

Abstract
Coiled-coil peptides have proven useful in a range of materials applications ranging from the formation of well-defined fibrils to responsive hydrogels. The ability to design from first principles their oligomerization and subsequent higher order assembly offers their expanded use in producing new materials. Toward these ends, homo-tetrameric, antiparallel, coiled-coil, peptide bundles have been designed computationally, synthesized via solid-phase methods, and their solution behavior characterized. Two different bundle-forming peptides were designed and examined. Within the targeted coiled coil structure, both bundles contained the same hydrophobic core residues. However, different exterior residues on the two different designs yielded sequences with different distributions of charged residues and two different expected isoelectric points of pI 4.4 and pI 10.5. Both coiled-coil bundles were extremely stable with respect to temperature (Tm > 80 C) and remained soluble in solution even at high (millimolar) peptide concentrations. The coiled-coil tetramer was confirmed to be the dominant species in solution by analytical sedimentation studies and by small-angle neutron scattering, where the scattering form factor is well represented by a cylinder model with the dimensions of the targeted coiled coil. At high concentrations (5–15 mM), evidence of interbundle structure was observed via neutron scattering. At these concentrations, the synthetic bundles form soluble aggregates, and interbundle distances can be determined via a structure factor fit to scattering data. The data support the successful design of robust coiled-coil bundles. Despite their different sequences, each sequence forms loosely associated but soluble aggregates of the bundles, suggesting similar dissociated states for each. The behavior of the dispersed bundles is similar to that observed for natural proteins.
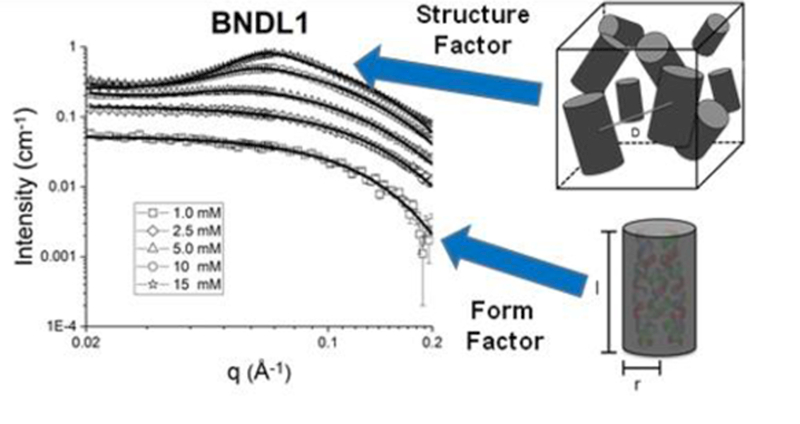
SANS was used to model the shape, size, and interactions of soluble, coiled-coil, peptide bundles.
Introduction
The design of self-assembled nanostructures with desired physical attributes such as specific shapes and sizes, as well as desired chemical properties such as the display of functional groups and charge, is important for creating model systems for studying self-assembly as well as the development of novel materials. Biopolymers such as DNA or proteins form highly specific and unique structures in nature by solution assembly. The monomer sequences of these biomolecules determine their self-assembled structures and, subsequently, how they function. Understanding and manipulating inter- and intramolecular interactions that cause these biomolecules to self-assemble in such intricate and specific fashion is paramount to the creation of new, synthetic materials with designed structures and functions formed via solution self-assembly. Many studies have reported the use of naturally occurring biomolecules and their analogues for the production of new materials, by virtue of naturally programmed and specific self-assembly; these methods also permit assembled architectures with physical properties afforded by natural biomolecules.1–6 The intramolecular folding of biomolecules can provide desired intermolecular interactions due to the specificity of spatial display of chemical functionality. This structured presentation of chemical functionality in peptide and protein folded structures has been exploited to generate designed intermolecular nanoscale assemblies such as cages,7,8 sheets,9 discs,10 tubes,11,12 and fibrils13–16.
In order to utilize the plethora of interactions responsible for the folding and assembly of biomolecules, computational methods, as compared to rational design methods, have been increasingly employed to identify suitable molecule candidates for building a desired structure. Related design strategies have been developed to create programmable nanostructures from DNA;17–21 once a desired intermolecular DNA structure has been targeted, computational methods identify the nucleotide sequences that mediate assembly of the nanostructure. This identification is based on utilizing complementary base pairing between nucleic acid chains as well as the topology of the design. Computational work has also been directed toward using natural proteins as building blocks for the creation of various symmetrical cages consisting of one or multiple protein sequences.22,23 These studies utilize naturally occurring proteins, and their inherent size and shape, to identify modifications to the exterior amino acids of the folded protein in order to create various cage-like particles from the induced intermolecular assembly. Similar strategies of altering naturally occurring proteins have been applied to design two-dimensional arrays through multiple noncovalent interactions,24 including metal binding-induced assembly.25
To provide a broad perspective on ordering and self-assembly, the dispersed state of assembling biomolecular structures needs to be examined. Computational methods have been applied to the design of peptides that, upon assembly in solution, yield desired nanostructures and behavior.26 An ultimate goal of this strategy is to design and control nanostructure and material function, unconstrained by the structures and sequences of naturally occurring proteins and therefore avoiding limitations in size, stability and possible chemical functionality. Herein we focus on the characterization of tetramerical helical bundles that do not aggregate or undergo further assembly. Previous studies have provided examples of success with de novo-designed nanostructures based on the coiled-coil bundle nanostructure.11,27–32 Coiled coils were selected for the present work due to their potential stability and specific arrangement of functionality on the interior and exterior surfaces of the bundles. Constituent peptides within a coiled-coil structure exhibit alpha helical secondary structure, intrahelical hydrogen bonds, and complementary hydrophobic interactions. The formation of helical bundles occurs when two or more peptides associate to bury hydrophobic groups within the interior of the bundle. A variety of studies have been performed on the rational design of coiled-coil peptides in order to understand the impact of various residues at different locations of the coiled coil33–37 as well as to create functional assemblies.11,15,31 Similar work on the design of stable protein structures has been conducted using computational methods.32,38,39 Herein, computation is used to guide the variation of peptide sequences, which form targeted tetrameric bundles through solution assembly.26,27 Our previous work showed that robust coiled coils could be computationally designed that form tetrameric bundles, which subsequently undergo interbundle assembly into targeted nanostructures (e.g. 2-D plates).26 Importantly, related coiled-coil bundles of the same length and same core hydrophobic amino acids, were designed specifically to not undergo assembly into any regular nanostructure but to remain as stable, dispersed bundles in solution. The current work explores the range of behaviors of a coiled-coil bundle design, where variants of this design have previously been shown to form regular lattices in solution. Understanding the dissociated (solubilized) state is a key element of understanding the self-assembly of such peptide systems, and herein we focus on sequences designed to remain soluble. We probe the concentration dependence of interbundle structure and the temperature stabilities of these bundles in solution. In combination with our previous work, these studies inform the picture of the dispersed state of such peptide bundles and the ability to manipulate assembly via modification of solution conditions and the exteriors of helical bundles with a common coiled-coil core.
A coiled-coil homo-tetramer with antiparallel helices (D2 symmetry) was chosen as the targeted nanostructure with two sequences predicted, BNDL1 and BNDL2 (Figure 1). As mentioned, the bundle core is designed to contain hydrophobic amino acid side chains, critical to the stability of the folded, coiled-coil state. The exterior amino acids of the bundles were designed to maintain solubility; this is in contrast to previous coiled-coil bundles that were computationally designed to undergo inter-bundle assembly into predetermined lattice nanomaterials.26,27 The coiled-coil peptide designs were experimentally investigated using circular dichroism (CD) spectroscopy to demonstrate their inherent alpha-helical character and thermal stabilities. The designed tetrameric oligomerization state was verified using analytical ultra-centrifugation (AUC). Finally, the peptide bundles were investigated in solution at various concentrations using small angle neutron scattering (SANS) to further assess their coiled-coil bundle nanostructure as well as the interbundle interactions present at higher concentrations. Importantly, a cylindrical form factor model is consistent with the scattering data and is in strong agreement with the designed size and shape of the targeted coiled-coil bundles. At higher peptide concentrations (~5 mM), an interparticle structure factor becomes apparent in the neutron scattering that reflects the soluble aggregate nature of the coiled coils when they become more crowded in solution and begin to interact in a reversible and dynamic fashion.
Figure 1 –
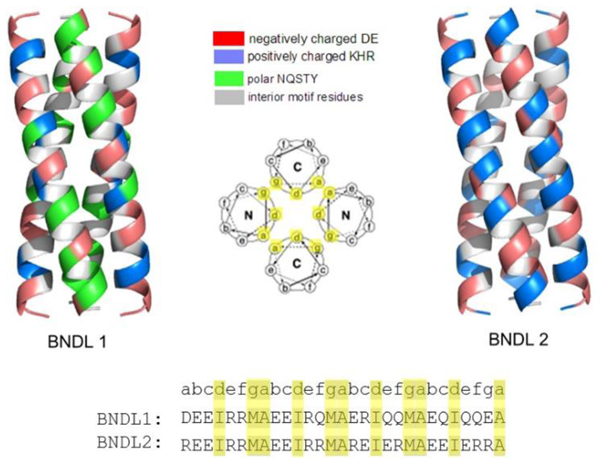
Computationally designed coiled-coil bundles BNDL1 and BNDL2. Residues are color-coded based on their chemical properties to show the chemical diversity on the surface of the bundles. The wheel diagram looking down the long axis shows the antiparallel design of the homo-tetramer bundle and indicates where residues will be located along the coiled-coil based on the heptad repeat. The residues that compose the hydrophobic core are highlighted in the wheel diagram and sequences.
Experimental
Equipment, instruments or materials are identified in this paper in order to adequately specify the experimental details. Such identification of vendors and manufacturers does not imply recommendation by the National Institute of Standards and Technology (NIST) nor does it imply the materials or equipment are necessarily the best available for the purpose.
Peptide Synthesis
Peptides were prepared at a 0.25 mM scale on Rink amide resin using a CEM Liberty Blue synthesizer. Standard microwave assisted Fmoc-based protocols were employed. Amino acids, resin and activator were purchased from ChemPep and CEM and used as received. All solvents were analytical grade (Fisher Scientific). Peptide cleavage was achieved by shaking peptide solutions for 2 hours in a cleavage cocktail comprising (by volume) 95% trifluoroacetic acid (TFA), 2.5 % triisoproylsilane, and 2.5 % Milli-Q water. The peptide was then precipitated by adding the cleavage cocktail and cleaved peptide to diethyl ether, and the mixture was then centrifuged and the supernatant discarded. The process of suspending in diethyl ether, centrifuging, and discarding the supernatant was repeated a total of three times. The resulting peptide was then dissolved in water and lyophilized.
Peptide Purification
Purification was performed via reverse-phase HPLC using a BEH130 Prep C18 10 μm column (XBridge, Waters Corporation, Milford, MA). Crude peptides were dissolved in Milli-Q water containing 0.1 % (by volume) TFA and were filtered (0.20 μm filter, Corning, Inc., Corning, NY) before HPLC injection. Products were subjected to an elution gradient (Quaternary Gradient Module (Waters 2545), Waters Corporation) of 100% Milli-Q water with 0.1 %-vol TFA to 40% Milli-Q water with 0.1 %-vol TFA and 60 % acetonitrile with 0.1 %-vol TFA within 60 min. Fractions were detected using UV-Vis detection at 214 nm (Waters 2489, Waters Corporation) and collected (Waters Fraction Collector III, Waters Corporation). The collected fractions were examined by ESI-mass spectrometry (LCQ Advantage Mass Spectrometer System, Thermo Finnigan, San Jose, CA) with an auto sampler system (Surveyor Autosampler, Thermo Finnigan). Pure fractions were combined and lyophilized followed by analytical UPLC-MS (Waters Xevo G2-S QTof, Waters Corporation) to demonstrate single species purity.
Solution Assembly of Coiled-Coil Bundles
All buffers were prepared at 50 mM concentration in Milli-Q water by dissolving the corresponding buffer salts and adjusting the pH to the desired value. The prepared buffers were then filtered using a 0.2 um nylon filter. The buffers used are sodium acetate buffer at pH 4.5 and borate buffer at pH 10. For every sample, purified, freeze-dried peptide for a desired concentration was weighed and dissolved in buffer at the desired pH and then heated to 80°C for 30 minutes in order to disrupt any irregular aggregates or bundles. The samples were then allowed to cool to room temperature before any experiments were performed.
Circular Dichroism (CD) Spectroscopy
Secondary structures and the temperature-dependent behavior of the synthetic peptides were analyzed using CD spectroscopy on a Jasco J-820 spectropolarimeter (JASCO, Inc., Easton, MD). Sample solutions were prepared at 1*10−1 mM concentration in a quartz cuvette with 1 mm path length (110-QS, Hellma, Inc.). Pure borate or sodium acetate buffer solutions at 50 mM were used to dissolve the samples and for the background correction. Sample spectra were recorded from (190–250) nm at desired temperatures. Temperature was ramped at 20°C / hour with 15 minutes of equilibration time at each temperature point before recording a spectra. CD spectra were recorded with a 1 nm bandwidth and a 4 second response time for each data point and averaged over three runs. The ellipticity at 222 nm was used to monitor the alpha helical character and therefore the temperature-dependent unfolding of the peptides. CD data is reported as the mean residue ellipticity, [θ]MRE (deg cm2 dmol−1) which is the molar ellipticity per residue of the peptide.40
Analytical Ultra Centrifugation (AUC)
Sedimentation velocity (SV) experiments were performed in a Beckman Coulter XL-I analytical ultracentrifuge using an AN-50 8 cell titanium rotor loaded with 12 mm optical path length, 2-sector, Epon-charcoal centerpieces at a centrifugation speed of 5236 rad/s (50 kRPM); experiments were performed at the National Institute of Standards and Technology, Gaithersburg, MD. All experiments were performed at 20.0°C, after a minimum 2 h equilibration time and with reference buffers of the same composition as the sample solutions. A filling volume of 400 mL was employed in each sector of appropriate reference or sample; reference and sample buffers comprised 50 mM sodium acetate with 50 mMNaCl. Density and viscosity values for the buffer solution were measured independently using an Anton Paar 5000 M densitometer/ Lovis ME viscometer combination instrument. Measured values were 1.001127 g/cm3 and 1.016 mPa s, respectively. Radial interference scans were measured once per minute for each cell (≈ 480 scans collected/cell).
Analysis of the interference data was conducted using the C(s) model in SEDFIT version 15.01b.41,42 The partial specific volume for the peptide was estimated using the center point of the value range given by the embedded calculator based on the peptide sequence (0.719 cm3/g as the center of (0.7147 – 0.7249) mL/g). The meniscus was determined, and ƒ/ƒ0 was fit, for each experiment independently. Both were consistent with visual identification and across all experiments; regularization used was P=0.68.
Small Angle Neutron Scattering (SANS)
SANS measurements were conducted at the NIST Center for Neutron Research (NCNR), National Institute of Standards and Technology, Gaithersburg, MD on the NG-B 30m SANS beamline. Samples were dissolved in borate or sodium acetate buffer prepared in D2O.
A neutron beam with a mean wavelength, λ, of 0.6 nm was defined using a mechanical velocity selector. The wavelength spread (Δλ/λ) was 0.15 at full width half max. The 640 mm x 640 mm 3He proportional counter used has a spatial resolution of 5.08 mm x 5.08 mm. Sample-to-detector distances of 1, 4, and 13 m were used to provide a q range of approximately 0.004 to 0.500 Å–1, where q is the scattering wave vector defined by q = (4π/λ)sin(θ) and 2θ is the angle between the scattered neutron and the incident beam. Data obtained were corrected for background noise and radiation, detector inhomogeneity, as well as empty cell scattering. Intensities were normalized to an absolute scale relative to the empty beam transmission. The uncertainties of individual data points were calculated statistically from the number of averaged detector counts. Data fitting was performed as described in the supplemental information.
Results and Discussion
Computational Design of BNDL1 and BNDL2
The design of the peptide sequences has been described previously.26 Based on a mathematical model43 of coiled coils, the coordinates of a peptide backbone atoms were calculated. This approach allows the creation of an ensemble of coiled-coil scaffolds by controlling five geometrical parameters associated with the coiled-coil bundle. Each peptide helix in a D2 symmetric tetramer contains 29 residues to allow the accommodation of four heptad repeats.
Monte-Carlo simulated annealing methods were used to sample the ensemble of coiled-coil scaffolds and identify candidate bundle structures and sequences as local energy minima. Initially, 11 interior sites were varied and constrained to be hydrophobic amino acids. The lowest-energy configurations were identified and the most probable sequences, calculated by the statistical-mechanics-based design theory,27,44–47 were selected as potential candidates.
The remaining 18 sites of the BNDL peptides were designed using the statistical-mechanics based design theory but in the context of an isolated tetrameric bundle. No information regarding interbundle interactions and a regular lattice nanostructure was included in the calculations.26 At the 18 exterior sites of each peptide, 18 natural amino acids (Pro and Cys were excluded) were allowed. The most probable sequences of BNDL1 and BNDL2 were designed using effective inverse temperatures of 0.5 mol/kcal and 1.5 mol/kcal respectively;27 BNDL2 has lower internal energy than BNDL1. This choice yields large numbers of complementary, low-energy interactions between residues on the the exterior of BNDL2. Electrostatic interactions play a prominent role in determining sequence identity, and the resulting sequence of BNDL2 has only ionizable residues on its exterior (Figure 1). Computational models of each designed structure were assessed using Molprobity.48 Both candidate sequences were synthesized, purified and characterized.
Solution Characterization of BNDL1 and BNDL2
Due to the hydrophobic nature of the residues designed to comprise the core of the designed bundles, close attention to the isoelectric point of the peptides was required to ensure solubility in aqueous solution. The isoelectric points of BNDL1 and BNDL2 were estimated to be pI = 4.4 and pI = 10.5, respectively, based upon the sequences and the pKa’s of the individual amino acids (S3). Correspondingly, BNDL1 was found to be soluble in basic conditions and BNDL2 was found to be soluble in acidic conditions. Subsequent studies were conducted with BNDL1 in 50 mM, pH 10 sodium borate buffer and BNDL2 in 50 mM, pH 4.5 sodium acetate buffer.
CD studies of the bundles were conducted to confirm their secondary structures. The acquired data (Figure 2) show the characteristic shape and minima expected for alpha helices; the ratios of the mean residue ellipticities (MRE) of the characteristic minima of 222 nm to 208 nm are 1.01 for BNDL1 and 1.04 for BNDL2 at 5°C, indicative of the presence of a coiled-coil tertiary structure.40,49,50 Performing CD measurements in solutions with trifluoroethanol (TFE), which is known to stabilize individual helices while being disruptive to coiled coils, also helps to demonstrate the presence of the coiled coil. Samples prepared in solutions of 50 % (by volume) TFE and 50 % (by volume) buffer exhibited a reduction in the ratio of the minima to 0.90 for both sequences, (S5) suggesting the disruption of the coiled coil by TFE.51,52
Figure 2 –
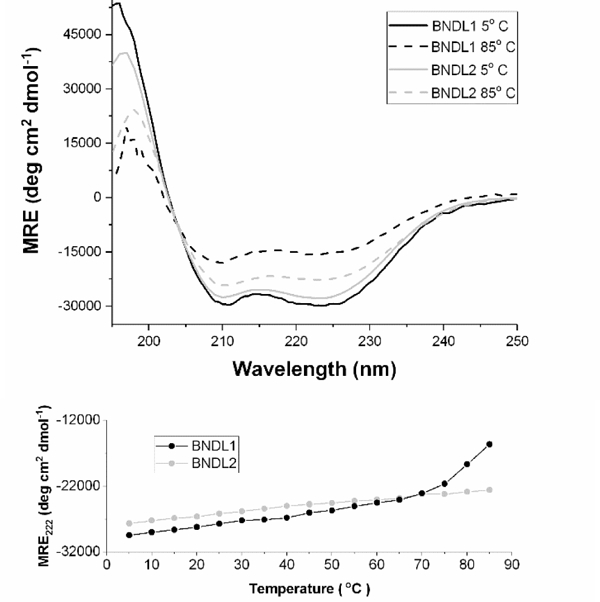
(Top) Circular dichroism spectra of BNDL1 (black) and BNDL2 (gray) upon heating from 5°C (solid) to 85°C (dashed). MRE: mean residue ellipticity. Alpha helical character is observable throughout. The solutions contained 0.1 mM peptide in 50 mM pH 10 sodium borate buffer (BNDL1) and 50 mM pH 4.5 sodium acetate buffer (BNDL2). (Bottom) The MRE at 222 nm is plotted as a function of temperature.
The thermal stability of each sequence was investigated by heating the peptide solutions. Both sequences display excellent thermal stability. At elevated temperature, this is apparent in the retention of both the characteristic shape and minima observed for an alpha helix as well as the ratio of the minima indicative of coiled-coil structure. BNDL1 and BNDL2 each retain CD spectra consistent with alpha-helical structures, even at 85°C, though there is a diminution in the absolute ellipticity for each sequence. The unfolding of each bundle was monitored using the mean residue ellipticity (MRE) at 222 nm as a function of the temperature (Figure 2B). For each sequence there is no clear inflection point apparent in this curve, i.e., there is no indication of cooperative loss of secondary structure over the accessible temperature range. The lack of such a transition suggests that BNDL1 and BNDL2 do not unfold completely at 85°C even after 12 hours. The increase in MRE at 222 nm that begins around 70°C for BNDL1 suggests the onset of unfolding; however, there is no plateau observed at higher temperature to indicate that the helices have fully unfolded within the timescale of the measurements. At 85°C the ratio of the MRE between the characteristic minima of an alpha helix, 222 nm and 208 nm, decreased to 0.90 for BNDL1 and 0.93 for BNDL2 indicating the coiled-coil structure has been slightly disrupted even though alpha helical structure is maintained at elevated temperatures. The net helical propensities of the residues in each sequence55 suggest that BNDL2 should have a higher helical propensity than BNDL1, which is in agreement with the experimental thermal stability suggested by the CD spectra, where BNDL2 appears to have greater helical content at high temperatures. For the circular dichroism thermal stability experiments 85°C was chosen as the maximum temperature due to the aqueous boiling point of the buffer. This temperature is a reasonable limit due to the primary solution condition for most experiments being room temperature. Given that the two sequences share interior residues, the high thermal stability of each is likely a direct result of the designed hydrophobic core. The coiled-coil character of both sequences is maintained even at high temperature. This core stability enables the peptides to tolerate variation of their exterior residues, allowing variation of a central structure and hydrophobic core to yield two distinct sequences with acidic and basic isoelectric points, respectively. The observations also confirm that designed, complementary electrostatic interactions on the exterior of the bundle can further enhance thermal stability.
AUC sedimentation velocity experiments also strongly indicate that the dominant morphology of the bundles is in the tetrameric form. For each sequence, the signal-weighted differential sedimentation coefficient distribution found that greater than 88 % of the signal was due to a component with sedimentation coefficient s(w,20) = 1.65 – 1.8 Sv. This observation is consistent with MWs for the bundles of MW(BNDL1) = 14.7 kDa and MW(BNDL2) = 16 kDa (S6). Based on the theoretical molecular weights of the peptide sequences, MW(BNDL1) = 3.4 kDa and MW(BNDL1) = 3.7 kDa, the data strongly suggest a tetrameric form for each peptide in solution. This theoretical MW of the tetrameric form is well within the uncertainty in MW values from the measurements due to shape factors and calculated parameters.
Having confirmed the computational design of the tetrameric coiled-coil bundles by AUC and CD, we sought to experimentally determine the shape and size of the bundles in solution and determine how they interact. Samples were therefore investigated using small angle neutron scattering (SANS) (Figure 3). Concentration series spanned 1 – 15 mM peptide (BNDL1 and BNDL2) in 50 mM sodium borate buffer or 50 mM sodium acetate buffer (prepared in D2O), respectively. All of the peptide solutions were visibly clear, suggesting that there was no large-scale or permanent aggregation.
Figure 3 –
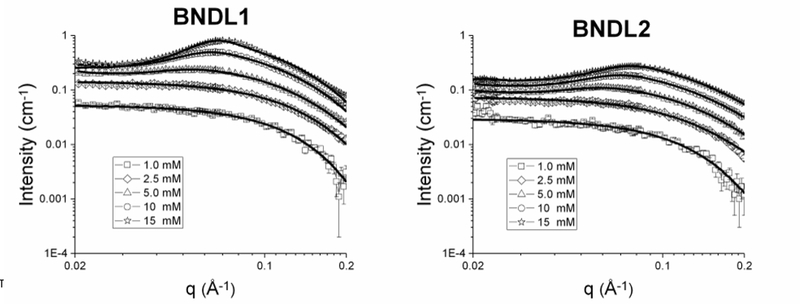
SANS concentration series of BNDL1 (left) and BNDL2 (right) from 1.0 – 15 mM with corresponding fitted models (solid black curves). All fits were performed using the same model that includes both form and structure factor components. Peptides were dissolved in buffers prepared in deuterium oxide, specifically 50 mM pH 10 sodium borate buffer for BNDL1 and pH 4.5 50 mM sodium acetate buffer for BNDL2.
The scattered intensity, I(q), is a function of the scattering vector, q, given by the expression:
The intensity is determined by the number density of scatterers, N, the volume, V, and the difference between the scattering length densities (SLD) of the scatterer and solvent, . The functions in Equation 1 include P(q), the form factor, which depends on the size and shape of individual scatterers while the interparticle interactions are described by the structure factor, S(q).
At low concentrations (1 mM), we may expect to see single particle form factor scattering which is representative of individual coiled-coil bundles.26 Increasing concentration will likely cause the bundles to become more crowded and to interact with each other nonspecifically. Neutron scattering can reveal such interparticle correlations present in the structure factor.56 The interaction of natural proteins in crowded, concentrated conditions has been widely studied, and such efforts have been performed to observe the stability of proteins in pharmaceutical formulations57 as well as intermolecular interactions among disease-causing amyloid proteins58. Such measurements also allow comparison of the solution behavior of the computationally designed system with that of natural proteins.
A cylindrical form factor alone, without a structure factor, well fit the low concentration, single particle scattering data from SANS (ST1).59 The goodness of the cylinder fit26 is not unexpected due to the anticipated, designed structure of the coiled-coil bundles and the use of this model to fit similarly sized and structured proteins.60,61 The average length and radius from all concentrations within a 95 % confidence interval are 4.37 + 0.19 nm and 1.26 + 0.04 nm, respectively, for BNDL1 and 4.37 + 0.25 nm and 1.10 + 0.05 nm, respectively, for BNDL2. We directly compared the cylinder model with the SASSIE PDB fit62,63 and found them to model the form factor similarly well. SASSIE is able to generate a scattering profile using a protein data base file (PDB) coordinates of the heavy atoms in a protein. This technique of fitting scattering data with models created from PDB files previously has succeeded in the study of structures of many protein systems, including monoclonal antibodies64 and DNA-binding proteins65. In fact, while the SASSIE fit to the SANS data for both coiled coil designs is good, the cylinder form factor better approximated the size of BNDL1 through a slightly better fit with the data at high q values (S7).62,63
At higher peptide concentrations, interparticle correlations are observed in the scattering structure factor. Assuming the nearest neighbor distance between coiled coils within an aggregate obeys a Gaussian distribution, the number of bundles present and the average distance between bundles within an aggregate can be approximated with a structure factor fit to the data (Figure 4).66,67 This kind of model has also been used to study concentrated behavior of naturally occurring lysozyme proteins66 as well as exchanged clay dispersions68.
Figure 4 –
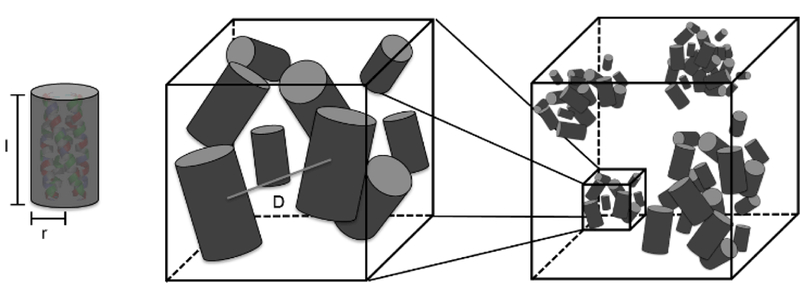
Representation of the solution behavior for SANS fitting. Each individual bundle is fit with a cylinder form factor. At high concentrations, the behavior of these bundles is fit using an aggregate model. The coiled-coil bundles loosely and nonspecifically associate to form soluble aggregates. The fit parameters indicated are the length of the cylinder, l, the radius of the cylinder, r, and the distance between bundles in an aggregate, D.
The structure factor contribution is apparent at concentrations of 5.0 mM and above, as indicated by the onset of a maximum between q values of 0.05 and 0.09 Å−1 (Figure 3); the maximum shifts to higher q with increasing concentration due to closer inter-bundle packing within the soluble aggregates. This trend can be modeled by the structure factor fits shown in Table 1; with an increased bundle concentration, the distance between coiled coils, D, in an aggregate decreased, as expected. Due to the convolution of the form factor and structure factor in the total scattering intensity, the real space distance corresponding to the maximum of the correlation peak does not match the separation distance determined by fitting the data. This explains why both peptides were fit to have a separation distance of 3.0 nm (Table 1) when the peak maxima are not located at the same q (Figure 3). The increased number of bundles per aggregate in BNDL1 over BNDL2 may also give rise to the sharper intensity of the correlation peak observed for BNDL1. The number of bundles per aggregate may be higher for BNDL1 due to the incorporation of polar residues, which can form hydrogen bonds, and also results in a lower surface charge for BNDL1 than for BNDL2. Fitting was performed over mid to high q in order to determine the size of the bundles and distance between coiled coils in the more concentrated samples. Importantly, the combination of optical clarity in the concentrated coiled coil suspensions and the presence of clear structure factor scattering is indicative of soluble aggregate behavior. Soluble aggregates are distinct from permanent aggregates or precipitates in that they are dynamic, loosely bound with a higher average local concentration of coiled coils than in bulk solution. The interactions causing the dynamic clustering of bundles are combinations of oppositely charged, mildly hydrophobic, and polar regions on the bundle surfaces (Figure 1). The soluble aggregate state is commonly seen in concentrated protein solutions, sometimes as a precursor state for a transition to a permanent aggregate56,58,69 while other times a stable suspension of dynamic aggregates70. In the coiled-coil system here, the soluble aggregated state observed in more highly concentrated bundle samples is stable for at least one month as observed via SANS experiments, therefore behaving in solution as designed and not further assembling or aggregating into a more regular, stable structure.
Table 1 -.
Fit parameters from SANS concentration series fits for BNDL1 and BNDL2 from 1.0 mM to 15 mM. The displayed parameters include the number of bundles per aggregate (N) and distance between bundles in an aggregate (D) in nanometers from the structure factor fit.
| Concentration | Sequence | N | D (nm) |
|---|---|---|---|
| 1.0 mM | BNDL1 | 1 | N/A |
| BNDL2 | 1 | N/A | |
| 2.5 mM | BNDL1 | 1 | N/A |
| BNDL2 | 1 | N/A | |
| 5.0 mM | BNDL1 | 69 | 4.7 |
| BNDL2 | 60 | 4.4 | |
| 10 mM | BNDL1 | 76 | 3.5 |
| BNDL2 | 62 | 3.4 | |
| 15 mM | BNDL1 | 79 | 3.0 |
| BNDL2 | 60 | 3.0 |
An upturn in SANS intensity at low q is observed for all concentrations of both sequences which is most likely the scattering from the overall soluble aggregate domains comprised of many bundles or long-range concentration fluctuations in dilute bundle solutions (S8). This upturn can be fit, without changing the fit parameters obtained using the mid and high q data, with an additional term that describes the distance between the soluble aggregates (S8, ST2). This type of large scale solution structure is seen in many solution assembled systems with micellar nanostructure such as casein micelles containing colloidal calcium phosphate nanoclusters71 as well as hydrogels comprised of beta hairpin fibrils72 or diblock copolymers73. The SANS data are consistent with the cylindrical structures expected for the designed bundles and reveal that the solution structure of the soluble aggregates is similar for each sequence, despite the large differences in their exterior amino acid residues.
Conclusions
The experimental confirmation, via CD, AUC, and SANS, of the formation of soluble coiled-coil bundles from computationally predicted sequences supports the successful preparation of robust, soluble, coiled-coil bundles in solution. The coiled-coil character apparent in the CD data, combined with AUC molecular weight observations, confirm that a peptide tetramer is dominant in solution. The hydrophobic interior core design of the coiled coil provides stability for two different bundle exteriors. In addition, SANS helped demonstrate that these synthetic, computationally derived bundles behave similarly to naturally occurring proteins in solution, forming soluble aggregates at higher concentrations. This further supports the robust character of these bundles that were designed with the primary goal of achieving a stable tetrameric unit. At the highest concentrations of peptide, the bundles remained soluble with an average interbundle separation distance of 3–5 nm within a soluble aggregate. Previous efforts have shown that specifically modifying the exterior residues of the bundle using computational design yielded a variety of unique, solution-assembled materials that form as a result of noncovalent, interbundle interactions.26 Herein we have characterized the structural features of the dissociated state and its interbundle correlations. Due to the design of the stable hydrophobic core, modifying the amino acids that lie on the exterior of the coiled coil did not drastically alter the formation of the bundle or the nature of the soluble aggregated state. The stability and vast possibility in amino acid sequence of these coiled coil bundles makes them excellent candidates for model colloidal systems of anisotropic particles, ideas typically reserved for rod-like assemblies of natural proteins56,74 and viruses73–75 as well as potential anisotropic building blocks for novel materials76.
Supplementary Material
Acknowledgements
We acknowledge the financial support from NSF DMREF (Designing Materials to Revolutionize and Engineer our Future) program under awards DMR-1234161 and DMR-1235084. J.G.S. acknowledges additional support from the Penn Laboratory for Research on the Structure of Matter (NSF DMR-1120901). Peptide modelling and design employed the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by NSF grant no. ACI-1053575, under grant no. TG-CHE110041. The National Institutes of Health (RO1 EB006006) are acknowledged for support and the UD COBRE NIH-COBRE 1P30 GM110758 and 1P20 RR017716 are acknowledged for instrument resources. The Delaware INBRE grant no. P20 GM103446 is acknowledged for support of the Delaware Biotechnology Institute. The publication was made possible by NIH grant no. P30 GM103519 from the National Institute for General Medical Sciences. The contents of the paper do not necessarily reflect the views of NIH. This manuscript was prepared under cooperative agreement # 370NANB17H302 from NIST, U.S. Department of Commerce. We acknowledge the support of the National Institute of Standards and Technology, U.S. Department of Commerce, in providing the neutron research facilities used in this work. This work utilized facilities supported in part by the National Science Foundation under Agreement No. DMR-0944772. The statements, findings, conclusions and recommendations are those of the authors and do not necessarily reflect the view of NIST or the U.S. Department of Commerce. Access to NGB 30m SANS was provided by the Center for High Resolution Neutron Scattering, a partnership between the National Institute of Standards and Technology and the National Science Foundation under Agreement No. DMR-1508249.
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Daamen WF, Veerkamp JH, van Hest JCM and van Kuppevelt TH, Biomaterials, 2007, 28, 4378–4398. [DOI] [PubMed] [Google Scholar]
- 2.Uchida M, Klem MT, Allen M, Suci P, Flenniken M, Gillitzer E, Varpness Z, Liepold LO, Young M and Douglas T, Adv. Mater, 2007, 19, 1025–1042. [Google Scholar]
- 3.Cherny I and Gazit E, Angew. Chemie - Int. Ed, 2008, 47, 4062–4069. [DOI] [PubMed] [Google Scholar]
- 4.Hardy JG, Romer LM and Scheibel TR, Polymer (Guildf), 2008, 49, 4309–4327. [Google Scholar]
- 5.Niemeyer CM, Angew. Chemie - Int. Ed, 2001, 40, 4128–4158. [DOI] [PubMed] [Google Scholar]
- 6.Dickerson MB, Sandhage KH and Naik RR, Chem. Rev, 2008, 108, 4935–4978. [DOI] [PubMed] [Google Scholar]
- 7.Fletcher JM, Harniman RL, Barnes FRH, Boyle AL, Collins A, Mantell J, Sharp TH, Antognozzi M, Booth PJ, Linden N, Miles MJ, Sessions RB, Verkade P and Woolfson DN, Science (80-. ), 2013, 340, 595–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ross J, Bridges A, Fletcher JM, Deborah S, Alibhai D, Bray H, Beesley JL, Dawson WM, Hodgson L, Mantell J, Verkade P, Edge C, Sessions RB, Tew D and Woolfson DN, ACS Nano, , DOI: 10.1021/acsnano.7b02368. [DOI] [PubMed] [Google Scholar]
- 9.Jiang T, Xu C, Liu Y, Liu Z, Wall JS, Zuo X, Lian T, Salaita K, Ni C, Pochan D and Conticello VP, J. Am. Chem. Soc, 2014, 136, 4300–4308. [DOI] [PubMed] [Google Scholar]
- 10.Przybyla DE and Chmielewski J, J. Am. Chem. Soc, 2010, 132, 7866–7. [DOI] [PubMed] [Google Scholar]
- 11.Xu C, Liu R, Mehta AK, Guerrero-Ferreira RC, Wright ER, Dunin-Horkawicz S, Morris K, Serpell LC, Zuo X, Wall JS and Conticello VP, J. Am. Chem. Soc, 2013, 135, 15565–15578. [DOI] [PubMed] [Google Scholar]
- 12.Valéry C, Paternostre M, Robert B, Gulik-Krzywicki T, Narayanan T, Dedieu J-C, Keller G, Torres M-L, Cherif-Cheikh R, Calvo P and Artzner F, Proc. Natl. Acad. Sci. U. S. A, 2003, 100, 10258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papapostolou D, Smith AM, Atkins EDT, Oliver SJ, Ryadnov MG, Serpell LC and Woolfson DN, Proc. Natl. Acad. Sci, 2007, 104, 10853–10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zimenkov Y, Dublin SN, Ni R, Tu RS, Breedveld V, Apkarian RP and Conticello VP, J. Am. Chem. Soc, 2006, 128, 6770–6771. [DOI] [PubMed] [Google Scholar]
- 15.Dublin SN and Conticello VP, J. Am. Chem. Soc, 2008, 130, 49–51. [DOI] [PubMed] [Google Scholar]
- 16.Przybyla DE and Chmielewski J, J. Am. Chem. Soc, 2008, 130, 12610–12611. [DOI] [PubMed] [Google Scholar]
- 17.Rothemund PWK, Nature, 2006, 440, 297–302. [DOI] [PubMed] [Google Scholar]
- 18.Seeman NC, Annu. Rev. Biochem, 2010, 79, 65–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han D, Pal S, Yang Y, Jiang S, Nangreave J, Liu Y and Yan H, Science, 2013, 339, 1412–5. [DOI] [PubMed] [Google Scholar]
- 20.Ke Y, Ong LL, Shih WM and Yin P, Science, 2012, 338, 1177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ke Y, Voigt NV, Gothelf KV and Shih WM, J. Am. Chem. Soc, 2012, 134, 1770–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.King NP, Sheffler W, Sawaya MR, Vollmar BS, Sumida JP, Andre I, Gonen T, Yeates TO and Baker D, Science, 2012, 336, 1171–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.King NP, Bale JB, Sheffler W, McNamara DE, Gonen S, Gonen T, Yeates TO and Baker D, Nature, 2014, 510, 103–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonen S, DiMaio F, Gonen T and Baker D, Science, 2015, 348, 1365–1368. [DOI] [PubMed] [Google Scholar]
- 25.Brodin JD, Ambroggio XI, Tang C, Parent KN, Baker TS and Tezcan FA, Nat. Chem, 2012, 4, 375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.V Zhang H, Polzer F, Haider MJ, Tian Y, Villegas JA, Kiick KL, Pochan DJ and Saven JG, Sci. Adv, 2016, 2, e1600307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lanci CJ, MacDermaid CM, Kang S. -g., Acharya R, North B, Yang X, Qiu XJ, DeGrado WF and Saven JG, Proc. Natl. Acad. Sci, 2012, 109, 7304–7309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gradišar H, Božič S, Doles T, Vengust D, Hafner-Bratkovič I, Mertelj A, Webb B, Šali A, Klavžar S and Jerala R, Nat. Chem. Biol, 2013, 9, 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jordan M, Harniman L, Frederick RH, Aimee L, Collins A, Antognozzi M, Booth PJ, Mervyn J, Sessions RB, Verkade P and Woolfson DN, Science (80-. ), 2013, 340, 595–599. [Google Scholar]
- 30.Dong H, Paramonov SE and Hartgerink JD, J. Am. Chem. Soc, 2008, 130, 13691–13695. [DOI] [PubMed] [Google Scholar]
- 31.Zimenkov Y, Conticello VP, Guo L and Thiyagarajan P, Tetrahedron, 2004, 60, 7237–7246. [Google Scholar]
- 32.Thomson A, Wood C, Burton A, Bartlett G, Sessions R, Brady R and Woolfson D, Science, 2014, 346, 485–488. [DOI] [PubMed] [Google Scholar]
- 33.Armstrong CT, Boyle AL, Bromley EHC, Mahmoud ZN, Smith L, Thomson AR and Woolfson DN, R. Soc. Chem, 2009, 143, 305–317. [DOI] [PubMed] [Google Scholar]
- 34.O’Shea EK, Lumb KJ and Kim PS, Curr. Biol, 1993, 3, 658–667. [DOI] [PubMed] [Google Scholar]
- 35.DeGrado WF, Wasserman ZR and Lear JD, Science, 1989, 243, 622–628. [DOI] [PubMed] [Google Scholar]
- 36.Lau SYM, Taneja AK and Hodges RS, J. Biol. Chem, 1984, 259, 13253–13261. [PubMed] [Google Scholar]
- 37.Litowski JR and Hodges RS, J. Biol. Chem, 2002, 277, 37272–37279. [DOI] [PubMed] [Google Scholar]
- 38.Koga N, Tatsumi-Koga R, Liu G, Xiao R, Acton TB, Montelione GT and Baker D, Nature, 2012, 491, 222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vincent TL, Woolfson DN and Adams JC, Int. J. Biochem. Cell Biol, 2013, 45, 2392–2401. [DOI] [PubMed] [Google Scholar]
- 40.Graddis TJ, Myszka DG and Chaiken IM, Biochemistry, 1993, 32, 12664–12671. [DOI] [PubMed] [Google Scholar]
- 41.Schuck P, Biophys. J, 2000, 78, 1606–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schuck P, Zhao H, Brautigam CA and Ghirlando R, Basic Principles of Analytical Ultracentrifugation, CRC Press, Boca Raton, FL, 2015. [Google Scholar]
- 43.Grigoryan G and Degrado WF, J. Mol. Biol, 2011, 405, 1079–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fry HC, Lehmann A, Sinks LE, Asselberghs I, Tronin A, Krishnan V, Blasie JK, Clays K, Degrado WF, Saven JG and Therien MJ, J. Am. Chem. Soc, 2013, 135, 13914–13926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McAllister KA, Zou H, Cochran FV, Bender GM, Senes A, Fry HC, Nanda V, Keenan PA, Lear JD, Saven JG, Therien MJ, Blasie JK and DeGrado WF, J. Am. Chem. Soc, 2008, 130, 11921–11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bender GM, Lehmann A, Zou H, Cheng H, Fry HC, Engel D, Therien MJ, Blasie JK, Roder H, Saven JG and DeGrado WF, J. Am. Chem. Soc, 2007, 129, 10732–10740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kono H and Saven JG, J. Mol. Biol, 2001, 306, 607–628. [DOI] [PubMed] [Google Scholar]
- 48.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS and Richardson DC, Acta Crystallogr. Sect. D Biol. Crystallogr, 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cooper TM and Woody RW, Biopolymers, 1990, 30, 657–676. [DOI] [PubMed] [Google Scholar]
- 50.Monera OD, Zhou NE, Kay CM and Hodges RS, J. Biol. Chem, 1993, 268, 19218–19227. [PubMed] [Google Scholar]
- 51.Monera OD, Kay CM and Hodges RS, Biochemistry, 1994, 33, 3862–3871. [DOI] [PubMed] [Google Scholar]
- 52.Zhou NE, Kay CM and Hodges RS, Biochemistry, 1992, 31, 5739–5746. [DOI] [PubMed] [Google Scholar]
- 53.Betz SF, Fairman R, O’Neil K, Lear JD and Degrado WF, Philos. Trans. R. Soc. Lond. B. Biol. Sci, 1995, 348, 81–88. [DOI] [PubMed] [Google Scholar]
- 54.Bolisetty S, Harnau L, Jung JM and Mezzenga R, Biomacromolecules, 2012, 13, 3241–3252. [DOI] [PubMed] [Google Scholar]
- 55.Kanai S, Liu J, Patapoff TW and Shire SJ, J. Pharm. Sci, 2008, 97, 4219–4227. [DOI] [PubMed] [Google Scholar]
- 56.Harper JD and Lansbury PT, Annu. Rev. Biochem, 1997, 66, 385–407. [DOI] [PubMed] [Google Scholar]
- 57.SasView, http://www.sasview.org.
- 58.Nayak A, Sorci M, Krueger S and Belfort G, Proteins, 2009, 74, 556–565. [DOI] [PubMed] [Google Scholar]
- 59.Oliveira CLP, Behrens MA, Pedersen JS, Erlacher K, Otzen D and Pedersen JS, J. Mol. Biol, 2009, 387, 147–161. [DOI] [PubMed] [Google Scholar]
- 60.Watson MC and Curtis JE, J. Appl. Crystallogr, 2013, 46, 1171–1177. [Google Scholar]
- 61.Curtis JE, Raghunandan S, Nanda H and Krueger S, Comput. Phys. Commun, 2012, 183, 382–389. [Google Scholar]
- 62.Yearley EJ, Godfrin PD, Perevozchikova T, Zhang H, Falus P, Porcar L, Nagao M, Curtis JE, Gawande P, Taing R, Zarraga IE, Wagner NJ and Liu Y, Biophys. J, 2014, 106, 1763–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krueger S, Shin JH, Raghunandan S, Curtis JE and Kelman Z, Biophys. J, 2011, 101, 2999–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krueger S, Ho D and Tsai A, in Proteins Misbehaving, eds. R. M. Murphy and A. M. Tsai, Springer, New York, NY, 2006, pp. 125–146. [Google Scholar]
- 65.Kratky O and Porod G, J. Colloid Sci, 1949, 4, 35–70. [DOI] [PubMed] [Google Scholar]
- 66.Hanley H, Muzny C, Ho D and Glinka C, Langmuir, 2003, 19, 5575–5580. [Google Scholar]
- 67.Azakami H, Mukai A and Kato A, J. Agric. Food Chem, 2005, 53, 1254–1257. [DOI] [PubMed] [Google Scholar]
- 68.Rajagopalan S, Kurt N and Cavagnero S, Biophys. J, 2011, 100, 747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mata JP, Udabage P and Gilbert EP, Soft Matter, 2011, 7, 3837. [Google Scholar]
- 70.Hule RA, Nagarkar RP, Altunbas A, Ramay HR, Branco MC, Schneider JP and Pochan DJ, Faraday Discuss, 2008, 139, 251–264-325, 419–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pochan DJ, Pakstis L, Ozbas B, Nowak AP and Deming TJ, Macromolecules, 2002, 35, 5358–5360. [Google Scholar]
- 72.Bolisetty S, Adamcik J and Mezzenga R, Soft Matter, 2011, 7, 493–499. [Google Scholar]
- 73.Dogic Z and Fraden S, Curr. Opin. Colloid Interface Sci, 2006, 11, 47–55. [Google Scholar]
- 74.Flynn CE, Lee SW, Peelle BR and Belcher AM, Acta Mater, 2003, 51, 5867–5880. [Google Scholar]
- 75.Lee SW, Mao C, Flynn CE and Belcher AM, Science, 2002, 296, 892–895. [DOI] [PubMed] [Google Scholar]
- 76.Glotzer SC and Solomon MJ, Nat. Mater, 2007, 6, 557–562. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.