Rab genes as determined by qRT-PCR. (B) Rab5b expression in the indicated lncRNA-APC1 stable cells or control cells as detected by Western blotting. Levels of Rab5b mRNA (C) and Rab5b protein (D) in the indicated cells. (E) Rab5b was efficiently knocked down by specific shRNAs. (F) Relative fold change in the protein concentration of exosome lysates as determined by BCA assay. (G) Images of the xenograft tumors formed in nude mice by injecting Rab5b–stably silenced or control cells. Data represent the mean ± SD of 3 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001, by independent Student’s t test (A) or 1-way ANOVA (C, F, and G).
Figure 9. lncRNA-APC1 silencing enhances the proliferation and migration of CRC cells through Rab5b.

(A) Cell proliferation rate induced by lncRNA-APC1 silencing and/or Rab5b knockdown as determined by CCK8 assay. (B) Representative images of foci formation. (C) Representative images of the Transwell invasion assay. Scale bars: 100 μm. Data represent the mean ± SD of 3 independent experiments. **P < 0.01, ***P < 0.001, and ****P < 0.0001, by 1-way ANOVA.
lncRNA-APC1 may interact with Rab5b mRNA and reduce its stability in CRC cells.
Our next goal was to explore the potential mechanisms by which lncRNA-APC1 regulates Rab5b in CRC. It has been established that the ability of lncRNA to recognize complementary sequences allows highly specific interactions that are able to regulate gene expression (4). Many lncRNAs have been reported to function as competing endogenous RNAs (ceRNAs) by competitively binding miRNAs or interacting with RNAs directly, thus affecting their stability (7, 8, 26, 27). To determine whether lncRNA-APC1 regulates Rab5b expression by directly interacting with its mRNA, we first compared the mRNA sequences of lncRNA-APC1 and Rab family genes using the Basic Local Alignment Search Tool (BLAST) (http://blast.ncbi.nlm.nih.gov/). Surprisingly, we identified 2 highly complementary regions between lncRNA-APC1 and Rab5b mRNA (Figure 10A) and 8 highly complementary regions between lncRNA-APC1 and Rab27b mRNA (Supplemental Figure 7A), however, this was isolated to Rab5b and Rab27b mRNA, and no such regions were observed for other RAB family genes or GAPDH mRNA (data not shown). To validate the direct interaction of lncRNA-APC1 with Rab5b mRNA, we mutated 2 binding sites in lncRNA-APC1 with Rab5b and performed affinity pulldown of endogenous Rab5b mRNA using in vitro–transcribed biotin-labeled lncRNA-APC1 and lncRNA-APC1-Mut (Rab5b). The results revealed that lncRNA-APC1 was significantly enriched in Rab5b mRNA compared with that seen with lncRNA-APC1-Mut (Rab5b), lncRNA-APC1 antisense control, and GAPDH mRNA (Figure 10B). The specific association between lncRNA-APC1 and Rab5b mRNA was further validated by our RNA immunoprecipitation (RIP) and qRT-PCR assays in both SW480 and DLD-1 cells (Figure 10, C and D). These findings indicate that lncRNA-APC1 might interact with Rab5b mRNA.
Figure 10. In CRC cells, lncRNA-APC1 interacts with and reduces the stability of Rab5b mRNA.

(A) Regions of putative binding between Rab5b mRNA (query) and lncRNA-APC1 (subject). (B) SW480 and DLD-1 cell lysates were incubated with biotin-labeled WT or mutant type (Mut) lncRNA-APC1. After pulldown, mRNA was extracted and measured by qRT-PCR. (C) Model of RIP assay. (D) RIP-derived RNA was examined by qRT-PCR. The levels of the qRT-PCR products were normalized relative to input RNA and IgG control. (E) The stability of Rab5b mRNA and Actb mRNA was measured by qRT-PCR relative to t0 after blocking new RNA synthesis with α-amanitin and normalized to 18S rRNA. Data represent the mean ± SD of 3 independent experiments. **P < 0.01, ***P < 0.001, and ****P < 0.0001, by 1-way ANOVA.
Next, to test whether lncRNA-APC1 regulates the stability of Rab5b mRNA, we treated DLD-1 and SW480 cells with α-amanitin to block RNA polymerase II–mediated new RNA synthesis and then measured the loss of Rab5b and β-actin over a 24-hour period. Ectopic overexpression of lncRNA-APC1, but not that of lncRNA-APC1-Mut (Rab5b), reduced the half-life of Rab5b mRNA (Figure 10E), whereas knockdown of lncRNA-APC1 clearly elongated the half-life of Rab5b mRNA (Supplemental Figure 7B). Moreover, enforced expression of WT APC significantly decreased the stability of Rab5b mRNA, which could be prevented by silencing lncRNA-APC1 (Supplemental Figure 7C). We then performed a rescue functional assay to verify the interaction among APC, lncRNA-APC1, and Rab5b in the CRC cell line HCT116 (with WT APC). As anticipated, ectopic overexpression of WT lncRNA-APC1 suppressed APC-knockdown–enhanced cell proliferation and migration as well as cytoskeletal remodeling. On the other hand, the plasmid containing mutated Rab5b binding sites in lncRNA-APC1 clearly prevented the suppressive effect of WT lncRNA-APC1 on CRC cell proliferation, cytoskeleton and migration, which was substantially rescued by induced knockdown of Rab5b (Supplemental Figure 8, A–D). In addition, a significant inverse correlation between the levels of lncRNA-APC1 and Rab5b mRNA was observed in CRC tissue samples from a cohort of 50 patients (Supplemental Figure 8E). These data collectively provided evidence that lncRNA-APC1 could specifically decrease the stability of Rab5b mRNA to suppress the malignant potential of CRC cells and that this process is dependent on the direct binding of lncRNA-APC1 with Rab5b mRNA.
Exosomes enhance tumor angiogenesis by activating the MAPK pathway in endothelial cells.
To analyze the mechanisms of lncRNA-APC1–regulated exosomes in controlling tumor angiogenesis, we extracted total RNA from HUVECs incubated with exosomes derived from lncRNA-APC1–silenced or control HCT116 cells. Next, we conducted a microarray-based gene expression profile analysis to identify the key genes and/or signaling pathways that regulate HUVEC functions. Consistent with the exosome-induced functions of HUVECs, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of genes potentially regulated by exosomes in our microarray assay revealed significant alternations in actin cytoskeletal regulation (Figure 11A), with the greatest enrichment occurring in the MAPK signaling pathway. Numerous studies have shown that p38 MAPK cascades are the major signaling pathways involved in regulating endothelial cell actin remodeling, migration, and angiogenesis during cancer progression and metastasis (28–30). To validate the result from the microarray, we first measured the expression of the factors HSPA6 and PPM1B, both of which can inhibit the MAPK pathway (31–34). qRT-PCR data confirmed a significant decrease in the expression of both HSPA6 and PPM1B in HUVECs treated with exosomes derived from lncRNA-APC1–silenced HCT116 cells (Figure 11B). Results from Western blotting showed that the p38 MAPK pathway was in fact activated by the exosomes (Figure 11C). Furthermore, knockdown of p38 MAPK largely inhibited tube formation and migration of HUVECs treated with those exosomes (Figure 11D and Supplemental Figure 9A). Actin remodeling is essential for the contractile motion of endothelial cells and angiogenesis (28, 29). Further F-actin staining revealed that stress fiber and lamellipodia were enhanced in CRC cells treated with exosomes and that this could be successfully halted by silencing of p38 (Supplemental Figure 9B).
Figure 11. Exosomes enhance tumor angiogenesis by activating MAPK signaling in HUVECs.

(A) Function (left) and pathway (right) enrichment analysis of the results from the gene expression profile microarray. (B) Relative expression of the indicated genes measured by qRT-PCR. (C) Western blot analysis shows that the MAPK pathway in HUVECs was activated by exosomes derived from lncRNA-APC1–silenced HCT116 cells. p-p44/42, phosphorylated p44/42; p-p38, phosphorylated p38. (D) Capillary tube formation (top) and Transwell invasion assay (bottom) of HUVECs treated with the indicated exosomes (P < 0.05, by 1-way ANOVA). Scale bars: 100 μm. The capillary tube formation experiment was performed at least 3 times, independently of the assay in Figure 7D. Data represent the mean ± SD of 3 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.001, by independent Student’s t test (B) or 1-way ANOVA (D).
Exosomal Wnt1 enhances the proliferation and migration of CRC cells through noncanonical Wnt signaling.
Our finding that ectopic lncRNA-APC1 expression can inhibit in vitro CRC cell proliferation and migration suggests that exosomes may exert their effects in an autocrine manner. A previous report showed that Wnt3A protein is secreted on exosomes and can induce canonical Wnt signaling (35). Therefore, we hypothesized that CRC cell–derived exosomes might function in an autocrine manner by activating the Wnt pathway. Our immunoblot analysis of CRC cell–derived exosomal lysates showed marked expression of Wnt1 (Figure 12A). Then, we silenced Wnt1 expression in CRC cells to further explore the function of exosomal Wnt1 (Figure 12B). Meanwhile, we observed no significant changes in lncRNA-APC1 expression with knockdown of Wnt1 (Figure 12C). As shown in the in vitro assay, exosomes derived from Wnt1-silenced CRC cells could partially abrogate the enhanced proliferation and migration of the cancer cells that were induced by the control exosomes, indicating that exosomal Wnt1 has signal-inducing functions (Figure 12, D and E). However, the levels of TCF/LEF reporter activity induced by exosomes derived from CRC cells or Wnt1-silenced CRC cells were comparable (Supplemental Figure 10A), suggesting that exosomal Wnt1 could act through noncanonical Wnt signaling. We next studied the Wnt/PCP signaling that regulates actin cytoskeletal and cellular movement (36, 37), as well as the Wnt/RTK signaling that activates the PI3K/AKT signaling cascade (38, 39). As anticipated, we found that CRC cell exosomes can largely activate the factors Rho, JNK (which is involved in Wnt/PCP signaling), and AKT and that such effects can be partly abrogated after depletion of Wnt1 (Supplemental Figure 10B).
Figure 12. Exosomal Wnt1 enhances CRC cell proliferation and migration through noncanonical Wnt signaling.

(A) Wnt1 expression in the indicated CRC cell–derived exosomes. (B) Wnt1 expression was effectively knocked down by specific siRNA-2 and siRNA-3. (C) Expression of lncRNA-APC1 detected by qRT-PCR. (D) Representative images of decreased foci formation induced by Wnt1-silencing exosomes. (E) Representative images of Transwell invasion assay. Scale bars: 50 μm. Data represent the mean ± SD of 3 independent experiments. **P < 0.01, ***P < 0.001, and ****P < 0.001, by 1-way ANOVA.
Discussion
Mutations in the APC gene were reported to occur in up to 80% of sporadic human CRCs (40). Inactivation of APC is sufficient to initiate colorectal adenoma in mice, and it is a well-established critical driver of the pathogenic process of CRC (1). Nevertheless, the abnormalities of certain lncRNAs and their roles in mediating the functions of APC in the tumorigenesis and/or progression of CRC have yet to be elucidated.
In the present study, we first used microarray screening to select and identify lncRNA-APC1 as a downstream factor of APC in CRC. We know that regulation of β-catenin stabilization is the most prominent function of APC, however, results from the β-catenin gene–silencing and ectopic expression assay showed that the expression of lncRNA-APC1 was clearly regulated by APC, independently of β-catenin. In addition, we found that WT, but not truncated, APC is mainly responsible for regulating lncRNA-APC1 expression. We performed further biological analysis with the lncRNA-APC1 promoter and the luciferase reporter assay, and our findings indicated that PPARα might bind with the promoter of lncRNA-APC1 and decrease its transcriptional activity. The PPAR family members play central roles in the regulation of glucose and lipid homeostasis, and they have been shown to be essential in CRC carcinogenesis (14), lending support to the theory that PPARα is involved in mediating the regulation that occurs between APC and lncRNA-APC1. Furthermore, our PPARα binding motif deletion reporter and ChIP assays confirmed that PPARα is responsible for APC-induced lncRNA-APC1 expression.
Subsequently, we sought to determine the clinical relevance of lncRNA-APC1 and its impact on patients with CRC. The results showed that low expression of lncRNA-APC1 in CRC tissues was positively correlated with lymph node and/or distant metastasis of tumors. Moreover, CRC patients with low lncRNA-APC1 expression had a shorter survival than did patients with normal lncRNA-APC1 expression levels. These findings suggest that lncRNA-APC1 might play an anticancer role in CRC pathogenesis. In order to further our understanding of these concepts, we performed a series of additional in vitro and in vivo and loss-of-function assays in this study. We verified that lncRNA-APC1, functioning as a mediator of APC, was a potent tumor suppressor that could dramatically inhibit the proliferation, growth, and migration of CRC cells in vitro and substantially suppress tumor formation and distant metastasis in vivo. Interestingly, in a subcutaneous xenograft mouse model, we observed that enforced expression of lncRNA-APC1 in CRC cells led to marked necrosis of tumor tissue. This suggests that, in addition to the moderate cell-cycle arrest effect of lncRNA-APC1 in CRC cells, its inhibitory effect on tumor growth in vivo could be the result of other factors such as the regulation of CRC angiogenesis in the tumor microenvironment. As anticipated, we further found that ectopic overexpression of lncRNA-APC1 in CRC cells markedly inhibited tumor angiogenesis both in vitro and in vivo, although certain angiogenic factors were not significantly affected by lncRNA-APC1. It has been reported that tumor-derived exosomes can efficiently induce angiogenesis, without the initial requirement for known angiogenic factors (22). We speculated that lncRNA-APC1 might exert its suppressive effect through exosomes. Unexpectedly, exosome lysate concentration measurement and quantitative testing by NanoSight showed that lncRNA-APC1 could substantially suppress the production of exosomes in CRC cells. Furthermore, we found that lncRNA-APC1 was sufficient to inhibit angiogenesis through the reduction of exosome levels, as determined by in vitro or in vivo assays. Taken together, these data provide sufficient evidence that lncRNA-APC1 reduces exosome production in CRC cells, ultimately resulting in the suppression of tumor angiogenesis.
It has been suggested that Ras-related Rab proteins control exosome biogenesis and release (23). Matias Ostrowski and colleagues previously identified 5 Rab proteins (Rab2b, Rab9a, Rab5a, Rab27a, and Rab27b) that could influence the secretion of exosomes in human HeLa cells (24). Other studies have shown that Rab11 is essential for Ca2+-regulated secretion exosomes in an erythroleukemia tumor cell line and that Rab35 is required for the secretion of proteolipoprotein-enriched (PLP-enriched) exosomes by oligodendrocytes (41, 42). Therefore, no consensus has been achieved on which intracellular machinery is involved in exosome production, although it does seem to be dependent on the cell type. Next, we further confirmed by a series of gain- and loss-of-function in vitro and in vivo assays that Rab5b, a small GTPase protein that regulates the motility and fusion of early endosomes, is a key target of lncRNA-APC1 and is important for the production of exosomes in CRC cells. Furthermore, RIP and RNA-pulldown assays verified that lncRNA-APC1 bound the mRNA of Rab5b and dramatically decreased its stability, an observation that was also confirmed by examining the correlation between lncRNA-APC1 and Rab5b mRNA expression in our clinical CRC tissues.
Despite all the latest research, the mechanisms by which exosomes induce angiogenesis in CRC remain largely unknown. In recent years, it has been suggested that p38 MAPK cascades are the major signaling pathways that regulate the actin remodeling, migration, and angiogenesis of endothelial cells (28, 30). Consistent with this, our gene expression profile microarray and KEGG analyses indicated that the MAPK pathway may play a critical role in the angiogenesis process induced by lncRNA-APC1–regulated exosomes. We further demonstrated through loss-of-function in vitro assays that exosomes derived from lncRNA-APC1–silenced CRC cells could enhance the migration, actin remodeling, and angiogenesis of endothelial cells through activation of the MAPK pathway. Exosomes exert their effects by transferring their contents, such as protein, miRNA, lncRNA, and circular RNA (circRNA), to recipient cells (18, 43). Further investigation is needed to elucidate the precise mechanisms by which lncRNA-APC1–regulated exosomes activate the MAPK pathway of endothelial cells.
Our in vitro data indicated that the exosomes of CRC cells function in an autocrine manner. Previous research has reported that Wnt protein is secreted on the surface of exosomes (35). Given the well-known role of APC as an essential component of the canonical Wnt pathway (Wnt/β-catenin signaling), we set out to further investigate whether lncRNA-APC1, as a key regulator of exosome production and downstream of APC, is involved in the Wnt pathway as it pertains to exosomes in CRC. We found that Wnt1 expression on CRC cell–derived exosomes was considerable and that exosomal Wnt1 substantially enhanced the capacity for CRC cell proliferation and migration through noncanonical Wnt1 signaling. Furthermore, we showed that CRC-derived total exosomes could largely activate the canonical Wnt pathway. However, neither lncRNA-APC1 nor exosomal Wnt1 had any significant effect on the canonical Wnt pathway. Collectively, these data suggested that lncRNA-APC1 might primarily act through the suppression of certain subgroups of exosomes, of which Wnt1+ exosomes could be an important member.
In summary, this report identifies for the first time to our knowledge that lncRNA-APC1 is an important mediator of APC function through the direct regulation of Rab5b mRNA stability, thereby reducing exosome production in CRC cells. Moreover, our findings reveal that CRC-derived exosomes exert a strong effect on the pathogenesis and/or angiogenesis of CRC regulated by the lncRNA-APC1/Rab5b axis, suggesting a mechanism for APC signaling that is independent of β-catenin and the canonical Wnt pathway (Figure 13). The importance of this result lies in its potential to provide additional targets for therapeutic intervention in human CRC.
Figure 13. Model of lncRNA-APC1 function and mechanisms during CRC pathogenesis.

lncRNA-APC1, an important mediator of APC, inhibits the pathogenesis and/or angiogenesis of CRC through directly reducing Rab5b mRNA stability and exosome production in CRC cells.
Methods
Cell culture.
The colorectal cell lines HCT116, DLD-1, SW480, LOVO, and SW1116 were obtained from the American Type Culture Collection (ATCC) and cultured in RPMI 1640 medium with 10% FBS (Invitrogen, Thermo Fisher Scientific). The human kidney HEK293T cell line was purchased from the ATCC and cultured in DMEM medium with 10% FBS. The HUVEC cell line was a gift of L.B. Song (Sun Yat-sen University Cancer Center) and was cultured in serum-free medium (SFM) supplemented with 10% FBS, 0.02 μg/ml basic fibroblast growth factor (bFGF), and 0.01 μg/ml EGF. Cells were cultured at 37°C in an incubator (Thermo Fisher Scientific) with 5% CO2 and 95% air.
Tissue specimens.
A total of 110 primary CRC tissue samples (including 30 adjacent healthy tissues) were collected between January 2005 and December 2005 at the Sun Yat-sen University Cancer Center. The tissue microarray (TAM) containing samples from 229 patients with CRC was constructed as previously reported (44). The CRC cases were selected on the basis of the following inclusion criteria: a clear pathological diagnosis, the presence of follow-up data, and the absence of previous local or systemic treatment. Two pathologists reassessed and confirmed all the pathologic diagnosis results. Tumor stage was defined according to the 2002 American Joint Committee on Cancer/International Union Against Cancer tumor-node-metastasis (TNM) classification system.
Microarray analysis.
SW480 and DLD-1 cells were transfected with WT APC, full-length CDS, or control vector. Then, total RNA was extracted and transcribed. Double-stranded cDNA was labeled using the Quick Amp Labeling Kit (Agilent Technologies) and hybridized to the Arraystar Human 8× 60K lncRNA Array, version 2.0. Following the washing steps, the arrays were scanned with the Agilent Scanner G2505B, and the array images were analyzed using Agilent Feature Extraction software, version 10.7.3.1. Quantile normalization and subsequent data processing were performed using GeneSpring GX software, version 11.5.1 (Agilent Technologies). Volcano plot filtering was used to identify the lncRNAs with statistically significant differences, and the threshold to screen upregulated or downregulated lncRNAs was identified at a fold change of 1.5 or greater and a P value of 0.05 or less.
The gene expression profiles of HUVECs incubated with lncRNA-APC1–silenced or control HCT116 cancer cell–derived exosomes were determined using Phalanx OneArray human microarrays (HOA 6.1) following the manufacturer’s instructions.
5′-RACE and 3′-RACE.
The 5′-RACE and 3′-RACE assays were performed to determine the transcriptional initiation and termination sites of lncRNA-APC1 using SuperScript IV Reverse Transcriptase (Invitrogen, Thermo Fisher Scientific) following the manufacturer’s instructions. The primers used for PCR of the RACE analysis were as follows: 5′-GATGTTCAAGGGCAGGAAGAA-3′ (5′-RACE) and 5′-TCACAGAAGGCTCTGCGACT-3′ (3′-RACE).
Northern blot analysis.
A biotin-16-dUTP–labeled lncRNA-APC1 complementary RNA probe (Sangon Biotech) and a NorthernMax Kit (Invitrogen, Thermo Fisher Scientific) were used according to the manufacturers’ instructions. Briefly, 10 μg total RNA was loaded into the wells of the gel and then run at approximately 5 V/cm. Following electrophoresis, RNA was transferred onto a nylon membrane (GE Healthcare) overnight. After 30 minutes of prehybridization, the membrane was hybridized for 16 hours at 52°C in ULTRAhyb buffer containing the denatured probe. After the blocking and washing steps, the membrane was probed using the Chemiluminescent Nucleic Acid Detection Module Kit (Thermo Fisher Scientific). The RNA probe sequence was as follows: 5′-biotin-GACUCUCAUACAGGUAGAAGACCCAGGACCCUACCAGUUUACUCAAGCC-3′.
FISH.
Paraffin-embedded tissues derived from CRC patients’ tumors and nontumor colonic tissue were used along with a digoxin-LNA–modified oligonucleotide probe (Exiqon) in order to determine the lncRNA location. The slides were mounted onto flow through slide chambers and placed in a hybridization instrument, and the following steps were performed: treatment with 15 μg/ml proteinase-K at 37°C for 8 minutes, prehybridization in hybridization buffer at 37°C for 30 minutes, and hybridization with LNA probe (5′-DigN-AGCGGGAGAGAAGAGTCACAT-3′-Dig_N). Stringent washes with 5× SSC, 1× SSC, and 0.2× SSC buffers at 37°C for 15 minutes each time were strictly executed. A biotin-SP IgG Fraction Monoclonal Mouse Anti-digoxin Antibody (Jackson ImmunoResearch Laboratories) was combined with the probe. An Alexa Fluor 488 Tyramide SuperBoost Kit (Invitrogen, Thermo Fisher Scientific) was used to enhance the expression signal of lncRNA-APC1 and stain the nuclei with DAPI (Beyotime).
ChIP.
ChIP assays were performed using anti-PPARα and anti-IgG antibodies (R&D Systems) and the EZ-Magna ChIP A/G Chromatin Immunoprecipitation Kit (Merck Millipore) according to the manufacturer’s instructions. Anti-Mouse IgG was used as a negative control. The strong binding of DNA fragments was detected by qRT-PCR using the specific primers.
Exosome isolation.
For exosome isolation, cells were maintained in their respective medium with exosome-free FBS, which was prepared by centrifugations to remove existing exosomes. Then, exosomes were collected through standard centrifugation steps, as previously described (45). Briefly, the collected culture medium was centrifuged at 300 g for 10 minutes, followed by 2000 g for 20 minutes, and then 10,000 g for 30 minutes. The supernatant was then filtered through a 0.2-μM filter (Pall Corp.). The resultant medium was centrifuged at 100,000 g for 70 minutes at 4°C to pellet the exosomes. The resultant supernatant was discarded without disturbing the pellet, which was washed with a large volume of PBS and then again ultracentrifuged under the same conditions, before final resuspension in a volume of PBS (usually 50 µl to 100 µl). Exosomes were examined by electron microscopy using negative staining and then quantified using a Micro BCA Protein Assay Kit (Thermo Fisher Scientific) and a NanoSight NS300 instrument equipped with NTA 3.0 analytical software (both from Malvern Instruments Ltd.).
RNA pulldown.
lncRNA-APC1-Mut (Rab5b), lncRNA-APC1, and the negative control lncRNA-APC1 antisense were transcribed in vitro from the vectors pSPT19-lncRNA-APC1-Mut (Rab5b) and pSPT19-lncRNA-APC1, respectively, using Biotin RNA Labeling Mix (Roche) and T7 and SP6 RNA polymerase (Promega). RNA was purified using the RNeasy Mini Kit (QIAGEN). Then, approximately 10 pmol biotinylated RNA from the previous step was added to 200 μg whole-cell lysate from DLD-1 cells and supplemented with tRNA (Ambion) to a final concentration of 0.1 μg/μl, followed by incubation at 4°C overnight with gentle rotation and then addition of 40 μl prewashed streptavidin magnetic beads (Invitrogen, Thermo Fisher Scientific) for 1 hour at room temperature. The RNA reserved in the beads was detected by qRT-PCR.
RIP experiments.
pSL-MS2-12x (Addgene) was digested with BamH I and Not I, and the MS2-12x fragment was subcloned into pcDNA3.1, pcDNA3.1-lncRNA-APC1-WT, and pcDNA3.1-lncRNA-APC1-Mut (Rab5b), which were named pcDNA3.1-MS2, pcDNA3.1-MS2-lncRNA-APC1-WT, and pcDNA3.1-MS2-lncRNA-APC1-Mut (Rab5b), respectively. DLD-1 cells were cotransfected with pcDNA3.1-MS2, pcDNA3.1-MS2-lncRNA-APC1-WT, pcDNA3.1-MS2-lncRNA-APC1-Mut (Rab5b), and pMS2-GFP (Addgene). After 48 hours, cells were harvested for use in RIP experiments. IgG and GFP antibodies (Abcam) were used along with the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (MilliporeSigma).
Xenograft mouse model.
Athymic nude mice were purchased from Vital River Laboratories, housed under standard conditions in the animal care facility at the Center of Animal Experiments of Sun Yat-sen University. Treated CRC cells (3 × 106 cells in 0.1 ml FBS-free culture) were injected subcutaneously into the dorsal flanks of 4- to 5-week-old male athymic nude mice (n = 8/group). After 4 weeks, mice were sacrificed, and tumors were excised and weighed. For the liver metastasis assays, stable cell lines (2 × 106 cells in 0.05 ml FBS-free culture) were injected into the spleens of 5- to 6-week-old male athymic nude mice (n = 6/group). The tumor metastatic and distant colonization abilities of the stable cells to metastasize and colonize distant sites was determined by intrasplenic injection and tail vein injection. For lung colonization assays, stable cell lines (2 × 106 cells in 0.1 ml FBS-free culture) were injected into the tail veins of 5- to 6-week-old male athymic nude mice (n = 6/group). After 6 to 8 weeks, mice were sacrificed, tissue from liver and/or lung was excised, and tumor nodules formed in the respective organs were counted and analyzed by H&E staining.
See the Supplemental Methods for further details on the experimental procedures.
Statistics.
For survival analysis, the median was used as the optimal cutpoint for lncRNA-APC1 expression. The correlation between lncRNA-APC1 and the clinicopathological features of patients with CRC was analyzed using the χ2 test or Fisher’s exact test. For univariate survival analysis, survival curves were obtained using the Kaplan-Meier method. The Cox proportional hazards regression model was used for multivariate survival analyses. Pearson’s correlation was used to analyze the relative expression of lncRNA-APC1 and Rab5b mRNA. Measurements were analyzed using an independent 2-tailed Student’s t test or 1-way ANOVA. Statistical significance was defined as a P value of less than 0.05.
Study approval.
The use of human CRC tissue specimens in this study was approved by the ethics committee of the Sun Yat-sen University Cancer Center, and written informed consent was obtained from the patients or their guardians before sample collection. All animal studies were approved by the IACUC of Sun Yat-sen University.
Accession numbers.
The lncRNA microarray data for APC-overexpressing cells as well as gene expression profile microarray data for HUVECs treated with exosomes were deposited in the NCBI’s Gene Expression Omnibus (GEO) database (GEO GSE113742 and GSE113739).
Author contributions
DX conceived and devised the study. DX and FWW designed the experiments and conducted analysis. CHC, KH, MYC, JXZ, JWC, ZCX, LPZ, YH, SFZ, and XHJ performed the experiments. MYC and ZCX performed bioinformatics and statistical analyses. DX and MYC reassessed and confirmed the pathologic diagnosis results. FWW, CHC, and KH analyzed the data. RHX, YXZ, XYG, and DX supervised the research and, together with FWW, CHC, and KH, wrote the manuscript.
Supplementary Material
Acknowledgments
This work was supported by the National Key R&D Program of China (grant nos. 2017YFC1309001 and 2016YFC1302305); the National Natural Science Foundation of China (grant nos. 81430055, 81572359, 81602063, 81730072, 81772595); the Natural Science Foundation of Guangdong (grant no. S2014030001589); the Doctor Start-up Project of the Natural Science Foundation of Guangdong (grant no. 2016A030310231); the Program for Changjiang Scholars and the Innovative Research Team of the Ministry of Education of China (grant no. IRT-15R13); and the Guangzhou Science and Technology Plan Project (Health Medical Collaborative Innovation Program of Guangzhou, grant no. 201803040019).
Version 1. 12/04/2018
In-Press Preview
Version 2. 01/14/2019
Electronic publication
Version 3. 02/01/2019
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
License: Copyright 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(2):727–743.https://doi.org/10.1172/JCI122478.
See the related Commentary at Colorectal cancer: the APC-lncRNA link.
Contributor Information
Feng-Wei Wang, Email: wangfengw@sysucc.org.cn.
Chen-Hui Cao, Email: caochenh@sysucc.org.cn.
Kai Han, Email: hankai@sysucc.org.cn.
Yong-Xiang Zhao, Email: yongxiang_zhao@126.com.
Mu-Yan Cai, Email: caimuyan@hotmail.com.
Zhi-Cheng Xiang, Email: xiangzhch@sysucc.org.cn.
Jia-Xing Zhang, Email: 375369141@qq.com.
Jie-Wei Chen, Email: chenjiew@sysucc.org.cn.
Li-Ping Zhong, Email: zhong_liping@163.com.
Yong Huang, Email: huangyong503@126.com.
Su-Fang Zhou, Email: zsf200000@163.com.
Xiao-Han Jin, Email: jinxh@sysucc.org.cn.
Xin-Yuan Guan, Email: xyguan@hkucc.hku.hk.
Rui-Hua Xu, Email: xurh@sysucc.org.cn.
Dan Xie, Email: xiedan@sysucc.org.cn.
References
- 1.Markowitz SD, Bertagnolli MM. Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med. 2009;361(25):2449–2460. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sjöblom T, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 3.Näthke I. Cytoskeleton out of the cupboard: colon cancer and cytoskeletal changes induced by loss of APC. Nat Rev Cancer. 2006;6(12):967–974. doi: 10.1038/nrc2010. [DOI] [PubMed] [Google Scholar]
- 4.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43(6):904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10(3):155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 6.Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29(4):452–463. doi: 10.1016/j.ccell.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan JH, et al. A long noncoding RNA activated by TGF-β promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell. 2014;25(5):666–681. doi: 10.1016/j.ccr.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 8.Cesana M, et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147(2):358–369. doi: 10.1016/j.cell.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawasaki Y, Sato R, Akiyama T. Mutated APC and Asef are involved in the migration of colorectal tumour cells. Nat Cell Biol. 2003;5(3):211–215. doi: 10.1038/ncb937. [DOI] [PubMed] [Google Scholar]
- 10.Jiang H, et al. Peptidomimetic inhibitors of APC-Asef interaction block colorectal cancer migration. Nat Chem Biol. 2017;13(9):994–1001. doi: 10.1038/nchembio.2442. [DOI] [PubMed] [Google Scholar]
- 11.Morin PJ, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275(5307):1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Zhang W, Evans PM, Chen X, He X, Liu C. Adenomatous polyposis coli (APC) differentially regulates beta-catenin phosphorylation and ubiquitination in colon cancer cells. J Biol Chem. 2006;281(26):17751–17757. doi: 10.1074/jbc.M600831200. [DOI] [PubMed] [Google Scholar]
- 13.Sérée E, et al. Evidence for a new human CYP1A1 regulation pathway involving PPAR-alpha and 2 PPRE sites. Gastroenterology. 2004;127(5):1436–1445. doi: 10.1053/j.gastro.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 14.Peters JM, Shah YM, Gonzalez FJ. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat Rev Cancer. 2012;12(3):181–195. doi: 10.1038/nrc3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Preitner N, et al. APC is an RNA-binding protein, and its interactome provides a link to neural development and microtubule assembly. Cell. 2014;158(2):368–382. doi: 10.1016/j.cell.2014.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2(8):569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 17.Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ. Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia. 2006;20(9):1487–1495. doi: 10.1038/sj.leu.2404296. [DOI] [PubMed] [Google Scholar]
- 18.Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 19.Iero M, et al. Tumour-released exosomes and their implications in cancer immunity. Cell Death Differ. 2008;15(1):80–88. doi: 10.1038/sj.cdd.4402237. [DOI] [PubMed] [Google Scholar]
- 20.Zhou W, et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell. 2014;25(4):501–515. doi: 10.1016/j.ccr.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoshino A, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527(7578):329–335. doi: 10.1038/nature15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nazarenko I, et al. Cell surface tetraspanin Tspan8 contributes to molecular pathways of exosome-induced endothelial cell activation. Cancer Res. 2010;70(4):1668–1678. doi: 10.1158/0008-5472.CAN-09-2470. [DOI] [PubMed] [Google Scholar]
- 23.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10(8):513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 24.Ostrowski M, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. 2010;12(1):19–30. doi: 10.1038/ncb2000. [DOI] [PubMed] [Google Scholar]
- 25.Yun HJ, et al. An early endosome regulator, Rab5b, is an LRRK2 kinase substrate. J Biochem. 2015;157(6):485–495. doi: 10.1093/jb/mvv005. [DOI] [PubMed] [Google Scholar]
- 26.Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146(3):353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tay Y, et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell. 2011;147(2):344–357. doi: 10.1016/j.cell.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rousseau S, Houle F, Landry J, Huot J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene. 1997;15(18):2169–2177. doi: 10.1038/sj.onc.1201380. [DOI] [PubMed] [Google Scholar]
- 29.Kayyali US, Pennella CM, Trujillo C, Villa O, Gaestel M, Hassoun PM. Cytoskeletal changes in hypoxic pulmonary endothelial cells are dependent on MAPK-activated protein kinase MK2. J Biol Chem. 2002;277(45):42596–42602. doi: 10.1074/jbc.M205863200. [DOI] [PubMed] [Google Scholar]
- 30.Yoshizuka N, et al. A novel function of p38-regulated/activated kinase in endothelial cell migration and tumor angiogenesis. Mol Cell Biol. 2012;32(3):606–618. doi: 10.1128/MCB.06301-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huot J, Houle F, Spitz DR, Landry J. HSP27 phosphorylation-mediated resistance against actin fragmentation and cell death induced by oxidative stress. Cancer Res. 1996;56(2):273–279. [PubMed] [Google Scholar]
- 32.Awano K, et al. Phosphorylation of protein phosphatase 2Czeta by c-Jun NH2-terminal kinase at Ser92 attenuates its phosphatase activity. Biochemistry. 2008;47(27):7248–7255. doi: 10.1021/bi800067p. [DOI] [PubMed] [Google Scholar]
- 33.Aburai N, Yoshida M, Ohnishi M, Kimura K. Sanguinarine as a potent and specific inhibitor of protein phosphatase 2C in vitro and induces apoptosis via phosphorylation of p38 in HL60 cells. Biosci Biotechnol Biochem. 2010;74(3):548–552. doi: 10.1271/bbb.90735. [DOI] [PubMed] [Google Scholar]
- 34.Shin SS, et al. HSPA6 augments garlic extract-induced inhibition of proliferation, migration, and invasion of bladder cancer EJ cells; Implication for cell cycle dysregulation, signaling pathway alteration, and transcription factor-associated MMP-9 regulation. PLoS One. 2017;12(2):e0171860. doi: 10.1371/journal.pone.0171860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gross JC, Chaudhary V, Bartscherer K, Boutros M. Active Wnt proteins are secreted on exosomes. Nat Cell Biol. 2012;14(10):1036–1045. doi: 10.1038/ncb2574. [DOI] [PubMed] [Google Scholar]
- 36.Katoh M. WNT/PCP signaling pathway and human cancer (review) Oncol Rep. 2005;14(6):1583–1588. [PubMed] [Google Scholar]
- 37.Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov. 2014;13(1):63–79. doi: 10.1038/nrd4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu J, et al. Wnt5a induces ROR1/ROR2 heterooligomerization to enhance leukemia chemotaxis and proliferation. J Clin Invest. 2016;126(2):585–598. doi: 10.1172/JCI83535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anastas JN, et al. WNT5A enhances resistance of melanoma cells to targeted BRAF inhibitors. J Clin Invest. 2014;124(7):2877–2890. doi: 10.1172/JCI70156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87(2):159–170. doi: 10.1016/S0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 41.Hales CM, Vaerman JP, Goldenring JR. Rab11 family interacting protein 2 associates with Myosin Vb and regulates plasma membrane recycling. J Biol Chem. 2002;277(52):50415–50421. doi: 10.1074/jbc.M209270200. [DOI] [PubMed] [Google Scholar]
- 42.Kouranti I, Sachse M, Arouche N, Goud B, Echard A. Rab35 regulates an endocytic recycling pathway essential for the terminal steps of cytokinesis. Curr Biol. 2006;16(17):1719–1725. doi: 10.1016/j.cub.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 43.Corrado C, Raimondo S, Chiesi A, Ciccia F, De Leo G, Alessandro R. Exosomes as intercellular signaling organelles involved in health and disease: basic science and clinical applications. Int J Mol Sci. 2013;14(3):5338–5366. doi: 10.3390/ijms14035338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu W, et al. Overexpression of EIF5A2 promotes colorectal carcinoma cell aggressiveness by upregulating MTA1 through C-myc to induce epithelial-mesenchymaltransition. Gut. 2012;61(4):562–575. doi: 10.1136/gutjnl-2011-300207. [DOI] [PubMed] [Google Scholar]
- 45.Théry C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006;Chapter 3:Unit 3.22. doi: 10.1002/0471143030.cb0322s30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.