

Abstract
A series of cationic heteroleptic iridium(III) complexes bearing tris-diimine ligands [Ir(phen)2(R-phen)]3+ (R-phen = phenanthroline (1), 3,8-diphenylphenanthroline (2), 3,8-dipyrenylphenanthroline (3), 3-phenylphenanthroline (4), 3-pyrenylphenanthroline (5), and 3,8-diphenylethynylphenanthroline (6)) were synthesized and characterized. These complexes possessed phen ligand-localized 1π,π* transitions below 300 nm, and charge transfer (1CT) and/or 1π,π* transitions between 300 and 520 nm. In 1, 2, 4, and 6, the low-energy bands were mixed 1CT/1π,π*. However, the increased π-donating ability of the pyrenyl substituent(s) in 3 and 5 split the low-energy bands into a pyrene-based 1π,π* transition at 300-380 nm and an intraligand charge transfer (1ILCT) transition at 380-520 nm. All complexes were emissive at room temperature in CH3CN, but the parentage of the emitting state varied depending on the R substituent(s). Complex 1 exhibited predominantly phen ligand-localized 3π,π* emission mixed with metal-to-ligand charge transfer (3MLCT) character, while the emission of 2, 4, and 6 was predominantly from the excited-state with 3π,π*/3ILCT/3MLCT character. The emission from 3 and 5 was dominated by pyrene-based 3π,π* states mixed with 3ILCT character. The different natures of the lowest triplet excited states were also reflected by the different spectral features and lifetimes of the triplet transient absorption of these complexes. Complexes 3 and 5 had singlet oxygen quantum yields as high as 81 and 72%, respectively. Both gave submicromolar phototoxicities toward cancer cells (SK-MEL-28 human melanoma) and bacteria (S. aureus and S. mutans) with visible light activation (and marginal to no photobiological activity with red light). Their visible-light phototherapeutic indices (PIs) toward SK-MEL-28 cells were 248 for 3 and >435 for 5; PIs were lower in bacteria (≤62) due to their inherent antimicrobial activities. Both complexes were shown to produce substantial amounts of intracellular reactive oxygen species (ROS), which may account for their photobiological activities.
Keywords: heteroleptic Ir(III) trisdiimine complexes, photophysics, photobiological activities, photodynamic therapy, antimicrobial, long-lived triplet excited state, reactive oxygen species
Table of Contents Graphic
INTRODUCTION
Metal-organic complexes have attracted broad interest from researchers in the past few decades due to their tunable optical properties associated with the interplay between the inorganic metal ion and the π-extended organic ligands.1 Ir(III) complexes featuring d6 transition-metal centers and octahedral coordination geometries are particularly promising because of their intriguing photophysical and photochemical properties.2-5 Until now, most of the reported work has focused on ortho-metalated neutral or monocationic Ir(III) complexes,6 which are good candidates as triplet emitters for organic light emitting devices because the heavy Ir(III) ion induced rapid intersystem crossing (ISC) facilitates the formation of the triplet excited states in these complexes.7-9 Additionally, most of these complexes exhibit strong absorption in the visible spectral region and possess relatively long-lived triplet excited states, which expands their application to the areas of triplet-triplet annihilation (TTA) upconversion,8 photovoltaics,10,11 bioimaging12 and photocatalysis.13
Although Ir(III) complexes have been widely exploited, their use as photosensitizers (PSs) for photobiological applications (e.g., photodynamic therapy, PDT) has been quite limited.14-16 PDT is of interest as an alternative cancer treatment modality due to its spatiotemporal selectivity, whereby, light activation of an otherwise nontoxic PS triggers its reaction with ground state oxygen to produce cytotoxic reactive oxygen species (ROS) through Type I (electron transfer) or Type II (energy transfer) mechanisms.17 PSs with long-lived triplet excited states are well-suited for PDT.18-20 It is known that the triplet excited state lifetime is mainly governed by the energy and electronic configuration of the lowest triplet excited state (T1). Possible excited state electronic configurations in metal complexes include: (i) metal-to-ligand charge transfer (MLCT), (ii) ligand-to-ligand charge transfer (LLCT), (iii) intraligand charge transfer (ILCT), (iv) metal-centered (MC) d,d transition, (v) ligand-to-metal charge transfer (LMCT), (vi) intraligand (IL, or π,π*), (vii) metal-to-metal charge transfer (MMCT), or (viii) mixtures of these aforementioned configurations.16 Triplet IL (3IL) states are of particular interest for their prolonged excited state lifetimes that arise from the increased organic character of the transition, reducing ISC rates and thus increasing the time allowed for bimolecular interactions (including those that would produce ROS). Metal complexes with low-lying 3IL states have proven to be excellent ROS generators, and thus attractive PSs for photobiological applications.
Previous studies on cyclometalated Ir(III) complexes of the type [Ir(C^N)2(N^N)]+ (where C^N and N^N refer to the cyclometalating ligand and the diimine ligand, respectively) have shown that extending the π-conjugation of the diimine ligand is an efficient strategy for harnessing long-lived, low-lying 3IL states.16,18,21 While well-developed, such systems present synthetic challenges and their stabilities and hydrophilicities decrease with the increased π-conjugation of the ligands.22 Thus, there is interest in exploring the analogous Ir(III) complexes, with higher charge, that are derived from neutral ligands, such as 2,2′-bipyridine (bpy) or 1,10-phenanthroline (phen). These [Ir(N^N)2(N^N)′]3+ (where N^N and (N^N)′ refer to different diimine ligands) complexes are relatively rare,6,23-26 possibly owing to the inertness of the Ir(III) ion that requires harsh reaction conditions and laborious purification procedures.2,6,24 Recently, there has been a growing interest in these Ir(III) complexes due to their possible application as antimicrobial agents23,27 and DNA intercalators.28 Nevertheless, little is known about the excited states of these systems, and their application as PSs for PDT have not been explored.
Herein we report the synthesis and exploration of the photophysical and photobiological properties of a series of heteroleptic [Ir(N^N)2(N^N)′]3+ complexes with neutral diimine ligands (Chart 1). We chose the rigid phen as the ancillary ligand, rather than bpy to prevent unwanted cyclometalation that would occur with rotation about the bpy coannular bond. The structures in the series were chosen to gradually increase the degree of π-conjugation on one of the phen ligands to explore - for the first time - the impact of extended π-conjugation on the triplet excited states of [Ir(N^N)2(N^N)′]3+ complexes. The photobiological activities of two of these complexes (3 and 5) with considerable ground-state absorption in the visible spectral region were investigated. We reveal that these complexes possess long-lived triplet excited states (ca. 4.3 - 33 μs), which facilitate the bimolecular interactions with ground-state oxygen to efficiently generate ROS. Complexes 3 and 5 exhibit high ROS production in vitro, which leads to strong PDT effects toward both cancer cells and bacteria when activated with visible light (400-700 nm).
Chart 1.
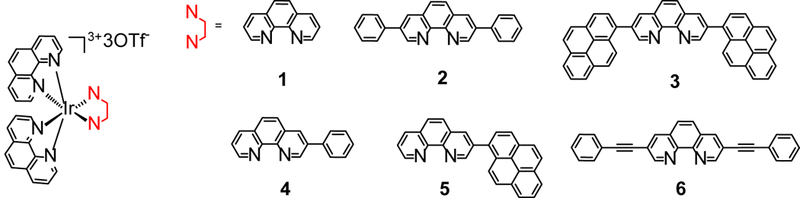
Molecular structures for complexes 1-6.
MATERIALS AND METHODS
Synthesis and Characterization.
All reagents and solvents were purchased from commercial sources and used as received unless otherwise mentioned. The solvents used for photophysical studies were spectroscopic grade and purchased from VWR International and used as received. 1H NMR spectra were recorded on a Bruker-400 spectrometer in CDCl3 with tetramethylsilane (TMS) as internal standard or DMSO-d6. High resolution mass (HRMS) analyses were performed on Waters Synapt G2-Si Mass Spectrometer. Elemental analyses were conducted by NuMega Resonance Laboratories, Inc. in San Diego, California. The diimine ligands 3-phenyl-1,10-phenanthroline, 3,8-diphenyl-1,10-phenanthroline,29 3,8-bis(phenylethynyl)-1,10-phenanthroline,30 3-(pyren-1-yl)-1,10-phenanthroline,31 3,8-di(pyren-1-yl)-1,10-phenanthroline32 were prepared according to the reported procedures, and the synthetic routes are depicted in ESI Scheme S1. The iridium precursor [Ir(phen)2OTf2]OTf was prepared following the reported method.6
General procedure for the synthesis of complexes 1-6: In a 100 mL round-bottom flask, [Ir(phen)2OTf2]OTf (86 mg, 0.086 mmol) and two equivalents of phenanthroline or substituted phenanthroline (0.172 mmol) were added successively. Then 10 mL 1,2-dichlorobenzene (DCB) was added as solvent. The suspension was vacuumed and back-filled with argon for three times before brought to reflux for 12 hours. The mixture was allowed to cool down to room temperature. Then the suspension was centrifuged to collect the solid, which was further washed with chloroform and ether. Several cycles of the washing and sonication were needed in some cases.
Complex 1.
The target complex was afforded as a white solid (65 mg, yield: 65%). 1H NMR (400 MHz, DMSO-d6): δ 9.09 (dd, J = 8.4, 1.1 Hz, 6H), 8.55 (s, 6H), 8.13 (dd, J = 5.4, 1.1 Hz, 6H), 7.95 (dd, J = 8.3, 5.4 Hz, 6H). ESI-HRMS calcd for [C36H24IrN6]3+: 244.3898, Found: 244.3893. Anal calcd (%) for C39H24IrN6S3O9F9•2H2O: C, 38.52; H, 2.32; N, 6.91. Found: C, 38.32; H, 2.57; N 7.08.
Complex 2.
Pale yellow solid was afforded (86 mg, yield: 76%). 1H NMR (400 MHz, DMSO-d6): δ 9.49 (d, J = 1.8 Hz, 2H), 9.08 (t, 4H), 8.57-8.51 (m, 6H), 8.25 (d, J = 4.0 Hz, 2H), 8.17 (d, J = 1.6 Hz, 2H), 8.12 (d, J = 4.0 Hz, 2H), 8.03-7.95 (m, 4H), 7.64-7.61 (m, 4H), 7.52-7.49 (m, 6H). ESI-HRMS calcd for [C48H32IrN6]3+: 295.0774, Found: 295.0777. Anal calcd (%) for C51H32IrN6S3O9F9•2H2O: C, 44.77; H, 2.65; N, 6.14. Found: C, 44.70; H, 2.80; N 6.13.
Complex 3.
Red solid was afforded (30 mg, yield: 22%). 1H NMR (400 MHz, DMSO-d6): δ 9.59 (s, 2H), 9.25 (d, J = 8.0 Hz, 2H),8.90 (d, J = 8.0 Hz, 2H), 8.76-8.73 (m, 4H), 8.63 (d, J = 8.4 Hz, 2H), 8.55 (d, J = 5.6 Hz, 2H), 8.46-8.40 (m, 6H), 8.32-8.19 (m, 14H), 7.80-7.76 (m, 4H), 7.33 (s, br, 2H). ESI-HRMS calcd for [C68H40IrN6]3+: 377.7650, Found: 377.7651. Anal calcd (%) for C71H40IrN6S3O9F9•1.5CH2Cl2•C4H10O: C, 51.56; H, 3.00; N, 4.72. Found: C, 51.42; H, 3.12; N 4.41.
Complex 4.
Pale yellow solid was afforded (43 mg, yield: 40%). 1H NMR (400 MHz, DMSO-d6): δ 9.48 (s, 1H), 9.12-9.07 (m, 5H), 8.59-8.53 (m, 6H), 8.26-8.20 (m, 3H), 8.15-8.11 (m, 2H), 8.04-7.94 (m, 6H), 7.63-7.61 (m, 2H), 7.51-7.49 (m, 3H). ESI-HRMS calcd for [C42H28IrN6]3+: 269.7336, Found: 269.7334. Anal calcd (%) for C45H28IrN6S3O9F9•2H2O: C, 41.54; H, 2.56; N, 6.46. Found: C, 41.27; H, 2.65; N 6.46.
Complex 5.
In addition to washing with CHCl3 and ether, further purification by recrystallization from hot 1,2-dichloroethane/acetone gave a brownish yellow solid (42 mg, yield: 35%). 1H NMR (400 MHz, DMSO-d6): δ 9.52 (s, 2H), 9.08-9.23 (m, 5H), 8.89 (d, J = 8.0 Hz, 1H), 8.75 (d, J = 8.0 Hz, 1H), 8.64-8.56 (m, 6H), 8.44-8.39 (m, 4H), 8.29 (d, J = 8.0 Hz, 1H), 8.28-8.08 (m, 7H), 8.03-7.94 (m, 4 H), 7.74-7.72 (m, 2H). ESI-HRMS calcd for [C52H32IrN6]3+: 311.0774, Found: 311.0773. Anal calcd (%) for C55H32IrN6S3O9F9•2CH2Cl2•H2O: C, 43.66; H, 2.44; N, 5.46. Found: C, 43.33; H, 2.06; N 5.88.
Complex 6.
Brown solid was afforded (60 mg, yield: 44%). 1H NMR (400 MHz, DMSO-d6): δ 9.36 (s, 2H), 9.13 (d, J = 8.4 Hz, 2H), 9.05 (d, J = 8.4 Hz, 2H), 8.59-8.51 (m, 6H), 8.25 (s, 4H), 8.04-7.90 (m, 6H), 7.52-7.48 (m, 10H). ESI-HRMS calcd for [C52H32IrN6]3+: 311.0774, Found: 311.0777. Anal calcd (%) for C55H32IrN6S3O9F9•CH2Cl2: C, 45.91; H, 2.34; N, 5.74. Found: C, 45.92; H, 1.99; N 5.94.
Photophysical Measurements.
The spectroscopic grade solvents used for photophysical studies were purchased from VWR International and used without further purification. The ultraviolet-visible (UV-vis) absorption spectra were recorded on a Varian Cary®50 spectrophotometer. Steady-state emission spectra were measured on a Jobin-Yvon FluoroMax-4 fluorometer/phosphorometer with a Hamamatsu photomultiplier tube (PMT) R928 as the detector. The emission lifetimes were obtained on an Edinburgh LP920 laser flash photolysis spectrometer excited at 355 nm. The emission quantum yields were determined by relative actinometry, in which [Ru(bpy)3]Cl2 in degassed CH3CN (λmax = 436 nm, Φem = 0.097)33 was used as the reference for complexes 3, 5 and 6, and a 1 N sulfuric acid solution of quinine bisulfate (λex = 347.5 nm, Φem = 0.546)34 was used as the reference for complexes 1, 2 and 4. All of the emission measurements were performed in deaerated solutions. Dynamic light scattering measurement was carried out on a Nano ZS Zetasizer from Malvern Instrument Ltd.
The nanosecond transient difference absorption (TA) spectra and decays were measured in degassed acetonitrile solutions on an Edinburgh LP920 laser flash photolysis spectrometer. The third harmonic output (355 nm) of a Nd:YAG laser (Quantel Brilliant, pulse width = 4.1 ns, repetition rate = 1 Hz) was used as the excitation source. Each sample was purged with argon for 45 min prior to measurement. The triplet excited-state absorption coefficients (εT) at the TA band maxima were estimated by the singlet depletion method,35 and the triplet quantum yields were determined by relative actinometry36 using silicon 2,3-naphthalocyanine bis(trihexylsilyloxide) (SiNc) in benzene (ε 590 nm = 70,000 L mol−1 cm−1, ΦT = 0.20)37 as the reference.
Singlet oxygen quantum yields (ΦΔ) for complexes 3 and 5 (5 μM in CH3CN) were determined from sensitized singlet oxygen emission centered at 1268 nm using a PTI Quantamaster equipped with a Hamamatsu R5509-42 near-infrared (NIR) PMT. The measurements were carried out under ambient oxygen concentration (21%) relative to the standard [Ru(bpy)3](PF6)2 (ΦΔ = 0.56 in aerated CH3CN)38 according to Eq 1, where I, A, and η are integrated emission intensity, absorbance at the excitation wavelength, and refractive index of the solvent, respectively. Values calculated for ΦΔ were reproducible to within <5%.
| Eq1 |
Computational Methods.
The ground state geometries of all iridium complexes were optimized at the level of density functional theory (DFT) using Gaussian09 software package.39 The excitation energies were calculated with linear response time dependent DFT (TD-DFT) formalism.40-43 A total of 60 excitations were calculated to reproduce the energy range of the experimental UV/Vis absorption spectra. The spectra were generated using inhomogeneous Gaussian line-broadening of 0.08 eV to match with experimental absorption spectra. To obtain the phosphorescence energies, the lowest triplet ground state geometry was optimized using unrestricted DFT (Δ-SCF method).44 Then using a combined scalar relativistic ZORA and TDDFT approach45-47 implemented in NWChem software package,48 the lowest triplet energies were calculated.
The hybrid PBE1PBE functional49 was used for both the ground and excited state calculations. LANL2DZ basis set50-52 was applied for Ir(III) ion, while 6-31G* basis set53 was used for the remaining atoms. Both geometry optimization and excited state calculations were performed in acetonitrile (CH3CN,), which was chosen for consistency with experiments. The solvent effect for the absorption spectra was modeled using conductor-like polarizable continuum model (CPCM)54,55 implemented in Gaussian 09, whereas the phosphorescence calculations were performed via COSMO continuum solvation56,57 implemented in NWChem software package.
Natural transition orbital (NTO) analysis was performed to obtain the hole-electron pairs that correspond to singlet excitation,43,58 where a hole-electron transition from a ground state to an excited state could be realized through unitary transformation of transition density matrix of a specific excited state.58 For visualizing the lowest-energy emitting state, we plotted the dominant molecular orbitals by performing the eigenvector analysis on the lowest excited state. Chemcraft-1.7 software59 was used for plotting the ground- and excited state charge densities by setting isovalue as 0.02 e-/au3. This value is typically used for the electron-density plots of different types of molecular systems, since it provides good resolution for the shape of molecular orbitals.
Photobiological Activity Studies.
The experimental details for cell culture, cellular assays, cytotoxicity and photocytotoxicity cell assays, confocal microscopy, bacterial survival assays, and measurement of ROS in SK-MEL-28 cells are provided in the Supporting Information.
RESULTS AND DISCUSSION
Synthesis.
The synthetic route for complexes 1-6 is depicted in Scheme 1. Reaction between IrCl3 and phenanthroline in refluxing glycerol gave dichloride intermediate P1 as a highly emissive yellow powder. The use of high temperature and short reaction time was reported to be important for the formation of P1 in high yield.6 P1 was precipitated out of the reaction mixture after adding excess amount of NH4PF6, which was then filtered and rinsed several times with water and ether. Then the inert chloride ligands were substituted by trifluoromethane sulfonate (¯OTf) using the method developed by Meyer.60 The PF6 counter anion could be more easily replaced by ¯OTf ion while it is more difficult to substitute the two chloride ligands with the OTf ligands.23 We found that the source of the reagent HOTf played a critical role than the solvent, with different batches of HOTf (even from the same manufacture) influencing the overall reaction yield drastically for unknown reasons. The intermediate P2 was collected by filtration and further purified by flash column chromatography on neutral Al2O3 with acetone as the eluent. For synthesizing the heteroleptic iridium complexes 1-6, different solvents such as 1,2-dichlorobenzene (o-DCB), methanol/dichloromethane, ethanol, isopropanol, ethylene glycol were used. However, only the reaction in refluxing 1,2-dichlorobenzene afforded the desired product with a high yield and purity. After cooling to room temperature, the crude product was collected by centrifuge and then further washed with chloroform and ether to afford the pure product. The structures of complexes 1-6 were characterized by 1H NMR, HRMS and elemental analysis (ESI Figures S1-S6). The solubility of these complexes in water was good enough for biological activity studies likely due to the numbers of charges in the complexes.28 However, the hydrophilicity of the complexes decrease with the increased π-conjugation of ligand.
Scheme 1.
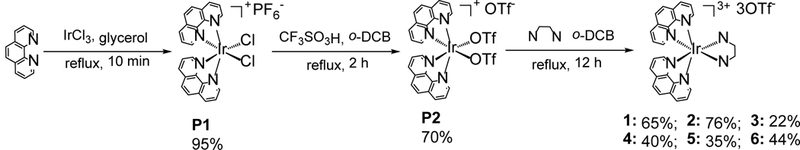
Synthetic routes for complexes 1-6.
Electronic Absorption.
For Ir(III) complexes to be used as PSs for photobiological applications, it is important that the complexes absorb strongly in the visible to the NIR regions. To evaluate whether the synthesized complexes have the potential for photobiological applications, the ground-state absorption of complexes 1-6 was measured in acetonitrile and compared to the calculated spectra in the same solvent (Figure 1 and Supporting Information Figures S7 and S8). The summarized absorption band maxima and molar extinction coefficients are compiled in Table 1. The absorption of all complexes obeyed the Beer’s law in the concentration range studied (5×10−6 - 2×10−4 mol L−1).
Figure 1.
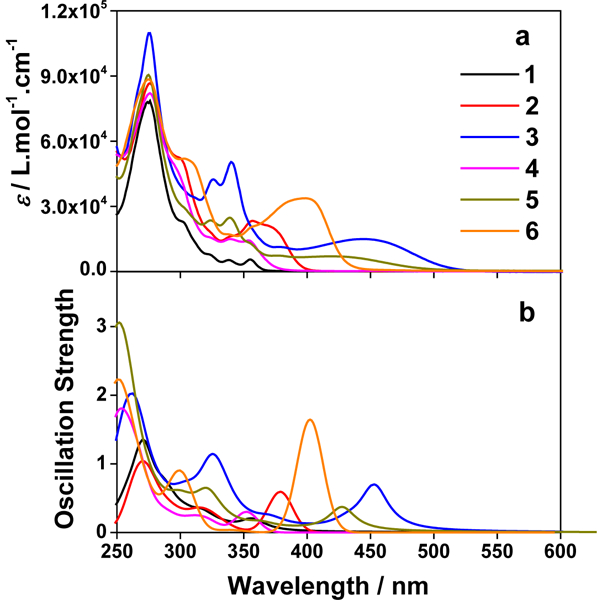
(a) Experimental and (b) calculated absorption spectra of complexes 1-6 in acetonitrile.
Table 1.
Photophysical Data for Complexes 1-6 in Acetonitrile.
|
λabs/nm (ε/104 L mol−1
cm−1)a |
λem/nm (τem/μs); Φemb |
kr/103 s−1 c | knr/105s−1 d |
theorλphos
/nme |
λT1-Tn /nm (τT/μs; εT1-Tn / 104 L mol−1 cm−1); ΦTf |
|
|---|---|---|---|---|---|---|
| 1 | 275 (7.99), 303 (2.30), 325 (0.77), 340 (0.50), 356 (0.58) | 455 (4.16), 485 (4.17), 520 (4.12); 0.0040 | 0.96 | 2.39 | 460 | 370 (4.27; -), 590 (4.52; -), 750 (5.08; -); -i |
| 2 | 277 (8.65), 302 (5.24), 354 (2.38), 377 (1.95) | 525 (17.6), 560 (18.3); 0.0178 | 1.09 | 0.60 | 499 | 410 (15.9; 1.07), 590 (15.6; 3.24); 0.93 |
| 3 | 276 (10.81), 325 (4.22), 341 (5.12), 382 (1.21), 448 (1.60, br) | 660 (−)g; 0.0065 | -h | -h | 689 | 539 (33.4; 5.37); 0.34 |
| 4 | 276 (8.10), 297 (4.70), 325 (1.52), 340 (1.44), 354 (1.32) | 510 (3.65), 542 (3.75); 0.0067 | 1.84 | 2.72 | 495 | 380 (3.62; -), 503 (3.32; -); - i |
| 5 | 275 (9.05), 302 (2.85), 324 (2.30), 341 (2.38), 357 (1.32), 429 (0.70, br) | 650 (−)g; 0.0058 | -h | -h | 680 | 497 (31.2; -); - i |
| 6 | 276 (8.73), 308 (5.08), 356 (2.06), 385 (3.20), 403 (3.32) | 565 (5.53), 608 (5.47); 0.0190 | 3.44 | 1.77 | 630 | 450 (5.28; 1.90), 610 (5.68; 3.76); 1 |
Absorption band maxima (λabs) and molar extinction coefficients (ε) at room temperature.
Emission wavelengths (λem), lifetimes (τem), and quantum yields (Φem) at room temperature. The lifetimes were measured in the same solution as used in the TA study. The concentration of sample solution was 7.2×10−5, 1.8×10−5, 1.9×10−5, 2.8×10−5, 3.1×10−5, and 2.0×10−5 mol•L−1 for 1-6, respectively. The emission quantum yields were determined by relative actinometry, in which [Ru(bpy)3]Cl2 in degassed CH3CN (λmax = 436 nm, Φem = 0.097)33 was used as the reference for complexes 3, 5 and 6, and a 1 N sulfuric acid solution of quinine bisulfate (λex = 347.5 nm, Φem = 0.546)34 was used as the reference for complexes 1, 2 and 4.
Radiative decay rates (kr) and nonradiative decay rates (knr) calculated by kr = Φem/(ΦTτem) and knr = (1-Φem)/(ΦTτem), respectively. For 2 and 6, the estimated triplet quantum yields (ΦT) from the TA measurement were used. For 1 and 4, ΦT was assumed to be 1
Radiative decay rates (kr) and nonradiative decay rates (knr) calculated by kr = Φem/(ΦTτem) and knr = (1-Φem)/(ΦTτem), respectively. For 2 and 6, the estimated triplet quantum yields (ΦT) from the TA measurement were used. For 1 and 4, ΦT was assumed to be 1.
Calculated phosphorescence energies by PBE1PBE for optimized triplet geometry.
Nanosecond transient absorption band maxima (λT1-Tn), triplet extinction coefficients (εT1-Tn), triplet excited-state lifetimes (τT) and quantum yields (ΦT) measured in CH3CN at room temperature. The concentration of sample solution was 7.2×10−5, 1.8×10−5, 1.9×10−5, 2.8×10−5, 3.1×10−5, and 2.0×10−5 mol•L−1 for 1-6, respectively. SiNc in benzene was used as the reference (ε Τ1−Τn = 70,000 L mol−1 cm−1 at 590 nm ΦT = 0.20).37
Emission signal was too weak to allow reliable lifetime to be measured.
Cannot be calculated because the lifetime was unable to be obtained.
No bleaching bands were detected, thus the εT1-Tn values were unable to be estimated using the singlet depletion method and the ΦT value was unable to be calculated.
All complexes possess a strong absorption band at ca. 275 nm (ε = 8 - 11 × 104 M−1 cm−1), which is essentially not influenced by the aryl substituent on the phenanthroline ligand. Based on the large molar extinction coefficients and the independency of its energy on the aryl substituents, we assign this band predominantly to the core phen ligand based 1π,π* transition. This assignment is supported by the NTOs listed in ESI Table S1 for the high-energy absorption band. In contrast, the low-energy absorption bands above 300 nm are quite distinct in these complexes.
Complex 1 showed structured weak absorption bands (≤ 5,000 M−1 cm−1) at 320-360 nm region, which is quite distinct from the broad 1MLCT band (ca. 350 - 500 nm) observed from the corresponding Ru(phen)32+ complex.61 The molar extinction coefficients of these bands and the NTOs shown in Table 2 and Supporting Information Table S1 point these bands to primarily 1MLCT/1LLCT transitions mixed with minor 1π,π* transitions. Increasing the π-conjugation of the diimine ligand via attaching aryl substituent dramatically impacted the features (i.e. transition energies and oscillation strengths) and natures of the low-energy absorption bands in these complexes. For example, addition of the phenyl rings to the phen ligands in 2 and 4 not only increased the molar extinction coefficients of these low-energy absorption bands but also red-shifted the lowest-energy absorption bands in these two complexes. Incorporation of two triple bonds between the phen ligand and the phenyl substituents in 6 further red-shifted the lowest-energy absorption band with enhanced ε values. Meanwhile, this band became broader and structureless, indicating the increased contribution from charge transfer (CT) transitions. With reference to the NTOs representing the lowest-energy transitions (Table 2) in 2, 4 and 6, the S1 states in these three complexes are dominated by intraligand charge transfer (1ILCT) and 1π,π* transitions.
Table 2.
Natural Transition Orbitals (NTOs) Representing the Major Transitions Contributing to the Low-Energy Absorption Band(s) of Complexes 1-6 in Acetonitrile.
| Excited state and properties | Holes | Electrons | |
|---|---|---|---|
| 1 |
S1 364 nm f = 0.02 |
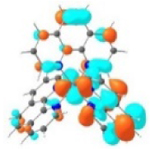 |
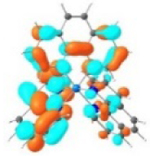 |
|
S5 356 nm f = 0.07 |
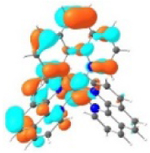 52% |
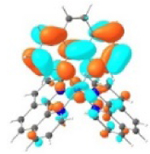 52% |
|
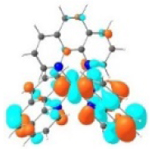 44% |
 44% |
||
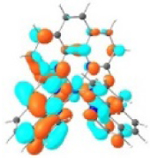
| 2 |
S1 379 nm f = 0.59 |
 |
 |
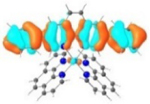
| 3 |
S1 453 nm f = 0.65 |
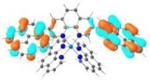 |
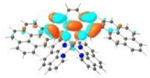 |
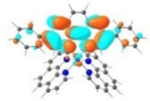
| 4 |
S1 352 nm f = 0.30 |
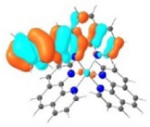 |
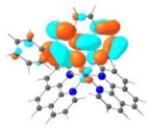 |
| 5 |
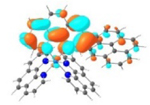
S1 428 nm f = 0.34 |
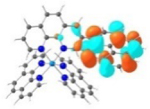 |
 |
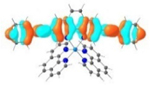
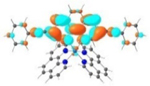
| 6 |
S1 402 nm f = 1.64 |
 |
 |
A similar phenomenon was observed from complexes 3 and 5 with pyrene-substituted phen ligands. In comparison to the corresponding phenyl substituted complexes 2 and 4, the red-shifts and broadening of the lowest-energy absorption bands in 3 and 5 are more pronounced but the ε values are smaller. These changes cannot be attributed to the increase of the ligand π-conjugation because the pyrenyl substituent(s) in complexes 3 and 5 cannot adopt a planar conformation with the phen plane due to steric hindrance although the pyrene motif contains a larger π-system. As shown in Figure 2, the DFT optimized geometries of the phenyl-substituted phen ligand for complex 4 and the pyrenyl-substituted phen ligand for complex 5 clearly manifested a larger dihedral angle (32.4°) between the pyrenyl substituent and the phen motif than that between the phenyl substituent and phen (0.2°). Although it is true that the ground-state conformation does not definitively dictate the minimum-energy conformation of the excited state S1, the geometries corresponding to the S1 states of 3 and 5 in Table 2 clearly show that the dihedral angle(s) between the pyrenyl substituent(s) and phen in 3 and 5 are larger than those between the phenyl substituent(s) and phen in 2 and 4.
Figure 2.
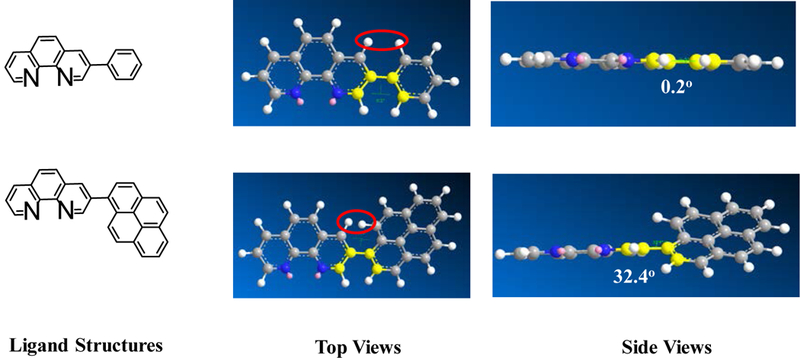
Optimized ground-state geometries of the substituted phen ligands in complexes 4 and 5 via DFT calculations.
It was observed that the lowest-energy absorption band of 5 also red-shifted by ca. 30 nm compared to that of 6. The NTOs in Table 2 clearly show that the holes (highest occupied transition orbitals) are exclusively localized on the pyrenyl substituent(s) in 3 and 5, while the electrons (lowest unoccupied transition orbitals) are predominantly on the phen core ligand in these two complexes. This led to intraligand charge transfer (1ILCT) as the lowest-energy transitions in 3 and 5. In contrast, the dominant transitions contributing to the S1 state of 1 are 1MLCT/1LLCT transitions while in 2, 4, and 6 are 1ILCT/1π,π* transitions. The different natures of the lowest-energy transitions resulted in the salient differences in the spectral features in these complexes.
The lower energies of the S1 states in 3 and 5 with respect to those in 2 and 4 are attributed to the larger dihedral angle between the pyrene ring(s) and phen than that between benzene and phen ring(s) (32.4o vs 0.2o) in the ground state (the difference in dihedral angles also holds in the S1 state although these angles differ from those in the ground state), which interrupted the π-conjugation in the ligand and increased the energies of HOMOs of 3 and 5. Although the energies of LUMOs of 3 and 5 were also increased due to the twisted conformation in the pyrenyl-substituted phen ligand, the increase of the LUMO energy was not as much as the HOMO energy (see Figure 3). This reduced the HOMO-LUMO gap and red-shifted the absorption bands corresponding to the S0→S1 transitions, in which the HOMO-LUMO transition made the dominant contribution to the S1 state, in these two complexes.
Figure 3.
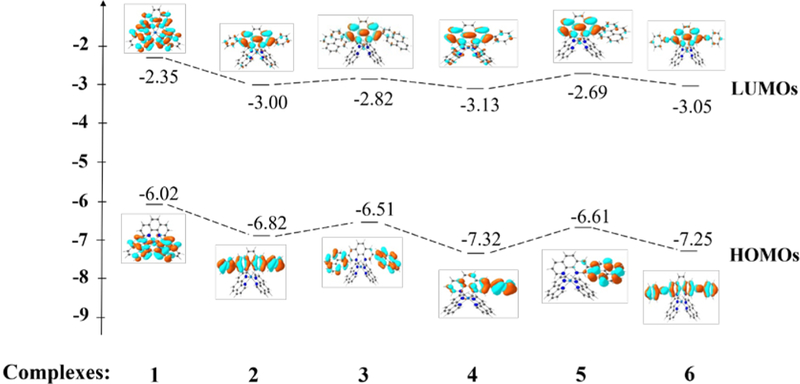
Ground-state energy level diagram for complexes 1-6.
Photoluminescence.
Room-temperature emission of complexes 1-6 were studied in acetonitrile, and the spectra and emission data are provided in Figure 4 and Table 1, respectively. In comparison to their excitation wavelengths, the emission energies of all six complexes are significantly red-shifted (125 - 220 nm). This feature, along with their long lifetimes (several to tens of microseconds) and being prone to oxygen quenching, suggest phosphorescence in nature. Incorporation of the aryl substituents to the phen ligand caused a pronounced red-shift of the emission, as evidenced by the emission energies of 1, 2, 4, and 6. The introduction of pyrene ring(s) in complexes 3 and 5 resulted in ca. 200 nm red-shift. For complexes with phenyl substituent(s) on the ligand (i.e. 2, 4 and 6), their emission spectra exhibited clear vibronic structures with relatively long lifetimes, suggesting 3π,π* nature of the emitting states in these complexes. In contrast, for complexes with pyrenyl substituent(s) (i.e. 3 and 5), much weaker, red-shifted, and featureless emission were detected, indicating a different parentage of the emitting triplet states.
Figure 4.
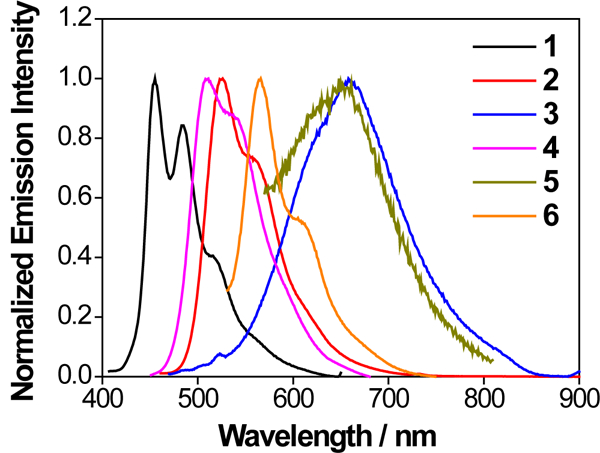
Normalized uncorrected emission spectra for complexes 1 (λex = 330 nm), 2 (λex = 365 nm), 3 (λex = 450 nm), 4 (λex = 350 nm), 5 (λex = 430 nm) and 6 (λex = 400 nm) in deaerated acetonitrile at room temperature. The detector used was a Hamamatsu PMT R928.
TDDFT calculations based on the optimized triplet excited state geometries of complexes 1-6 support the aforementioned assessment. The MOs shown in Table 3 clearly indicate that the emitting triplet states of all complexes have predominant 3π,π* character. However, for complexes bearing phenyl substituents, i.e. 2, 4 and 6, the 3π,π* transitions are based on the phenanthroline ligand, mixed with 3ILCT/3MLCT characters. As the ligands π-conjugation increases in 6 due to the insertion of the two C≡C bonds between phen and the phenyl substituents and also the increased coplanarity in this ligand, the contribution of 3MLCT diminished but the 3ILCT character increased. While for complexes 3 and 5 with pyrenyl substituent(s), the major contributing transitions to the emitting triplet states are switched to pyrene localized 3π,π* state mixed with some 3ILCT character from pyrene to phen motif. The change of the nature of the emitting state is the results of different degrees of π-conjugation and π-donating ability of the phenyl substituent vs. the pyrenyl substituent.
Table 3.
Molecular Orbitals (MOs) Representing Major Transitions Contributing to the Triplet Emission of 1-6 in Acetonitrile. The Emission Was Calculated via Optimization of the Triplet Excited State Geometries.
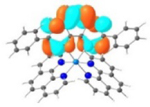
| Triplet Emission | Unoccupied MOs | Occupied MOs | |
|---|---|---|---|
| 1 |
T1 460 nm |
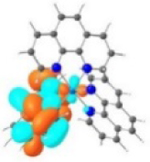 LUMO |
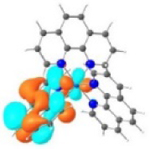 HOMO |
| 2 |
T1 499 nm |
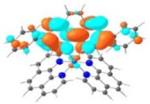 LUMO (74%) 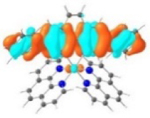 LUMO+3 (21%) |
 HOMO (74%) 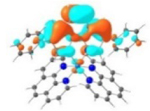 HOMO-1 (21%) |
| 3 |
T1 689 nm |
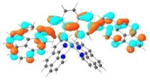 LUMO (66%) 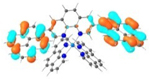 LUMO+6 (28%) |
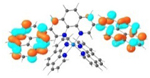 HOMO (66%) 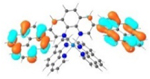 HOMO-1 (28%) |
| 4 |
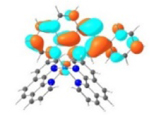
T1 495 nm |
 LUMO |
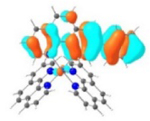 HOMO |
| 5 |
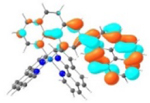
T1 680 nm |
 LUMO |
 HOMO |
| 6 |
T1 630 nm |
 LUMO |
 HOMO |
An interesting phenomenon arose in complexes 2, 4 and 6 that contain the phenyl substituent(s). The emission lifetimes increased dramatically as the solution concentration increased (see Figure 5 and ESI Figure S9). For complexes 3 and 5, the emission was too weak to allow for a reliable lifetime to be measured. Such a phenomenon is opposite to the self-quenching effect typically observed with increased solution concentration. In addition, no new emission bands were observed when the concentration was increased. Therefore, the possibility of forming excimer in the concentration range studied was excluded. We speculate that the increased lifetime at higher concentrations could arise from the formation of unobservable ground-state aggregates with increased concentrations, within which the rotation of phenyl group(s) was limited. This hypothesis was directly supported by the dynamic light scattering (DLS) measurement of complex 2 in acetonitrile solutions with different concentrations, which revealed a concentration-dependent assembly behavior. The number-averaged hydrodynamic diameter of assemblies of 2 was approximately 45, 110, and 260 nm at the concentration of 5×10−5, 1×10−4, and 2×10−4 mol.L−1, respectively. The size of assembly was too small to be measured at the concentration of 1×10−5 mol.L−1 due to the instrument resolution (0.2 nm). The lack of absorption and emission spectral change may reflect either the very small association constant within the aggregates to be detected or the formed aggregates being spectrally too similar to the monomers to be distinguished.
Figure 5.
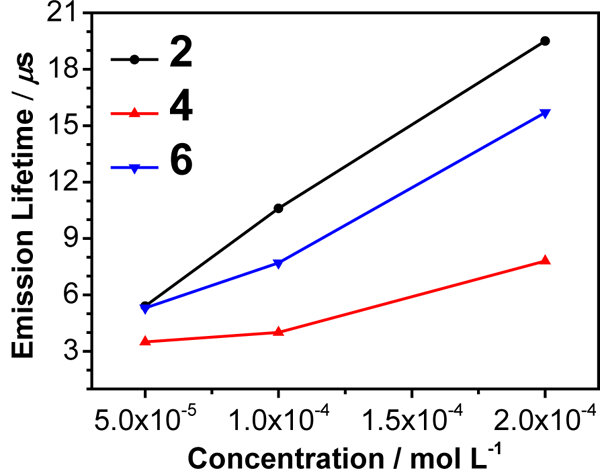
Concentration-dependent emission lifetimes for complexes 2, 4, and 6 in acetonitrile at room temperature.
Because the complexes studied in this work are all ionic, formation of aggregates will involve the anions. Variations of different anions would dramatically impact the aggregation. Our study on the Cl- salt of complex 2 revealed that both the emission spectral feature and the emission lifetime remained the same at concentrations of 5×10−5, 1×10−4, and 2×10−4 mol•L−1, which drastically differed from the concentration-dependent behavior of the emission lifetime for the -OTf salt of 2.
Another piece of evidence to support the aforementioned hypothesis is the aggregation induced/enhanced emission (AIE/AEE) test by adding varied amount of poor solvent CH2Cl2 to a dilute CH3CN solution (1×10−5 mol•L−1) of 2 to intentionally induce aggregation. As shown in the inset of Figure 6, with the increased percentage of CH2Cl2, the UV-vis absorption spectrum of 2 gradually red-shifted and an isosbestic point appeared at 380 nm, demonstrating the formation of aggregates. Accompanying the absorption changes, the emission intensity in 1:9 (v/v) CH3CN/CH2Cl2 solution was approximately one order of magnitude higher than that in CH3CN. Meanwhile, the emission lifetimes increased from 4.4 μs in CH3CN solution, to 17.7 μs in 1:1 (v/v) CH3CN/CH2Cl2 solution, and to 36.0 μs in 1:9 (v/v) CH3CN/CH2Cl2 solution; while the emission band maximum only exhibited a 6-nm red-shift. The AIE/AEE phenomenon has been well studied in recent years in organic molecules with rotating substituents,62 in which the restricted motion (rotation or vibration) in the formed aggregates attenuates the nonradiative decay pathway while switching on the radiative decay pathway.
Figure 6.
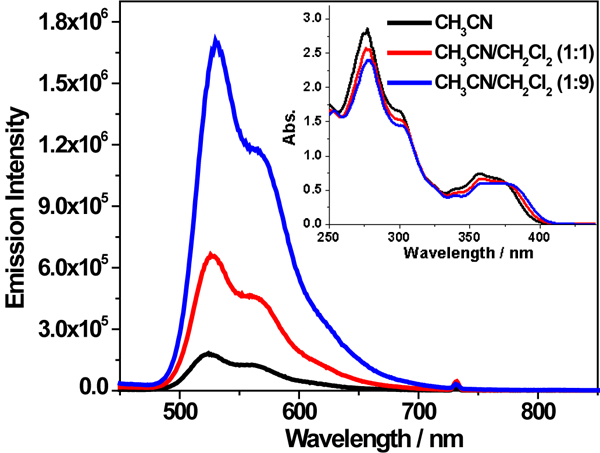
Emission spectra of complex 2 in CH3CN/CH2Cl2 solutions with varied percentage of CH2Cl2. The concentration of each solution was 1×10−5 mol/L and λex = 370 nm. The inset is the UV-vis absorption spectra of the solutions used for the emission measurements.
The experiment that validates the hypothesis of restricted rotation enhancing emission is investigating the emission of complex 2 in alcohols with different viscosity. As Figure 7 displays, when the viscosity increased from ethanol, 1-butanol, 1-hexanol, to 1-octanol (the dynamic viscosity (μ) is 1.09, 2.53, 4.59, and 7.59 mPa.s for ethanol, 1-butanol, 1-hexanol, and 1-octanol, respectively),63 the emission intensity increased while the absorption remained to be almost the same. Because the polarities of these alcohols are quite similar (the ET(30) value is 51.9, 49.7, 48.8, and 48.1 kcal.mol−1 for ethanol, 1-butanol, 1-hexanol, and 1-octanol, respectively),64 it should not impact the emission characteristics pronouncedly. We assume that the viscosity dependence observed is not due to changes in the oxygen quenching rate constant since this parameter should be well below the diffusion limit. Thus, the observed emission intensity increase should mainly be attributed to the increased viscosity of the alcohol solvent, which limited the rotation of the phenyl substituents and resulted in the increased emission intensity. Attempt to remove oxygen from these solvents to the same degree in order to get accurate emission lifetime under air-free condition was unsuccessful because degassing in viscous solvent was quite slow and difficult. Nonetheless, this experiment manifested that the reduced rotation of the phenyl rings increased the emission intensity.
Figure 7.
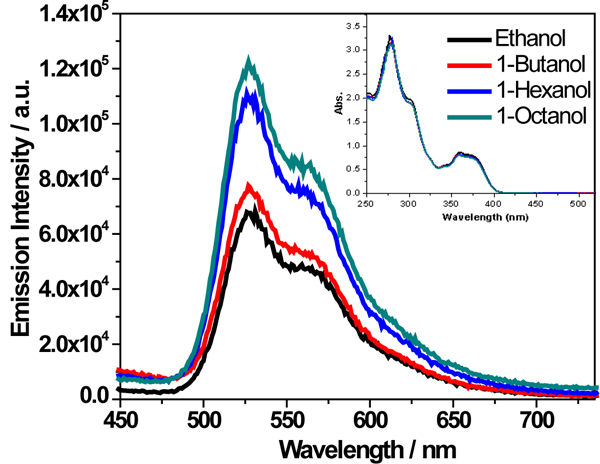
Emission spectra of complex 2 in air-saturated alcohols with different viscosity. The concentration of each solution was 1×10−5 mol/L and λex = 370 nm. The inset is the UV-vis absorption spectra of the four alcoholic solutions of 2 used for the emission measurements.
The hypothesis of limited rotation in high-concentration solutions due to formation of unmeasurable aggregates was further supported by the concentration-dependency emission study of complex 2 in butyronitrile glassy matrix at 77 K. As displayed in Supporting Information Figure S9 (panel f), the emission lifetime of complex 2 remained to be the same in glassy matrixes with different concentrations, which is in contrast to the increased lifetime in high concentration fluid solutions (Figure S9 panel d) at room temperature. This phenomenon should be attributed to the restricted rotation of the phenyl substituents in confined environment at low temperature even if at low concentration. Therefore, the concentration-dependency emission lifetime phenomenon disappeared at 77 K.
Transient Absorption.
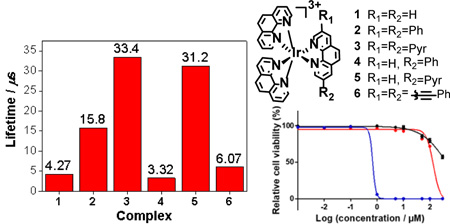
The triplet excited-state characteristics of complexes 1-6 were further studied by nanosecond (ns) transient absorption (TA) spectroscopy in acetonitrile. The time-resolved nanosecond TA spectra were given in Figure 8. For complex 1 with three identical phenanthroline ligands, the TA spectrum was characterized with positive signals in the entire scanned wavelength range (360 - 820 nm). This is quite different from its ruthenium analog with the same set of ligands, which exhibited strong bleaching at 434 nm.65 The triplet lifetime deduced from the decay of TA signals was 4.27 μs, almost one order of magnitude longer than that of the corresponding ruthenium complex.65 Introducing one phenyl ring to the phen ligand in complex 4 essentially kept the overall spectral feature but significantly increased the intensity of the TA band maximum at 500 nm in comparison to that in 1. Incorporation of another phenyl ring to the phen ligand in complex 2 clearly red-shifted the TA band maximum to ca. 560 nm. In addition, obvious bleaching occurred at ca. 370 nm, which is in line with the strong ground-state absorption at 377 nm. Meanwhile, the lifetime was increased to 15.8 μs (Figure 9). When two C≡C bonds were introduced between the phenyl and phen components in complex 6, the entire TA spectrum was red-shifted with respect to that of 2. However, the triplet lifetime of 6 was decreased to 6.07 μs. For complexes 1, 2, 4, and 6, the lifetimes obtained from the TA decay profiles were essentially the same as those measured in the same solution for the emission measurements, indicating the same origin of the transient absorbing excited state as the emitting excited states, i.e. the phenanthroline 3π,π* state or the substituted phenanthroline ligand-localized 3π,π*/3ILCT states. Introduction of more π-conjugated pyrene ring(s) in complexes 3 and 5 changed the TA spectra and the triplet lifetime dramatically. While the lifetime for the corresponding ruthenium complex with the same (or similar) ligands was only 2.49 μs,32 the triplet lifetimes of 3 and 5 were longer than 30 μs. Considering the long triplet lifetime and the similarity to the reported TA spectrum of a Ru(II) polypyridine complex with pyrene covalently attached,66 we attribute the observed TA spectra for 3 and 5 to the pyrene-localized 3π,π* state.
Figure 8.
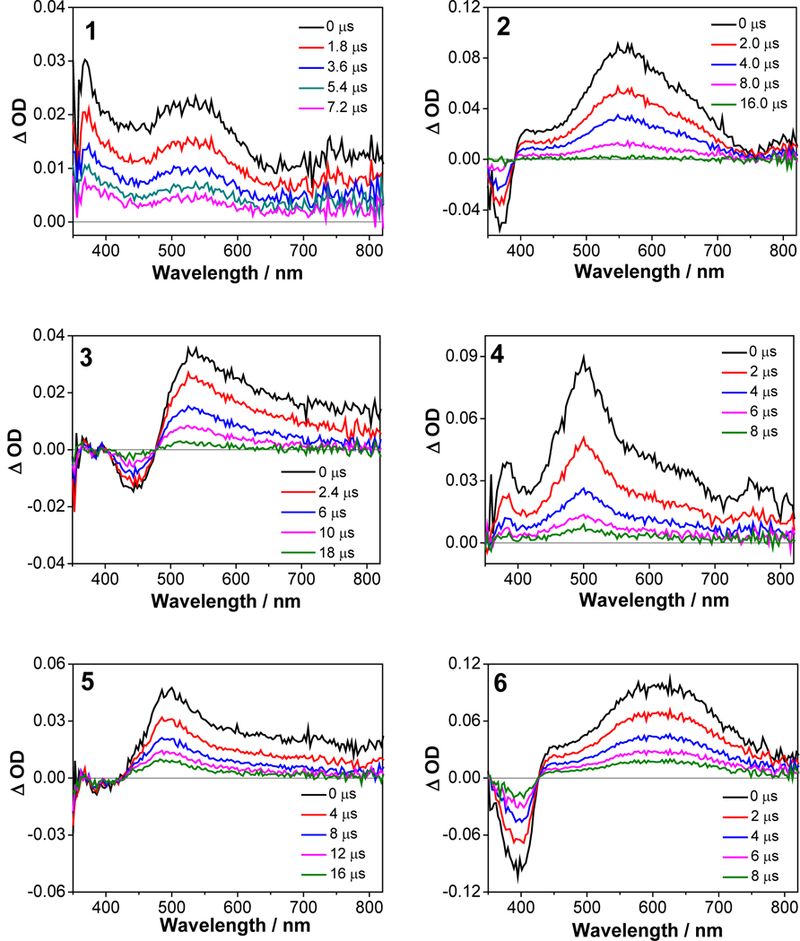
Nanosecond time-resolved TA spectra of 1-6 in acetonitrile solution. λex = 355 nm, A355 = 0.4 in a 1-cm cuvette. The concentration of each sample solution was 7.2×10−5, 1.8×10−5, 1.9×10−5, 2.8×10−5, 3.1×10−5, and 2.0×10−5 mol•L−1 for 1-6, respectively.
Figure 9.
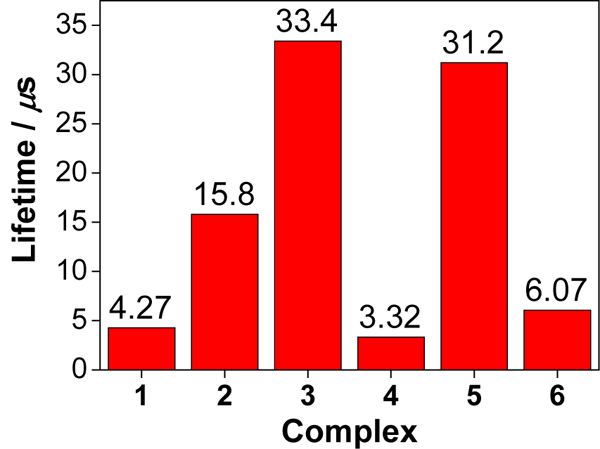
Triplet lifetimes of 1-6 in acetonitrile deduced from the decay of TA signals at the corresponding TA band maxima of each complex.
Singlet Oxygen Sensitization.
The longest wavelength absorbing complexes in the series were the two pyrenyl-containing Ir(III) complexes 3 and 5. Their longest wavelength absorption maxima extended out near 450 nm, while the maxima for the other family members occurred below 400 nm (except for 6, which was centered near 400 nm). Therefore, only complexes 3 and 5 were evaluated for their singlet oxygen quantum yields, since visible light absorption is a desirable property for photobiological applications.
The singlet oxygen quantum yields (ΦΔ) for 3 and 5 were determined in CH3CN relative to [Ru(bpy)3](PF6)2 as the standard (ΦΔ = 56%) by measuring singlet oxygen luminescence centered at 1268 nm. The reason that ΦΔ was determined for the ¯OTf salt of the complex in CH3CN (rather than for the Cl¯ salt in water) is because the phosphorescence lifetime of 1O2 is quenched in pure water, and thus ΦΔ is greatly attenuated and does not reflect the actual amount of 1O2 formed.67 Pure water is not representative of the biological milieu, and it is expected that the complexes will be bound to biological macromolecules and thus experience a less polar environment relative to pure water. For these reasons, as well as to be able to compare to previously reported values of ΦΔ for other metal complexes, we chose CH3CN as the solvent. Since the Cl¯ salt is sparingly soluble in CH3CN, researchers generally use the more soluble ¯OTf complex with this solvent. These conditions give us a convenient way to compare ΦΔ for different complexes and report on the potential of the compounds to form 1O2, although we cannot predict the actual yields in vitro or in vivo from this measurement.
With excitation at 450 and 462 nm, Φem for 3 was calculated to be 67% and 81%, respectively. The efficiency of 5 for singlet oxygen generation was slightly lower, with ΦΔ = 66% (λex = 430 nm) and 72% (λex = 450 nm). These values for ΦΔ were similar to or larger than those for known singlet oxygen sensitizers (e.g., rose bengal, methylene blue, [Ru(bpy)3]2+).38 Since 3 and 5 differ only in the number of pyrenyl units appended to 1,10-phenanthroline (two for 3 versus one for 5), the extra pyrenyl chromophore in 5 is likely responsible for the modest enhancement of ΦΔ observed for 3.
Photobiological Activity.
The efficient singlet oxygen generation detected for 3 and 5 in cell free conditions was anticipated to yield phototoxic effects in vitro. To confirm this, SK-MEL-28 cells were dosed with 3 or 5 in the range of 1 nM to 300 μM and allowed to incubate for 16 h before receiving a dark treatment or a light treatment. For cell-dosing, the compounds were dissolved in water supplemented with 10% DMSO (v/v) and serially diluted with phosphate buffered saline (PBS). Two light treatments were tested: broadband visible (100 J cm−2, 33 mW cm−2) or monochromatic red (625 nm, 100 J cm−2, 43 mW cm−2). The cells were incubated for 48 h followed by addition of the resazurin cell viability indicator68 and a further incubation of 2-4 h. The relative cell viability was quantified under the different conditions according to the metabolic reduction of resazurin to its fluorescent product resorufin (Figure 10, Table 4).
Figure 10.
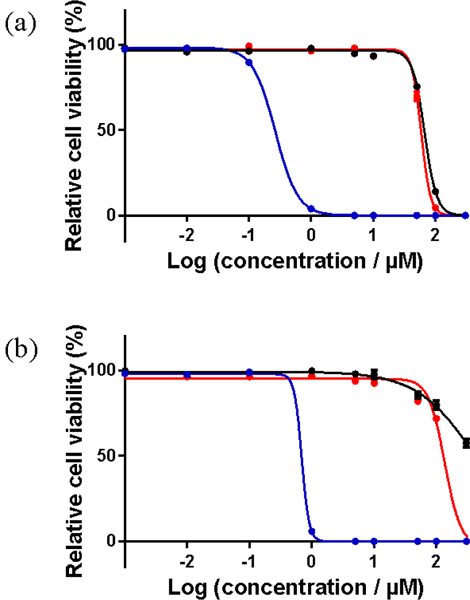
In vitro dose-response curves for complexes 3 (a) and 5 (b) in SK-MEL-28 cells treated in the dark (black) and with visible (blue) or red (red) light activation.
Table 4.
Comparison of (Photo)toxicities of Complexes 3 and 5 Toward SK-MEL-28 Cancer Cells.
| Complex | Dark | Vis lighta | Red lightc | ||
|---|---|---|---|---|---|
| EC50 / μM | EC50 / μM | PIb | EC50 / μM | PId | |
| 3 | 67.0±0.91 | 0.27±0.01 | 248 | 58.9±0.69 | 1 |
| 5 | > 300 | 0.69±0.01 | > 435 | 136±5.73 | > 2 |
16 hours drug-to-light interval followed by 100 J·cm−2 broadband visible light irradiation,
PI = phototherapeutic index (ratio of dark EC50 to visible-light EC50),
16 hours drug-to-light interval followed by 100 J·cm−2 light irradiation with 625-nm LEDs,
PI = phototherapeutic index (ratio of dark EC50 to red-light EC50).
Complex 5 was nontoxic toward SK-MEL-28 cells, while 3 was only slightly cytotoxic at relatively high concentrations (EC50 = 67 μM). With broadband visible light activation, both complexes were photobiologically active. Their photocytotoxicities were submicromolar, with EC50 values of 270 nM for 3 and 690 nM for 5, and PIs of 248 for 3 and more than 435 for 5. The larger PI for 5 was due to its lower dark cytotoxicity combined with a higher light potency. The relatively potent photocytotoxicies for these two complexes were attributed to long-lived 3π,π* states contributed by the pyrenyl groups. The intrinsically long lifetimes of ligand-localized triplet states allow ample time for bimolecular processes such as singlet oxygen sensitization, and have been implicated in the potent photobiological effects observed for other metal complexes that incorporate the π-expansive pyrenyl group.18,69
Because we have previously observed in vitro photobiological effects with red light for π-expansive Ru(II) complexes that have very little absorption at 625 nm,19 we also tested these pyrenyl-based Ir(III) complexes for their abilities to be activated with red light, and there was virtually no photobiological activity (Figure 10, Table 4). Although there was a marginal two-fold PI for 5 with red light activation, the EC50 was too large and the PI too small to be therapeutically useful.
The qualitative effects of 3 and 5 on SK-MEL-28 cells were observed by confocal microscopy in the dark (sham) and with a visible light treatment of 50 J cm−2 (Figure 11). A concentration of 50 μM was chosen because a marked difference in the dark cytotoxicity of 3 and 5 at this concentration was anticipated (since 50 μM is close to the EC50 value of 3, but not of 5). The light treatment was chosen to be 50 J cm−2 (half the dose of the cell cytotoxicity assays) and imaging was done at 15 min post-treatment to ensure that cells were imaged under sub-lethal conditions, where a comparison of cellular morphologies and uptake might be made.
Figure 11.
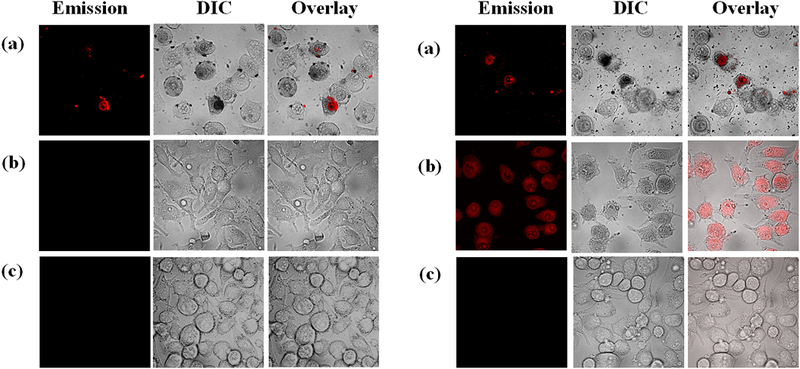
Confocal luminescence images of SK-MEL-28 cells dosed with 50 μM of 3 (a) or 5 (b) in the dark (left) and with visible light (50 J cm−2) (right) compared to cells that were not treated with any complex (c). The images were captured 15 min post-treatment.
As expected from the cell cytotoxicity studies, 3 and 5 differed in their effects on SK-MEL-28 cells when dosed at 50 μM in the dark. Treatment with 3 led to significant changes in cellular morphology relative to healthy control cells, which are generally elongated and spindle-shaped. Cells treated with 3 in the dark were detached and spherical, and the images also showed cellular debris from dead/dying cells. Compound 3 also showed luminescence in some fraction of cells in the dark. By contrast, cells treated with compound 5 at the same concentration appeared relatively healthy with no intracellular luminescence from the photosensitizer.
When cells treated with 3 were exposed to a visible light treatment, a larger number of dead/dying cells were present in the general cell population alongside a significant increase in cellular debris. However, there appeared to be no substantial difference in the fraction of cells or cell particles exhibiting luminescence from 3 when compared to the dark sample. While the dark- and light-treated cells dosed with compound 5 looked similar in terms of morphology (with light-treated cells slightly less elongated) at the imaging time point, there was a substantial difference in photosensitizer uptake. No luminescence was detectable in the images of the dark-treated cells, whereas, all light-treated cells were highly luminescent (Figure 11b, right panel).
This photoactivated uptake could be responsible for the low dark toxicity associated with compound 5 and its much larger PI compared to 3. The lack of a significant difference in the luminescence profiles and morphologies between the dark- and light-treated cells dosed with 3 supports the notion that 3 is readily taken up by cells in the dark and that this may lead to higher dark toxicity. The increased cellular uptake of 3 relative to 5 in the absence of a light trigger could be due to the increased lipophilicity imparted by the additional pyrenyl substituent.
It is interesting to note that both 3 and 5 exhibited weak phosphorescence (too weak to establish an emission lifetime) centered near 650-660 nm at room temperature in fluid solution, yet only 5 showed appreciable luminescence in the confocal images. Both compounds had excited state lifetimes slightly longer than 30 μs in the cell-free TA measurements, arising from a triplet state with a large amount of pyrene-localized 3π,π* character. Despite these similarities, clearly the additional pyrenyl group influences not only biological properties such as cellular uptake and dark cytotoxicity, but also the photoluminescence properties in biological environments.
Measurement of ROS Generation in SK-MEL-28 Cells.
The highly sensitive DCFDA fluorogenic dye was used to probe whether the production of reactive oxygen species (ROS) was a possible source of the photocytotoxic activity against SK-MEL-28 cells (Figure 12). The most common ROS include superoxide anion (O2•-), hydrogen peroxide (H2O2), hydroxyl radical (HO•), and singlet oxygen (1O2), which react directly or indirectly with DCFDA.70 The premise behind this assay is that the cell permeable DCFDA is deacetylated by cellular esterases to a nonfluorescent compound, which is then oxidized by ROS to the highly fluorescent DCF product that can be detected by its characteristic emission.71 To probe for increased ROS production in SK-MEL-28 cells upon photoactivation of complexes 3 and 5, the cells were pre-incubated with DCFDA before performing the photocytotoxicity assay. The sublethal light dose used for the confocal experiments was also used for the ROS assays to ensure that the cells were intact for intracellular ROS measurements.
Figure 12.
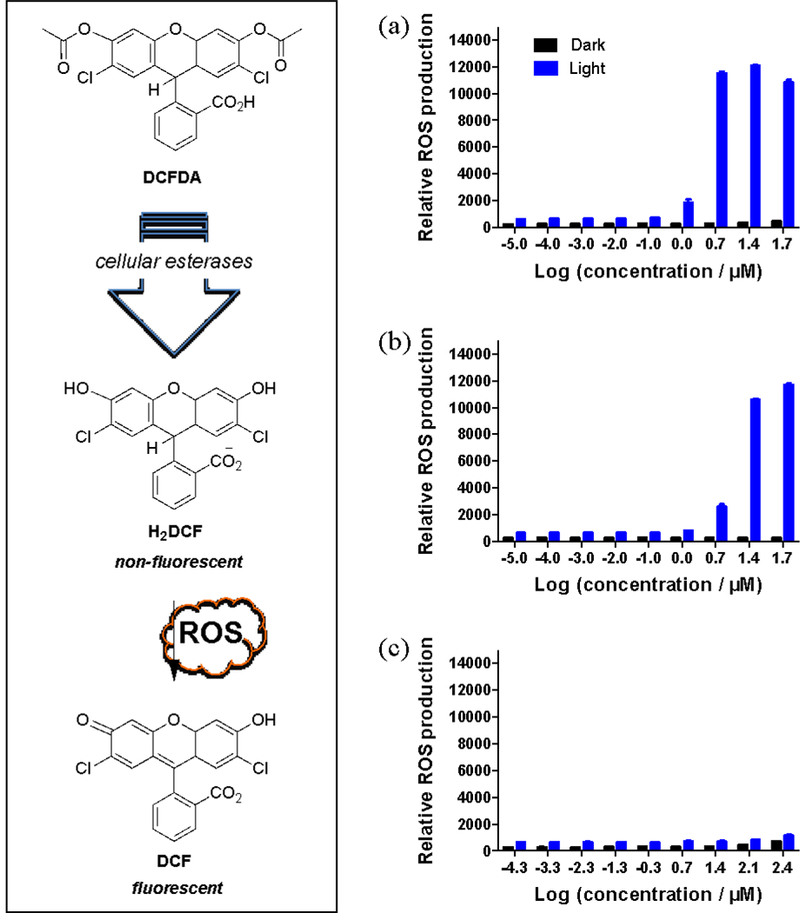
ROS assay results for SK-MEL-28 cells treated with 3 (a), 5 (b), or the positive control tert-butyl hydrogen peroxide TBHP (c) using DCFH-DA as a ROS probe. Cells were treated in the dark (black bars) or with 50 J cm−1 visible light (blue bars).
TBHP was used as a positive control, and dark treatments were included to quantify baseline ROS levels under the assay conditions. ROS generation with 3 and 5 in the dark was minimal, but a light treatment of 50 J cm−2 was sufficient to increase the detected fluorescence from the DCF product in a dose-dependent manner. Light-treated complexes 3 and 5 substantially increased ROS production over the dark controls and also in comparison to the positive control THBP, which was used at a much higher concentration in the assay. Complex 3 produced slightly more fluorescence from DCF at some of the higher concentrations, but the difference was marginal and thus both compounds appeared equally effective in generating phototoxic ROS. Therefore, the photobiological activity observed in SK-MEL-28 cells may stem from an increase in ROS production in cells dosed with 3 or 5 and treated with visible light. Given that there was no photocytotoxicity observed with red light for 3 and very little with 5, the blue wavelengths of the visible spectrum are likely the most important in activating the photosensitizer for ROS production. The observation agrees well with the UV-Vis absorption profiles of the two complexes.
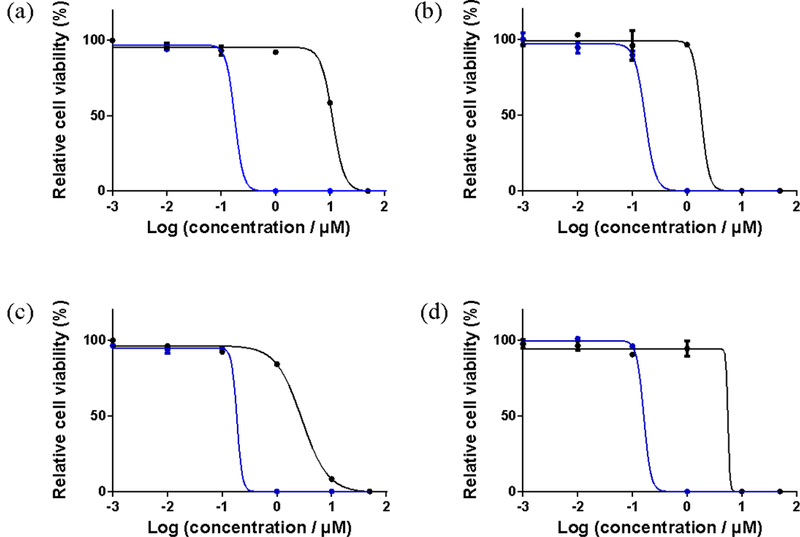
Bacterial Survival Assays.
Complexes 3 and 5 were also evaluated for photoinactivation (PDI) of microorganisms using two different bacterial strains, Streptococcus mutans (Figure 13a and 13c) and Streptococcus aureus (Figure 13b and 13d). The experiments were carried out on planktonic cultures at photosensitizer concentrations between 1 nM and 50 μM. Light treatments were visible light of approximately 35 J cm−2 delivered at a rate of 9.7 mW cm−2, and the EC50 values were determined from dose-response curves constructed from measuring sample turbidy (as absorbance at 562 nm). Both 3 and 5 exhibited antibiotic activity in the absence of a light trigger, with 5 being more potent toward S. mutans and 3 being more potent toward S. aureus. Both 3 and 5 also yielded PDI effects, with submicromolar EC50 values ranging from 160 to 190 nM and PIs between 11 and 62 (Table 5). Neither compound stood out as being the better PDI agent since their photocytotoxicities were virtually the same against both bacteria, but 3 had the larger PI for S. mutans and 5 had the larger PI for S. aureus. The source is these minor differences in activity is unknown, but the conclusion is that the compounds act as light-responsive agents against both cancer and bacterial cells (and possibly through similar mechanisms such as the generation of ROS).
Figure 13.
Bacterial cell survival dose-response curves for complexes 3 (a,b) and 5 (c,d) in Streptococcus mutans (left) and Streptococcus aureus (right) treated in the dark (black lines) or 35 J cm−1 visible (blue lines) light activation.
Table 5.
Comparison Bacterial Cell Survival EC50 Values (μM) for Streptococcus mutans and Streptococcus aureus Dosed with Complexes 3 and 5.
| Complex | Streptococcus mutans | Streptococcus aureus | ||||
|---|---|---|---|---|---|---|
| Dark | Visa | PIb | Dark | Visa | PIb | |
| EC50 / μM | EC50 / μM | EC50 / μM | EC50 / μM | |||
| 3 | 11.2±2.74 | 0.18±0.01 | 62 | 1.81±0.37 | 0.17±0.02 | 11 |
| 5 | 2.87±0.28 | 0.19±0.07 | 15 | 5.59±1.92 | 0.16±0.01 | 35 |
35 J·cm−2 broadband visible light irradiation,
PI = phototherapeutic index (ratio of dark EC50 to visible-light EC50).
CONCLUSIONS
We have synthesized a series of new [Ir(phen)2(R-phen)]3+ (R = H, phenyl, pyrenyl, phenylethynyl) complexes bearing tris-diimine ligands. The UV-vis absorption, emission, and transient absorption studies demonstrated that the lowest singlet and triplet excited state characteristics of these complexes were drastically impacted by the degree of π-conjugation and the π-donating ability of the R substituent(s). With the increased π-conjugation of the R-phen ligand, the low-energy absorption bands in the UV-vis absorption spectra of 1-6 gradually red-shifted, as were the bands in the emission and the TA spectra. Moreover, the increased π-donating ability of the R substituent induced more intraligand charge transfer character to the lowest singlet excited states in 3 and 5, and more pyrene-based 3π,π* character in the triplet excited states of these two complexes. Therefore, the triplet excited states lifetimes of 3 and 5 were quite long (> 30 μs), which resulted in high singlet oxygen quantum yields: of 81 and 72%, respectively.
Ir(III) complexes 3 and 5, with their longest wavelength absorption maxima in the visible region, also gave photobiological effects toward both cancer cells and bacteria when activated with visible light (400-700 nm). This activity was diminished with red light in cancer cells, highlighting that the bluer wavelengths of the visible spectrum were most likely responsible for the photocytotoxic effects. Substantial ROS production was confirmed in SK-MEL-28 cells, which could be the source of their photobiological activity toward both cancer cells and microorganisms. Complex 5, with one fewer pyrenyl groups compared to 3, was less dark cytotoxic in SK-MEL-28 cells, leading to a PI >435. However, even 3 yielded a PI of 248.
Confocal microscopy confirmed that the dark cytotoxicity of 3 was higher, evidenced by substantial morphological changes to the cells without a light treatment. The additional pyrenyl group was implicated in this dark cytotoxicity given that cells exposed to 5 in the dark were relatively healthy in appearance. Complex 5 appeared to exhibit photoactivated uptake that could be visualized by its relatively bright luminescence in light-treated cells relative to dark controls. The photoactivated uptake for 5 led to a larger PI. Considering structure-activity correlations, the reduced hydrophobic character of 5 relative to 3 resulted in enhanced water solubility and reduced dark cytotoxicity. Future improvements would shift the absorption farther into the red, although for sterilization applications, this shift is not necessary.
Overall, we have developed a facile method for synthesizing heteroleptic [Ir(phen)2(R-phen)]3+ complexes in 1,2-dichlorobenzene with easy separation and purification of the desired product. The synthesized complexes are air- and photo-stable, and have better water solubility than their analogous monocationic cyclometalating Ir(III) complexes. These complexes possessed long-lived triplet excited states and exhibited very low or no dark toxicity towards SK-MEL-28 cancer cells. However, they showed very high phototoxicity towards both the SK-MEL-28 cancer cells and the bacteria cells upon visible light excitation. The straightforward reaction route and structure-property correlations gained from this work would allow for a rational design and synthesis of second-generation derivatives of these complexes with predetermined properties. With appropriate ligand modifications to shift the ground-state absorption to the red / NIR region, these trischelated Ir(III) complexes could be promising new photosensitizers for photodynamic therapy.
Supplementary Material
ACKNOWLEDGMENTS
W. Sun and S. Kilina acknowledges the financial support from the National Science Foundation (DMR-1411086 and CNS-1229316) for materials synthesis, characterization and computational simulation of the optical spectra. For computational resources and administrative support, authors thank the Center for Computationally Assisted Science and Technology (CCAST) at North Dakota State University. The photobiological research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number R01CA222227. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. S. A. M. also acknowledges financial support for this work from the University of North Carolina at Greensboro, the Natural Sciences and Engineering Council of Canada, the Canadian Institutes of Health Research, the Canadian Foundation for Innovation, the Nova Scotia Research and Innovation Trust, and Acadia University. We are also grateful to Dr. Yanxiong Pan at the North Dakota State University for his help in the dynamic light scattering measurement.
Footnotes
Supporting Information. Synthetic scheme for the substituted phenanthroline ligands; NMR spectra and high-resolution mass spectra (HRMS); comparison of the experimental and calculated UV-vis absorption spectra of complexes 1-6 in CH3CN; the expanded UV-vis absorption spectra of complexes 1-6 in CH3CN at 360-600 nm; natural transition orbitals (NTOs) representing transitions contributing to the major absorption bands of 1-6 in CH3CN; the emission spectra and decay curves at different concentrations for complexes 1 and 2 in butyronitrile at room temperature and at 77 K. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Xu H; Chen R; Sun Q; Lai W; Su Q; Huang W; Liu X Recent Progress in Metal-organic Complexes for Optoelectronic Applications. Chem. Soc. Rev. 2014, 43 (10), 3259–3302. [DOI] [PubMed] [Google Scholar]
- (2).Dixon IM; Collin J-P; Sauvage J-P; Flamigni L; Encinas S; Barigelletti F A Family of Luminescent Coordination Compounds: Iridium(III) Polyimine Complexes. Chem. Soc. Rev. 2000, 29 (6), 385–391. [Google Scholar]
- (3).Lamansky S; Djurovich P; Murphy D; Abdel-Razzaq F; Kwong R; Tsyba I; Bortz M; Mui B; Bau R; Thompson ME Synthesis and Characterization of Phosphorescent Cyclometalated Iridium Complexes. Inorg. Chem. 2001, 40 (7), 1704–1711. [DOI] [PubMed] [Google Scholar]
- (4).Lamansky S; Djurovich P; Murphy D; Abdel-Razzaq F; Lee H-E; Adachi C; Burrows PE; Forrest SR; Thompson ME Highly Phosphorescent Bis-Cyclometalated Iridium Complexes: Synthesis, Photophysical Characterization, and Use in Organic Light Emitting Diodes. J. Am. Chem. Soc. 2001, 123 (18), 4304–4312. [DOI] [PubMed] [Google Scholar]
- (5).Li Z; Cui P; Wang C; Kilina S; Sun W Nonlinear Absorbing Cationic Bipyridyl Iridium(III) Complexes Bearing Cyclometalating Ligands with Different Degrees of π-Conjugation: Synthesis, Photophysics, and Reverse Saturable Absorption. J. Phys. Chem. C 2014, 118 (49), 28764–28775. [Google Scholar]
- (6).Soman S; Manton JC; Inglis JL; Halpin Y; Twamley B; Otten E; Browne WR; De Cola L; Vos JG; Pryce MT New Synthetic Pathways to the Preparation of Near-blue Emitting Heteroleptic Ir(III)N6 Coordinated Compounds with Microsecond Lifetimes. Chem. Commun. 2014, 50 (49), 6461–6463. [DOI] [PubMed] [Google Scholar]
- (7).Zhao Q; Yu M; Shi L; Liu S; Li C; Shi M; Zhou Z; Huang C; Li F Cationic Iridium(III) Complexes with Tunable Emission Color as Phosphorescent Dyes for Live Cell Imaging. Organometallics 2010, 29 (5), 1085–1091. [Google Scholar]
- (8).Zhao J; Wu W; Sun J; Guo S Triplet Photosensitizers: From Molecular Design to Applications. Chem. Soc. Rev. 2013, 42 (12), 5323–5351. [DOI] [PubMed] [Google Scholar]
- (9).Wang L; Cui P; Kilina S; Sun W Toward Broadband Reverse Saturable Absorption: Investigating the Impact of Cyclometalating Ligand π-Conjugation on the Photophysics and Reverse Saturable Absorption of Cationic Heteroleptic Iridium Complexes. J. Phys. Chem. C 2017, 121 (10), 5719–5730. [Google Scholar]
- (10).Nazeeruddin MK; Grätzel M Transition Metal Complexes for Photovoltaic and Light Emitting Applications In Photofunctional Transition Metal Complexes; Yam VWW, Ed.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2007; pp 113–175. [Google Scholar]
- (11).Schulz GL; Holdcroft S Conjugated Polymers Bearing Iridium Complexes for Triplet Photovoltaic Devices. Chem. Mater. 2008, 20 (16), 5351–5355. [Google Scholar]
- (12).Lo KK-W Luminescent Rhenium(I) and Iridium(III) Polypyridine Complexes as Biological Probes, Imaging Reagents, and Photocytotoxic Agents. Acc. Chem. Res. 2015, 48 (12), 2985–2995. [DOI] [PubMed] [Google Scholar]
- (13).Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113 (7), 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Majumdar P; Yuan X; Li S; Le Guennic B; Ma J; Zhang C; Jacquemin D; Zhao J Cyclometalated Ir(iii) Complexes with Styryl-BODIPY Ligands Showing Near IR Absorption/emission: Preparation, Study of Photophysical Properties and Application as Photodynamic/luminescence Imaging Materials. J. Mater. Chem. B. 2014, 2 (19), 2838–2854. [DOI] [PubMed] [Google Scholar]
- (15).He L; Li Y; Tan C-P; Ye R-R; Chen M-H; Cao J-J; Ji L-N; Mao Z-W Cyclometalated Iridium(iii) Complexes as Lysosome-targeted Photodynamic Anticancer and Real-time Tracking Agents. Chem. Sci. 2015, 6 (10), 5409–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wang C; Lystrom L; Yin H; Hetu M; Kilina S; McFarland SA; Sun W Increasing the Triplet Lifetime and Extending the Ground-state Absorption of Biscyclometalated Ir(III) Complexes for Reverse Saturable Absorption and Photodynamic Therapy Applications. Dalton Trans. 2016, 45 (41), 16366–16378. [DOI] [PubMed] [Google Scholar]
- (17).Mari C; Pierroz V; Ferrari S; Gasser G Combination of Ru(II) Complexes and Light: New Frontiers in Cancer Therapy. Chem. Sci. 2015, 6 (5), 2660–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lincoln R; Kohler L; Monro S; Yin H; Stephenson M; Zong R; Chouai A; Dorsey C; Hennigar R; Thummel RP; McFarland SA Exploitation of Long-Lived 3IL Excited States for Metal-Organic Photodynamic Therapy: Verification in a Metastatic Melanoma Model. J. Am. Chem. Soc. 2013, 135 (45), 17161–17175. [DOI] [PubMed] [Google Scholar]
- (19).Yin H; Stephenson M; Gibson J; Sampson E; Shi G; Sainuddin T; Monro S; McFarland SA In Vitro Multiwavelength PDT with 3IL States: Teaching Old Molecules New Tricks. Inorg. Chem. 2014, 53 (9), 4548–4559. [DOI] [PubMed] [Google Scholar]
- (20).Wang L; Yin H; Jabed MA; Hetu M; Wang C; Monro S; Zhu X; Kilina S; McFarland SA; Sun W π-Expansive Heteroleptic Ruthenium(II) Complexes as Reverse Saturable Absorbers and Photosensitizers for Photodynamic Therapy. Inorg. Chem. 2017, 56 (6), 3245–3259. [DOI] [PubMed] [Google Scholar]
- (21).Sun Y; Joyce LE; Dickson NM; Turro C Efficient DNA Photocleavage by [Ru(bpy)2(dppn)]2+ with Visible Light. Chem. Commun. 2010, 46 (14), 2426–2428. [DOI] [PubMed] [Google Scholar]
- (22).Stępień M; Gońka E; Żyła M; Sprutta N Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds: Synthetic Routes, Properties, and Applications. Chem. Rev. 2017, 117 (4), 3479–3716. [DOI] [PubMed] [Google Scholar]
- (23).Pandrala M; Li F; Wallace L; Steel PJ; Moore II B; Autschbach J; Collins JG; Keene FR Iridium(III) Complexes Containing 1,10-Phenanthroline and Derivatives: Synthetic, Stereochemical, and Structural Studies, and Their Antimicrobial Activity. Aust. J. Chem. 2013, 66 (9), 1065–1073. [Google Scholar]
- (24).Baschieri A; Monti F; Matteucci E; Mazzanti A; Barbieri A; Armaroli N; Sambri L A Mesoionic Carbene as Neutral Ligand for Phosphorescent Cationic Ir(III) Complexes. Inorg. Chem. 2016, 55 (16), 7912–7919. [DOI] [PubMed] [Google Scholar]
- (25).Ayala NP; Flynn CM Jr.; Sacksteder L; Demas JN; DeGraff BA Synthesis, Luminescence, and Excited-State Complexes of the Tris(1,10-phenanthroline)- and Bis(terpyridine)iridium(III) Cations. J. Am. Chem. Soc. 1990, 112 (10), 3837–3844. [Google Scholar]
- (26).Krausz E; Higgins J; Riesen H The Apparent “Dual Emitter” Characteristics of [Ir(bpy)x(phen)3-x]3+ for x = 1,2. Inorg. Chem. 1993, 32 (19), 4053–4056. [Google Scholar]
- (27).Pandrala M; Li F; Feterl M; Mulyana Y; Warner JM; Wallace L; Keene FR; Collins JG Chlorido-containing Ruthenium(II) and Iridium(III) Complexes as Antimicrobial Agents. Dalton Trans. 2013, 42 (13), 4686–4694. [DOI] [PubMed] [Google Scholar]
- (28).Stimpson S; Jenkinson DR; Sadler A; Latham M; Wragg DA; Meijer AJHM; Thomas JA Tuning the Excited State of Water-Soluble IrIII-Based DNA Intercalators that are Isostructural with [RuII(NN)2(dppz)] Light-Switch Complexes. Angew. Chem. Int. Ed. 2015, 54 (10), 3000–3003. [DOI] [PubMed] [Google Scholar]
- (29).Nishikawa Y; Yamamoto H Iron-Catalyzed Asymmetric Epoxidation of β,β-Disubstituted Enones. J. Am. Chem. Soc. 2011, 133 (22), 8432–8435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tzalis D; Tor Y Tuning the Electronic Properties of Phenanthroline Ligands: 3,8-Bis(arylethynyl)-1,10-phenanthrolines and Their Ru(II) Complexes. Tetrahedron Lett. 1995, 36 (34), 6017–6020. [Google Scholar]
- (31).Takahashi A; Hirose Y; Kusama H; Iwasawa N Chelation-assisted Electrocyclic Reactions of 3-Alkenyl-2,2’-bipyridines: An Efficient Method for the Synthesis of 5,6-Dihydro-1,10-phenanthroline and 1,10-Phenanthroline Derivatives. Chem. Commun. 2008, (5), 609–611. [DOI] [PubMed] [Google Scholar]
- (32).Ji S; Wu W; Wu W; Song P; Han K; Wang Z; Liu S; Guo H; Zhao J Tuning the Luminescence Lifetimes of Ruthenium(II) Polypyridine Complexes and Its Application in Luminescent Oxygen Sensing. J. Mater. Chem. 2010, 20 (10), 1953–1963. [Google Scholar]
- (33).Suzuki K; Kobayashi A; Kaneko S; Takehira K; Yoshihara T; Ishida H; Shiina Y; Oishi S; Tobita S Reevaluation of Absolute Luminescence Quantum Yields of Standard Solutions Using a Spectrometer with an Integrating Sphere and a Back-Thinned CCD Detector. Phys. Chem. Chem. Phys. 2009, 11 (42), 9850–9860. [DOI] [PubMed] [Google Scholar]
- (34).Melhuish WH Quantum Efficiencies of Fluorescence of Organic Substances: Effect of Solvent and Concentration of the Fluorescent Solute. J. Phys. Chem. 1961, 65 (2), 229–235. [Google Scholar]
- (35).Carmichael I; Hug GL Triplet-Triplet Absorption Spectra of Organic Molecules in Condensed Phases. J. Phys. Chem. Ref. Data. 1986, 15 (1), 1–250. [Google Scholar]
- (36).Kumar CV; Qin L; Das PK Aromatic Thioketone Triplets and Their Quenching Behaviour Towards Oxygen and Di-t-butylnitroxy Radical. A Laser-Flash-Photolysis Study. J. Chem. Soc., Faraday Trans. 2 1984, 80 (7), 783–793. [Google Scholar]
- (37).Firey PA; Ford WE; Sounik JR; Kenney ME; Rodgers MAJ Silicon Naphthalocyanine Triplet State and Oxygen. A Reversible Energy-Transfer Reaction. J. Am. Chem. Soc. 1988, 110 (23), 7626–7630. [Google Scholar]
- (38).DeRosa MC; Crutchley RJ Photosensitized Singlet Oxygen and Its Applications. Coord. Chem. Rev. 2002, 233–234, 351-371. [Google Scholar]
- (39).van Lenthe E; Baerends EJ; Snijders JG Relativistic Total Energy using Regular Approximations. J. Chem. Phys. 1994, 101 (11), 9783–9792. [Google Scholar]
- (40).Casida ME Time-Dependent Density Functional Response Theory for Molecules. In Recent Advances in Density Functional Methods. 1995, pp 155–192. [Google Scholar]
- (41).Davidson ER The Iterative Calculation of a Few of the Lowest Eigenvalues and Corresponding Eigenvectors of Large Real-Symmetric Matrices. J. Comput. Phys. 1975, 17 (1), 87–94. [Google Scholar]
- (42).Bauernschmitt R; Ahlrichs R Treatment of Electronic Excitations within the Adiabatic Approximation of Time Dependent Density Functional Theory. Chem. Phys. Lett. 1996, 256 (4), 454–464. [Google Scholar]
- (43).Künne LD Recent Developments and Applications of Modern Density Functional Theory. Z. Phys. Chem. 1998, 204, 263–264. [Google Scholar]
- (44).Van Caillie C; Amos RD Geometric Derivatives of Excitation Energies Using SCF and DFT. Chem. Phys. Lett. 1999, 308 (3-4), 249–255. [Google Scholar]
- (45).Chang C; Pelissier M; Durand P Regular Two-Component Pauli-like Effective Hamiltonians in Dirac Theory. Phys. Scr. 1986, 34 (5), 394–404. [Google Scholar]
- (46).Faas S; Snijders J; Van Lenthe J; Van Lenthe E; Baerends E The ZORA Formalism Applied to the Dirac-Fock Equation. Chem. Phys. Lett. 1995, 246 (6), 632–640. [Google Scholar]
- (47).Nichols P; Govind N; Bylaska EJ; De Jong WA Gaussian Basis Set and Planewave Relativistic Spin− Orbit Methods in NWChem. J. Chem. Theor. Comput. 2009, 5 (3), 491–499. [DOI] [PubMed] [Google Scholar]
- (48).Valiev M; Bylaska EJ; Govind N; Kowalski K; Straatsma TP; Van Dam HJ; Wang D; Nieplocha J; Apra E; Windus TL NWChem: a Comprehensive and Scalable Open-Source Solution for Large Scale Molecular Simulations. Comput. Phys. Commun. 2010, 181 (9), 1477–1489. [Google Scholar]
- (49).Perdew JP; Burke K; Ernzerhof M Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77 (18), 3865–3868. [DOI] [PubMed] [Google Scholar]
- (50).Hay PJ; Wadt WR Ab initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82 (1), 270–283. [Google Scholar]
- (51).Wadt WR; Hay PJ Ab initio Effective Core Potentials for Molecular Calculations. Potentials for Main Group Elements Na to Bi. J. Chem. Phys. 1985, 82 (1), 284–298. [Google Scholar]
- (52).Hay PJ; Wadt WR, Ab initio Effective Core Potentials for Molecular Calculations. Potentials for K To Au Including The Outermost Core Orbitals. J. Chem. Phys. 1985, 82 (1), 299–310. [Google Scholar]
- (53).Rassolov VA; Ratner MA; Pople JA; Redfern PC; Curtiss LA 6‐31G* Basis Set for Third‐Row Atoms. J. Comput. Chem. 2001, 22 (9), 976–984. [Google Scholar]
- (54).Barone V; Cossi M Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102 (11), 1995–2001. [Google Scholar]
- (55).Cossi M; Rega N; Scalmani G; Barone V Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comput. Chem. 2003, 24 (6), 669–681. [DOI] [PubMed] [Google Scholar]
- (56).Klamt A; Schuurmann G COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. J. Chem. Soc., Perkin Trans. 2 1993, (5), 799–805. [Google Scholar]
- (57).York DM; Karplus M A Smooth Solvation Potential Based on the Conductor-Like Screening Model. J. Phys. Chem. A 1999, 103 (50), 11060–11079. [Google Scholar]
- (58).Martin RL Natural Transition Orbitals. J. Chem. Phys. 2003, 118 (11), 4775–4777. [Google Scholar]
- (59).Zhurko G, and Zhurko D ChemCraft, version 1.6, Build. 2009. [Google Scholar]
- (60).Sullivan BP; Meyer TJ Synthesis of Hydrido-iridium(III) Complexes from a Trifluoromethanesulphonato Intermediate. J. Chem. Soc., Chem. Commun. 1984, (7), 403–405. [Google Scholar]
- (61).Nakamaru K Synthesis, Luminescence Quantum Yields, and Lifetimes of Trischelated Ruthenium(II) Mixed-ligand Complexes Including 3,3′-Dimethyl-2,2′-bipyridyl. Bull. Chem. Soc. Jpn. 1982, 55 (9), 2697–2705. [Google Scholar]
- (62).Hong Y; Lam JWY; Tang BZ Aggregation-Induced Emission. Chem. Soc. Rev. 2011, 40 (11), 5361–5388. [DOI] [PubMed] [Google Scholar]
- (63).García B; Alcalde R; Aparicio S; Leal JM The N-Methylpyrrolidone-(C1-C10) Alkan-1-ols Solvent Systems. Phys. Chem. Chem. Phys. 2002, 4 (7), 1170–1177. [Google Scholar]
- (64).Reichardt C Solvents and Solvent Effects in Organic Chemistry. 3rd ed., Wiley-VCH, Weinheim, Germany, 2003, pp. 419–420. [Google Scholar]
- (65).Wang L; Cui P; Liu B; Kilina S; Sun W Novel N6 Trisbidentate Ligand Coordinated Ir(III) Complexes and Their Ru(II) Analogs. Dalton Trans. 2018, 47 (39), 13776–13780. [DOI] [PubMed] [Google Scholar]
- (66).Ford WE; Rodgers MAJ Reversible Triplet-Triplet Energy Transfer within a Covalently Linked Bichromophoric Molecule. J. Phys. Chem. 1992, 96 (7), 2917–2920. [Google Scholar]
- (67).Ogilby PR; Foote CS Chemistry of Singlet Oxygen. 42. Effect of Solvent, Solvent Isotopic Substitution, and Temperature on the Lifetime of Singlet Molecular Oxygen (1Δg). J. Am. Chem. Soc. 1983, 105 (11), 3423–3430. [Google Scholar]
- (68).O’Brien J; Wilson I; Orton T; Pognan F Investigation of the Alamar Blue (resazurin) Fluorescent Dye for the Assessment of Mammalian Cell Cytotoxicity. Eur. J. Biochem. 2000, 267 (17), 5421–5426. [DOI] [PubMed] [Google Scholar]
- (69).Stephenson M; Reichardt C; Pinto M; Wächtler M; Sainuddin T; Shi G; Yin H; Monro S; Sampson E; Dietzek B; McFarland SA Ru(II) Dyads Derived from 2-(1-Pyrenyl)-1H-imidazo[4,5-f][1,10]phenanthroline: Versatile Photosensitizers for Photodynamic Applications. J. Phys. Chem. A 2014, 118 (45), 10507–10521. [DOI] [PubMed] [Google Scholar]
- (70).Daghastanli NA; Itri R; Baptista MS Singlet Oxygen Reacts with 2′,7′-Dichlorodihydrofluorescein and Contributes to the Formation of 2′,7′-Dichlorofluorescein. Photochem. Photobiol. 2008, 84 (5), 1238–1243. [DOI] [PubMed] [Google Scholar]
- (71).Rosenkranz AR; Schmaldienst S; Stuhlmeier KM; Chen W; Knapp W; Zlabinger GJ A Microplate Assay for the Detection of Oxidative Products Using 2′,7′-Dichlorofluorescin-diacetate. J. Immunol. Methods 1992, 156 (1), 39–45. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.