

Abstract
Tuberculosis (TB) remains a leading cause of death globally. Dissemination of tuberculosis to the brain results in the most severe form of extra-pulmonary TB, tuberculous meningitis (TBM), which represents a medical emergency associated with high rates of mortality and disability. Via various mechanisms the Mycobacterium tuberculosis (M.tb) bacillus disseminates from the primary site of infection and overcomes protective barriers to enter the central nervous system. There it induces an inflammatory response involving both the peripheral and resident immune cells, which initiates a cascade of pathological mechanisms that may either contain the disease or result in significant brain injury. Here we review the steps from primary infection to cerebral disease, factors which contribute to the virulence of the organism and the vulnerability of the host and discuss the immune response and the clinical manifestations arising. Priorities for future research directions are suggested.
Summary sentence
Much of the morbidity and mortality caused by tuberculous meningitis is mediated by a dysregulated immune response
Graphical Abstract
INTRODUCTION
Tuberculous Meningitis (TBM) is the most serious extrapulmonary manifestation of tuberculosis with mortality rates up to 50% observed in the context of HIV-1 co-infection[1]. The pathogenesis of TBM is incompletely understood and further understanding is required in order to develop effective vaccines, optimal antibiotic and host directed therapies. We present an overview of the pathogenesis of TBM following the pathogen from site of primary infection to dissemination to and within the brain. We have included observations from important studies that have contributed to understanding pathogenic mechanisms in TBM. We also highlight areas important for future research.
FROM PRIMARY INFECTION TO THE CENTRAL NERVOUS SYSTEM
The systemic immune response to tuberculous infection
Tuberculosis (TB) infection occurs through the inhalation of infectious droplets of aerosolised Mycobacterium tuberculosis (M.tb), which cross the lung epithelium and infect lung alveolar macrophages, neutrophils and dendritic cells (DCs). This leads to the secretion of antimicrobial peptides, cytokines (including interleukin-1 alpha and beta, tumour necrosis factor alpha (TNF-α), interleukin (IL)-6 and −12, chemokines, lipoxins that may stimulate necrosis and contribute to immune protection, and prostaglandins that may induce apoptosis[2]. Under the influence of IL-12 and chemokines CCL-19 and −21, infected DCs migrate to the local draining lymph nodes to stimulate the differentiation of T-helper I (Th1) cells. Th1 cell release of interferon gamma (IFN-γ) and TNF-α at the site of infection activates macrophages and DCs to produce cytokines and antimicrobial factors that contribute to containment of the TB bacillus[2]. This inflammatory process results in the formation of a granuloma, which encapsulates the infected cells and retards the replication of TB bacilli and can lead to latent infection. However, in the elderly, immune compromised or very young, the ongoing immune response may progress to active primary TB disease associated with tissue destruction in the lung and dissemination of the TB bacillus to other organ systems[3, 4].
Dissemination to the brain
Dissemination of TB involves seeding of M.tb to other sites including the central nervous system (CNS). Various mechanisms by which the bacilli migrate into the lymphatic system or blood stream have been suggested[5–7]. Bacterial proteins early secretory antigenic target 6kDa (ESAT-6) and culture filtrate protein 10kDA (CFP-10) are involved in cell lysis, while heparin binding haemagglutinin adhesion (HBHA) aids M.tb translocation across the epithelium without lysis[5]. M.tb may also invade and traverse vascular endothelial cells [6], replicate in lymphatic endothelial cells [8], and be trafficked to distant locations in phagocytes[5]. Furthermore, mycobacteria are able to survive and replicate in infected macrophages and lymphatic endothelial cells (LEC) surrounding granulomas in the lymph nodes. Research on LECs demonstrates that although M.tb bacilli are initially phagocytosed upon infecting the cell, through their genetic locus termed the region of difference 1 (RD1), the bacilli are able to escape from the phagosomes into the cytosol, where they more readily replicate. For those bacteria remaining in the phagosome they are able to prevent fusion of lysosomes with the phagosome, also in an RD1 dependent manner, thereby allowing bacterial replication in the phagosome and contributing to lymphatic tuberculosis[8]. The activation of LEC by IFN-γ is key to restricting these RD1 mechanisms of replication[8]. Additionally, host immunity and M.tb strain variation may play a role; polymorphisms in the genes encoding for antigen recognition and macrophage activation[9] or impaired pro-inflammatory cytokine release may influence the ability of the initial innate response to control infection[5]. TB strains assigned greater virulence, like the lineage 2 strain, which is postulated to subvert the innate immune response, promoting its survival and replication and thereby more severe disease[10–12].
The CNS is protected from influx of potentially harmful blood-borne bacteria by 2 vascular barriers; the blood-brain barrier (BBB) and the blood–cerebrospinal fluid barrier (BCSFB)[13] (Figure 1). The BBB is mainly formed by brain microvascular endothelial cells that are characterised by intercellular tight junctions and a paucity of endocytotic vesicles and fenestrae, and exhibit dedicated transport mechanisms to regulate influx and efflux across the CNS and blood compartments[13, 14]. Pericytes, embedded within a basement membrane, and astrocytes’ end-feet support the endothelial cells and also make an indispensable contribution to BBB integrity. In contrast, the BCSFB is composed of choroid plexus epithelial cells joined together by tight junctions and the arachnoid membrane. However, despite these protective mechanisms, M.tb bacilli migrate across these barriers. In vitro and animal models demonstrate that M.tb invades and traverses brain endothelial cells in the microvasculature through rearrangement of their actin[6, 14]. Further, the M.tb gene Rv0931c (pknD) has been identified as a potential virulence factor promoting CNS infection in certain TB strains as it enables the bacilli to interact with extracellular factors on the brain endothelium facilitating bacillary endothelial adhesion[14]. Another potential route of entry is the ‘Trojan horse’ mechanism by which M.tb are trafficked in infected macrophages and neutrophils across the BBB[7].
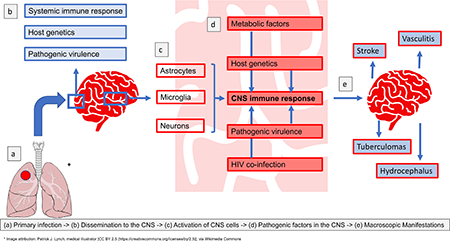
Figure 1: Summary of the pathogenesis of TBM.

A: Mycobacterium tuberculosis bacilli (M.tb) disseminate from the primary site of infection in the lung to seed the brain. The bacilli are able to traverse the blood brain barrier (BBB) and blood cerebrospinal fluid barrier (BCSFB) through various virulence factors that enable the invasion of and migration through cerebral vascular endothelial cells, or are carried into the central nervous system (CNS) by infected peripheral innate immune cells. B: In the CNS antigen recognition and internalisation by microglia, neurons and astrocytes occurs, mediated by numerous host genetic factors. C: The resulting immune response stimulates the release of pro-inflammatory cytokines and chemokines and other immune mediators that contribute to the breakdown of the BBB and the influx of innate and adaptive immune cells from the periphery. D: A prolific inflammatory response ensues. The inflammatory exudate in the basal cisterns contributes to cerebral vascular pathology and the development of hydrocephalus and raised intracranial pressure. Vasogenic oedema due to an influx of proteins through the leaky BBB, and cytotoxic oedema as a result of cellular damage contribute to the raised pressure. The overall decrease in cerebral blood flow puts the brain at risk of ischaemia, infarction and poor patient outcomes. In some cases the infection is controlled in discrete tuberculomas or abscesses, which resolve with treatment and time. Extension of the disease into the spinal canal manifests as spinal arachnoiditis, tuberculomas, or collection of exudate.
Once the TB bacilli gain access to the brain, limited local innate immunity allows their survival and replication and the development of silent tuberculous lesions. Based on postmortem studies, Rich and McCordock suggested that TBM is initiated by the rupture of one of these lesions, the Rich focus, located under the cortical pia or adjacent to the meninges or ventricles, which releases M.tb bacilli into the sub-arachnoid space causing a granulomatous infection of the meninges[15–18] and the subsequent induction of inflammation. Recently the relationship between the Rich focus and the onset of miliary tuberculosis has been reviewed. Rich and McCordock did not acknowledge a role of miliary tuberculosis in the pathogenesis of TBM, however, several studies since have suggested that the bacteraemia seen in these cases increased the likelihood that a meningeal of sub-cortical focus is established with subsequent rupture giving rise to TBM (reviewed in [16]).
PATHOGENIC AND PATHOPHYSIOLOGICAL MECHANISMS WITHIN THE BRAIN (Figure 1)
The host immune response to tuberculosis in the brain
Microglia within the cerebral parenchyma are the principal CNS cells infected by M.tb [19, 20] and are involved in immune regulation. Other CNS cells that have potential roles in this process are astrocytes and neurons[20, 21] (Figure 1). Randall et al have demonstrated direct infection of neurons with M.tb, however, the effect on neuronal function and implications for intercellular interactions is not clear[21]. Although not as prominent as microglia in their role, astrocytes have also been implicated with a study by Rock and colleagues demonstrating 15% of astrocytes having cell associated bacilli (average of 1.3 bacilli per cell) compared to 76% of microglia (average of 4.2 bacilli/cell) under the same conditions[20].
M.tb is recognized by microglial cells via innate immune and neuro-specific receptors, including pattern recognition receptors. The toll-like receptors (TLRs), a family of ten pattern recognition molecules, play a crucial role in innate immunity. Internalisation of M.tb by human microglia is dependent on CD14, a monocyte differentiation antigen, which binds to lipopolysaccharide with TLR-4[22]. This was demonstrated in a study in which uptake of non-opsonized tubercle bacilli by microglia was reduced by 64% and 62% in the presence of anti-CD14 monoclonal antibodies and soluble CD14 ligand respectively. This is in contrast to peripheral mononuclear phagocytes where CD14 neutralising anti-CD14 antibodies did not affect bacillary uptake[23], but interestingly the presence of CD14 led to upregulation of CD14 expression on these cells perhaps facilitating pathogenic immune responses. This receptor, along with the β2-integrin CD18 and TNF-α, is also involved in the formation of histologically characteristic multinucleated giant cells seen at autopsy and experimentally identified in porcine microglia infected with Mycobacterium bovis[24].
Activation of microglia leads to secretion of a number of cytokines (Figure 1). In murine microglia the intracellular signalling pathways leading to cytokine release are M.tb inducible leading to a pro-inflammatory response through NAPDH oxidase-dependant reactive oxygen species (ROS) generation[25]. Although cytokines play a critical role in the host defence to infection with M.tb, they can also mediate inflammation. Tumour Necrosis Factor (TNF) is central to the pathogenesis of central nervous system tuberculosis[26–28]. It has a protective role in the immune response to mycobacteria[29]; but is also associated with pathology in vivo via induction of fever, activation of the hypothalamo-adrenal axis and by triggering the release of other cytokines[30–32]. Local TNF-α production in the CNS also increases permeability of the BBB and thus influx of other immune mediators to the CNS[31]. In a murine CNS TB model, Tsenova et al demonstrated a correlation between levels of TNF-α and the extent of cerebral pathology as measured by CSF leukocytosis, protein accumulation, meningeal inflammation, persistence of bacillary load and clinical deterioration[28]. TNF-α antagonists such as thalidomide[33] and analogues thereof[34], used in rabbit models of TBM, downregulated the production of TNF-α and subsequently improved survival. This finding was not replicated in human studies where a clinical trial of thalidomide used in conjunction with standard antituberculous therapy and corticosteroids in children with TBM was stopped early due to adverse events associated with thalidomide use[35]. Since then there has been some suggestion of benefit with the use of thalidomide in the context of tuberculous mass lesions [36].
CSF concentrations of IL-6 independently associate with more severe presentation of TBM[37]. In this context it is unclear whether this is due to its pro-inflammatory or anti-inflammatory effects. In murine models of TB IL-6 has been implicated in the stimulation of IFN-γ production but not necessarily essential for the protective immunity to M.tb[38]. It may also have an anti-inflammatory role by suppressing gene expression of pro-inflammatory cytokines[39]. In a study by Rock et al, pro-inflammatory cytokines other than TNF-α, IFN-γ and IL-6 found to be secreted by microglia in response to TBM included IL-1β, CCL2, CCL5 and CXCL-10. In contrast to microglia, astrocytes produced only moderate amounts of CXCL10[20]. Other cytokines, confirmed experimentally, to be secreted by microglia following M.tb stimulation include: IL-1α, IL-10, IL-12p40, G-CSF, and GM-CSF[25, 40].
Recent focus has turned to the pathogenic role of inflammatory mediators such as DAMPs (Damage Associated Molecular Patterns) and PAMPs (Pathogen Associated Molecular Patterns)[41], their validity as biomarkers of cerebral injury[42], and as potential targets for novel host directed therapies in TBM[43]. PAMPs are by-products released from pathogens that are recognised by host cell receptors subsequently leading to activation of the innate immune response. DAMPs, which are released by damaged host immune cells, interact with PAMPs leading to an accelerated cycle of cell death and injury. Host poly(ADP-ribose) polymerase 1 (PARP1; also known as ARTD1) is an ADP-ribosylating enzyme essential for initiating various forms of DNA repair[44]. Recent studies have suggested a role for PARP1 in the pathogenesis of TBM via its potential to modulate the release and activation of DAMPs. This includes high mobility group box-1 (HMGB1), a non-histone nuclear DNA binding protein expressed in all mammalian cells, and S100B, a protein synthesised by astrocytes, oligodendrocytes and Schwann cells known to be involved in cell-to-cell communication, cell growth, and intracellular signal transduction, as well as the development and maintenance of the CNS[45]. These biomarkers of cerebral injury are known to increase in TBM [58, [42], and therefore suggest that PARP1 may be a potential new target in the development of host directed therapies [46]. S100A8/9, also from the S100 family has a role in neutrophil chemoattraction and stimulation [47] and is implicated in the pathogenesis of tuberculosis in pulmonary disease. In HIV-1 uninfected patients raised S100A8/9 in serum correlated with increased radiographic disease severity[48, 49]. In murine models IL-17 induced S100A8/9 was a key factor in neutrophil accumulation and led to exacerbated lung inflammation due to increased levels of pro-inflammatory cytokines[50]. In TBM Marais et al demonstrated that in patients with TBM and HIV infection, levels of S100A8/9 were significantly elevated two weeks after the initiation of antiretroviral therapy in those who developed Immune Reconstitution Inflammatory Syndrome (IRIS) defined as a paradoxical worsening of infection despite adequate treatment following the initiation of antiretroviral drugs, compared to those who did not. This observation may explain in part the ongoing paradoxical inflammation observed in IRIS discussed in more detail later in this review[51].
In addition to the inflammatory milieu described above, there are several other factors implicated in the pathogenesis of TBM, in particular the subsequent increasing permeability of the BBB and influx of inflammatory mediators and cells (Figure 1). Vascular Endothelial Growth factor (VEGF) is a potent endothelial growth factor playing diverse roles in vasculogenesis and angiogenesis. In tuberculosis it is now considered a useful biomarker of disease, where it may be used as an indicator of active vs latent disease activity or a marker of extrapulmonary vs primary lung disease. In several types of cancer, VEGF inhibitors such as Bevacizumab are well established as an effective therapeutic approach. In ischaemic conditions of the brain, VEGF has a pathological and protective role depending on pathogenic stage due to either effect on permeability of the microvasculature or subsequent reparative angiogenesis. In age related neurological conditions such as Alzheimer’s disease, Parkinson’s disease and motor neuron disease however, VEGF is thought to be pathogenic due to its effect on BBB dysfunction[52]. In these conditions the effect of VEGF on increasing endothelial permeability is clear, however the mechanism by which this happens remains less understood with possibilities including its effect on cell-cell junctions including tight junctions and adherens junctions as well as on transcytosis[53]. In TBM VEGF disrupts the permeability of the BBB[54, 55], which has been proposed as a mechanism by which dexamethasone exerts efficacy as a host directed therapy in TBM. Also interesting is its neuroprotective effects which have been more thoroughly explored in the context of amyotrophic lateral sclerosis where low VEGF has been reported with BBB dysfunction and the therapeutic use of a VEGF analogue (VEGF-A165) is currently under investigation in a clinical trial (NCT02269436). The release of intercellular and vascular adhesion molecules (ICAMs and VCAMs) as well as matric metalloproteinases (MMPs) from inflammatory cells within the CNS have also been shown to increase the permeability of the BBB[56].
Metabolic factors in the host
Neuroendocrine associated metabolic abnormalities are common in TBM. In an observational study of patients newly diagnosed with TBM, neuroendocrine dysfunction occurred in half[57]. This is likely due to the tendency for TBM to affect the basal structures such as the interpeduncular fossa, cisterna ambiens and cisterna pontis, which surround the pituitary gland, pituitary stalk and hypothalamus (Figure 4). Exudates here lead to oedema, perivascular infiltration and subsequent microglial reactions known collectively as ‘borderzone encephalitis’. Subsequent metabolic abnormalities include gonadotropin deficiency, hyperprolactinemia, thyrotropin deficiency, corticotropin deficiency and somatotropic hormone deficiency[57]. Hyponatraemia is also common, although the mechanism by which is occurs is debated. Cerebral salt wasting (CSW) and a syndrome of inappropriate anti-diuretic hormone (SIADH) are possible explanations. In a prospective study of 76 TBM patients conducted in India, 34 patients had hyponatraemia, which was due to CSW in 17, SIADH in 3 and miscellaneous causes in 14 [58]. Although the mechanism of hyponatraemia is unclear, its role in the pathophysiology TBM is important as it independently associates with poor outcome[59, 60], and also more recently as a possible predictor for stroke in the context of CSW[61]. Corticotropin deficiency may be modulated by treatment with dexamethasone, which at present remains the only host directed therapy of proven benefit in TBM. Pituitary dysfunction and its role in the pathogenesis of TBM remains an area of interest, particularly given the observations of worsening outcomes and possible benefit of cortisol replacement.
Figure 4: Basal cisterns and pituitary anatomy.

A: Basal cisterns affected in TBM are represented here in a sagittal view of the brain. Note the quadrigeminal cistern which extends laterally to become a thin sheet like cistern surrounding the midbrain and posterior thalamus, named the ambient cistern shown in C. B: Anatomy of the pituitary gland and surrounding structures.
Glucose and its metabolic effects are also of interest. In a pivotal study where adjunctive dexamethasone was found to decrease short term mortality in HIV-1 uninfected patients with TBM, low glucose at presentation predicted poor outcome[62]. In more recent studies, including those in children, CSF glucose, lactate and protein levels have been linked to poor outcome[37, 63]. If these metabolic markers are simply markers of disease activity as in most forms of neurological infection, then this finding is unsurprising. Mason et al demonstrated that the increase in lactate levels commonly observed in the CSF of patients who go on to develop poor outcomes is of the L-form and therefore solely a response from the host to the infection, rather than being of microbial origin[64]. This finding contributes to this groups’ hypothesis that in the context of neuroinflammatory-inducing infection, energy flow in brain metabolism is shifted away from the neurons and shunted towards the microglia. They theorise that this leads to lactic acid production by glycolysis in astrocytes, which subsequently participates in the activated immune response by contributing to oxidative phosphorylation and hence production of high levels of adenosine triphosphate (ATP) and forms of ROS required for degradation of the invading pathogen.
The metabolism of tryptophan is also of interest in TBM. Tryptophan is a key amino acid required for protein biosynthesis, and a precursor for various metabolites, including serotonin and melatonin (serotonin pathway) and kynurenine and quinolinic acid (kynurenine pathway) [65] (Figure 2). Pro-inflammatory cytokines such as IL-6, TNF-α and IFN-γ are known to trigger the kynurenine pathway by stimulating indoleamine 2, 3-dioxygenase (IDO) [66, 67]. Once IDO is activated, the kynurenine pathway is promoted at the expense of the serotonin pathway. Microglia and astrocytes then secrete quinolinic acid and kynurenic acid, respectively. Both products have opposite roles, while kynurenic acid acts as an antagonist of the glutamate N-methyl-D-aspartate receptor (NMDAr), quinolinic acid acts as its agonist and leads to neurotoxicity [66]. Activation of the kynurenine pathway is also known to exacerbate progression of neurodegenerative diseases and has been described in HIV associated dementia [68].
Figure 2: Mycobacterium tuberculosis and vitamin D impact on the tryptophan metabolism.

M.tb infection activates the inflammatory response resulting in pro-inflammatory cytokines secretion. Along with M.tb, these cytokines can trigger the kynurenine pathway by stimulating IDO. Once the pathway is activated, astrocytes and microglia respectively produce kynurenic acid and quinolinic acid, the latter is neurotoxic. Thus, an imbalance in the synthesis of these two products may result in neurotoxicity. Vitamin D, cofactor of TPH, promotes serotonin production and neurotransmission. Alone or when bound with one of its receptor, VDR, vitamin D attenuates the inflammatory response. M.tb: Mycobacterium tuberculosis; IDO: indoleamine 2, 3-dioxygenase; TPH: tryptophan hydrolase; KAT: kynurenine aminotransferase; KMO: kynurenine-3-monooxygenase; 3-HAO: 3-hydroxyanthranilic acid dioxygenase; NMDA: N-methyl-d-aspartate; VDR: vitamin D receptor. Blue: neuroprotection; red: neurotoxicity; green: influential factors.
Recently, Van Laarhoven et al conducted a metabolomic study examining serum and CSF of TBM patients. Their results showed that while most metabolites demonstrated elevated concentrations in TBM patients compared to controls, concentrations of tryptophan were low in TBM and further reduced in patients who survived. They further demonstrated upregulation of the gene IDO1 and identified 11 trait loci that correlated with tryptophan concentrations and were prognostic of survival when combined with sex and age. The prognostic potential of these genetic correlates was demonstrated in a validation cohort. These data suggest that tryptophan metabolism may play an important role in TBM outcome, and that further investigation into this metabolic pathway is warranted[69].
While M.tb may directly stimulate of IDO at the site of infection[70], vitamin D on the other hand is an essential cofactor of the tryptophan hydrolase (TPH) and promotes serotonin production and thus neurotransmission. Furthermore, Vitamin D alone or bound to the vitamin D receptor (VDR) expressed on various cell types, including astrocytes and microglia, leads to a decrease in inflammatory response [71]. Thus tryptophan metabolism could be a modality by which the vitamin D status of an individual modulates both susceptibility to M.tb infection and TBM pathogenesis. By restoring a non-inflammatory environment and restricting M.tb replication, vitamin D could promote the serotonin pathway at the expense of the kynurenine one and protect the brain from neurotoxicity.
Host genetic factors
A number of immune response genes encoding the pathways described are under genetic influence. For instance, mutations and polymorphisms within the genes involved in the TLR pathway have been associated with susceptibility to infection in tuberculosis. A recent study of patients with TBM in Vietnam was the first to demonstrate an association between single nucleotide polymorphisms (SNPs) in the TLR9 gene region and TBM[72]. Further, a Vietnamese study found an association between TLR2 SNP T597C and the development of TBM and millary TB, suggesting that TLR2 influences the dissemination of M.tb[73]. Toll Interleukin 1 receptor domain containing adaptor protein (TIRAP) mediates signals from TLR1, −2, −4 and −6 to activate macrophages and dendritic cells. Hawn et al investigated the association of the TIRAP SNP C558T with i) susceptibility to TB (odds ratio, 2.25, p < 0.001) and ii) susceptibility to meningeal TB (OR, 3.02; p < 0.001) vs pulmonary TB (OR, 1.55; p = 0.22). They also demonstrated that compared to the to the 558CC genotype, the 558TT genotype was associated with decreased whole-blood interleukin-6 production, suggesting that TIRAP influences disease susceptibility by modulating the inflammatory response [9].
Another gene of interest is leukotriene A4 hydrolase (LTA4H). LTA4H influences the balance of pro- and anti- inflammatory eicosanoids and subsequent TNF-α regulation through either reduced inflammation due to excess lipoxins or augmented inflammation due to excess leukotriene B4. In a study of zebrafish and humans, mutations in the gene encoding LTA4H led to immunosuppressive Lipoxin A4 (LXA4) accumulation and increased susceptibility to mycobacteria[27]. In this study, heterozygosity at several LTA4H SNPs was associated with protection against meningeal tuberculosis (TBM) [27]. These findings supported a hypothesis that excess LTA4H activity leads to a ‘hyperinflammatory state’, whereas lack of LTA4H activity leads to an inadequate host response[74]. In a later study by the same group the LTA4H promoter region SNP rs17525495, defining 3 genotypes—TT, CT, and CC— was identified as a likely molecular determinant of genetic susceptibility. In this study lymphoblastoid cell lines with SNP genotype CC conferred a hypoinflammatory and TT a hyperinflammatory phenotype[27]. It was also demonstrated that i) genotype correlated with pre-treatment CSF leukocytosis and survival and ii) those benefiting from adjunctive dexamethasone were carriers of the ‘hyperinflammatory’ TT genotype[27]. Several follow-up studies to further investigate the relevance of this finding have tended to reproduce an association with susceptibility to disease, but not necessarily outcome [75–78]. The two largest of these both published in 2017 investigated the association in Vietnamese [78] and Indonesian [77] populations. The first showed that individuals with the LTA4H CC genotype had a higher risk of early death, whereas the second did not find an association between genotype and mortality. Possible explanations may include differences in linkage disequilibrium as well as the observed overall differences in patient characteristics such as mortality (40.7% in Indonesia vs 18.9% in Vietnam), frequency of culture confirmed diagnosis (55.3% vs 42.8% in Vietnam), severity of disease (BMC grade I severity in Vietnam 37% vs 11% in Indonesia) and median age of patients (41 years vs 29 years in Vietrnam) (reviewed in [79]). Nonetheless this remains an area of great interest and a clinical trial is under way to determine whether LTA4H genotype, defined at randomisation, influences dexamethasone’s clinical efficacy when added to the first 6–8 weeks of anti-tuberculosis treatment of TBM (NCT03100786).
There are a number of other polymorphisms documented in the literature to relate to the pathogenesis of TBM. Polymorphisms in CD43 encoding a surface glycoprotein involved in M.tb adhesion and pro-inflammatory cytokine induction has been associated with decreased survival from TBM[80]. The mannose binding protein (MBP) binds mycobacteria and acts as an opsonin in vitro. Although MBP plays a role in the first line of defence against M.tb, it may also facilitate the spread of the intracellular pathogen[81]. A hypothesis, was therefore advanced that phenotypes in which levels of MBP are low may result in protection from TBM. In a study to test this, a mutation in the MBP B allele (G54D), which leads to disruption of the collagen region of the MBP protein, was found to be associated with reduced dissemination to the brain and suggested protection against TBM[82]. Polymorphism in the PKP3-SIGIRR-TMEM16J gene region encoding a negative regulator of toll-like receptor/IL-1R signaling has been associated with reduced survival in both pulmonary TB (PTB) and TBM[83]. Lastly, Vitamin D Receptor gene polymorphisms with heterozygous (TC) and mutant (CC) genotypes of Taq1 VDR SNP associate with TBM[84], although further research is required to investigate whether this is universal across all populations[85].
Pathogen virulence factors and their effect on pathogenesis
M.tb was formerly regarded as an organism exhibiting little genetic variation. More recent studies using genetic typing techniques to analyse M.tb isolates from diverse geographic populations have revealed a cladal phylogeographic distribution with variation between different lineages [86, 87]. Seven lineages are now identified classified as “ancient” (lineages 1, 5, 6 and 7) or “modern” (2, 3 and 4)[88]. One lineage of particular interest has been lineage 2, which is highly prevalent in East Asia (and therefore known as ‘Beijing’) and has been ascribed hypervirulence in a rabbit model[89] as well as some human studies demonstrating increased risk of disseminated disease[90]. This has been attributed to an intact polyketide synthase (pks 15/1) gene and the production of a phenolic glycoprotein (PGL), which is thought to attenuate the early host immune response leading to a reduced production of pro-inflammatory cytokines[91]. A later study by Caws et al comparing bacterial and host genotype across two groups of Vietnamese adults with pulmonary or meningeal TB found that disease caused by the Euro-American lineage was significantly more likely in pulmonary disease, however, by contrast found no association between the lineage 2 and disease phenotype[10].
Epidemiological studies have reported several differences in disease phenotype between M.tb lineages in terms of pathogen virulence[92, 93], transmission of disease[87, 94, 95], progression from latent to active disease [96] and in response to treatment [97, 98]. In vitro studies have explored whether the host immune response is specific to genotype. A study in South Africa found differential mycobacterial growth and levels of early pro-inflammatory cytokines including TNF and IL-12p40 between three modern lineages[99]. Others have found difference in human macrophage responses between lineages and have hypothesised that the lack of early pro-inflammatory response observed with modern lineages may contribute to more rapid disease progression and transmission and therefore confers survival advantage for these strains of M.tb[100]. In Madagascar differences between ancient and modern lineages were characterised by contrasting IFN-γ responses[101]. Specifically, comparison of the IFN-γ responses with the spoligotype of the infecting clinical strains showed that modern M.tb strains tended to induce lower IFN-gamma responses than ancient strains in index cases and their household contacts[101]. The aforementioned study by Caws et al was the first to demonstrate an interaction between pathogen and host genetic factors as a predictor of disease phenotype by showing that individuals with the C allele of the TLR02-T597C genotype were more likely to have tuberculosis caused by the Beijing genotype (OR 1.57; 95% CI 1.15 – 2.15)[10]. The most recent study in this field performed whole genome sequencing of M.tb strains from 322 HIV-1 uninfected patients with TBM (n=106) and PTB (n=216). Unlike the previous studies[10] [90] there was no association with disease phenotype and lineage, however using a homoplasy based association analysis they identified three M.tb genes associated with disease phenotype. This included Rv0218, a secretome gene encoding a protein that influences pathogen recognition and host-pathogen interaction. They hypothesise that a SNP in the region of Rv0218 would alter the appearance of the surface of M.tb therefore allowing it to evade recognition and the immune response, enabling dissemination to extrapulmonary sites.
DIFFERENCES IN THE INTRACEREBRAL IMMUNE RESPONSE IN THE HIV-1 INFECTED HOST
There is a relative paucity of data directly comparing the intracerebral immune response to TBM in HIV-1 infected and uninfected hosts. The impact of HIV-1 infection on the clinical presentation of TBM is debated, some studies report that HIV-1 infection does not alter the cerebral features of TBM [102, 103], while others suggest a higher rate of extra-meningeal and extra-pulmonary disease associated with HIV-1 infection [104–106]. These studies consistently report that HIV-1 infection associates with death [102] and decreased efficacy of treatment against M.tb [105]. A better characterisation of the effect of HIV-1 in the pathogenesis of tuberculous meningitis is important.
A study conducted in Vietnam demonstrated a small reduction in IFN-γ yet more significant reduction in the anti-inflammatory cytokine IL-10 in HIV-1 infected patients, the latter being 6-fold decreased in the context of HIV. This skewed the CSF IFN-γ: IL-10 ratio towards excess IFN-γ with subsequent analysis that was independently associated with HIV co-infection (p=−.009, odds ratio (95% CI), 2.47 (1.25 – 4.88)).
Torok et al [107] described a higher percentage of CSF neutrophils and suggested this may be due to prior HIV-1 infection. Indeed, this elevated percentage of CSF neutrophils in HIV-1 infected patients may be caused by the microglial response to HIV-1 infection and HIV-1 proteins gp120, Tat (Trans-activator of transcription), Nef (Negative regulatory factor) and Vpr (Viral protein R). Atluri et al demonstrated that these four proteins disrupt the BBB integrity by triggering the decline of tight proteins expression on brain microvascular endothelial cells, MMP expression and apoptosis, and leading to infiltration of leukocytes[108]. Moreover, it has been reported in vitro that microglia secrete numerous pro-inflammatory cytokines and chemokines when infected by HIV-1, including the pro-inflammatory neutrophil chemotactic factor IL-8 [20]. Those cells also secrete TNF, a cytokine implicated in BBB permeability and whose role in pathogenesis of M.tb is critical, indicating the influence of HIV-1 in the promotion of TBM and M.tb spread in the CNS.
Further insight into the mechanisms underlying immunopathogenesis of TBM have been observed through the study of paradoxical immune reactions. A paradoxical reaction in tuberculosis is characterised by worsening of pre-existing tuberculous lesions, or the appearance of new tuberculous lesions despite effective anti-tuberculosis treatment in patients who have demonstrated initial improvement to therapy. In HIV-1 co-infection, paradoxical worsening often occurs following the introduction of antiretroviral therapy (ART): a phenomenon known as immune reconstitution inflammatory syndrome (IRIS). Although initiation of ART during treatment of TBM is associated with a reduced six month mortality (HR 0.3, 95% CI=0.08–0.82)[1] IRIS in this context carries a poor prognosis with one study reporting a mortality rate of 25% with severe disability at 9 months observed in 23% of those who survived[109]. The role of inflammasomes, immune complexes of receptors and sensors that mediate innate immune responses and lead to inflammation, have been described in this setting[110–112].
In TBM specifically, IRIS is characterised by high CSF neutrophil counts and M.tb positivity at presentation[109]. In those who develop IRIS CSF TNF was found to be high whereas CNF IFN gamma was low[109]. Further analysis of predictive factors has demonstrated the role of neutrophil mediators in particular S100A8/9 which two weeks after the initiation of ART was found to be significantly higher in the CSF of those who develop TBM-IRIS compared to those who do not[51]. Using longitudinal microarray analysis of blood from patients with HIV and TBM Marais et al have also demonstrated that neutrophil abundant transcripts were significantly higher in those who develop IRIS from before ART initiation until the onset of TBM-IRIS[113]. After initiation of ART a significantly higher number of transcripts associated with canonical and non-canonical inflammasome activation were found in patients who went on to develop IRIS than in those who did not[113]. These observations together with the finding that inflammasome activation can contribute to pyroptosis [114] suggest that tissue injury observed in TBM may be partly induced by inflammasome-mediated pyroptosis. It is unclear as to whether similar mechanisms exist in the absence of HIV.
HIV-1 infiltrates the CNS soon after systemic infection and around half of HIV-1 infected people develop HIV-associated neurocognitive disorders [115]. It has been reported that IRIS may worsen established HIV related cognitive impairment[116]. Little is known of the pathological mechanisms which lead to worsening cognition in the context of IRIS but some have postulated a role of the HIV-1 Trans-Activator of Transcription (Tat) protein[117]. They described the presence of Tat in the CSF of the virologically controlled ART-treated population and proposed that Tat would contribute to neuroinflammation and IL-17 production by infiltrating Th17 cells. Given that an increase of IL-17 secretion predisposes to IRIS in HIV-1 infected individuals in the context of cryptococcal meningitis[118], it is possible that Tat is a key player in the pathogenesis of HIV related cognitive impairment in the context of IRIS.
MACROSCOPIC MANIFESTATIONS OF THE DISEASE IN RELATION TO THE IMMUNE RESPONSE
The characteristic feature of TBM in post-mortem studies is the presence of a thick, gelatinous inflammatory exudate in the basal cisterns and subarachnoid spaces of the brain (Figure 3), which may extend into the spinal canal[18, 119, 120]. The predominantly basal location has important implications; firstly, the major cerebral vessels originating from the base of the brain become encased with exudate; secondly, exudate blocks the circulation of CSF; and thirdly, it envelopes and compresses the local cranial nerves resulting in cranial nerve palsies[18].
Figure 3: Radiological features of TBM.

Top left: Exudate
Contrast-enhanced T1-weighted MRI scan images: A: Normal scan showing cerebrospinal fluid in the cisterns (interpeduncular cistern in front of the midbrain, black arrow) and vessels at the base of the brain in normal cisterns (posterior cerebral artery, white arrow); B: Scan of a patient with TBM showing exudate in the basal cisterns of the brain (interpeduncular cistern, anterior to the brainstem and beneath the hypothalamus, black arrow) and vessels coursing through the exudate in the cisterns (posterior cerebral artery, white arrow).
Middle left: Hydrocephalus
A: Initial head CT scan images of a patient with TBM showing acute hydrocephalus with dilated ventricles and a compressed brain; B: Head CT scan of the same patient after 3 weeks of medical therapy showing resolution of the hydrocephalus.
Lower left: Tuberculomas
MRI scans demonstrating different patterns and imaging characteristics of brain tuberculomas: A: Contrast-enhanced T1-weighted MRI scan showing multiple ring-enhancing small tuberculomas (arrowed); B: T2-weighted MRI scan showing a large tuberculoma in the cerebellum compressing the brainstem, surrounding oedema, and hydrocephalus from obstruction of the cerebral aqueduct.
Right: Infarcts
Demonstration of infarction patterns in TBM, A: T2-weighted MRI scan showing normal ventricular size and no infarcts; B: T2-weighted axial MRI scan of a patient with TBM showing discrete unilateral small perforator vessel infarcts (arrowed); C: T2-weighted MRI scan showing more extensive infarcts in the thalami and basal ganglia (arrowed); D: Head CT scan of a patient with TBM showing global infarction with hypodense hemispheres bilaterally and a swollen brain.
Vasculitis
As the exudate spreads along the cisternal extensions from the base of the brain it coats the large arteries and their small perforators, although it has a predilection for extension into the Sylvian fissures. Therefore, the middle cerebral artery (MCA) and its perforators around the floor of the 3rd ventricle are most commonly involved, resulting in ischemic damage to the sub-thalamic nuclei and lower internal capsule[18] (Figure 5). Involvement of small vessels supplying the mid-brain may lead to infarction[119] and exudate in the interpeduncular cistern can compromise the vessels supplying the hypothalamic region and precipitate hypothalamic symptoms in these patients[119]. Vascular pathology includes infiltration of inflammation from the adventitia of arteries and veins inwards, resulting in peri- or pan-arteritis that involves segments or the full thickness of the vascular wall tissue[121]. Evolution of vascular inflammation may involve thickening of the vessel intima resulting in vessel stenosis or occlusion[121]. Further, vasospasm is also an important contributor to brain ischaemia 21. Both cerebral and spinal vessels may be affected by any or all of these processes and this may reflect the duration of illness[121]. Infarcts (Figure 3) and changes on angiography are reported in the majority of TBM patients[122] and are associated with post-mortem cerebral and vascular pathology[18].
Figure 5: Lenticulostriate arteries.

Lenticulostriate arteries branching from the M1 segment of the middle cerebral artery supply the basal ganglia and internal capsule.
Hydrocephalus (Figure 3)
Extension of exudative material into the basal cisterns results in a collar of exudate around the midbrain and a blockage in CSF flow around the upper brain stem. Exudate may also collect around the cerebral aqueduct of the 3rd and 4th ventricles hindering the flow of CSF through the ventricular system (Figure 4). Consequently, hydrocephalus is common, occurring in 75–80% of TBM patients[122–124]. The pressure of expanding ventricles and the opposing pressure of brain oedema due to pathological processes can negatively impact the grey and white matter, leading to pallor and diffuse loss of myelin[18], and the raised intracranial pressure can severely compromise cerebral blood flow.
Tuberculomas and Tuberculous brain abscess (Figure 3)
Tuberculomas may coexist with TBM or occur independently and are thought to originate from expanding tubercles in the parenchyma. These lesions are typically granulomatous consisting of a necrotic centre surrounded by epithelioid cells (which may merge to form Langhans giant cells), lymphocytes and a border of astrocytes, and are associated with peripheral oedema and vascular proliferation[15, 18]. With time the tubercles become discretely organised and bound by a rim of connective tissue of reticulin fibres, which enhances on contrasted brain imaging. They may occur throughout the brain in the cerebrum, cerebellum and brainstem[18, 122].
TB brain abscesses may follow TBM or develop independently[125, 126], however, they occur infrequently[127]. Abscesses manifest as encapsulated collections of pus that contain tubercular bacilli in absence of typical TB granuloma features including epithelioid and giant cells and mononuclear cell infiltrates[127]. These abscesses have been reported in the cerebrum[125, 127] and posterior fossa[125, 128, 129], occur infrequently in the brain stem[126], and may be singular or multiple[15].
Spinal TB
Spinal TB may develop as a primary tuberculous lesion, from the downward extension of intracranial TBM, or secondary to vertebral TB[18, 130, 131]. It involves the cord, meninges and nerve roots and manifests as spinal arachnoiditis, intradural (extramedullary) tuberculomas or rarely as intramedullary tuberculomas[122, 132–134]. Spinal nerve roots may become entrapped and matted in exudate filling the subarachnoid space, and complete obliteration of the thecal space can occur in severe cases[122]. Exudate involvement of the lower lumbar segments is associated with difficulty in performing lumbar punctures and a high CSF protein[18, 122]. The microscopic appearance of spinal exudate is identical to intracranial exudate, consisting of giant cells and caseous areas, and causing vasculitis of the spinal veins and arteries, which may lead to cord ischemia and infarction [18, 135].
RESEARCH GAPS AND THE PATH FORWARD
Intracerebral and spinal pathology in TBM is mediated by a dysregulated inflammatory response that contributes to meningitis, tuberculoma formation, arteritis, obstruction of cerebrospinal fluid (CSF) flow, and vascular complications including stroke. The development of animal or in vitro cellular models could aid understanding of the underlying immune mechanisms and point the way to adjunctive anti-inflammatory and improved antibiotic therapies. In human studies high-resolution MRI or CT imaging to assess TBM disease activity could provide a more detailed clinical phenotype for TBM than is currently available. Unbiased application of ‘omics’ technologies (particularly transcriptomics) in particular analysis of cells and fluid arising from the site of disease is likely to be particularly valuable. Genes encoding proteins involved in metabolism, cell growth, transport, immune response, cell communication and signal transduction are particularly of interest. Longitudinal sampling of blood or CSF during observational studies or randomized controlled trials is highly informative regarding the dynamics of the disease, such as disease progression and drug response.
Table 1:
Cell types within the central nervous system
| Cell Types | Description | |
|---|---|---|
| Neuron | Primary cell type within the central nervous system (CNS) and peripheral nervous system (PNS) distinct from other cell types due to their electrical excitability. Responsible for receiving, processing and transmitting electrical and chemical signals. A typical neuron consists of a cell body (soma), dendrites and an axon. | |
| Neuroglia | Non-neuronal cells within the peripheral and central nervous system. | |
| •Macroglia | Cells derived from the ectoderm. | |
| CNS | ○Astrocytes | Most abundant macroglia in the CNS. Provide a vascular supply to neurons and in doing so form an important part of the blood brain barrier. They affect potassium influx and efflux and influence neurotransmitters to regulate the external environment of neurons. |
| ○Oligodendrocytes | These cells create the ‘myelin sheath’; a protective coating around the axon which enables effective propagation of electrical impulses along the neuron. In addition they provide trophic support by the production of glial cell line-derived neurotrophic factor (GDNF), brain-derived neurotrophic factor (BDNF), or insulin-like growth factor-1 (IGF-1). |
|
| ○Ependymal cells | These cells whch comprise the blood-CSF barrier line the ventricular system and spinal cord and producing cerebrospinal fluid and enabling its flow throughout the CNS. |
|
| PNS | ○Schwann cells | Responsible for the production of the myelin sheath in peripheral nerve axons are known to have phagocytic properties which allows for ongoing production of PNS neurons. |
| ○Satellite cells | These cells comprise sensory, parasympathetic and sympathetic ganglia and are responsible for regulation of the external chemical environment. |
|
| ○Enteric glial cells | Within the intrinsic ganglia of the digestive system these cells contribute to regulation of the enteric system. |
|
| •Microglia | Located within the brain and spinal cord these cells are the resident macrophages within the CNS. | |
Acknowledgments
RJW receives support from the Francis Crick Institute which is supported by Cancer Research UK (10218), Wellcome (10218) and UK Research and Innovation (10218). He also receives support from Wellcome (104803, 203135) and the National Institutes of Health (U01AI115940). AGD is supported by a Wellcome Clinicians PhD grant to University College London. AAF is supported by the National Research Foundation of South Africa through the SARChI Chair of Clinical Neurosciences. The funders had no role in the writing of the review or decision to publish it.
Abbreviations page
- ART
Antiretroviral therapy
- BBB
Blood brain barrier
- BCSFB
Blood–cerebrospinal fluid barrier
- CSF
Cerebro-spinal fluid
- DC
Dendritic cell
- G-CSF
Granulocyte colony stimulating factor
- GM-CSF
Granulocyte-monocyte colony stimulating factor
- IFN
Interferon
- IL-
Interleukin
- IRIS
Immune reconstitution inflammatory syndrome
- LEC
Lymphoid endothelial cell
- M.tb.
Mycobacterium tuberculosis
- PGL
Phenolic glycolipid
- RD
Region of difference
- TB
Tuberculosis
- TBM
Tuberculous meningitis
- TNF
Tumor necrosis factor
- TPH
Trypophan hydrolase
- VDR
Vitamin D receptor
- VEGF
Vascular endothelial growth factor
Footnotes
Conflict of Interest Disclosure
The authors have no relevant financial or other conflicts of interest.
References
- 1.Marais S, et al. , Presentation and outcome of tuberculous meningitis in a high HIV prevalence setting. PLoS One, 2011. 6(5): p. e20077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Garra A, et al. , The immune response in tuberculosis. Annu Rev Immunol, 2013. 31: p. 475–527. [DOI] [PubMed] [Google Scholar]
- 3.Kumar V and Kumar V, Robbins and Cotran’s pathologic basis of disease. 2010, Philadelphia, Pa.; London: Saunders; xiv, 1450 [Google Scholar]
- 4.Coico R and Sunshine G, Immunology : a short course. 2009, Hoboken, N.J: Wiley-Blackwell; xx, 391 : [Google Scholar]
- 5.Krishnan N, Robertson BD, and Thwaites G, The mechanisms and consequences of the extra-pulmonary dissemination of Mycobacterium tuberculosis. Tuberculosis (Edinb), 2010. 90(6): p. 361–6. [DOI] [PubMed] [Google Scholar]
- 6.Jain SK, et al. , Mycobacterium tuberculosis invasion and traversal across an in vitro human blood-brain barrier as a pathogenic mechanism for central nervous system tuberculosis. J Infect Dis, 2006. 193(9): p. 1287–95. [DOI] [PubMed] [Google Scholar]
- 7.Nguyen L and Pieters J, The Trojan horse: survival tactics of pathogenic mycobacteria in macrophages. Trends Cell Biol, 2005. 15(5): p. 269–76. [DOI] [PubMed] [Google Scholar]
- 8.Lerner TR, et al. , Lymphatic endothelial cells are a replicative niche for Mycobacterium tuberculosis. J Clin Invest, 2016. 126(3): p. 1093–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hawn TR, et al. , A polymorphism in Toll-interleukin 1 receptor domain containing adaptor protein is associated with susceptibility to meningeal tuberculosis. J Infect Dis, 2006. 194(8): p. 1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caws M, et al. , The influence of host and bacterial genotype on the development of disseminated disease with Mycobacterium tuberculosis. PLoS Pathog, 2008. 4(3): p. e1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fernando SL, et al. , A polymorphism in the P2X7 gene increases susceptibility to extrapulmonary tuberculosis. Am J Respir Crit Care Med, 2007. 175(4): p. 360–6. [DOI] [PubMed] [Google Scholar]
- 12.Caws M, et al. , Beijing genotype of Mycobacterium tuberculosis is significantly associated with human immunodeficiency virus infection and multidrug resistance in cases of tuberculous meningitis. J Clin Microbiol, 2006. 44(11): p. 3934–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pulzova L, Bhide MR, and Andrej K, Pathogen translocation across the blood-brain barrier. FEMS Immunol Med Microbiol, 2009. 57(3): p. 203–13. [DOI] [PubMed] [Google Scholar]
- 14.Be NA, et al. , Pathogenesis of central nervous system tuberculosis. Curr Mol Med, 2009. 9(2): p. 94–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rock RB, et al. , Central nervous system tuberculosis: pathogenesis and clinical aspects. Clin Microbiol Rev, 2008. 21(2): p. 243–61, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donald PR, Schaaf HS, and Schoeman JF, Tuberculous meningitis and miliary tuberculosis: the Rich focus revisited. J Infect, 2005. 50(3): p. 193–5. [DOI] [PubMed] [Google Scholar]
- 17.Rich A MH, The Pathogenesis of Tuberculous Meningitis. Bull John Hopkins Hosp, 1933. 52: p. 5–37. [Google Scholar]
- 18.Dastur DK, Manghani DK, and Udani PM, Pathology and pathogenetic mechanisms in neurotuberculosis. Radiol Clin North Am, 1995. 33(4): p. 733–52. [PubMed] [Google Scholar]
- 19.Peterson PK, et al. , CD14 receptor-mediated uptake of nonopsonized Mycobacterium tuberculosis by human microglia. Infect Immun, 1995. 63(4): p. 1598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rock RB, et al. , Mycobacterium tuberculosis-induced cytokine and chemokine expression by human microglia and astrocytes: effects of dexamethasone. J Infect Dis, 2005. 192(12): p. 2054–8. [DOI] [PubMed] [Google Scholar]
- 21.Randall PJ, et al. , Neurons are host cells for Mycobacterium tuberculosis. Infect Immun, 2014. 82(5): p. 1880–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wright SD, et al. , CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science, 1990. 249(4975): p. 1431–3. [DOI] [PubMed] [Google Scholar]
- 23.Shams H, et al. , The CD14 receptor does not mediate entry of Mycobacterium tuberculosis into human mononuclear phagocytes. FEMS Immunol Med Microbiol, 2003. 36(1–2): p. 63–9. [DOI] [PubMed] [Google Scholar]
- 24.Peterson PK, et al. , Multinucleated giant cell formation of swine microglia induced by Mycobacterium bovis. J Infect Dis, 1996. 173(5): p. 1194–201. [DOI] [PubMed] [Google Scholar]
- 25.Yang CS, et al. , Reactive oxygen species and p47phox activation are essential for the Mycobacterium tuberculosis-induced pro-inflammatory response in murine microglia. J Neuroinflammation, 2007. 4: p. 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mastroianni CM, et al. , Cerebrospinal fluid cytokines in patients with tuberculous meningitis. Clin Immunol Immunopathol, 1997. 84(2): p. 171–6. [DOI] [PubMed] [Google Scholar]
- 27.Tobin DM, et al. , Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell, 2012. 148(3): p. 434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsenova L, et al. , Tumor necrosis factor alpha is a determinant of pathogenesis and disease progression in mycobacterial infection in the central nervous system. Proc Natl Acad Sci U S A, 1999. 96(10): p. 5657–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaplan G and Freedman VH, The role of cytokines in the immune response to tuberculosis. Res Immunol, 1996. 147(8–9): p. 565–72. [DOI] [PubMed] [Google Scholar]
- 30.Hashimoto M, et al. , Action site of circulating interleukin-1 on the rabbit brain. Brain Res, 1991. 540(1–2): p. 217–23. [DOI] [PubMed] [Google Scholar]
- 31.de Vries HE, et al. , The blood-brain barrier in neuroinflammatory diseases. Pharmacol Rev, 1997. 49(2): p. 143–55. [PubMed] [Google Scholar]
- 32.Ramilo O, et al. , Tumor necrosis factor alpha/cachectin and interleukin 1 beta initiate meningeal inflammation. J Exp Med, 1990. 172(2): p. 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsenova L, et al. , A combination of thalidomide plus antibiotics protects rabbits from mycobacterial meningitis-associated death. J Infect Dis, 1998. 177(6): p. 1563–72. [DOI] [PubMed] [Google Scholar]
- 34.Tsenova L, et al. , Use of IMiD3, a thalidomide analog, as an adjunct to therapy for experimental tuberculous meningitis. Antimicrob Agents Chemother, 2002. 46(6): p. 1887–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schoeman JF, et al. , Adjunctive thalidomide therapy for childhood tuberculous meningitis: results of a randomized study. J Child Neurol, 2004. 19(4): p. 250–7. [DOI] [PubMed] [Google Scholar]
- 36.van Toorn R, et al. , Clinicoradiologic response of neurologic tuberculous mass lesions in children treated with thalidomide. Pediatr Infect Dis J, 2015. 34(2): p. 214–8. [DOI] [PubMed] [Google Scholar]
- 37.Simmons CP, et al. , Pretreatment intracerebral and peripheral blood immune responses in Vietnamese adults with tuberculous meningitis: diagnostic value and relationship to disease severity and outcome. J Immunol, 2006. 176(3): p. 2007–14. [DOI] [PubMed] [Google Scholar]
- 38.Saunders BM, et al. , Interleukin-6 induces early gamma interferon production in the infected lung but is not required for generation of specific immunity to Mycobacterium tuberculosis infection. Infect Immun, 2000. 68(6): p. 3322–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xing Z, et al. , IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest, 1998. 101(2): p. 311–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Curto M, et al. , Inhibition of cytokines expression in human microglia infected by virulent and non-virulent mycobacteria. Neurochem Int, 2004. 44(6): p. 381–92. [DOI] [PubMed] [Google Scholar]
- 41.Chen Y, et al. , HMGB1 level in cerebrospinal fluid as a complimentary biomarker for the diagnosis of tuberculous meningitis. Springerplus, 2016. 5(1): p. 1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rohlwink UK, et al. , Biomarkers of Cerebral Injury and Inflammation in Pediatric Tuberculous Meningitis. Clin Infect Dis, 2017. 65(8): p. 1298–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berger NA, et al. , Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. Br J Pharmacol, 2018. 175(2): p. 192–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ko HL and Ren EC, Functional Aspects of PARP1 in DNA Repair and Transcription. Biomolecules, 2012. 2(4): p. 524–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heizmann CW, Fritz G, and Schafer BW, S100 proteins: structure, functions and pathology. Front Biosci, 2002. 7: p. d1356–68. [DOI] [PubMed] [Google Scholar]
- 46.Mahon RN and Hafner R, Immune Cell Regulatory Pathways Unexplored as Host-Directed Therapeutic Targets for Mycobacterium tuberculosis: An Opportunity to Apply Precision Medicine Innovations to Infectious Diseases. Clin Infect Dis, 2015. 61 Suppl 3: p. S200–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ryckman C, et al. , Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol, 2003. 170(6): p. 3233–42. [DOI] [PubMed] [Google Scholar]
- 48.Pechkovsky DV, et al. , Calprotectin (MRP8/14 protein complex) release during mycobacterial infection in vitro and in vivo. FEMS Immunol Med Microbiol, 2000. 29(1): p. 27–33. [DOI] [PubMed] [Google Scholar]
- 49.Gopal R, et al. , S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med, 2013. 188(9): p. 1137–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tadokera R, et al. , Hypercytokinaemia accompanies HIV-tuberculosis immune reconstitution inflammatory syndrome. Eur Respir J, 2011. 37(5): p. 1248–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marais S, et al. , Neutrophil-associated central nervous system inflammation in tuberculous meningitis immune reconstitution inflammatory syndrome. Clin Infect Dis, 2014. 59(11): p. 1638–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lange C, et al. , Vascular endothelial growth factor: a neurovascular target in neurological diseases. Nat Rev Neurol, 2016. 12(8): p. 439–54. [DOI] [PubMed] [Google Scholar]
- 53.Ayata C and Ropper AH, Ischaemic brain oedema. J Clin Neurosci, 2002. 9(2): p. 113–24. [DOI] [PubMed] [Google Scholar]
- 54.van der Flier M, et al. , Vascular endothelial growth factor and blood-brain barrier disruption in tuberculous meningitis. Pediatr Infect Dis J, 2004. 23(7): p. 608–13. [DOI] [PubMed] [Google Scholar]
- 55.Kim H, et al. , Dexamethasone coordinately regulates angiopoietin-1 and VEGF: a mechanism of glucocorticoid-induced stabilization of blood-brain barrier. Biochem Biophys Res Commun, 2008. 372(1): p. 243–8. [DOI] [PubMed] [Google Scholar]
- 56.Rai D, et al. , Cerebrospinal fluid cytokines and matrix metalloproteinases in human immunodeficiency seropositive and seronegative patients of tuberculous meningitis. Ann Indian Acad Neurol, 2014. 17(2): p. 171–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.More A, et al. , A study of neuroendocrine dysfunction in patients of tuberculous meningitis. J Neurol Sci, 2017. 379: p. 198–206. [DOI] [PubMed] [Google Scholar]
- 58.Misra UK, et al. , A study of hyponatremia in tuberculous meningitis. J Neurol Sci, 2016. 367: p. 152–7. [DOI] [PubMed] [Google Scholar]
- 59.Cotton MF, et al. , Plasma arginine vasopressin and the syndrome of inappropriate antidiuretic hormone secretion in tuberculous meningitis. Pediatr Infect Dis J, 1991. 10(11): p. 837–42. [DOI] [PubMed] [Google Scholar]
- 60.Singh BS, Patwari AK, and Deb M, Serum sodium and osmolal changes in tuberculous meningitis. Indian Pediatr, 1994. 31(11): p. 1345–50. [PubMed] [Google Scholar]
- 61.Misra UK, et al. , Hypovolemia due to cerebral salt wasting may contribute to stroke in tuberculous meningitis. QJM, 2018. [DOI] [PubMed] [Google Scholar]
- 62.Thwaites GE, et al. , Dexamethasone for the treatment of tuberculous meningitis in adolescents and adults. N Engl J Med, 2004. 351(17): p. 1741–51. [DOI] [PubMed] [Google Scholar]
- 63.Bang ND, et al. , Clinical presentations, diagnosis, mortality and prognostic markers of tuberculous meningitis in Vietnamese children: a prospective descriptive study. BMC Infect Dis, 2016. 16(1): p. 573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mason S, et al. , Cerebrospinal fluid in tuberculous meningitis exhibits only the L-enantiomer of lactic acid. BMC Infect Dis, 2016. 16: p. 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lesniak WG, et al. , Concurrent quantification of tryptophan and its major metabolites. Anal Biochem, 2013. 443(2): p. 222–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Campbell BM, et al. , Kynurenines in CNS disease: regulation by inflammatory cytokines. Front Neurosci, 2014. 8: p. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Connor JC, et al. , Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J Neurosci, 2009. 29(13): p. 4200–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Majlath Z, Tajti J, and Vecsei L, Kynurenines and other novel therapeutic strategies in the treatment of dementia. Ther Adv Neurol Disord, 2013. 6(6): p. 386–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van Laarhoven A, et al. , Cerebral tryptophan metabolism and outcome of tuberculous meningitis: an observational cohort study. Lancet Infect Dis, 2018. 18(5): p. 526–535. [DOI] [PubMed] [Google Scholar]
- 70.Weiner J 3rd, et al. , Biomarkers of inflammation, immunosuppression and stress with active disease are revealed by metabolomic profiling of tuberculosis patients. PLoS One, 2012. 7(7): p. e40221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wobke TK, Sorg BL, and Steinhilber D, Vitamin D in inflammatory diseases. Front Physiol, 2014. 5: p. 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Graustein AD, et al. , TLR9 gene region polymorphisms and susceptibility to tuberculosis in Vietnam. Tuberculosis (Edinb), 2015. 95(2): p. 190–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thuong NT, et al. , A polymorphism in human TLR2 is associated with increased susceptibility to tuberculous meningitis. Genes Immun, 2007. 8(5): p. 422–8. [DOI] [PubMed] [Google Scholar]
- 74.Tobin DM, et al. , The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell, 2010. 140(5): p. 717–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang J, et al. , Relationship between human LTA4H polymorphisms and extra-pulmonary tuberculosis in an ethnic Han Chinese population in Eastern China. Tuberculosis (Edinb), 2014. 94(6): p. 657–63. [DOI] [PubMed] [Google Scholar]
- 76.Dunstan SJ, et al. , LTA4H genotype is associated with susceptibility to bacterial meningitis but is not a critical determinant of outcome. PLoS One, 2015. 10(3): p. e0118789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van Laarhoven A, et al. , Clinical Parameters, Routine Inflammatory Markers, and LTA4H Genotype as Predictors of Mortality Among 608 Patients With Tuberculous Meningitis in Indonesia. J Infect Dis, 2017. 215(7): p. 1029–1039. [DOI] [PubMed] [Google Scholar]
- 78.Thuong NTT, et al. , Leukotriene A4 Hydrolase Genotype and HIV Infection Influence Intracerebral Inflammation and Survival From Tuberculous Meningitis. J Infect Dis, 2017. 215(7): p. 1020–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fava VM and Schurr E, Evaluating the Impact of LTA4H Genotype and Immune Status on Survival From Tuberculous Meningitis. J Infect Dis, 2017. 215(7): p. 1011–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Campo M, et al. , Common polymorphisms in the CD43 gene region are associated with tuberculosis disease and mortality. Am J Respir Cell Mol Biol, 2015. 52(3): p. 342–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hoppe HC, et al. , Identification of phosphatidylinositol mannoside as a mycobacterial adhesin mediating both direct and opsonic binding to nonphagocytic mammalian cells. Infect Immun, 1997. 65(9): p. 3896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hoal-Van Helden EG, et al. , Mannose-binding protein B allele confers protection against tuberculous meningitis. Pediatr Res, 1999. 45(4 Pt 1): p. 459–64. [DOI] [PubMed] [Google Scholar]
- 83.Horne DJ, et al. , Common polymorphisms in the PKP3-SIGIRR-TMEM16J gene region are associated with susceptibility to tuberculosis. J Infect Dis, 2012. 205(4): p. 586–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rizvi I, et al. , Vitamin D status, vitamin D receptor and toll like receptor-2 polymorphisms in tuberculous meningitis: a case-control study. Infection, 2016. 44(5): p. 633–40. [DOI] [PubMed] [Google Scholar]
- 85.Areeshi MY, et al. , Vitamin D Receptor ApaI (rs7975232) Polymorphism Confers Decreased Risk of Pulmonary Tuberculosis in Overall and African Population, but not in Asians: Evidence from a Meta-analysis. Ann Clin Lab Sci, 2017. 47(5): p. 628–637. [PubMed] [Google Scholar]
- 86.Gagneux S and Small PM, Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect Dis, 2007. 7(5): p. 328–37. [DOI] [PubMed] [Google Scholar]
- 87.Gagneux S, et al. , Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A, 2006. 103(8): p. 2869–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Comas I, et al. , Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet, 2013. 45(10): p. 1176–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tsenova L, et al. , Virulence of selected Mycobacterium tuberculosis clinical isolates in the rabbit model of meningitis is dependent on phenolic glycolipid produced by the bacilli. J Infect Dis, 2005. 192(1): p. 98–106. [DOI] [PubMed] [Google Scholar]
- 90.Kong Y, et al. , Association between Mycobacterium tuberculosis Beijing/W lineage strain infection and extrathoracic tuberculosis: Insights from epidemiologic and clinical characterization of the three principal genetic groups of M. tuberculosis clinical isolates. J Clin Microbiol, 2007. 45(2): p. 409–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dormans J, et al. , Correlation of virulence, lung pathology, bacterial load and delayed type hypersensitivity responses after infection with different Mycobacterium tuberculosis genotypes in a BALB/c mouse model. Clin Exp Immunol, 2004. 137(3): p. 460–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guerra-Assuncao JA, et al. , Recurrence due to relapse or reinfection with Mycobacterium tuberculosis: a whole-genome sequencing approach in a large, population-based cohort with a high HIV infection prevalence and active follow-up. J Infect Dis, 2015. 211(7): p. 1154–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Reed MB, et al. , A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature, 2004. 431(7004): p. 84–7. [DOI] [PubMed] [Google Scholar]
- 94.Coscolla M and Gagneux S, Consequences of genomic diversity in Mycobacterium tuberculosis. Semin Immunol, 2014. 26(6): p. 431–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Guerra-Assuncao JA, et al. , Large-scale whole genome sequencing of M. tuberculosis provides insights into transmission in a high prevalence area. Elife, 2015. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.de Jong BC, et al. , Progression to active tuberculosis, but not transmission, varies by Mycobacterium tuberculosis lineage in The Gambia. J Infect Dis, 2008. 198(7): p. 1037–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.van Crevel R, et al. , Mycobacterium tuberculosis Beijing genotype strains associated with febrile response to treatment. Emerg Infect Dis, 2001. 7(5): p. 880–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Parwati I, et al. , Mycobacterium tuberculosis Beijing genotype is an independent risk factor for tuberculosis treatment failure in Indonesia. J Infect Dis, 2010. 201(4): p. 553–7. [DOI] [PubMed] [Google Scholar]
- 99.Sarkar R, et al. , Modern lineages of Mycobacterium tuberculosis exhibit lineage-specific patterns of growth and cytokine induction in human monocyte-derived macrophages. PLoS One, 2012. 7(8): p. e43170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Portevin D, et al. , Human macrophage responses to clinical isolates from the Mycobacterium tuberculosis complex discriminate between ancient and modern lineages. PLoS Pathog, 2011. 7(3): p. e1001307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rakotosamimanana N, et al. , Variation in gamma interferon responses to different infecting strains of Mycobacterium tuberculosis in acid-fast bacillus smear-positive patients and household contacts in Antananarivo, Madagascar. Clin Vaccine Immunol, 2010. 17(7): p. 1094–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Katrak SM, et al. , The clinical, radiological and pathological profile of tuberculous meningitis in patients with and without human immunodeficiency virus infection. J Neurol Sci, 2000. 181(1–2): p. 118–26. [DOI] [PubMed] [Google Scholar]
- 103.Karande S, et al. , Tuberculous meningitis and HIV. Indian J Pediatr, 2005. 72(9): p. 755–60. [DOI] [PubMed] [Google Scholar]
- 104.Thwaites GE, et al. , The influence of HIV infection on clinical presentation, response to treatment, and outcome in adults with Tuberculous meningitis. J Infect Dis, 2005. 192(12): p. 2134–41. [DOI] [PubMed] [Google Scholar]
- 105.Karstaedt AS, et al. , Tuberculous meningitis in South African urban adults. QJM, 1998. 91(11): p. 743–7. [DOI] [PubMed] [Google Scholar]
- 106.Azuaje C, et al. , [Tuberculous meningitis: a comparative study in relation to concurrent human immunodeficiency virus infection]. Enferm Infecc Microbiol Clin, 2006. 24(4): p. 245–50. [DOI] [PubMed] [Google Scholar]
- 107.Torok ME, et al. , Clinical and microbiological features of HIV-associated tuberculous meningitis in Vietnamese adults. PLoS One, 2008. 3(3): p. e1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Atluri VS, et al. , Effect of human immunodeficiency virus on blood-brain barrier integrity and function: an update. Front Cell Neurosci, 2015. 9: p. 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Marais S, et al. , Frequency, severity, and prediction of tuberculous meningitis immune reconstitution inflammatory syndrome. Clin Infect Dis, 2013. 56(3): p. 450–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lai RP, et al. , HIV-tuberculosis-associated immune reconstitution inflammatory syndrome is characterized by Toll-like receptor and inflammasome signalling. Nat Commun, 2015. 6: p. 8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tan HY, et al. , Plasma interleukin-18 levels are a biomarker of innate immune responses that predict and characterize tuberculosis-associated immune reconstitution inflammatory syndrome. AIDS, 2015. 29(4): p. 421–31. [DOI] [PubMed] [Google Scholar]
- 112.Tan HY, et al. , Aberrant Inflammasome Activation Characterizes Tuberculosis-Associated Immune Reconstitution Inflammatory Syndrome. J Immunol, 2016. 196(10): p. 4052–63. [DOI] [PubMed] [Google Scholar]
- 113.Marais S, et al. , Inflammasome Activation Underlying Central Nervous System Deterioration in HIV-Associated Tuberculosis. J Infect Dis, 2017. 215(5): p. 677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bergsbaken T, Fink SL, and Cookson BT, Pyroptosis: host cell death and inflammation. Nat Rev Microbiol, 2009. 7(2): p. 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tozzi V, et al. , Prevalence and risk factors for human immunodeficiency virus-associated neurocognitive impairment, 1996 to 2002: results from an urban observational cohort. J Neurovirol, 2005. 11(3): p. 265–73. [DOI] [PubMed] [Google Scholar]
- 116.Miller RF, et al. , Cerebral CD8+ lymphocytosis in HIV-1 infected patients with immune restoration induced by HAART. Acta Neuropathol, 2004. 108(1): p. 17–23. [DOI] [PubMed] [Google Scholar]
- 117.Johnson TP, et al. , Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc Natl Acad Sci U S A, 2013. 110(33): p. 13588–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Boulware DR, et al. , Clinical features and serum biomarkers in HIV immune reconstitution inflammatory syndrome after cryptococcal meningitis: a prospective cohort study. PLoS Med, 2010. 7(12): p. e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Daniel PM, Gross morbid anatomy of the central nervous system of cases of tuberculous meningitis treated with streptomycin. Proc R Soc Med, 1949. 42(3): p. 169–74. [PMC free article] [PubMed] [Google Scholar]
- 120.Shinoyama M, Suzuki M, and Nomura S, Fulminant tuberculous meningitis--autopsy case report. Neurol Med Chir (Tokyo), 2012. 52(10): p. 761–4. [DOI] [PubMed] [Google Scholar]
- 121.Lammie GA, et al. , Tuberculous cerebrovascular disease: a review. J Infect, 2009. 59(3): p. 156–66. [DOI] [PubMed] [Google Scholar]
- 122.Rohlwink UK, et al. , Imaging Features of the Brain, Cerebral Vessels and Spine in Pediatric Tuberculous Meningitis With Associated Hydrocephalus. Pediatr Infect Dis J, 2016. 35(10): p. e301–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.van Well GT, et al. , Twenty years of pediatric tuberculous meningitis: a retrospective cohort study in the western cape of South Africa. Pediatrics, 2009. 123(1): p. e1–8. [DOI] [PubMed] [Google Scholar]
- 124.Thwaites GE, et al. , Serial MRI to determine the effect of dexamethasone on the cerebral pathology of tuberculous meningitis: an observational study. Lancet Neurol, 2007. 6(3): p. 230–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schoeman JF, et al. , Intractable intracranial tuberculous infection responsive to thalidomide: report of four cases. J Child Neurol, 2006. 21(4): p. 301–8. [DOI] [PubMed] [Google Scholar]
- 126.Kumar R SV, Tuberculous brain stem abscesses in children. Journal of Pediatric Neurology, 2004. 2(2): p. 101–106. [Google Scholar]
- 127.Kumar R, et al. , Tuberculous brain abscess: clinical presentation, pathophysiology and treatment (in children). Childs Nerv Syst, 2002. 18(3–4): p. 118–23. [DOI] [PubMed] [Google Scholar]
- 128.Schoeman JF, et al. , Massive posterior fossa tuberculous abscess developing in a young child treated for miliary tuberculosis. Possible role of very rapid acetylation of isoniazid. Pediatr Neurosurg, 1998. 29(2): p. 64–8. [DOI] [PubMed] [Google Scholar]
- 129.Saini AG, et al. , Primary tuberculous cerebellar abscess: case report. Ann Trop Paediatr, 2011. 31(4): p. 367–9. [DOI] [PubMed] [Google Scholar]
- 130.Moghtaderi A and Alavi Naini R, Tuberculous radiculomyelitis: review and presentation of five patients. Int J Tuberc Lung Dis, 2003. 7(12): p. 1186–90. [PubMed] [Google Scholar]
- 131.Hernandez Pando R, et al. , Specific bacterial genotypes of Mycobacterium tuberculosis cause extensive dissemination and brain infection in an experimental model. Tuberculosis (Edinb), 2010. 90(4): p. 268–77. [DOI] [PubMed] [Google Scholar]
- 132.Srivastava T and Kochar DK, Asymptomatic spinal arachnoiditis in patients with tuberculous meningitis. Neuroradiology, 2003. 45(10): p. 727–9. [DOI] [PubMed] [Google Scholar]
- 133.Skendros P, et al. , Intradural, eextramedullary tuberculoma of the spinal cord as a complication of tuberculous meningitis. Infection, 2003. 31(2): p. 115–7. [DOI] [PubMed] [Google Scholar]
- 134.Lim YS, et al. , Disseminated tuberculosis of central nervous system : spinal intramedullary and intracranial tuberculomas. J Korean Neurosurg Soc, 2013. 54(1): p. 61–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kato M, et al. , [Magnetic resonance imaging of a case of central nervous system tuberculosis with tuberculous arachnoiditis and multiple tuberculomas]. Nihon Ronen Igakkai Zasshi, 1997. 34(10): p. 818–24. [DOI] [PubMed] [Google Scholar]