

Abstract
The Hedgehog pathway is critical for the development of diverse organs. Misactivation of the Hedgehog pathway can cause developmental abnormalities and cancers, including medulloblastoma, the most common pediatric brain tumor, and basal cell carcinoma, the most common cancer in the United States. Here, we review how basic, translational, and clinical studies of the Hedgehog pathway have helped reveal how cells communicate, how intercellular communication controls development, how signaling goes awry to cause cancer, and how to use targeted molecular agents to treat both inherited and sporadic cancers.
Introduction
Cancer is a genetic disease caused by activation of oncogenes, inactivation of tumor suppressors, or a combination thereof (1). The genetic and epigenetic changes underlying cancer can be induced by infectious agents (e.g., human papilloma and hepatitis viruses), environmental carcinogens (e.g., tobacco, asbestos, and ultraviolet radiation), and mistakes in DNA replication or repair. Most cancers are sporadic, meaning they occur in individuals without a family history of cancer or an inherited genetic risk of cancer (2). However, some individuals inherit genetic changes that predispose them to cancer. Inherited mutations can cooperate with acquired mutations to cause malignancies with greater frequency, earlier age of onset, and/or more aggressive clinical behavior than sporadic cancers. For instance, individuals with germline mutations in the retinoblastoma (RB) tumor suppressor may develop multiple aggressive tumors early in life, including ocular retinoblastomas, pineoblastomas, and sarcoma.
Identification of genes and mutations underlying cancer is complicated by the fact that most human malignancies harbor many mutations, only some of which contribute to the cancer phenotype. Functional mutations can be distinguished from passenger mutations by their recurrence in distinct sporadic cancers as well as their ability to dysregulate cell proliferation (3, 4). In contrast to sporadic cancers, inherited cancer predisposition syndromes allow population genetics to identify drivers of malignancy. For example, identifying mutations that segregate with highly penetrant phenotypes in cancer-prone pedigrees provides important clues to the genetic changes that cause cancer. Because the molecular pathways underlying rare inherited cancers and their more common sporadic counterparts are often the same, the identification of inherited cancer-predisposing mutations can illuminate the etiology of sporadic cancers.
As most current cancer treatment modalities (e.g., surgery, cytotoxic chemotherapy, and ionizing radiation) are agnostic to the molecular causes of cancer, much of diagnosis and management of cancer proceeds with limited or no investigation into underlying genetic changes. However, improved understanding of cancer genotypes has distinguished subgroups of tumors that display different sensitivities to traditional or targeted therapies, suggesting that genetic profiling of cancers is likely to facilitate more effective and less toxic treatments for cancer patients. Moreover, genetic profiling of normal tissue from cancer patients may provide insights into whether they are at risk of additional cancers. Identification of genetic cancer risk factors may guide medical surveillance, and prophylactic or therapeutic medical interventions. For example, individuals with inherited BRCA1 or BRCA2 mutations may opt for regular imaging surveillance or surgical prophylaxis to mitigate their increased risk of breast, ovarian, or prostate cancer.
Here, we review how insights into a pathway involved in inherited cancer predisposition, the Hedgehog (HH) pathway, have informed our knowledge of human malignancy. This curious and ancient pathway is essential for embryonic development and adult tissue homeostasis (5, 6). Molecular studies of the HH pathway have helped reveal how cells communicate, how intercellular communication controls cell growth, how signaling goes awry to cause malignancy, and how to use targeted molecular agents to treat both inherited and sporadic cancers.
Hedgehog signaling controls tissue patterning and growth
HH ligands are secreted lipoproteins (7). Mammals have at least three HH genes, the best studied of which encodes Sonic hedgehog (SHH), a crucial regulator of embryonic development and adult tissue homeostasis (8, 9). HH proteins are synthesized, often by regionally defined epithelial cells, as precursors that are activated by autocatalytic cleavage of the C-terminal intein domain (10). Once active, HH ligands stimulate the HH pathway, often in nearby mesenchymal cells, by binding to their transmembrane receptors, Patched 1 (PTCH1) and Patched 2 (PTCH2) (11–13). Vertebrate HH signaling is transduced by the primary cilium, a cellular antenna that projects from the surface of most cells (Figure 1 and refs. 14–16). When HH binds to PTCH proteins at the cilium, a π-cation lock is broken in another transmembrane protein, Smoothened (SMO), and SMO accumulates at the cilium to activate the downstream pathway (Figure 1 and refs. 17, 18).
Figure 1. A model of ciliary Hedgehog signaling.

(A) In the absence of Hedgehog ligands such as SHH, PTCH1 localizes to the primary cilium and, through an unknown mechanism, prevents SMO from entering the cilium. GLI proteins bind SUFU, a negative regulator, and are phosphorylated by kinases, such as PKA, to generate transcriptional repressors that enter the nucleus and silence the Hedgehog transcriptional program. (B) In the presence of SHH, PTCH1 leaves the cilium, allowing SMO to accumulate at the primary cilium membrane. At the cilium, SMO inhibits the formation of GLI3 repressor and activates GLI2, which enters the nucleus to promote transcription of Hedgehog target genes. (C) Inactivating mutations in PTCH1, PTCH2, or SUFU; activating mutations in SMO (denoted here as an asterisk); or amplification of GLI2 can activate expression of Hedgehog target genes in an unregulated way, leading to cancer.
In vertebrates, SMO activates GLI2, the primary activator of the HH transcriptional program (19). Activated SMO also suppresses the formation of a transcriptional repressor form of GLI3, which — through repression of a pathway inhibitor — contributes to the activation of the HH transcriptional program. GLI2 and GLI3 regulate the third member of the GLI family, GLI1, a feed-forward amplifier of transcriptional activation (6). In the absence of HH pathway activation, GLI family members are restrained by binding to Suppressor of Fused (SUFU) (20–22). Upon HH pathway activation, SUFU-GLI complexes accumulate in cilia and dissociate, and the GLI proteins regulate HH target gene expression in the nucleus (23–25).
Despite abiding interest in understanding mechanisms of HH signal transduction, the processes by which HH inhibits PTCH, PTCH inhibits SMO, SMO is activated, and SMO activates GLI transcription factors, as well as the full diversity of transcriptional targets of HH signaling, remain active areas of investigation. Adding to the difficulty of understanding HH signal transduction, many aspects of the HH pathway display overlapping or tissue-specific behavior. For example, HH signaling adaptation occurs during neural tube patterning, but not in other tissues (26). The tissue-specific aspects of HH signaling during development may help elucidate the different ways in which HH signaling functions in different cancer types.
Misactivation of the Hedgehog pathway causes cancer
Basal cell carcinoma.
HH signals are critical for the development of diverse organs, such as the central nervous system, limbs, skeleton, eyes, lungs, muscles, pancreas, testes, and skin (7). In the developing skin, SHH produced by epidermal placodes signals to the underlying dermal condensates to form hair follicles (27, 28). SHH signals promote cell proliferation, and some of the genomic targets of SHH signaling in the skin, such as E2F2, are key activators of the cell cycle (29, 30). Consequently, in the absence of SHH, epidermal placodes form but fail to grow down into the dermis (27).
After embryogenesis, the HH pathway maintains tissue homeostasis, in part by regulating stem cells and tissue growth (31–33). In the postnatal skin, hair follicle growth recapitulates much of embryonic hair growth, including the involvement of HH signaling. For example, SHH is expressed during anagen, the hair follicle phase during which there is downward growth of the follicle into the dermis, and is important for maintaining epidermal stem cells and regulating epidermal growth (34–37).
Affecting about 2 million people per year, basal cell carcinoma (BCC) is the most common cancer in the United States (38). BCC is a skin cancer that typically arises in adult patients as a result of ultraviolet radiation (Figure 2A). Ultraviolet radiation causes many genomic mutations, and BCCs typically harbor about 65 mutations per megabase, making them the most mutated human cancer (39). Despite these many mutations, BCCs are unified by a common molecular defect, misactivation of the HH pathway.
Figure 2. Hedgehog pathway–associated cancers.

(A) Basal cell carcinomas (BCCs) of the preauricular space (top left), temple with preoperative markings (bottom left), and upper extremity (right) of adult patients demonstrate the morphologic diversity of BCC, which may present as an open sore, red patch, pearly papule, or scar. (B) T2 magnetic resonance 3-D fast-recovery fast-spin echo cube image of medulloblastoma in the cerebellar vermis (white arrow) of a pediatric patient. (C) Coronal PET (top) and CT (bottom) images of testicular rhabdomyosarcoma (white asterisk) with regional metastases to the left renal hilum (black arrow) in a young adult patient; the bladder in both images is denoted by a black asterisk.
The most common HH pathway–activating mutations in BCC include biallelic loss-of-function mutations in genes encoding negative pathway regulators (i.e., PTCH1, PTCH2, and SUFU); amplification of GLI2, the gene encoding the principal HH pathway transcriptional activator; and monoallelic activating substitutions in positive pathway regulators, such as SMO (Table 1 and refs. 39–42). A frequent activating mutation in SMO is W535R, also known as SMOM2, a substitution within the seventh transmembrane domain (42, 43). Consistently, SMOM2 or other activating substitutions in this region activate the unregulated, SHH-independent ciliary accumulation of SMO, emphasizing the intimate association between SMO ciliary accumulation and downstream pathway activation (18).
Table 1. Cancer-predisposing Hedgehog pathway mutations.

Despite their extraordinary mutation burden, BCCs are typically slow-growing and often amenable to cure by local excision. However, BCC size, number, or proximity to critical structures such as the eye, lip, or nose can preclude complete excision. In such instances, nonsurgical treatments such as superficial radiotherapy, photodynamic therapy, and topical cytotoxic agents can inhibit the growth of BCC. For patients with localized disease who fail to respond to conventional therapies, or for rare patients with metastatic BCC, systemic therapy with SMO antagonists is superior to traditional systemic chemotherapy (44–46). Although the proportion of tumors that achieve long-term control with SMO antagonists remains to be established, responses to SMO antagonists may be transient (47–49), and the combination of HH pathway inhibition with radiation may increase the durability of molecular therapy for BCC patients (although it may also promote additional resistance mechanisms in recalcitrant BCCs) (50, 51). BCC cells may survive HH pathway inhibition through chromatin remodeling and enhancer reprogramming, but synergy between Wnt and SMO antagonists, or Notch agonists and SMO antagonists, may be a clinically relevant strategy to prevent relapse (52–54).
BCC arises from multiple skin compartments, including hair follicles, the interfollicular epidermis, and mechanosensory niches in the epidermis (55). Many skin cells are ciliated, but as cilia are deconstructed before the cell’s entry into mitosis, it might be expected that actively cycling cancer cells would not have cilia. However, many BCC cells do possess cilia (56). During development and cancer alike, cilia are required to transduce HH signals from SMO to GLI transcription factors. In mouse genetic models of BCC, the ability of SMOM2 to trigger oncogenesis depends on the presence of cilia (56, 57). In contrast, oncogenic HH pathway activation downstream of SMO, such as genomic amplification of GLI2 or loss of SUFU, is independent of cilia (56, 57). Thus, HH signal transduction can become deregulated to cause unrestrained growth and cancer in both cilium-dependent and cilium-independent fashions.
Medulloblastoma.
The functions of HH signaling are perhaps best understood in the patterning of the embryonic central nervous system’s dorsoventral axis. During early brain development, SHH produced at the ventral midline helps specify the cerebellum in the hindbrain (58). Later in brain development, SHH is secreted by the choroid plexus into the ventricular space (59), and later still, SHH is produced by Purkinje cells within the developing cerebellum (60, 61). SHH from the choroid plexus and Purkinje cells stimulates the proliferation of cerebellar progenitors in the rhombic lip and the fourth ventricle (59–61). Around birth, the rhombic lip and ventricular progenitors migrate to form the external granular layer and dive into the prospective white matter, where they give birth to a remarkable number of neurons. As in the skin, SHH acts as a mitogen in the developing cerebellum by inducing expression of cell cycle regulators including N-Myc, Ccnd1, and Ccnd2 (Figure 3 and ref. 60). At the end of cerebellar development, the number of neurons in the cerebellum dwarfs even the number in the cortex (62); in the mouse, the granule cell precursors in the external granule layer give rise to 108 cerebellar granule neurons (63, 64).
Figure 3. A model of the Hedgehog transcriptional program in cancer.

Misactivation of the Hedgehog pathway in cancer through upstream mutations, gene amplification, atypical protein kinase C (aPKC) signaling, or epigenetic misregulation of the SWI/SNF chromatin remodeling complex or BET bromodomain proteins culminates in activation of GLI transcription factors. GLI transcription factors induce the expression of N-MYC, CDK6, CCND1, and CCND2 to promote cancer cell proliferation and tumor growth. Parallel activation of Cdk5 and PI3K/mTOR signaling may facilitate evasion of immune elimination and resistance to SMO antagonists, respectively.
Consistent with their intimate involvement with HH signaling, primary cilia are also critical for HH-mediated patterning of the central nervous system (65, 66). Defects in HH signaling disrupt cerebellar growth and alter foliation (67). Similarly, inherited defects in ciliary structure can compromise developmental HH signaling and result in defective cerebellar growth as part of Joubert syndrome, a congenital syndrome characterized by agenesis of the cerebellar vermis (68–70).
A few weeks after birth, HH signaling and cerebellar neurogenesis cease (71–73). How HH signaling is turned off at this and other time points during development remains an active area of investigation, but in the cerebellum, cessation of HH signaling may involve feedback from the granule cell precursors to the Purkinje cells or cessation of granule cell response to HH (74).
As in the skin, persistent misactivation of the HH pathway with concomitant loss of control over cell division leads to cancer in the cerebellum (61). Although not every cell that misactivates the HH pathway in the cerebellum generates a tumor, some do give rise to medulloblastoma, the most common malignant brain cancer in children (Figure 2B and ref. 75). Concurrent mutations in other oncogenic pathways or genes, such as p53, may predispose certain cells with misactivated HH signaling in the cerebellum to form tumors (76). The likelihood of these events appears to decrease with age and may be less common in girls, as the incidence of medulloblastoma decreases from 0.53 per 100,000 in children aged 0–4 years, to 0.33 per 100,000 in adolescents aged 15–19 years, and medulloblastoma is more common in boys than in girls (75). Medulloblastomas are mostly treated as a uniform clinical entity, but they constitute molecular subgroups with drastic differences in genomic, demographic, and clinical features (77, 78). Of the 12 or more distinct molecular variants of medulloblastoma, misactivation of the HH pathway underlies approximately one-third of cases (76, 79). In relation to the other principal subgroups of medulloblastoma tumors, HH pathway–associated medulloblastomas display bimodal distribution, with most cases occurring in infants and adults (80).
The current standard of care for medulloblastoma involves maximal safe resection followed by ionizing radiotherapy to the entire craniospinal axis with concurrent and adjuvant cytotoxic chemotherapy (81). Although this well-established regimen cures approximately 75% of pediatric medulloblastoma patients, long-term survival for some patients within particular medulloblastoma molecular subgroups can be as low as 25% (77, 78). Moreover, survivors are debilitated by significant therapy-associated toxicity, chief among which are neurocognitive deficits, endocrine and growth abnormalities, and an increased risk of secondary malignancy (82–84). To reduce the burden of treatment-associated toxicity of radiotherapy, high-dose chemotherapy with hematopoietic stem cell rescue and other risk-adapted therapeutic strategies have been used, particularly in young patients (85–88). However, outcomes remain suboptimal and patients are frequently afflicted with significant therapy-associated sequelae.
As in BCC, biallelic inactivating PTCH and SUFU loss-of-function mutations, amplification of GLI transcription factors, or activating SMO substitutions are common in HH-associated medulloblastoma (Table 1 and ref. 76). Also like BCC, many HH pathway–associated medulloblastoma cells are ciliated, and primary cilia are critical for misactivated SMO to drive medulloblastoma formation and growth (57). GLI2 amplification and, presumably, SUFU mutations underlie cilia-independent oncogenic HH pathway activation, and, when they co-occur with mutations compromising p53 function, these GLI2 and SUFU aberrations portend particularly poor prognoses for pediatric medulloblastoma patients (57, 76).
Treatment of HH pathway–associated medulloblastoma with SMO antagonists such as vismodegib can dramatically shrink tumors, but recurrence and development of acquired resistance are common and can be rapid (47, 48). Importantly, SMO antagonists have only been trialed in medulloblastoma patients with recurrent tumors previously treated with DNA-damaging agents, and prior genotoxic therapy may attenuate the efficacy of SMO antagonism. Moreover, only half of pediatric HH pathway–associated medulloblastomas respond to SMO inhibition, limiting the proportion of patients who may achieve even a transient benefit from HH pathway inhibition (89). Thus, novel therapies are required to improve outcomes for patients with HH pathway–associated medulloblastoma.
An alternative pharmacologic target of oncogenic HH signaling in medulloblastoma is cyclin-dependent kinase 6 (CDK6), a mitogenic kinase and direct transcriptional target of GLI2 (Figure 3 and refs. 90, 91). When activated through interaction with cyclin D1, CDK6 induces cell cycle progression by phosphorylating the tumor suppressor RB and relieving repression of E2F transcription factors. Potent small-molecule inhibitors of CDK6 and the related kinase CDK4 have been tested in diverse human malignancies, and are now being investigated in pediatric medulloblastoma patients (92). Another possible pharmacologic target in medulloblastoma is CDK5, a serine-threonine kinase that promotes expression of the immune exhaustion marker programmed death–ligand 1 (PD-L1), thereby allowing tumor cells to evade immune elimination (Figure 3 and ref. 93). The efficacy of CDK6 or CDK5 inhibition for medulloblastoma remains to be established by clinical trials, but intra- and intermedulloblastoma heterogeneity, including divergent clonal selection at the time of recurrence, suggests that molecular monotherapy will not be a durable therapeutic strategy for patients with HH pathway–associated medulloblastoma (94).
Another possible pharmacologic strategy for HH pathway–associated medulloblastoma is to block the formation of lipids that are required for oncogenic HH signaling. The ciliary membrane is enriched in specific lipids, such as phosphatidylinositol 4-phosphate (PtdIns4P) (16). Ciliary PtdIns4P is generated by inositol polyphosphate 5-phosphatase (INPP5E) to restrain the ciliary accumulation of GPR161, an inhibitor of HH signaling (95, 96). In the absence of INPP5E, ciliary PtdIns4P is replaced with phosphatidylinositol 4,5-bisphosphate (PtdIns4,5P), which increases ciliary GPR161 and dampens HH signaling. In medulloblastoma, INPP5E constrains oncogenic HH signaling, and INPP5E copy number and expression are reduced in HH pathway–associated medulloblastoma (97, 98). Thus, the development and implementation of pharmacologic INPP5E inhibitors may increase ciliary GPR161 and restrain oncogenic HH signaling, although, as with SMO inhibition, amplification of GLI2 or loss of SUFU would be expected to circumvent the effect of INPP5E inhibition.
In addition to PtdIns4P, lipids such as oxysterols can be enriched in cilia (16). Sterol biosynthesis is required for vertebrate HH signaling, and both synthetic oxysterols and cholesterol can bind SMO to activate the downstream pathway (17, 99–109). Consistently, statin medications, which inhibit cholesterol biosynthesis, may synergize with HH pathway inhibition for the treatment of medulloblastoma (110). Endogenous, cilia-associated oxysterols activate the HH pathway by interacting with two separate domains of SMO (111). An enzyme that participates in the production of SMO-activating oxysterols, HSD11β2, is dramatically enriched in HH-associated medulloblastoma, raising the possibility that inhibiting HSD11β2 may also be a strategy for inhibiting HH pathway activity in cancer (111). In support of this hypothesis, genetic inhibition of HSD11β2 in a mouse model of HH-associated medulloblastoma attenuates HH signal transduction and the growth of cancer, and blocking HSD11β2 with an active ingredient from licorice similarly inhibits medulloblastoma growth in mouse models (111). These data suggest that, like INPP5E inhibition, inhibition of ciliary oxysterol biosynthesis may be a tractable alternative means of inhibiting oncogenic HH pathway activation in medulloblastoma.
Rhabdomyosarcoma.
Rhabdomyosarcoma (RMS), the most common pediatric soft tissue sarcoma, has two major histologic subtypes, alveolar and embryonal (Figure 2C). Genetically, alveolar RMS often harbors reciprocal chromosomal translocations between either of two myogenic transcription factor genes, PAX3 or PAX7, and FOXO1, a promitotic transcription factor. Embryonal RMS and alveolar RMS that is not associated with PAX-FOXO1 translocations are instead characterized by HH pathway misactivation (112–114). In mouse models, HH pathway activity is implicated in RMS cell self-renewal, chemoresistance, motility, and differentiation (115).
Identifying the RMS cell of origin remains an ongoing area of investigation, but RMS may arise from a mesodermal progenitor (116). Many skeletal muscles are derived from somites, paired, segmentally organized blocks of paraxial mesoderm. Subdomains of somites give rise to bone and cartilage (the sclerotome), or skeletal muscle and connective tissue (the epaxial dermomyotome and hypaxial myotome). The epaxial dermomyotome develops into the deep muscles of the back, whereas the hypaxial myotome develops into limb muscles and diaphragm (117, 118). During mouse development, HH signals are critical for the specification of muscle progenitors specifically in the epaxial domain of the somite (119). One possibility is that embryonal RMS arises from persistent epaxial dermomyotome–like progenitors that depend on HH signals, whereas alveolar RMS arises from HH-independent hypaxial myotome–like progenitors. Multiple origins of RMS may help explain how this cancer can originate throughout the body, even in organs without skeletal muscle such as the urogenital and biliary tracts. Consistently, the effects of SMO inhibitors on RMS cell lines are variable, suggesting that HH pathway inhibition may be efficacious in only a subset of RMS patients (120).
Nevoid basal cell carcinoma syndrome.
Considering that activating HH pathway mutations cause sporadic BCC and medulloblastoma, it is perhaps unsurprising that inherited mutations in genes encoding negative HH pathway regulators predispose individuals to the same cancers. More specifically, inheritance of inactivating heterozygous mutations in key negative regulators of the HH pathway cause nevoid basal cell carcinoma syndrome (NBCCS), also known as basal cell nevus syndrome or Gorlin syndrome (121–123). Individuals with NBCCS may develop between several and several thousand BCCs (Figure 2A). A subset of individuals with NBCCS are also predisposed to developing medulloblastoma (Figure 2B) or RMS (Figure 2C and refs. 124, 125).
NBCCS is a rare disorder, with an estimated prevalence of 2–4 in 100,000, and with equal incidence in males and females (126). In addition to increased cancer risk, individuals with NBCCS can display developmental abnormalities such as macrocephaly, intracranial calcification, and skeletal changes (i.e., kyphoscoliosis, bifid ribs, craniofacial bossing, hypertelorism, odontogenic jaw keratocysts, and mandibular prognathism). The inheritance pattern for NBCCS is autosomal dominant with complete penetrance, although oncogenesis is caused by a second hit to the wild-type allele, making it recessive at the cellular level. This syndrome probably has affected humans throughout our existence, as ancient Egyptian mummies with NBCCS characteristics have been identified (5).
The cancers and developmental abnormalities that characterize NBCCS can be caused by inactivating mutations in PTCH1 or PTCH2, leading to misactivation of the downstream pathway (12, 122, 123). Inherited mutations in SUFU can also cause NBCCS, albeit without apparent odontogenic jaw keratocysts and with possibly higher risk of medulloblastoma (127, 128). The rarity of NBCCS complicates precise estimates of the prevalences of PTCH1, PTCH2, and SUFU mutations in affected individuals, but mutations in one of these three genes are present in all or almost all cases that have been reported (127).
Hedgehog signaling in other cancers
The increased incidence of BCC, medulloblastoma, and RMS in individuals with NBCCS provides clues about the molecular causes of both the syndromic and the sporadic forms of these tumors. However, beyond these three cancer types, there are limited mechanistic data linking HH pathway mutations to other human malignancies. Consequently, investigations of HH signaling have not translated to new clinical therapies or improved outcomes in prostate, breast, and gastrointestinal cancers — some of the most common cancers — or in lung and pancreatic cancers — some of the deadliest cancers. Thus, it is possible that many cancer-associated mutations affecting HH pathway components are passengers that do not substantively contribute to the cancer phenotype (4). Alternatively, misactivation of aspects of the HH pathway may contribute to oncogenesis through mechanisms that are distinct from those elucidated in BCC, medulloblastoma, and RMS (129). For example, atypical teratoid rhabdoid tumor (ATRT) is a cancer that may, in part, depend on activation of transcriptional effectors of the HH pathway through a mechanism that does not require activation of the upstream pathway.
Atypical teratoid rhabdoid tumor.
Only several hundred cases of ATRT are diagnosed in the United States each year, but this enigmatic cancer is the most common malignant brain tumor in children younger than 6 months (130). Survival from ATRT is generally poor, and because of the rarity of diagnosis, a standard of care has yet to be defined. ATRT is associated with inactivating somatic mutations in SMARCB1 or, rarely, SMARCA4, both of which encode essential components of the chromatin remodeling SWI/SNF complex (131, 132). The SWI/SNF complex epigenetically regulates diverse genes involved in lineage specification, stem cell pluripotency, and signal transduction.
Despite sharing underlying mutated genes, ATRT displays three distinct gene expression profiles, one of which is associated with misactivation of the HH pathway (133). The mechanism by which SMARCB1 or SMARCA4 mutations cause HH pathway activation occurs via epigenetic derepression of the genes encoding the HH transcriptional effectors GLI1 and GLI2 (Figure 3 and refs. 133–135). Activation of a GLI1- and GLI2-dependent HH transcriptional program in ATRTs therefore circumvents any requirement for HH ligands, SMO, or cilia.
Bromodomain proteins bind to acetylated lysines in histones that mark transcription factor–accessible chromatin. BET bromodomain proteins are critical regulators of GLI1 and GLI2 that directly occupy the promoter of both genes (Figure 3 and ref. 136). Inhibiting GLI transcription factors by antagonizing BET bromodomain proteins is effective in mouse models of diverse HH-associated cancers, including BCC, medulloblastoma, and ATRT (136). Moreover, BET bromodomain inhibition is effective for HH pathway–associated cancers even in the presence of genetic lesions that confer resistance to SMO inhibition, and therefore represents a promising therapeutic target for other HH-associated cancers. CDK6 is also enriched in HH-subgroup ATRT tumors, and pharmacologic inhibition of CDK6 may be similarly useful for treating patients with ATRT (133, 137).
Beyond BCC, medulloblastoma, RMS, and ATRT, aberrant HH signaling has been implicated in diverse human malignancies, but mechanistic connections are often sparsely understood or confusing. For example, in meningioma, the most common intracranial tumor in the United States, rare HH pathway mutations are found in tumors with excellent clinical outcomes (138, 139). Nevertheless, constitutive activation of SMO in the meninges fails to cause meningioma in most mice, suggesting that HH misactivation is not sufficient to induce or drive meningioma growth (140).
Pancreatic ductal adenocarcinoma.
Pancreatic ductal adenocarcinoma (PDAC) is associated with upregulation of SHH, but in mice, SMO is dispensable for PDAC growth, suggesting that misactivation of the HH pathway is not a critical driver of this cancer either (141, 142). Moreover, unlike BCC and medulloblastoma, PDAC is associated with loss of cilia (143, 144), providing ancillary evidence that the normal HH pathway is not hyperactivated in PDAC. However, PDAC stromal cells, such as fibroblasts and endothelial cells, respond to SHH produced by tumor cells, suggesting that paracrine signaling may modulate PDAC progression (145–147). SHH and tumor-associated stromal cells induce the development of desmoplastic PDAC (144, 148), suggesting that the HH pathway may function non–cell-autonomously in PDAC. Consistently, depleting PDAC-associated stromal cells accelerates tumor growth, suggesting that pharmacologic inhibition of HH signaling will not be an effective strategy for patients with PDAC (149–152).
The observation that stromal HH signaling restrains PDAC growth suggests that paracrine signaling may modulate the progression of other cancers (147, 150–152). Indeed, HH signaling also restrains bladder cancer progression (153), and HH signals from prostate cancer signal to tumor-associated stroma (154, 155). These data have important implications for the clinical implementation of HH pathway antagonists, and suggest that HH pathway inhibition could accelerate the growth of some cancers. In support of that hypothesis, clinical trials of SMO antagonists for PDAC have failed to demonstrate a benefit or have been halted early for poor outcomes (156). Although a subset of individuals with genetically defined HH pathway–associated PDAC may benefit from targeted therapy, the lessons learned from PDAC preclinical models and clinical testing demonstrate that HH pathway antagonists cannot be broadly applied.
Lung cancer.
Lung cancer is the leading cause of cancer mortality in the United States, and HH signaling has been implicated in small-cell, non–small-cell, and mesothelioma lung tumors. In pleural mesothelioma, SHH and GLI1 are expressed, and SMO antagonists attenuate growth in vitro and in vivo (157). However, HH pathway mutations in mesothelioma are rare, and whether HH signaling drives growth in human patients is not known (158). GLI1 is expressed in most non–small-cell lung cancers, but responses to SMO antagonists in vitro are heterogeneous, raising the possibility that a subset of these tumors may benefit from HH pathway inhibition (159). In a mouse model of small-cell lung cancer, genetic deletion of Smo suppresses tumor initiation and progression, suggesting that misactivation of the HH pathway contributes to tumor growth (160). Thus, given the poor clinical outcomes with the current standard of care for small-cell lung cancer, HH pathway inhibitors such as SMO and BET bromodomain antagonists are of clinical interest (161).
Prostate cancer.
With the exception of BCC, prostate cancer is the most common malignancy in adult males in the United States. During prostate development, HH signaling regulates epithelial tubule formation (162). HH signaling also mediates regeneration of prostatic epithelial cells, and in prostate cancer, the HH pathway is implicated in dedifferentiation, invasiveness, and metastasis (163). Consistently, HH pathway mutations are identified in prostate cancer, and SMO inhibitors and GLI1 RNA interference both block the growth of cultured prostate cancer cells in vitro (164, 165). In prostate cancer, androgen receptors bind GLI2 and GLI3 to inhibit the proteolytic generation of GLI3 repressor and activate the HH transcriptional program (166). Breast cancer, another hormone-driven malignancy, displays a strong correlation between GLI1 expression in the nucleus and estrogen receptor staining; furthermore, elevated SHH expression delineates a subpopulation of breast cancer patients with aggressive pathologic features (167, 168). Despite these preclinical data, and clinical evidence that SMO antagonists suppress HH signaling in localized high-risk and metastatic prostate cancer, whether HH pathway inhibition would be effective in either prostate and breast cancer remains unclear (169).
Hedgehog pathway–directed therapeutics
The discovery of cyclopamine, a teratogenic plant-derived alkaloid that inhibits HH signal transduction, provided a foundation for developing HH pathway inhibitors for use in cancer therapy (170, 171). Cyclopamine binds to SMO and inhibits the unregulated activity caused by oncogenic mutations (172, 173). Some agents modeled on cyclopamine inhibit HH pathway–associated BCC, but other HH pathway modulators, such as inhibitors of SHH and GLI transcription factors, have not successfully transitioned to clinical use because of short half-life, poor pharmacokinetic or pharmacodynamic properties, or lack of efficacy in human patients (174–177).
The most commonly used HH pathway antagonists in cancer are vismodegib and sonidegib (Table 2). These agents have pharmacokinetics and pharmacodynamics compatible with systemic administration in humans and have been extensively studied in BCC and medulloblastoma (178, 179). Both BCC and medulloblastoma can regress below the threshold of detection by visual, histopathologic, or radiologic examination in response to vismodegib or sonidegib, but a significant proportion of BCC and HH-associated medulloblastoma patients fail to respond to SMO inhibition (44, 45, 180–182). Responses in BCC can be durable, but adult patients receiving vismodegib for BCC report adverse events such as nausea, muscle cramps, loss of taste, weight loss, and alopecia (45, 182). Although typically mild, these symptoms can cause patients to discontinue therapy, which may lead to recurrence of BCC in some patients. Pediatric medulloblastoma patients experience similar symptoms with SMO antagonists, but can suffer premature and irreversible growth plate fusion from on-target inhibition of HH signaling (183).
Table 2. FDA-approved molecular therapies affecting the Hedgehog pathway in cancer.
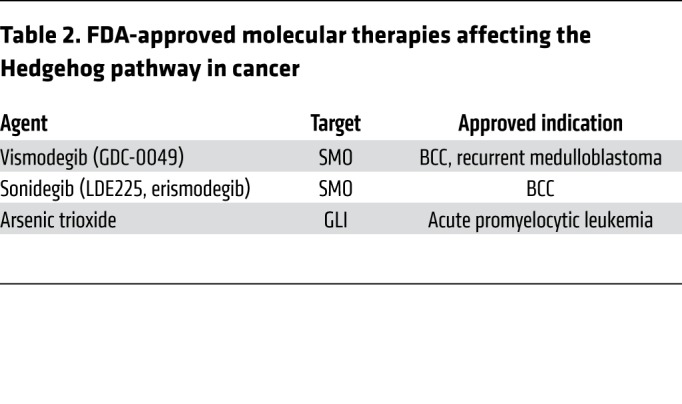
Among patients with HH pathway–associated cancers that respond to HH pathway antagonists, tumors can acquire resistance to molecular therapy. Despite remarkable clinical responses in metastatic HH-associated medulloblastoma, targeted HH pathway inhibition is associated only with transient tumor regression followed by recurrence of resistant cancers (47–49). One way in which tumors evade SMO inhibition to maintain constitutively high levels of HH pathway activity is through SMO mutations that disrupt drug binding (48, 184). Additionally, genomic amplification of GLI transcription factors or HH target genes such as CCND1, as well as loss-of-function SUFU mutations, can bypass SMO inhibition to maintain uncontrolled growth in both medulloblastoma and BCC (Figure 3 and refs. 49, 185, 186). Other means of circumventing SMO inhibition in BCC can include increased expression of the transcription factor SRF, which cooperates with its partner MKL1 to increase the transcriptional activity of GLI1 (187). Medulloblastomas that are resistant to SMO inhibitors may also display activation of phosphoinositide-3-kinase and AKT signaling, and inhibiting these pathways may delay the development of resistance (Figure 3 and ref. 49).
Arsenic trioxide, which can inhibit HH signal transduction by preventing ciliary accumulation and DNA binding of GLI transcription factors, as well as destabilizing of GLI2, may provide an approach to blocking the growth of tumors with acquired resistance to SMO antagonists (188–190). Arsenic trioxide also blocks the JNK, NF-κB, thioredoxin reductase, and MAPK pathways, and effects on pathways other than HH may underlie its efficacy in the treatment of promyelocytic leukemia (Table 2 and ref. 191).
Taken together, these data suggest that molecular monotherapy may not be an effective strategy for patients with HH pathway–associated cancers. Rather, concurrent inhibition of (a) pathway activators, such as GLI transcription factors or INPP5E and HSD11β2, which regulate cilia-associated lipids that modulate HH signaling intensity; (b) pathway mediators, such as BET bromodomain proteins, which participate in GLI transcriptional activation; and (c) pathway effectors, such as CDK5 or CDK6, which are in involved in suppressing immune clearance and activating the cell cycle, respectively, may improve the durability of clinical responses and decrease acquired resistance. Elucidation of effective molecular antagonists against these targets will likely require a partnership of foundational biology and deep analysis of human tumors, involving the collaborative efforts of basic, translational, and clinical scientists.
Acknowledgments
We thank Sarah Arron for providing the BCC images, and Steve Braunstein for referring the rhabdomyosarcoma patient featured in Figure 2. This work was supported by grants from the NIH (CA212279-01 to DRR and AR054396 and GM095941 to JFR).
Version 1. 02/01/2019
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2019;129(2):465–475.https://doi.org/10.1172/JCI120850.
References
- 1.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lichtenstein P, et al. Environmental and heritable factors in the causation of cancer — analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343(2):78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 3.Lawrence MS, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao J, Ciriello G, Sander C, Schultz N. Collection, integration and analysis of cancer genomic profiles: from data to insight. Curr Opin Genet Dev. 2014;24:92–98. doi: 10.1016/j.gde.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Satinoff MI, Wells C. Multiple basal cell naevus syndrome in ancient Egypt. Med Hist. 1969;13(3):294–297. doi: 10.1017/S0025727300014563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ingham PW, Nakano Y, Seger C. Mechanisms and functions of Hedgehog signalling across the metazoa. Nat Rev Genet. 2011;12(6):393–406. doi: 10.1038/nrg2984. [DOI] [PubMed] [Google Scholar]
- 7.McMahon AP, Ingham PW, Tabin CJ. Developmental roles and clinical significance of hedgehog signaling. Curr Top Dev Biol. 2003;53:1–114. doi: 10.1016/s0070-2153(03)53002-2. [DOI] [PubMed] [Google Scholar]
- 8.Echelard Y, et al. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75(7):1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- 9.Chiang C, et al. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383(6599):407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- 10.Perler FB. Protein splicing of inteins and hedgehog autoproteolysis: structure, function, and evolution. Cell. 1998;92(1):1–4. doi: 10.1016/S0092-8674(00)80892-2. [DOI] [PubMed] [Google Scholar]
- 11.Marigo V, Davey RA, Zuo Y, Cunningham JM, Tabin CJ. Biochemical evidence that patched is the Hedgehog receptor. Nature. 1996;384(6605):176–179. doi: 10.1038/384176a0. [DOI] [PubMed] [Google Scholar]
- 12.Smyth I, et al. Isolation and characterization of human patched 2 (PTCH2), a putative tumour suppressor gene inbasal cell carcinoma and medulloblastoma on chromosome 1p32. Hum Mol Genet. 1999;8(2):291–297. doi: 10.1093/hmg/8.2.291. [DOI] [PubMed] [Google Scholar]
- 13.Stone DM, et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature. 1996;384(6605):129–134. doi: 10.1038/384129a0. [DOI] [PubMed] [Google Scholar]
- 14.Bangs F, Anderson KV. Primary cilia and mammalian Hedgehog signaling. Cold Spring Harb Perspect Biol. 2017;9(5):a028175. doi: 10.1101/cshperspect.a028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rohatgi R, Scott MP. Cell biology. Arrestin’ movement in cilia. Science. 2008;320(5884):1726–1727. doi: 10.1126/science.1160448. [DOI] [PubMed] [Google Scholar]
- 16.Garcia G, Raleigh DR, Reiter JF. How the ciliary membrane is organized inside-out to communicate outside-in. Curr Biol. 2018;28(8):R421–R434. doi: 10.1016/j.cub.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang P, et al. Structural basis of smoothened activation in Hedgehog signaling. Cell. 2018;175(1):295–297. doi: 10.1016/j.cell.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437(7061):1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 19.Kim J, Kato M, Beachy PA. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc Natl Acad Sci U S A. 2009;106(51):21666–21671. doi: 10.1073/pnas.0912180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kogerman P, et al. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol. 1999;1(5):312–319. doi: 10.1038/13031. [DOI] [PubMed] [Google Scholar]
- 21.Stone DM, et al. Characterization of the human suppressor of fused, a negative regulator of the zinc-finger transcription factor Gli. J Cell Sci. 1999;112(pt 23):4437–4448. doi: 10.1242/jcs.112.23.4437. [DOI] [PubMed] [Google Scholar]
- 22.Svärd J, et al. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev Cell. 2006;10(2):187–197. doi: 10.1016/j.devcel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 23.Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1(4):e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santos N, Reiter JF. A central region of Gli2 regulates its localization to the primary cilium and transcriptional activity. J Cell Sci. 2014;127(pt 7):1500–1510. doi: 10.1242/jcs.139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tukachinsky H, Lopez LV, Salic A. A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol. 2010;191(2):415–428. doi: 10.1083/jcb.201004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohen M, et al. Ptch1 and Gli regulate Shh signalling dynamics via multiple mechanisms. Nat Commun. 2015;6:6709. doi: 10.1038/ncomms7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chiang C, et al. Essential role for Sonic hedgehog during hair follicle morphogenesis. Dev Biol. 1999;205(1):1–9. doi: 10.1006/dbio.1998.9103. [DOI] [PubMed] [Google Scholar]
- 28.St-Jacques B, et al. Sonic hedgehog signaling is essential for hair development. Curr Biol. 1998;8(19):1058–1068. doi: 10.1016/S0960-9822(98)70443-9. [DOI] [PubMed] [Google Scholar]
- 29.Mill P, et al. Sonic hedgehog-dependent activation of Gli2 is essential for embryonic hair follicle development. Genes Dev. 2003;17(2):282–294. doi: 10.1101/gad.1038103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mill P, Mo R, Hu MC, Dagnino L, Rosenblum ND, Hui CC. Shh controls epithelial proliferation via independent pathways that converge on N-Myc. Dev Cell. 2005;9(2):293–303. doi: 10.1016/j.devcel.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 31.Lai K, Kaspar BK, Gage FH, Schaffer DV. Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat Neurosci. 2003;6(1):21–27. doi: 10.1038/nn983. [DOI] [PubMed] [Google Scholar]
- 32.Levy V, Lindon C, Harfe BD, Morgan BA. Distinct stem cell populations regenerate the follicle and interfollicular epidermis. Dev Cell. 2005;9(6):855–861. doi: 10.1016/j.devcel.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 33.Petrova R, Joyner AL. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development. 2014;141(18):3445–3457. doi: 10.1242/dev.083691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oro AE, Higgins K. Hair cycle regulation of Hedgehog signal reception. Dev Biol. 2003;255(2):238–248. doi: 10.1016/S0012-1606(02)00042-8. [DOI] [PubMed] [Google Scholar]
- 35.Sato N, Leopold PL, Crystal RG. Induction of the hair growth phase in postnatal mice by localized transient expression of Sonic hedgehog. J Clin Invest. 1999;104(7):855–864. doi: 10.1172/JCI7691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang LC, et al. Regular articles: conditional disruption of hedgehog signaling pathway defines its critical role in hair development and regeneration. J Invest Dermatol. 2000;114(5):901–908. doi: 10.1046/j.1523-1747.2000.00951.x. [DOI] [PubMed] [Google Scholar]
- 37.Brownell I, Guevara E, Bai CB, Loomis CA, Joyner AL. Nerve-derived sonic hedgehog defines a niche for hair follicle stem cells capable of becoming epidermal stem cells. Cell Stem Cell. 2011;8(5):552–565. doi: 10.1016/j.stem.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Asgari MM, Moffet HH, Ray GT, Quesenberry CP. Trends in basal cell carcinoma incidence and identification of high-risk subgroups, 1998–2012. JAMA Dermatol. 2015;151(9):976–981. doi: 10.1001/jamadermatol.2015.1188. [DOI] [PubMed] [Google Scholar]
- 39.Bonilla X, et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat Genet. 2016;48(4):398–406. doi: 10.1038/ng.3525. [DOI] [PubMed] [Google Scholar]
- 40.Xie J, et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391(6662):90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 41.Gailani MR, et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet. 1996;14(1):78–81. doi: 10.1038/ng0996-78. [DOI] [PubMed] [Google Scholar]
- 42.Lam CW, et al. A frequent activated smoothened mutation in sporadic basal cell carcinomas. Oncogene. 1999;18(3):833–836. doi: 10.1038/sj.onc.1202360. [DOI] [PubMed] [Google Scholar]
- 43.Xie J, et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391(6662):90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 44.Sekulic A, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366(23):2171–2179. doi: 10.1056/NEJMoa1113713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang JY, et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N Engl J Med. 2012;366(23):2180–2188. doi: 10.1056/NEJMoa1113538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jacobsen AA, Aldahan AS, Hughes OB, Shah VV, Strasswimmer J. Hedgehog pathway inhibitor therapy for locally advanced and metastatic basal cell carcinoma: a systematic review and pooled analysis of interventional studies. JAMA Dermatol. 2016;152(7):816–824. doi: 10.1001/jamadermatol.2016.0780. [DOI] [PubMed] [Google Scholar]
- 47.Rudin CM, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361(12):1173–1178. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yauch RL, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326(5952):572–574. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buonamici S, et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med. 2010;2(51):51ra70. doi: 10.1126/scitranslmed.3001599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raleigh DR, Algazi A, Arron ST, Neuhaus IM, Yom SS. Induction Hedgehog pathway inhibition followed by combined-modality radiotherapy for basal cell carcinoma. Br J Dermatol. 2015;173(2):544–546. doi: 10.1111/bjd.13748. [DOI] [PubMed] [Google Scholar]
- 51.Li C, Athar M. Ionizing radiation exposure and basal cell carcinoma pathogenesis. Radiat Res. 2016;185(3):217–228. doi: 10.1667/RR4284.S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Biehs B, et al. A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition. Nature. 2018;562(7727):429–433. doi: 10.1038/s41586-018-0596-y. [DOI] [PubMed] [Google Scholar]
- 53.Sánchez-Danés A, et al. A slow-cycling LGR5 tumour population mediates basal cell carcinoma relapse after therapy. Nature. 2018;562(7727):434–438. doi: 10.1038/s41586-018-0603-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eberl M, et al. Tumor architecture and Notch signaling modulate drug response in basal cell Carcinoma. Cancer Cell. 2018;33(2):229–243.e4. doi: 10.1016/j.ccell.2017.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peterson SC, et al. Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell Stem Cell. 2015;16(4):400–412. doi: 10.1016/j.stem.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wong SY, et al. Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat Med. 2009;15(9):1055–1061. doi: 10.1038/nm.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han YG, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez-Buylla A. Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med. 2009;15(9):1062–1065. doi: 10.1038/nm.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim JJ, et al. Suppressor of fused controls mid-hindbrain patterning and cerebellar morphogenesis via GLI3 repressor. J Neurosci. 2011;31(5):1825–1836. doi: 10.1523/JNEUROSCI.2166-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang X, et al. Transventricular delivery of Sonic hedgehog is essential to cerebellar ventricular zone development. Proc Natl Acad Sci U S A. 2010;107(18):8422–8427. doi: 10.1073/pnas.0911838107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dahmane N, Ruiz i Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126(14):3089–3100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- 61.Corrales JD, Rocco GL, Blaess S, Guo Q, Joyner AL. Spatial pattern of sonic hedgehog signaling through Gli genes during cerebellum development. Development. 2004;131(22):5581–5590. doi: 10.1242/dev.01438. [DOI] [PubMed] [Google Scholar]
- 62.Andersen BB, Korbo L, Pakkenberg B. A quantitative study of the human cerebellum with unbiased stereological techniques. J Comp Neurol. 1992;326(4):549–560. doi: 10.1002/cne.903260405. [DOI] [PubMed] [Google Scholar]
- 63.Miale IL, Sidman RL. An autoradiographic analysis of histogenesis in the mouse cerebellum. Exp Neurol. 1961;4:277–296. doi: 10.1016/0014-4886(61)90055-3. [DOI] [PubMed] [Google Scholar]
- 64.Fujita S, Shimada M, Nakamura T. H3-thymidine autoradiographic studies on the cell proliferation and differentiation in the external and the internal granular layers of the mouse cerebellum. J Comp Neurol. 1966;128(2):191–208. doi: 10.1002/cne.901280206. [DOI] [PubMed] [Google Scholar]
- 65.Spassky N, et al. Sonic hedgehog-dependent emergence of oligodendrocytes in the telencephalon: evidence for a source of oligodendrocytes in the olfactory bulb that is independent of PDGFRα signaling. Development. 2001;128(24):4993–5004. doi: 10.1242/dev.128.24.4993. [DOI] [PubMed] [Google Scholar]
- 66.Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426(6962):83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- 67.Corrales JD, Blaess S, Mahoney EM, Joyner AL. The level of sonic hedgehog signaling regulates the complexity of cerebellar foliation. Development. 2006;133(9):1811–1821. doi: 10.1242/dev.02351. [DOI] [PubMed] [Google Scholar]
- 68.Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19(9):813–825. doi: 10.1212/WNL.19.9.813. [DOI] [PubMed] [Google Scholar]
- 69.Shi X, et al. Super-resolution microscopy reveals that disruption of ciliary transition-zone architecture causes Joubert syndrome. Nat Cell Biol. 2017;19(10):1178–1188. doi: 10.1038/ncb3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aguilar A, et al. Analysis of human samples reveals impaired SHH-dependent cerebellar development in Joubert syndrome/Meckel syndrome. Proc Natl Acad Sci U S A. 2012;109(42):16951–16956. doi: 10.1073/pnas.1201408109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Choi Y, Borghesani PR, Chan JA, Segal RA. Migration from a mitogenic niche promotes cell-cycle exit. J Neurosci. 2005;25(45):10437–10445. doi: 10.1523/JNEUROSCI.1559-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Durand B, Fero ML, Roberts JM, Raff MC. p27Kip1 alters the response of cells to mitogen and is part of a cell-intrinsic timer that arrests the cell cycle and initiates differentiation. Curr Biol. 1998;8(8):431–440. doi: 10.1016/S0960-9822(98)70177-0. [DOI] [PubMed] [Google Scholar]
- 73.Miyazawa K, Himi T, Garcia V, Yamagishi H, Sato S, Ishizaki Y. A role for p27/Kip1 in the control of cerebellar granule cell precursor proliferation. J Neurosci. 2000;20(15):5756–5763. doi: 10.1523/JNEUROSCI.20-15-05756.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Argenti B, et al. Hedgehog antagonist REN(KCTD11) regulates proliferation and apoptosis of developing granule cell progenitors. J Neurosci. 2005;25(36):8338–8346. doi: 10.1523/JNEUROSCI.2438-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ostrom QT, et al. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro-oncology. 2017;19(suppl 5):v1–v88. doi: 10.1093/neuonc/nox158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cavalli FMG, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell. 2017;31(6):737–754.e6. doi: 10.1016/j.ccell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Taylor MD, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123(4):465–472. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thompson MC, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24(12):1924–1931. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- 79.Schwalbe EC, et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol. 2017;18(7):958–971. doi: 10.1016/S1470-2045(17)30243-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Northcott PA, et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol. 2011;122(2):231–240. doi: 10.1007/s00401-011-0846-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kline CN, et al. Case-based review: pediatric medulloblastoma. Neurooncol Pract. 2017;4(3):138–150. doi: 10.1093/nop/npx011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bhakta N, et al. The cumulative burden of surviving childhood cancer: an initial report from the St Jude Lifetime Cohort Study (SJLIFE) Lancet. 2017;390(10112):2569–2582. doi: 10.1016/S0140-6736(17)31610-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Perkins SM, Fei W, Mitra N, Shinohara ET. Late causes of death in children treated for CNS malignancies. J Neurooncol. 2013;115(1):79–85. doi: 10.1007/s11060-013-1197-0. [DOI] [PubMed] [Google Scholar]
- 84.Kieran MW, Walker D, Frappaz D, Prados M. Brain tumors: from childhood through adolescence into adulthood. J Clin Oncol. 2010;28(32):4783–4789. doi: 10.1200/JCO.2010.28.3481. [DOI] [PubMed] [Google Scholar]
- 85.Rutkowski S, et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med. 2005;352(10):978–986. doi: 10.1056/NEJMoa042176. [DOI] [PubMed] [Google Scholar]
- 86.Raleigh DR, et al. Survival after chemotherapy and stem cell transplant followed by delayed craniospinal irradiation is comparable to upfront craniospinal irradiation in pediatric embryonal brain tumor patients. J Neurooncol. 2017;131(2):359–368. doi: 10.1007/s11060-016-2307-6. [DOI] [PubMed] [Google Scholar]
- 87.Gajjar A, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7(10):813–820. doi: 10.1016/S1470-2045(06)70867-1. [DOI] [PubMed] [Google Scholar]
- 88.Robinson GW, et al. Risk-adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol. 2018;19(6):768–784. doi: 10.1016/S1470-2045(18)30204-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kool M, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25(3):393–405. doi: 10.1016/j.ccr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cook Sangar ML, et al. Inhibition of CDK4/6 by palbociclib significantly extends survival in medulloblastoma patient-derived xenograft mouse models. Clin Cancer Res. 2017;23(19):5802–5813. doi: 10.1158/1078-0432.CCR-16-2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Raleigh DR, Choksi PK, Krup AL, Mayer W, Santos N, Reiter JF. Hedgehog signaling drives medulloblastoma growth via CDK6. J Clin Invest. 2018;128(1):120–124. doi: 10.1172/JCI92710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14(2):130–146. doi: 10.1038/nrd4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dorand RD, et al. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science. 2016;353(6297):399–403. doi: 10.1126/science.aae0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Morrissy AS, et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature. 2016;529(7586):351–357. doi: 10.1038/nature16478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Garcia-Gonzalo FR, et al. Phosphoinositides regulate ciliary protein trafficking to modulate Hedgehog signaling. Dev Cell. 2015;34(4):400–409. doi: 10.1016/j.devcel.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chávez M, Ena S, Van Sande J, de Kerchove d’Exaerde A, Schurmans S, Schiffmann SN. Modulation of ciliary phosphoinositide content regulates trafficking and sonic Hedgehog signaling output. Dev Cell. 2015;34(3):338–350. doi: 10.1016/j.devcel.2015.06.016. [DOI] [PubMed] [Google Scholar]
- 97.Conduit SE, et al. A compartmentalized phosphoinositide signaling axis at cilia is regulated by INPP5E to maintain cilia and promote Sonic Hedgehog medulloblastoma. Oncogene. 2017;36(43):5969–5984. doi: 10.1038/onc.2017.208. [DOI] [PubMed] [Google Scholar]
- 98.Shimada IS, et al. Basal suppression of the sonic Hedgehog pathway by the G-protein-coupled receptor Gpr161 restricts medulloblastoma pathogenesis. Cell Rep. 2018;22(5):1169–1184. doi: 10.1016/j.celrep.2018.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Byrne EFX, et al. Structural basis of Smoothened regulation by its extracellular domains. Nature. 2016;535(7613):517–522. doi: 10.1038/nature18934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cooper MK, et al. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat Genet. 2003;33(4):508–513. doi: 10.1038/ng1134. [DOI] [PubMed] [Google Scholar]
- 101.Corcoran RB, Scott MP. Oxysterols stimulate Sonic hedgehog signal transduction and proliferation of medulloblastoma cells. Proc Natl Acad Sci U S A. 2006;103(22):8408–8413. doi: 10.1073/pnas.0602852103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dwyer JR, Sever N, Carlson M, Nelson SF, Beachy PA, Parhami F. Oxysterols are novel activators of the hedgehog signaling pathway in pluripotent mesenchymal cells. J Biol Chem. 2007;282(12):8959–8968. doi: 10.1074/jbc.M611741200. [DOI] [PubMed] [Google Scholar]
- 103.Huang P, et al. Cellular cholesterol directly activates smoothened in Hedgehog signaling. Cell. 2016;166(5):1176–1187.e14. doi: 10.1016/j.cell.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Luchetti G, et al. Cholesterol activates the G-protein coupled receptor Smoothened to promote Hedgehog signaling. Elife. 2016;5:e20304. doi: 10.7554/eLife.20304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Myers BR, et al. Hedgehog pathway modulation by multiple lipid binding sites on the smoothened effector of signal response. Dev Cell. 2013;26(4):346–357. doi: 10.1016/j.devcel.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nachtergaele S, et al. Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat Chem Biol. 2012;8(2):211–220. doi: 10.1038/nchembio.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nachtergaele S, et al. Structure and function of the Smoothened extracellular domain in vertebrate Hedgehog signaling. Elife. 2013;2:e01340. doi: 10.7554/eLife.01340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nedelcu D, Liu J, Xu Y, Jao C, Salic A. Oxysterol binding to the extracellular domain of Smoothened in Hedgehog signaling. Nat Chem Biol. 2013;9(9):557–564. doi: 10.1038/nchembio.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xiao X, et al. Cholesterol modification of Smoothened is required for Hedgehog signaling. Mol Cell. 2017;66(1):154–162.e10. doi: 10.1016/j.molcel.2017.02.015. [DOI] [PubMed] [Google Scholar]
- 110.Gordon RE, et al. Statins synergize with Hedgehog pathway inhibitors for treatment of medulloblastoma. Clin Cancer Res. 2018;24(6):1375–1388. doi: 10.1158/1078-0432.CCR-17-2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Raleigh DR, et al. Cilia-associated oxysterols activate Smoothened. Mol Cell. 2018;72(2):316–327.e5. doi: 10.1016/j.molcel.2018.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pressey JG, Anderson JR, Crossman DK, Lynch JC, Barr FG. Hedgehog pathway activity in pediatric embryonal rhabdomyosarcoma and undifferentiated sarcoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2011;57(6):930–938. doi: 10.1002/pbc.23174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zibat A, et al. Activation of the hedgehog pathway confers a poor prognosis in embryonal and fusion gene-negative alveolar rhabdomyosarcoma. Oncogene. 2010;29(48):6323–6330. doi: 10.1038/onc.2010.368. [DOI] [PubMed] [Google Scholar]
- 114.Tostar U, Malm CJ, Meis-Kindblom JM, Kindblom LG, Toftgård R, Undén AB. Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J Pathol. 2006;208(1):17–25. doi: 10.1002/path.1882. [DOI] [PubMed] [Google Scholar]
- 115.Satheesha S, et al. Targeting hedgehog signaling reduces self-renewal in embryonal rhabdomyosarcoma. Oncogene. 2016;35(16):2020–2030. doi: 10.1038/onc.2015.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Charytonowicz E, Cordon-Cardo C, Matushansky I, Ziman M. Alveolar rhabdomyosarcoma: is the cell of origin a mesenchymal stem cell? Cancer Lett. 2009;279(2):126–136. doi: 10.1016/j.canlet.2008.09.039. [DOI] [PubMed] [Google Scholar]
- 117.Tajbakhsh S, Bober E, Babinet C, Pournin S, Arnold H, Buckingham M. Gene targeting the myf-5 locus with nlacZ reveals expression of this myogenic factor in mature skeletal muscle fibres as well as early embryonic muscle. Dev Dyn. 1996;206(3):291–300. doi: 10.1002/(SICI)1097-0177(199607)206:3<291::AID-AJA6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 118.Ott MO, Bober E, Lyons G, Arnold H, Buckingham M. Early expression of the myogenic regulatory gene, myf-5, in precursor cells of skeletal muscle in the mouse embryo. Development. 1991;111(4):1097–1107. doi: 10.1242/dev.111.4.1097. [DOI] [PubMed] [Google Scholar]
- 119.Borycki AG, Brunk B, Tajbakhsh S, Buckingham M, Chiang C, Emerson CP. Sonic hedgehog controls epaxial muscle determination through Myf5 activation. Development. 1999;126(18):4053–4063. doi: 10.1242/dev.126.18.4053. [DOI] [PubMed] [Google Scholar]
- 120.Ridzewski R, Rettberg D, Dittmann K, Cuvelier N, Fulda S, Hahn H. Hedgehog inhibitors in Rhabdomyosarcoma: a comparison of four compounds and responsiveness of four cell lines. Front Oncol. 2015;5:130. doi: 10.3389/fonc.2015.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gorlin RJ, Goltz RW. Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib. A syndrome. N Engl J Med. 1960;262:908–912. doi: 10.1056/NEJM196005052621803. [DOI] [PubMed] [Google Scholar]
- 122.Johnson RL, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272(5268):1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 123.Hahn H, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85(6):841–851. doi: 10.1016/S0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 124.Herzberg JJ, Wiskemann A. [The fifth phakomatosis. Basal cell nevus with hereditary malformation and medulloblastoma] Dermatologica. 1963;126:106–123. [PubMed] [Google Scholar]
- 125.Schweisguth O, Gerard-Marchant R, Lemerle J. [Basal cell nevus syndrome. Association with congenital rhabdomyosarcoma] Arch Fr Pediatr. 1968;25(9):1083–1093. [PubMed] [Google Scholar]
- 126.Lo Muzio L. Nevoid basal cell carcinoma syndrome (Gorlin syndrome) Orphanet J Rare Dis. 2008;3:32. doi: 10.1186/1750-1172-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Smith MJ, et al. Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J Clin Oncol. 2014;32(36):4155–4161. doi: 10.1200/JCO.2014.58.2569. [DOI] [PubMed] [Google Scholar]
- 128.Pastorino L, et al. Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am J Med Genet A. 2009;149A(7):1539–1543. doi: 10.1002/ajmg.a.32944. [DOI] [PubMed] [Google Scholar]
- 129.Jenkins D. Hedgehog signalling: emerging evidence for non-canonical pathways. Cell Signal. 2009;21(7):1023–1034. doi: 10.1016/j.cellsig.2009.01.033. [DOI] [PubMed] [Google Scholar]
- 130.Frühwald MC, Biegel JA, Bourdeaut F, Roberts CW, Chi SN. Atypical teratoid/rhabdoid tumors-current concepts, advances in biology, and potential future therapies. Neuro-oncology. 2016;18(6):764–778. doi: 10.1093/neuonc/nov264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11(7):481–492. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- 132.Kieran MW, et al. Absence of oncogenic canonical pathway mutations in aggressive pediatric rhabdoid tumors. Pediatr Blood Cancer. 2012;59(7):1155–1157. doi: 10.1002/pbc.24315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Johann PD, et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell. 2016;29(3):379–393. doi: 10.1016/j.ccell.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 134.Jeibmann A, et al. Identification of genes involved in the biology of atypical teratoid/rhabdoid tumours using Drosophila melanogaster. Nat Commun. 2014;5:4005. doi: 10.1038/ncomms5005. [DOI] [PubMed] [Google Scholar]
- 135.Jagani Z, et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat Med. 2010;16(12):1429–1433. doi: 10.1038/nm.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tang Y, et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat Med. 2014;20(7):732–740. doi: 10.1038/nm.3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hashizume R, et al. Inhibition of DNA damage repair by the CDK4/6 inhibitor palbociclib delays irradiated intracranial atypical teratoid rhabdoid tumor and glioblastoma xenograft regrowth. Neuro-oncology. 2016;18(11):1519–1528. doi: 10.1093/neuonc/now106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Clark VE, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. 2013;339(6123):1077–1080. doi: 10.1126/science.1233009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sahm F, et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol. 2017;18(5):682–694. doi: 10.1016/S1470-2045(17)30155-9. [DOI] [PubMed] [Google Scholar]
- 140.Boetto J, Apra C, Bielle F, Peyre M, Kalamarides M. Selective vulnerability of the primitive meningeal layer to prenatal Smo activation for skull base meningothelial meningioma formation. Oncogene. 2018;37(36):4955–4963. doi: 10.1038/s41388-018-0328-7. [DOI] [PubMed] [Google Scholar]
- 141.Nolan-Stevaux O, et al. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009;23(1):24–36. doi: 10.1101/gad.1753809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Berman DM, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425(6960):846–851. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 143.Liu H, Kiseleva AA, Golemis EA. Ciliary signalling in cancer. Nat Rev Cancer. 2018;18(8):511–524. doi: 10.1038/s41568-018-0023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Seeley ES, Carrière C, Goetze T, Longnecker DS, Korc M. Pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions are devoid of primary cilia. Cancer Res. 2009;69(2):422–430. doi: 10.1158/0008-5472.CAN-08-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Thayer SP, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425(6960):851–856. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bailey JM, et al. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res. 2008;14(19):5995–6004. doi: 10.1158/1078-0432.CCR-08-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yauch RL, et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455(7211):406–410. doi: 10.1038/nature07275. [DOI] [PubMed] [Google Scholar]
- 148.Olive KP, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Özdemir BC, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25(6):719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rhim AD, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25(6):735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lee JJ, et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci U S A. 2014;111(30):E3091–E3100. doi: 10.1073/pnas.1411679111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mathew E, et al. Dosage-dependent regulation of pancreatic cancer growth and angiogenesis by hedgehog signaling. Cell Rep. 2014;9(2):484–494. doi: 10.1016/j.celrep.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Shin K, et al. Hedgehog signaling restrains bladder cancer progression by eliciting stromal production of urothelial differentiation factors. Cancer Cell. 2014;26(4):521–533. doi: 10.1016/j.ccell.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhang J, Lipinski R, Shaw A, Gipp J, Bushman W. Lack of demonstrable autocrine hedgehog signaling in human prostate cancer cell lines. J Urol. 2007;177(3):1179–1185. doi: 10.1016/j.juro.2006.10.032. [DOI] [PubMed] [Google Scholar]
- 155.Yang Z, Peng YC, Gopalan A, Gao D, Chen Y, Joyner AL. Stromal hedgehog signaling maintains smooth muscle and hampers micro-invasive prostate cancer. Dis Model Mech. 2017;10(1):39–52. doi: 10.1242/dmm.027417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Curran T. Reproducibility of academic preclinical translational research: lessons from the development of Hedgehog pathway inhibitors to treat cancer. Open Biol. 2018;8(8):180098. doi: 10.1098/rsob.180098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Shi Y, et al. Role of hedgehog signaling in malignant pleural mesothelioma. Clin Cancer Res. 2012;18(17):4646–4656. doi: 10.1158/1078-0432.CCR-12-0599. [DOI] [PubMed] [Google Scholar]
- 158.Lim CB, et al. Mutational analysis of hedgehog signaling pathway genes in human malignant mesothelioma. PLoS One. 2013;8(6):e66685. doi: 10.1371/journal.pone.0066685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yuan Z, et al. Frequent requirement of hedgehog signaling in non-small cell lung carcinoma. Oncogene. 2007;26(7):1046–1055. doi: 10.1038/sj.onc.1209860. [DOI] [PubMed] [Google Scholar]
- 160.Park KS, et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat Med. 2011;17(11):1504–1508. doi: 10.1038/nm.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kaur G, et al. Bromodomain and hedgehog pathway targets in small cell lung cancer. Cancer Lett. 2016;371(2):225–239. doi: 10.1016/j.canlet.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lim A, Shin K, Zhao C, Kawano S, Beachy PA. Spatially restricted Hedgehog signalling regulates HGF-induced branching of the adult prostate. Nat Cell Biol. 2014;16(12):1135–1145. doi: 10.1038/ncb3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Karhadkar SS, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431(7009):707–712. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 164.Sheng T, et al. Activation of the hedgehog pathway in advanced prostate cancer. Mol Cancer. 2004;3:29. doi: 10.1186/1476-4598-3-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sanchez P, et al. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc Natl Acad Sci U S A. 2004;101(34):12561–12566. doi: 10.1073/pnas.0404956101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Li N, et al. Non-canonical activation of hedgehog in prostate cancer cells mediated by the interaction of transcriptionally active androgen receptor proteins with Gli3. Oncogene. 2018;37(17):2313–2325. doi: 10.1038/s41388-017-0098-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kubo M, et al. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004;64(17):6071–6074. doi: 10.1158/0008-5472.CAN-04-0416. [DOI] [PubMed] [Google Scholar]
- 168.Noman AS, et al. Overexpression of sonic hedgehog in the triple negative breast cancer: clinicopathological characteristics of high burden breast cancer patients from Bangladesh. Sci Rep. 2016;6:18830. doi: 10.1038/srep18830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ross AE, et al. Pharmacodynamic and pharmacokinetic neoadjuvant study of hedgehog pathway inhibitor Sonidegib (LDE-225) in men with high-risk localized prostate cancer undergoing prostatectomy. Oncotarget. 2017;8(61):104182–104192. doi: 10.18632/oncotarget.22115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Incardona JP, Gaffield W, Kapur RP, Roelink H. The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development. 1998;125(18):3553–3562. doi: 10.1242/dev.125.18.3553. [DOI] [PubMed] [Google Scholar]
- 171.Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 1998;280(5369):1603–1607. doi: 10.1126/science.280.5369.1603. [DOI] [PubMed] [Google Scholar]
- 172.Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002;16(21):2743–2748. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Taipale J, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406(6799):1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 174.Berman DM, et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science. 2002;297(5586):1559–1561. doi: 10.1126/science.1073733. [DOI] [PubMed] [Google Scholar]
- 175.Athar M, et al. Inhibition of smoothened signaling prevents ultraviolet B-induced basal cell carcinomas through regulation of Fas expression and apoptosis. Cancer Res. 2004;64(20):7545–7552. doi: 10.1158/0008-5472.CAN-04-1393. [DOI] [PubMed] [Google Scholar]
- 176.Kim J, et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell. 2010;17(4):388–399. doi: 10.1016/j.ccr.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Xin M, Ji X, De La Cruz LK, Thareja S, Wang B. Strategies to target the Hedgehog signaling pathway for cancer therapy. Med Res Rev. 2018;38(3):870–913. doi: 10.1002/med.21482. [DOI] [PubMed] [Google Scholar]
- 178.Williams JA, et al. Identification of a small molecule inhibitor of the hedgehog signaling pathway: effects on basal cell carcinoma-like lesions. Proc Natl Acad Sci U S A. 2003;100(8):4616–4621. doi: 10.1073/pnas.0732813100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Romer JT, et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/–)p53(–/–) mice. Cancer Cell. 2004;6(3):229–240. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 180.Robinson GW, et al. Vismodegib exerts targeted efficacy against recurrent sonic Hedgehog-subgroup medulloblastoma: results from phase II Pediatric Brain Tumor Consortium studies PBTC-025B and PBTC-032. J Clin Oncol. 2015;33(24):2646–2654. doi: 10.1200/JCO.2014.60.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kieran MW. Targeted treatment for sonic hedgehog-dependent medulloblastoma. Neuro Oncol. 2014;16(8):1037–1047. doi: 10.1093/neuonc/nou109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Von Hoff DD, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361(12):1164–1172. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- 183.Robinson GW, et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget. 2017;8(41):69295–69302. doi: 10.18632/oncotarget.20619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Atwood SX, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27(3):342–353. doi: 10.1016/j.ccell.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Metcalfe C, de Sauvage FJ. Hedgehog fights back: mechanisms of acquired resistance against Smoothened antagonists. Cancer Res. 2011;71(15):5057–5061. doi: 10.1158/0008-5472.CAN-11-0923. [DOI] [PubMed] [Google Scholar]
- 186.Atwood SX, Chang AL, Oro AE. Hedgehog pathway inhibition and the race against tumor evolution. J Cell Biol. 2012;199(2):193–197. doi: 10.1083/jcb.201207140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Whitson RJ, et al. Noncanonical hedgehog pathway activation through SRF-MKL1 promotes drug resistance in basal cell carcinomas. Nat Med. 2018;24(3):271–281. doi: 10.1038/nm.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Beauchamp EM, et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J Clin Invest. 2011;121(1):148–160. doi: 10.1172/JCI42874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kim J, Lee JJ, Kim J, Gardner D, Beachy PA. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc Natl Acad Sci U S A. 2010;107(30):13432–13437. doi: 10.1073/pnas.1006822107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kim J, et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell. 2013;23(1):23–34. doi: 10.1016/j.ccr.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Antman KH. Introduction: the history of arsenic trioxide in cancer therapy. Oncologist. 2001;6(suppl 2):1–2. doi: 10.1634/theoncologist.6-suppl_2-1. [DOI] [PubMed] [Google Scholar]