

The fully human monoclonal antibody bezlotoxumab is indicated for preventing the recurrence of Clostridioides difficile (formerly Clostridium difficile) infection (CDI) in adults who receive antibacterial treatment for CDI and who are at high risk for a CDI recurrence. The efficacy and safety of 10-mg/kg of body weight bezlotoxumab were demonstrated in two phase 3 trials: the MODIFY I (ClinicalTrials.gov registration number NCT01241552) and MODIFY II (ClinicalTrials.gov registration number NCT01513239) trials.
KEYWORDS: Clostridium difficile, bezlotoxumab, population pharmacokinetics
ABSTRACT
The fully human monoclonal antibody bezlotoxumab is indicated for preventing the recurrence of Clostridioides difficile (formerly Clostridium difficile) infection (CDI) in adults who receive antibacterial treatment for CDI and who are at high risk for a CDI recurrence. The efficacy and safety of 10-mg/kg of body weight bezlotoxumab were demonstrated in two phase 3 trials: the MODIFY I (ClinicalTrials.gov registration number NCT01241552) and MODIFY II (ClinicalTrials.gov registration number NCT01513239) trials. Here, a population pharmacokinetic (popPK) analysis, performed using data from the MODIFY I and II trials (n = 1,515) and from three phase 1 trials (n = 72) to characterize bezlotoxumab pharmacokinetics (PK) in phase 3 clinical trial participants and in healthy subjects, is reported. A stepwise covariate search was conducted to identify factors influencing PK. Post hoc-estimated bezlotoxumab exposures from the popPK model were used to conduct an exposure-response analysis for CDI recurrence. Bezlotoxumab PK were described by a two-compartment model with linear elimination and allometric scaling for clearance and the volume of distribution by body weight. Although the final popPK model included gender, ethnicity (Japanese descent), race (black versus nonblack), and albumin level as significant covariates, the impact of these factors was not clinically meaningful, based on the totality of the PK and clinical experience. Exposure-response analysis of CDI recurrence demonstrated a similar low rate of CDI recurrence over the entire range of exposures achieved in the phase 3 trials, indicating that exposures were on the maximal response plateau of the exposure-response curve. Overall, the analyses confirmed the appropriateness of the 10-mg/kg dose across the phase 3 trial population with no dose adjustments necessary over a broad demographic background.
INTRODUCTION
Clostridioides difficile (formerly Clostridium difficile), an anaerobic, spore-forming, Gram-positive bacillus, is the most common cause of infectious diarrhea in hospitalized patients given broad-spectrum antibiotics (1, 2). C. difficile infection (CDI) is mediated by the actions of two toxins produced by C. difficile, toxin A and toxin B. These toxins target gut epithelium, causing cellular morphological changes that result in disruption of the intestinal barrier function (3, 4). The symptoms of CDI include mild to severe diarrhea and abdominal pain that can progress to severe manifestations, including fulminant colitis, sepsis, and death (3, 5). Although most cases of CDI resolve after initial antibacterial drug treatment for CDI, approximately 25% of individuals will experience recurrent CDI (rCDI) (6, 7). Furthermore, the likelihood of recurrence increases with each subsequent episode, such that approximately 40% of individuals with a first recurrence will experience a second recurrence (6, 8). An inadequate immune response to toxin A and/or B, manifested as low levels of antibodies against the toxins, is associated with an increased risk of recurrence (9, 10).
Actoxumab (MK-3415) and bezlotoxumab (MK-6072) are fully human IgG monoclonal antibodies (MAbs) that bind to and neutralize toxin A and toxin B, respectively. Two randomized, double-blind, phase 3 trials, the MODIFY I and MODIFY II trials, assessed the safety and efficacy of single 10-mg/kg of body weight infusions of bezlotoxumab, actoxumab (MODIFY I only), and actoxumab plus bezlotoxumab compared with placebo in adults receiving antibacterial drug treatment for primary or recurrent CDI. The primary endpoint was the proportion of participants with rCDI during 12 weeks of follow-up. Bezlotoxumab was associated with a statistically significant lower rate of rCDI than placebo and had a safety profile similar to that of placebo; the addition of actoxumab did not improve efficacy (11). In a post hoc analysis, participants at high risk of recurrence, as defined by the presence of at least one prespecified risk factor (≥1 CDI episode in the past 6 months; severe CDI at baseline; age ≥ 65 years; infection with ribotype 027, 078, or 244 at the baseline; and/or immunocompromise), had a statistically significant and clinically important reduction in rCDI with bezlotoxumab compared with placebo, while only a modest, nonsignificant reduction was seen in participants without any risk factors for recurrence (12). Based on these findings, bezlotoxumab at a dose of 10 mg/kg is now indicated for the prevention of rCDI in adults receiving antibacterial drug treatment for CDI who are at a high risk for rCDI (13, 14).
As bezlotoxumab is an intravenously administered MAb, its bioavailability is 100% with a limited extravascular distribution; no hepatic pathways would be expected to have a role in bezlotoxumab metabolism. Elimination is expected to occur primarily by protein catabolism (15), and thus, intrinsic factors, such as organ dysfunction or age, which typically have only a limited effect on the exposure of therapeutic antibodies (16), are not anticipated to affect the exposure of bezlotoxumab to a clinically meaningful extent.
Despite a low likelihood of pharmacokinetic (PK) variability, bezlotoxumab phase 3 trials included intensive PK sampling (≥6 samples/participant) of a large, diverse population. The richness of the final data set provided an opportunity to robustly assess covariate effects on bezlotoxumab PK using a population pharmacokinetic (popPK) modeling approach, thereby reducing the need for dedicated clinical studies in special populations.
The current analysis describes the development and qualification of a bezlotoxumab popPK model based on data from the phase 3 MODIFY trials and three phase 1 trials. The objectives of this analysis were to identify covariate factors influencing bezlotoxumab PK and to support an exposure-response (E-R) analysis between bezlotoxumab exposure and rCDI to confirm the appropriateness of the 10-mg/kg dose across the demography of the target population.
RESULTS
Pooled study population.
Clinical data were available from a total of 2,631 evaluable participants (MODIFY I, n = 1,396; MODIFY II, n = 1,163; phase 1 trials, n = 72) (Table 1). Demographic information and disease characteristics for all participants are summarized in Table 2.
TABLE 1.
Clinical trial data included in the popPK model and exposure-response analysisb
| Trial | Study population | Design | Treatment(s) | na | PK sampling |
|---|---|---|---|---|---|
| Phase 3 trials | |||||
| MODIFY I | Participants with CDI | Single dose adjunctive to antibacterial treatment for CDI | 10-mg/kg actoxumab | 232 | For all treatments, predose; 1 h postinfusion; and 3, 10, 28, 56, and 84 days postdose; also unscheduled if CDI recurred |
| 10-mg/kg bezlotoxumab | 384 | ||||
| 10-mg/kg actoxumab + 10-mg/kg bezlotoxumab | 382 | ||||
| Placebo | 398 | ||||
| MODIFY II | Participants with CDI | Single dose adjunctive to antibacterial treatment for CDI | 10-mg/kg actoxumab | 1 | For all treatments, predose; 1 h postinfusion; and 3, 10, 28, 56, 84, and 168 days postdose; also unscheduled if CDI recurred |
| 10-mg/kg bezlotoxumab | 395 | ||||
| 10-mg/kg actoxumab + 10 mg/kg bezlotoxumab | 389 | ||||
| Placebo | 378 | ||||
| Phase 1 trials | |||||
| PN004 | Healthy subjects | Two sequential doses | 10-mg/kg actoxumab + 10 mg/kg bezlotoxumab (repeated after 12 wk) | 30 | Predose; 0.5, 1, 2, 4, and 8 h and 2, 8, and 43 days after the first dose; predose on day 85 and then 0.5, 1, 2, 4, and 8 h and 86, 92, 127, 169, and 253 days postdose |
| PN005 | Healthy subjects | Single dose | 10-mg/kg actoxumab + 10-mg/kg bezlotoxumab | 29 | Predose and 0.5, 1, 2, 4, 6, 8, 24, and 72 h and 7, 10, 20, 28, 56, and 84 days postdose |
| PN006 | Japanese healthy subjects | Single dose | 10-mg/kg actoxumab + 10 mg/kg bezlotoxumab | 6 | For both treatments, predose and 0.5, 1, 2, 4, 6, 8, 24, and 72 h and 7, 10, 20, 28, 56, 84, and 168 days postdose |
| 20-mg/kg actoxumab + 20 mg/kg bezlotoxumab | 7 |
n, number of participants with evaluable PK data.
CDI, Clostridium difficile infection; PK, pharmacokinetics; popPK, population pharmacokinetics.
TABLE 2.
Pooled demographic information and disease characteristicsc
| Characteristic | Value for: |
|
|---|---|---|
| All participants | PopPK population | |
| No. (%) of participants | ||
| Total pooled participants | 2,631 (100) | 1,587 (100) |
| MODIFY I trial | 1,396 (53.1) | 752 (47.4) |
| MODIFY II trial | 1,163 (44.2) | 763 (48.1) |
| Phase 1 trials | 72 (2.7) | 72 (4.5) |
| Continuous covariates [mean (SD)] | ||
| Age (yr) | 62.6 (18) | 61.8 (18.4) |
| Body wt (kg) | 72.4 (20.3) | 72.3 (20.1) |
| BMI (kg/m2) | 26 (6.6) | 25.9 (6.4) |
| eGFR (ml/min/1.73 m2) | 87.5 (52.8) | 89.5 (54.5) |
| Albumin concn (g/dl) | 3.4 (0.8) | 3.5 (0.8) |
| WBC (103 cells/μl) | 8.7 (5) | 8.7 (5.1) |
| Categorical covariates [no. (%) of participants] | ||
| Gender | ||
| Male | 1,167 (44.4) | 721 (45.4) |
| Female | 1,464 (55.6) | 866 (54.6) |
| Race | ||
| White | 2,229 (84.7) | 1,313 (82.7) |
| Black | 140 (5.3) | 94 (5.9) |
| Asian | 212 (8.1) | 147 (9.3) |
| Other | 50 (1.9) | 33 (2.1) |
| Japanese descent | ||
| Non-Japanese | 2,524 (95.9) | 1,507 (95.0) |
| Japanese | 107 (4.1) | 80 (5.0) |
| Ethnicity | ||
| Non-Hispanic | 2,266 (86.1) | 1,382 (87.1) |
| Hispanic | 293 (11.1) | 159 (10.0) |
| Missing | 72 (2.7) | 46 (2.9) |
| Hospitalization status | ||
| Healthy subject | 72 (2.7) | 72 (4.5) |
| Outpatient | 828 (31.5) | 491 (30.9) |
| Inpatient | 1,731 (65.8) | 1,024 (64.5) |
| Antibiotic therapy for CDI | ||
| None | 72 (2.7) | 72 (4.5) |
| Metronidazole | 1,245 (47.3) | 738 (46.5) |
| Vancomycin | 1,227 (46.6) | 724 (45.6) |
| Fidaxomicin | 87 (3.3) | 53 (3.3) |
| Concomitant systemic antibiotics | ||
| No antibiotics | 1,566 (59.5) | 960 (60.5) |
| Antibiotics | 1,065 (40.5) | 627 (39.5) |
| Concomitant PPI use | ||
| No PPI use | 1,349 (51.3) | 833 (52.5) |
| PPI use | 1,282 (48.7) | 754 (47.5) |
| Modified Horn’s index | ||
| <3a | 1,898 (72.1) | 1,173 (73.9) |
| ≥3 | 733 (27.9) | 414 (26.1) |
| Charlson comorbidity index score | ||
| <3a | 1,577 (59.9) | 963 (60.7) |
| ≥3 | 1,054 (40.1) | 624 (39.3) |
| Zar scoreb | ||
| <2a | 2,239 (85.1) | 1,341 (84.5) |
| ≥2 | 392 (14.9) | 246 (15.5) |
| History of CDI in past 6 mo | ||
| Yes | 704 (26.8) | 408 (25.7) |
| No + unknown | 1927 (73.2) | 1,179 (74.3) |
| Hepatic impairment | ||
| Liver disease | 163 (6.2) | 101 (6.4) |
| No liver disease + missing | 2,468 (93.8) | 1,486 (93.6) |
Including healthy subjects.
Disease severity score, as described by Zar et al. (33).
BMI, body mass index; CDI, Clostridium difficile infection; eGFR, estimated glomerular filtration rate; popPK, population pharmacokinetics; PPI, proton pump inhibitor; SD, standard deviation; WBC, white blood cell count.
Population pharmacokinetic modeling.
In total, 8,784 evaluable bezlotoxumab concentrations from 1,587 participants (MODIFY I and II trials, n = 1,515; phase 1 trials, n = 72) who received either bezlotoxumab alone or bezlotoxumab in combination with actoxumab were included in the popPK analysis (Table 2). The popPK analysis population was comprised predominantly of female (55%), white (83%), non-Hispanic (87%) participants. Black and Asian participants comprised 6% and 9% of the population, respectively, with Japanese participants as a subset of Asian participants comprising 5% of the total population (Table 2). The overall median age and weight of the population were 65 years (range, 18 to 100 years) and 69.8 kg (range, 30 to 194 kg), respectively. Baseline albumin levels ranged from approximately 1 to 6 g/dl, with a median level of 3.4 g/dl. Normal albumin levels (≥3.5 g/dl) were observed in 49% of participants with CDI and all healthy subjects. Based upon the estimated glomerular filtration rate (eGFR), assessed by the modification of diet in renal disease formula, 43% of participants were classified as having normal renal function (eGFR ≥ 90 ml/min/1.73 m2). In total, 30% of participants had mild renal impairment, 18% had moderate renal impairment, and 8% had severe renal impairment or end-stage renal disease. In addition, 6% of participants were classified as having hepatic impairment, based on having two or more of the following criteria: an albumin concentration of ≤3.1 g/dl, an alanine aminotransferase (ALT) level ≥2 times the upper limit of normal (ULN), a bilirubin level ≥1.3 times the ULN, or mild, moderate, or severe liver disease, as reported in the Charlson comorbidity index score (17).
The PK of bezlotoxumab were characterized by a two-compartment model with linear elimination. As body weight is known to influence MAb clearance (CL) and volume of distribution (V) (18, 19), allometric scaling by body weight for CL and intercompartmental clearance (Q), and for the central volume of distribution (Vc) and peripheral volume of distribution (Vp), was included in the PK structural model. An allometric exponent of 0.48 was estimated for CL and Q and for Vc and Vp (Table 3). The inclusion of body weight as a covariate on these parameters resulted in an 800.1-point decrease in the objective function value (OFV) for the bezlotoxumab popPK model, indicating the strong relationship between body weight and bezlotoxumab PK and the importance of accounting for weight effects in the model. Interindividual variability (IIV) random effects on CL and Vc were also included. Residual variability was described using a log-additive error term to account for the high degree of variability observed in some participants.
TABLE 3.
PK parameter estimates for the final bezlotoxumab popPK modela
| Parameter or covariate | Estimate (CVb) | 95% confidence intervalc (CVb) | % relative standard errord | Shrinkage (%) |
|---|---|---|---|---|
| PK parameter | ||||
| CL (liter/day) | 0.281 | 0.275, 0.288 | 0.257 | |
| Vc (liters) | 3.43 | 3.37, 3.50 | 0.793 | |
| Q (liter/day) | 0.552 | 0.518, 0.588 | 0.851 | |
| Vp (liters) | 3.57 | 3.48, 3.68 | 1.14 | |
| α for CL and Q | 0.477 | 0.409, 0.542 | 7.04 | |
| α for Vc or Vp | 0.477 | 0.426, 0.524 | 5.11 | |
| Covariates on CL | ||||
| Albumin level (power) | −0.897 | −0.968, −0.825 | 4.07 | |
| Japanese descent | −0.0947 | −0.141, −0.0347 | 28.8 | |
| Black race | 0.154 | 0.0801, 0.231 | 25.1 | |
| Male gender | 0.219 | 0.18, 0.261 | 9.56 | |
| Covariates on Vc | ||||
| Albumin level (linear) | −0.124 | −0.141, −0.107 | 7.03 | |
| Japanese descent | −0.144 | −0.195, −0.0846 | 20.1 | |
| Male gender | 0.243 | 0.209, 0.277 | 7.23 | |
| Random effect | ||||
| IIV CL | 0.0791 (28.7) | 0.0717, 0.0867 (27.3, 30.1) | 3.1 | |
| IIV Vc | 0.0111 (10.6) | 0.0061, 0.0164 (7.8, 12.9) | 26.1 | |
| IIV residual error | 0.328 (62.3) | 0.286, 0.384 (57.5, 68.4) | −1.1 | |
| Residual error | ||||
| Log-additive error | 0.182 | 0.174, 0.191 | 3.9 |
CL, clearance; PK, pharmacokinetics; popPK, population pharmacokinetics; Q, intercompartmental flow rate; Vc, central volume of distribution; Vp, peripheral volume of distribution; α, power value for weight-based allometric scaling; IIV, interindividual variability.
Percent coefficient of variation (CV) was calculated as sqrt{[exp(omega) − 1]·100}, where omega is variance.
The 95% confidence interval was taken from bootstrap analysis.
Percent relative standard error was derived from bootstrap analysis and was calculated as 100·(standard error/median).
In total, 14 covariates, selected for physiological plausibility to potentially impact exposure, were evaluated on the CL and Vc of bezlotoxumab using the stepwise covariate modeling algorithm. No significant effect on CL or Vc was found for coadministration with actoxumab, age, ethnicity, hepatic function, antibiotic treatment for CDI, or concomitant proton pump inhibitor (PPI) use. Covariates retained in the model following backwards elimination were: albumin level, Charlson comorbidity index score, concomitant use of systemic antibiotics, eGFR, Japanese descent, race, and gender on CL; and albumin, Charlson comorbidity index score, Japanese descent, gender, and participant type (healthy subject versus patient with CDI) on Vc. The inclusion of the race effect was primarily driven by the black population, while the effect of the Asian population was nonsignificant. The Asian population was estimated to have a <10% change in CL and Vc compared with the values for the white population, while the Japanese covariate corresponded to a 14% decrease in CL and Vc. Removal of the Asian race effect from the model resulted in a 4-point increase in OFV, while removal of the Japanese effect resulted in a 23-point increase. Hence, the covariate for Japanese descent appeared to be a stronger predictor than the overall Asian covariate. Consequently, model refinement for the popPK model included removal of the Asian race effect from CL and Vc. Participant type, Charlson comorbidity index score, eGFR, and concomitant use of systemic antibiotics were reevaluated for inclusion in the final model, as they were correlated with albumin levels, were estimated with a relatively low precision (relative standard error, 30 to 45%), and had small magnitudes of effect. Inclusion of these covariates did not visibly improve the model fit or reduce variability, and, therefore, none were retained in the final model.
Parameter estimates for the final PK model are shown in Table 3. Significant covariates included in the final popPK model were gender, Japanese descent, race (black versus nonblack), and albumin level. Importantly, coadministration with actoxumab did not have a significant effect on bezlotoxumab PK in this analysis, supporting the pooling of data from the bezlotoxumab and bezlotoxumab plus actoxumab treatment arms in subsequent E-R analyses. Scatter plots of individual and population predicted concentrations determined using the final model versus observed concentrations showed good agreement and an adequate fit of the bezlotoxumab PK model to the data (Fig. 1a and b). Visual predictive checks confirmed the robustness of the final popPK model (Fig. 1c).
FIG 1.

Goodness-of-fit plots (a, b) and visual predictive check of phase 3 trial concentrations in participants receiving bezlotoxumab only (c) for the final bezlotoxumab popPK model. In panels a and b, dots are individual data; solid lines are the locally estimated scatterplot smoothing (LOESS) lines. In panel a, dashed lines are lines of unity; in panel b, dashed lines are thresholds for outliers (|CWRES| = 6); data for 5 samples from the MODIFY I and MODIFY II trials taken beyond day 200 are not shown in the plots. In panel c, circles represent observed data; light solid lines and light dashed lines are the observed median and observed 95% interval, respectively. Dark lines and dark dashed lines are the predicted median and predicted 95% interval, respectively. conc, concentration; CWRES, conditional weighted residuals; popPK, population pharmacokinetics.
Exposure predictions.
The final model, together with the observed concentrations of bezlotoxumab, was utilized to obtain individual post hoc estimates of bezlotoxumab PK parameters for each participant in the phase 3 trials. In phase 3 trial participants with CDI who received a single 10-mg/kg intravenous dose of bezlotoxumab, the geometric mean (GM) bezlotoxumab area under the serum concentration-time curve (AUC) from time zero to infinity (AUC0–inf) and maximum (peak) serum concentration (Cmax) were 53,000 μg·h/ml and 185 μg/ml, respectively. Consistent with the PK profiles of other human MAbs (15, 18, 19), bezlotoxumab had a low CL (0.317 liter/day) and a limited Vd (7.33 liters). With an elimination half-life (t1/2) of approximately 19 days, bezlotoxumab was present in serum at measurable concentrations during the first 12 weeks after treatment, when the risk of recurrence is greatest, and remained detectable until 24 weeks, based on limited sampling in some participants. Bezlotoxumab had moderate PK variability (coefficient of variation [CV] for AUC0–inf and Cmax, 40.2% and 20.7%, respectively).
All covariate effects on bezlotoxumab AUC0–inf in the phase 3 trial participant population were associated with a <50% change in exposure (Fig. 2). While body weight is one of the primary covariates influencing bezlotoxumab PK, differences in bezlotoxumab exposure due to body weight were moderate. At the 5th (45.5 kg) and 95th (109.6 kg) percentiles of the body weight distribution of the popPK data set, body weight resulted in a 20% decrease and a 27% increase in the bezlotoxumab AUC0–inf, respectively, when all other covariates were held constant. In addition to this univariate analysis, the impact of body weight, including the effect of correlated covariates observed among the phase 3 trial participants, was also considered. Based on the stratification of the exposures associated with the phase 3 trial participants by body weight, the impact of body weight across the intended target population was more limited, with an 18% decrease in AUC0–inf for participants weighing <60 kg and a 20% increase in AUC0–inf for participants weighing ≥100 kg compared with the value for the participants in the 60- to 80-kg weight range.
FIG 2.
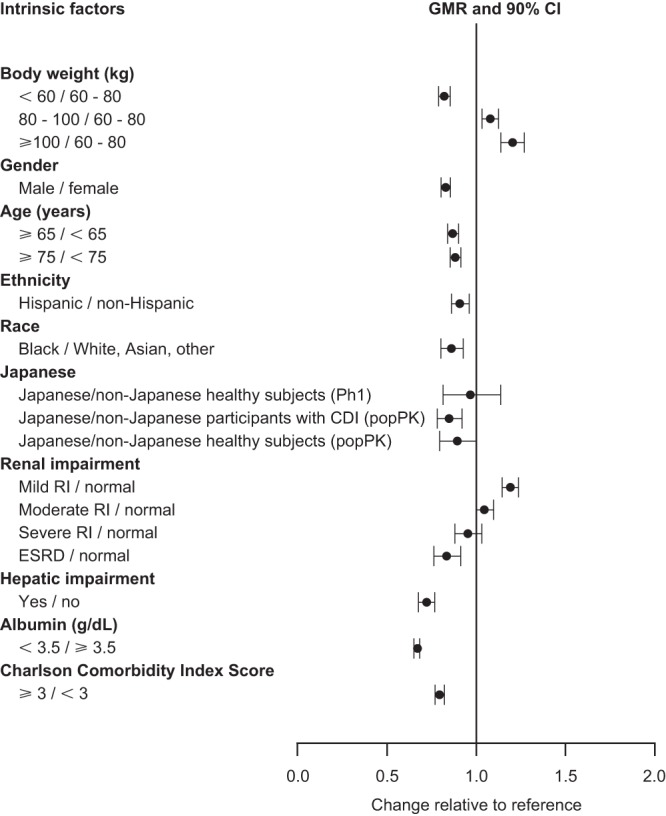
Impact of intrinsic factors on bezlotoxumab AUC0–inf in the phase 3 trial population. AUC0–inf, area under the concentration-time curve from time zero to infinity; CDI, Clostridium difficile infection; GMR, geometric mean ratio; CI, confidence interval; Ph1, phase 1; popPK, population pharmacokinetics; RI, renal impairment; ESRD, end-stage renal disease.
Bezlotoxumab exposure was positively correlated with albumin levels (Fig. 3a and b), such that at the 5th percentile (2.2 g/dl) and 95th percentile (4.7 g/dl) of the albumin distribution in the popPK data set, the effects of the albumin level resulted in a 47.7% increase and a 25.2% decrease in CL, respectively, relative to the CL at the median albumin level (3.4 g/dl) (Fig. 3c). Similarly, Vc was increased by 14.9% and decreased by 16.1% at the 5th percentile and 95th percentile of the albumin distribution, respectively, compared with the Vc at the median albumin level (Fig. 3d). To summarize the effect of albumin levels in the phase 3 trial population, bezlotoxumab exposure estimates were stratified by albumin levels. This showed that bezlotoxumab exposures were 33% lower in participants with albumin levels of <3.5 g/dl than in participants with normal albumin levels (AUC0–inf geometric mean ratio, 0.67; 90% confidence interval [CI], 0.65 to 0.69).
FIG 3.

Association of bezlotoxumab exposure with the albumin level. (a) Bezlotoxumab AUC0–inf by albumin levels in phase 3 trial participants and healthy subjects. (b) Bezlotoxumab AUC0–inf by albumin levels in phase 3 trial participants and healthy subjects after albumin normalization. (c) Effect of albumin on bezlotoxumab CL in phase 3 trial participants and healthy subjects. (d) Effect of albumin on the bezlotoxumab Vc in phase 3 trial participants and healthy subjects. The line with a shaded area represents the change in the pharmacokinetic parameter (CL or Vc) as a function of the covariate value; the shaded area represents the 90% confidence interval based on uncertainty; thick dashed lines represent body weight effects in the case of a scaling coefficient of 0.75 and 1 for CL and Vc, respectively. AUC0–inf, area under the concentration-time curve from time zero to infinity; CL, clearance; P05, 5th percentile; P95, 95th percentile; Vc, central volume of distribution.
Exposure-response analysis.
Data from a total of 2,559 participants administered bezlotoxumab (n = 779), bezlotoxumab plus actoxumab (n = 771), actoxumab (n = 233), or placebo (n = 776) in the phase 3 trials were included in the E-R analysis for rCDI. The primary statistical analysis indicated that rCDI rates following actoxumab treatment alone were similar to those following placebo treatment, and use of a combination of actoxumab and bezlotoxumab did not improve efficacy compared with that achieved with bezlotoxumab alone (11). Based on these findings, data for the the placebo and actoxumab arms, and the bezlotoxumab and bezlotoxumab plus actoxumab arms were pooled for subsequent assessment of placebo and exposure effects, respectively.
Based on graphical exploration, without consideration of covariate effects on PK or response, there was an apparent positive trend in E-R, with increasing exposure being associated with increasing rCDI rates (Fig. 4a). Of note, while a robust reduction in CDI recurrence relative to that with placebo was observed for participants treated with bezlotoxumab across all albumin quartiles, a similar trend of increasing rCDI with increasing levels of albumin was also observed in participants who received placebo (Fig. 4b). As bezlotoxumab exposure was demonstrated to be strongly correlated with the albumin level (Fig. 3a), the apparent trend between bezlotoxumab exposure and rCDI may reflect the relationship between rCDI and the albumin level rather than bezlotoxumab exposure. Thus, assessment of covariates to understand how patient factors may influence rCDI is an important step in characterizing the E-R relationship.
FIG 4.

Association of CDI recurrence rate by bezlotoxumab exposure quartile (a) and CDI recurrence rate by albumin level quartile (b). ACT, actoxumab; AUC0–inf, area under the concentration-time curve from time zero to infinity; BEZ, bezlotoxumab; CDI, Clostridium difficile infection.
Following characterization of the placebo response, the E-R relationship for bezlotoxumab AUC0–inf and rCDI rate was described with a sigmoid maximum effect (Emax) function (P < 0.05). The AUC at which 50% of the effect was obtained (EAUC50) could not be accurately estimated; hence, EAUC50 was arbitrarily fixed to 100 μg·h/ml, which falls below the exposure range observed following treatment with 10-mg/kg bezlotoxumab and represents an EAUC50 value that cannot be identified from the range of exposures tested in the phase 3 trials. Log-likelihood profiling showed no improvement in model fit for assumed EAUC50 values ranging from 1 × 10−3 to 1 × 105 μg·h/ml; this exposure range spans essentially undetectable concentrations well below the lowest exposures observed at 10 mg/kg in the phase 3 trials to the higher end of exposures observed at this dose level. In particular, EAUC50 values of >1,000 μg·h/ml (the lowest exposures observed in the phase 3 trials) corresponded to a poorer model fit than lower values, indicating that EAUC50 likely falls well below the exposures achieved at the 10-mg/kg dose in the phase 3 trials, and that the exposures achieved in the phase 3 trials were on the plateau of the E-R curve. Based on a stepwise covariate assessment, age, albumin level, Charlson comorbidity index score, and history of CDI in the past 6 months were identified as significant covariates impacting the placebo response and were included in the final E-R model.
The final model was parameterized as follows:
where p is probability; covpl,histCDI, covpl,alb, covpl,age, and covpl,Chrix are covariate effects for a history of CDI in the past 6 months, albumin level, age, and Charlson comorbidity index score on the placebo effect, respectively; Alb is the albumin concentration, and b0 is the placebo treatment effect.
The parameter estimates of the final E-R model are summarized in Table 4. To illustrate the E-R relationship, simulations of rCDI in the phase 3 trial participant population were conducted. Simulated and observed rCDI rates were plotted after accounting for the two covariates with the strongest association with rCDI rate: a history of CDI in the past 6 months and albumin level. To account for the correlation between bezlotoxumab exposure and the albumin level, individual exposures were normalized by individual albumin level (i.e., AUC0–inf/albumin) (Fig. 5a). A history of CDI in the past 6 months (yes or no) was accounted for by stratification of the patient responses by this covariate. After adjusting for the effects of the history of CDI in the past 6 months and albumin levels, there were no trends between bezlotoxumab exposure and rCDI rates over the range of exposures achieved with 10 mg/kg in phase 3 trials, based on plotting of both observed and simulated rCDI rates (Fig. 6), indicating that bezlotoxumab exposure over the investigated exposure range was not a predictor for rCDI.
TABLE 4.
Parameter estimates of the final bezlotoxumab exposure-response modeld
| Parameter | Estimatea | 95% confidence intervalb | % relative standard errorc |
|---|---|---|---|
| Placebo treatment effect (b0) | −1.15 | −1.34, −0.953 | 8.37 |
| Covariates affecting placebo | |||
| Albumin level, continuous | 0.318 | 0.172, 0.471 | 24.0 |
| Age, continuous | 0.0103 | 0.00391, 0.0166 | 31.7 |
| History of CDI in the past 6 mo, yes | 0.707 | 0.513, 0.919 | 15.1 |
| Charlson comorbidity index score ≥ 3 | −0.267 | −0.485, −0.0423 | 42.2 |
| Bezlotoxumab | |||
| Emax | −0.643 | −0.844, −0.452 | 16.0 |
| EAUC50 (μg·h/ml) | 100 (fixed) |
Final model estimates.
Bootstrap estimates.
Percent relative standard error was derived from bootstrap analysis and was calculated as 100·(standard error/median).
CDI, Clostridium difficile infection; EAUC50, area under the concentration-time curve at which 50% of the effect is obtained; Emax, maximum effect.
FIG 5.

Sensitivity analysis of CDI recurrence rate by bezlotoxumab exposure quartile (a) and CDI recurrence rate by albumin level quartile (b). Participants who discontinued or died were considered as having a CDI recurrence, in addition to participants already classified as having a CDI recurrence according to the primary endpoint definition. ACT, actoxumab; AUC0–inf, area under the concentration-time curve from time zero to infinity; BEZ, bezlotoxumab; CDI, Clostridium difficile infection.
FIG 6.

Observed and model-predicted maximal response in CDI recurrence rate across albumin-corrected bezlotoxumab AUC0–inf by decile following a single intravenous dose of 10-mg/kg bezlotoxumab. AUC0–inf, area under the concentration-time curve from time zero to infinity; CDI, Clostridium difficile infection; CI, confidence interval.
The apparent trend of increasing rCDI rates across exposure quartiles that was initially observed reflects the different levels of albumin in each exposure quartile and the imputation of participants who failed to complete the 12-week follow-up period (i.e., participants who discontinued the study) as having no CDI recurrence. Participants who discontinued, most of whom discontinued due to death, tended to have lower albumin levels, as low albumin levels are associated with poor patient health. Thus, the imputation of study-discontinued participants as having no rCDI results in an artifact that suggests a worse outcome with increasing albumin level. Due to the association between albumin levels and the bezlotoxumab AUC0–inf, study-discontinued participants who also tended to have lower albumin levels disproportionately fell into the lowest bezlotoxumab exposure quartile. Albumin levels rose and study discontinuation rates decreased across increasing exposure quartiles. As a result, the imputation of study-discontinued participants as having no CDI recurrence, together with the association between albumin level and exposure, resulted in the apparent trend of increasing rCDI with increasing exposure. To visualize the impact of study-discontinued participants on the rCDI E-R relationship, a graphical sensitivity analysis was conducted, in which study-discontinued participants were imputed as having rCDI. In this sensitivity analysis, treatment with bezlotoxumab resulted in a robust decrease in the CDI recurrence rate and the apparent trend for an increased rate of rCDI with increased exposure was no longer evident (Fig. 5a). As would be expected, a trend of decreasing CDI recurrence rate with increasing albumin level was also observed across albumin quartiles in participants who received placebo, as well as in those receiving bezlotoxumab (Fig. 5b).
DISCUSSION
The PK of bezlotoxumab were well characterized by a two-compartment model with linear elimination. Concomitant medications were not anticipated to affect the PK of bezlotoxumab, as MAbs are not eliminated by the metabolic or transporter pathways that are typically affected by concomitant medications. Consistent with this, the popPK analysis showed that the effects of antibiotic drug treatments for CDI, in addition to systemic antibiotics and PPIs, on bezlotoxumab PK were small (≤20%) and not considered to be clinically meaningful.
In popPK analyses, body size covariates are frequently identified as clinically relevant covariates and are well established as predictors of systemic exposure for small-molecule drugs and therapeutic MAbs (18, 20), reflecting the relationship between CL, V, and body size (21). As a result, most MAbs are administered on the basis of body weight. In this study, bezlotoxumab CL increased with body weight. The resulting exposure differences were adequately addressed by the administration of a weight-based dose. The incremental impact of body weight across the intended target population was limited, with an 18% decrease in AUC0–inf for participants weighing <60 kg and a 20% increase in AUC0–inf for participants weighing ≥100 kg compared with the value for participants in the 60- to 80-kg weight range. This assessment of body weight demonstrated that the comparative change in AUC is modest and supports the appropriateness of the weight-based dosing paradigm.
Gender, race (black versus nonblack), and Japanese descent were identified as significant covariates on CL or Vc and, thus, were included in the final popPK model. However, comparisons of bezlotoxumab AUC0–inf values between covariate groups for gender, Japanese descent, and race indicated that these factors were associated with a <20% change in bezlotoxumab exposure. The effects of Japanese descent on CL and Vc were driven largely by differences in body weight between Japanese and non-Japanese participants. These findings are consistent with the characteristics of other MAbs approved for use in Japan (22). Gender has been identified to be a predictor of CL and/or V for other MAbs, even after adjustment for body size, although this association is not always clinically significant (18). The cause(s) of gender differences in the apparent CL (CL/F) and apparent V (V/F) of MAbs has not been well-defined. However, altered lymphatic flow and interstitial fluid volume are possible mechanisms that may result in a change of MAb PK (23). As expected from a MAb that is cleared by catabolism, neither hepatic nor renal impairment affected bezlotoxumab PK.
In contrast, albumin was identified as a major predictor of bezlotoxumab exposure. This was reflected in the correlation between albumin levels and exposures; participants with albumin levels of <3.5 g/dl had bezlotoxumab exposure 33% lower than that for participants with normal albumin levels. The mechanism of the relationship between the albumin level and the clearance of MAbs has been described: both are protected from lysosomal degradation by the same neonatal Fc receptor (FcRn) recycling system, though they interact with different binding sites (24–26). Factors that affect the recycling capacity of FcRn influence the concentrations of albumin and IgG in a similar fashion, so that the serum levels of albumin and IgG increase and decrease concomitantly, and, hence, albumin may be a predictor of IgG PK (24). Albumin levels were also correlated to indicators of participant health, such that albumin levels (and, therefore, exposures) were lower in participants with poor health. Low serum albumin concentrations have been linked with poor outcomes (27, 28) and may be associated with advanced age (29).
The phase 2 and phase 3 trials evaluated a single 10-mg/kg dose of bezlotoxumab (registration dose) (11, 30), limiting the E-R analysis to data from a single dose level. Despite this, a considerable range of exposures was observed due to intersubject variability. The 10th and 90th percentiles of exposures at the 10-mg/kg dose in the phase 3 trials were 31,700 μg·h/ml and 85,600 μg·h/ml, respectively. These values are approximately 40% lower and 60% higher than the median exposure of 54,700 μg·h/ml, respectively, and encompass the range of covariate effects. This rich PK data set facilitated exploration of the E-R relationship between bezlotoxumab PK and the probability of rCDI.
The E-R relationship for the bezlotoxumab AUC0–inf and the rCDI rate was characterized with an Emax relationship, where the responses in phase 3 trial participants with CDI to 10-mg/kg bezlotoxumab were on the maximal response plateau of the E-R curve. AUC0–inf was selected as the PK measure for E-R assessment, as it reflects an integration of concentrations over the total time period in which participants were exposed to bezlotoxumab in the serum. The bezlotoxumab area under the serum concentration-time curve from time zero to day 84 (AUC0–84) and Cmax were also evaluated as predictors for rCDI, but no improvement in model fit compared with that achieved with AUC0–inf was found, indicating that these parameters are not more predictive of rCDI than AUC0–inf. This analysis included participants who received bezlotoxumab alone or bezlotoxumab with actoxumab; repeating the analysis using participants who received bezlotoxumab monotherapy resulted in no difference in outcome.
Patient covariates are known to impact the probability of rCDI (31, 32). Therefore, the E-R analysis evaluated the effect of covariates on the rCDI rates achieved with placebo. Given the potential correlation between bezlotoxumab exposure and covariates (i.e., albumin level) that may influence the rCDI rate, the same criteria used for the assessment of an E-R relationship (P ≤ 0.05) were also applied to the identification of covariates to ensure an unbiased assessment of covariates, in addition to exposure, that may influence rCDI rates. Based on this assessment, covariates related to patient health, rather than bezlotoxumab exposure, were found to be the strongest determinants of rCDI. After adjusting for the effects of a history of CDI in the past 6 months and albumin level, there was no relationship between bezlotoxumab exposure and observed rCDI rates over the range of exposures achieved with 10 mg/kg in the phase 3 trials. The apparent trend of increasing rCDI across exposure quartiles was also influenced by the relatively high number of study-discontinued participants in the first exposure quartile. As study-discontinued participants were imputed as having no rCDI, rates of recurrence were disproportionately low in the first exposure quartile, resulting in an artifact that suggested a worse outcome with increasing exposures. When the impacts of the albumin level and study discontinuation on trends in E-R were considered, treatment with bezlotoxumab resulted in robust decreases in rCDI.
Thus, there is no significant dependence of rCDI on bezlotoxumab exposures over the range of exposures achieved in phase 3 trials, and the entire range of exposures achieved with the 10-mg/kg dose is associated with a similar low rate of rCDI. Therefore, covariate-associated changes in exposure, which were all modest based on the popPK analysis, do not impact bezlotoxumab efficacy in a clinically meaningful way. Of the significant covariates identified in the popPK analysis, serum albumin level had the largest effect on bezlotoxumab exposure. Nevertheless, participants with albumin levels of <3.5 g/dl, which is associated with a 33% lower bezlotoxumab exposure, benefit from bezlotoxumab treatment to a similar extent as participants with albumin levels in the normal range.
In conclusion, this popPK and E-R analysis showed that intrinsic and extrinsic factors do not affect bezlotoxumab exposure to a clinically meaningful extent based on the totality of PK and clinical experience, and therefore, dose adjustments for these factors are not required. These data support the use of the current clinical dose of 10-mg/kg bezlotoxumab in all adult individuals with CDI. Similar reductions in rCDI at this dose are expected regardless of the variability in exposure due to patient covariates.
MATERIALS AND METHODS
Study populations.
Source data were obtained from the phase 3 trials MODIFY I (ClinicalTrials.gov registration number NCT01241552) and MODIFY II (ClinicalTrials.gov registration number NCT01513239) and from three phase 1 trials (protocols PN004, PN005, and PN006). MODIFY I and MODIFY II were randomized, double-blind, placebo-controlled trials that assessed the efficacy and safety of single infusions of bezlotoxumab and actoxumab in participants receiving antibiotic therapy for CDI (Table 1). Participants received a single infusion of 10-mg/kg bezlotoxumab, 10-mg/kg actoxumab (MODIFY I only), 10-mg/kg actoxumab plus 10-mg/kg bezlotoxumab, or placebo (0.9% saline) over 1 h during treatment with a CDI-active antibiotic. The primary efficacy endpoint for the phase 3 trials was the proportion of participants with rCDI at week 12, defined as the development of a new episode of diarrhea (defined as the passage of three or more loose stools in 24 h or less) associated with a positive stool test for toxigenic C. difficile following an initial clinical cure of the baseline CDI episode. Participants in MODIFY I and MODIFY II were heterogeneous, due to few exclusion criteria being applied, and are therefore representative of the intended patient population.
The phase 1 trials assessed the safety and PK of bezlotoxumab and actoxumab in healthy male and female adult subjects and included one study specifically conducted in a Japanese population (Table 1). Phase 1 study PN004 administered two sequential doses of 10-mg/kg actoxumab plus 10-mg/kg bezlotoxumab to healthy subjects (n = 30), with a 12-week interval between doses. Phase 1 study PN005 administered a single infusion of 10-mg/kg actoxumab plus 10-mg/kg bezlotoxumab to healthy subjects (n = 29) and placebo to 6 subjects over a 1-h period. Study PN006 recruited 19 healthy Japanese subjects; of these, 6 were administered 10-mg/kg actoxumab plus 10-mg/kg bezlotoxumab, 7 were administered 20-mg/kg actoxumab plus 20-mg/kg bezlotoxumab, and 6 received placebo.
All protocols were approved by the institutional review board or independent ethics committee at each study site. Written informed consent was provided by all participants before each trial began.
Sampling.
Intensive PK sampling data (≥6 samples/participant) were obtained from participants in all included phase 3 and phase 1 trials (for details, see Table 1). Serum concentrations of bezlotoxumab were quantified using validated idiotypic assays with a lower limit of quantification of 100 ng/ml (D. L. Montgomery, R. P. Matthews, K. L. Yee, L. M. Tobias, M. B. Dorr, and R. E. Wrishko, submitted for publication).
Population pharmacokinetic modeling.
Data were pooled from participants who received a single infusion of 10-mg/kg bezlotoxumab or 10-mg/kg actoxumab plus 10-mg/kg bezlotoxumab. PopPK analysis was performed using a nonlinear mixed-effects modeling approach with the NONMEM (version VII) program (Globomax, Hanover, MD, USA) running under Perl-speaks-NONMEM (PsN; version 3.4.2) software. Postprocessing of the data was conducted using R (version 2.15.2) software.
The three stages of base model development consisted of development of a structural model, exploration of interindividual random effects, and addition of residual random effects. One- and two-compartment models, with and without allometric scaling for body weight, were selected as the starting point for base structural model development. Runs were compared using goodness-of-fit plots and likelihood ratio testing. Stochastic models for IIV were evaluated for CL, Q, Vc, and Vp, and IIV on residual error was evaluated to address phase 3 trial participants with highly variable PK data (conditional weighted residuals [CWRES] > 6).
A stepwise covariate assessment was conducted with the stepwise covariate modeling functionality in PsN. This procedure involved stepwise testing of covariate relationships in a forward inclusion (change of OFV [ΔOFV], 3.84; P < 0.05 for 1 degree of freedom [DF]) and backward exclusion (ΔOFV, 7.88; P < 0.005 for 1 DF) procedure. The following covariates were evaluated: age, albumin level, gender, race, ethnicity, Japanese descent, eGFR, hepatic impairment (defined as two or more of the following: albumin level ≤3.1 g/dl; ALT level ≥2 times the ULN; bilirubin level ≥1.3 times the ULN; or mild, moderate, or severe liver disease, as determined by the Charlson comorbidity index score), hospitalization status, Charlson comorbidity index score (<3 versus ≥3), coadministration of actoxumab, type of anti-CDI antibiotic treatment at randomization, concomitant systemic antibiotic use, and concomitant use of PPIs. Significant covariates with marginal effects were further evaluated for improvement of model fit. Reductions in interindividual variability deviation (ETA) and residual variability, shifts in ETA distributions, and diagnostic plots with and without the covariates were investigated. Rejection of marginal-effect covariates was based on small effect sizes on CL and Vc (<10%), relative standard errors of >25%, and a lack of improvement in model fit and diagnostics. Structural model development indicated that shrinkage was high for IIV terms associated with peripheral compartment PK parameters Q and Vp, so covariate effects were not tested for these parameters.
The reliability of the final model was checked with diagnostic plots, a visual predictive check, and bootstrap analysis. The final popPK model together with the observed concentrations of bezlotoxumab were utilized to obtain individual post hoc estimates of PK parameters. For each participant from the MODIFY I and MODIFY II trials for whom measurable bezlotoxumab concentrations were available, the bezlotoxumab AUC0–inf, AUC0–84, Cmax, and t1/2 were estimated based on post hoc compartmental PK parameters.
Exposure-response analysis.
E-R analysis was based on data from the MODIFY I and MODIFY II trials for participants receiving placebo, actoxumab, bezlotoxumab, and bezlotoxumab plus actoxumab and individual participant PK parameter estimates obtained from the final popPK model. Data for participants receiving placebo and actoxumab were pooled for the assessment of the placebo response, and data for participants receiving bezlotoxumab and bezlotoxumab plus actoxumab were pooled for the assessment of exposure effects. Logistic regression analysis was used to characterize the E-R relationship for the primary endpoint of the phase 3 trials (rCDI rate) in NONMEM (version VII; Globomax, Hanover, MD, USA) running under PsN (version 3.4.2). The PK endpoints examined were the bezlotoxumab AUC0–inf, AUC0–84, and Cmax. Model development for rCDI was performed in a three-step approach: in the first step, the baseline incidence of rCDI was quantified (placebo effect), followed by inclusion of an E-R term and, subsequently, a covariate analysis using stepwise covariate modeling to identify factors that affect the baseline incidence of rCDI. The criterion for both the forward addition and backward elimination steps of the stepwise covariate modeling was a P value of <0.05, the same criterion applied to test for exposure effects. Given the complex correlation between exposure and covariates that may influence the rCDI rate, use of criteria during a backward elimination step more stringent than those applied to the E-R assessment may result in the rejection of influential covariates during the stepwise covariate modeling. By relaxing the criteria in the backwards step, an unbiased assessment of influential covariates, in addition to exposure, could be conducted. The covariates assessed were age, weight, gender, race, albumin level, white blood cell count, the presence of clinically severe CDI at the time of infusion (yes or no), the Charlson comorbidity index score, modified Horn’s index, hospitalization status, compromised immunity status (yes or no), a history of CDI in the past 6 months (yes or no), the presence of endogenous IgG to toxin A or B, antibacterial drug therapy for CDI, and the use of systemic antibiotics or PPIs (yes or no).
Simulations were used to characterize the probability of rCDI across the range of bezlotoxumab exposures achieved in the phase 3 trials and the influence of any significant covariates in the final Emax PK/PD model. The probability of a CDI recurrence event was calculated for each participant in the phase 3 trial population utilizing the post hoc-estimated PK exposures, each participant’s vector of covariates, and the final PK/PD model. The individual probability of rCDI was then utilized to simulate the occurrence of a CDI recurrence event (yes or no) in each participant by random draw from a binomial distribution with the individual’s probability. Subsequently, the incidence of rCDI for the population was derived. This procedure was repeated 1,000 times by incorporating parameter uncertainty for the estimated fixed-effect parameters of the PK/PD model. The median and the 5th and 95th percentiles of the rCDI rate from these 1,000 runs were reported. All model predictions were in agreement with the observed rCDI rates. The simulated results were also stratified by significant covariates to explore the effect of covariates on the association between exposures and rCDI.
Exploration of potential EAUC50 estimates was also performed with log likelihood profiling. Specifically, EAUC50 values were fixed over a range of 1 × 10−3 and 1 × 105 μg·h/ml. For each fixed EAUC50 value, the Emax model for the bezlotoxumab AUC0–inf was run to obtain the OFV associated with each EAUC50 value’s model fit.
ACKNOWLEDGMENTS
We acknowledge Lori Tobias, who provided the bezlotoxumab serum concentration data, and Diana Montgomery, who contributed to the interpretation of the popPK analysis.
Medical writing support, under the direction of the authors, was provided by Paul O’Neill of CMC Affinity, a division of Complete Medical Communications Ltd., Glasgow, United Kingdom, funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, in accordance with good publication practice (GPP3) guidelines.
Funding for this research was provided by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
K.L.Y., R.P.M., M.B.D., and R.E.W. are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and may own stock and/or stock options in the company. H.J.K. and T.K. are paid consultants for Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and do not own stock or stock options in the company. K.W.G. has previously received research support from Merck & Co., Inc., Kenilworth, NJ, USA.
All authors contributed to the writing and reviewing of the manuscript. H.J.K. conducted the modeling analysis. T.K. and K.L.Y. were involved in modeling analysis and interpretation. M.B.D. and R.P.M. were involved in the selection of covariates for inclusion in the analyses.
REFERENCES
- 1.McDonald LC, Killgore GE, Thompson A, Owens RC Jr, Kazakova SV, Sambol SP, Johnson S, Gerding DN. 2005. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med 353:2433–2441. doi: 10.1056/NEJMoa051590. [DOI] [PubMed] [Google Scholar]
- 2.Rupnik M, Wilcox MH, Gerding DN. 2009. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 7:526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 3.Carter GP, Rood JI, Lyras D. 2010. The role of toxin A and toxin B in Clostridium difficile-associated disease: past and present perspectives. Gut Microbes 1:58–64. doi: 10.4161/gmic.1.1.10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riegler M, Sedivy R, Pothoulakis C, Hamilton G, Zacherl J, Bischof G, Cosentini E, Feil W, Schiessel R, LaMont JT. 1995. Clostridium difficile toxin B is more potent than toxin A in damaging human colonic epithelium in vitro. J Clin Invest 95:2004–2011. doi: 10.1172/JCI117885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamont JT, Kelly CP, Bakken JS. 2018. Clostridium difficile infection in adults: clinical manifestations and diagnosis. UpToDate, Waltham, MA. [Google Scholar]
- 6.Sheitoyan-Pesant C, Abou Chakra CN, Pepin J, Marcil-Héguy A, Nault V, Valiquette L. 2016. Clinical and healthcare burden of multiple recurrences of Clostridium difficile infection. Clin Infect Dis 62:574–580. doi: 10.1093/cid/civ958. [DOI] [PubMed] [Google Scholar]
- 7.Cornely OA, Crook DW, Esposito R, Poirier A, Somero MS, Weiss K, Sears P, Gorbach S. 2012. Fidaxomicin versus vancomycin for infection with Clostridium difficile in Europe, Canada, and the USA: a double-blind, non-inferiority, randomised controlled trial. Lancet Infect Dis 12:281–289. doi: 10.1016/S1473-3099(11)70374-7. [DOI] [PubMed] [Google Scholar]
- 8.Cornely OA, Miller MA, Louie TJ, Crook DW, Gorbach SL. 2012. Treatment of first recurrence of Clostridium difficile infection: fidaxomicin versus vancomycin. Clin Infect Dis 55:S154–S161. doi: 10.1093/cid/cis462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kyne L, Warny M, Qamar A, Kelly CP. 2001. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet 357:189–193. doi: 10.1016/S0140-6736(00)03592-3. [DOI] [PubMed] [Google Scholar]
- 10.Bauer MP, Nibbering PH, Poxton IR, Kuijper EJ, van Dissel JT. 2014. Humoral immune response as predictor of recurrence in Clostridium difficile infection. Clin Microbiol Infect 20:1323–1328. doi: 10.1111/1469-0691.12769. [DOI] [PubMed] [Google Scholar]
- 11.Wilcox MH, Gerding DN, Poxton IR, Kelly C, Nathan R, Birch T, Cornely OA, Rahav G, Bouza E, Lee C, Jenkin G, Jensen W, Kim YS, Yoshida J, Gabryelski L, Pedley A, Eves K, Tipping R, Guris D, Kartsonis N, Dorr MB, MODIFY I and MODIFY II Investigators. 2017. Bezlotoxumab for prevention of recurrent Clostridium difficile infection. N Engl J Med 376:305–317. doi: 10.1056/NEJMoa1602615. [DOI] [PubMed] [Google Scholar]
- 12.Gerding DN, Kelly CP, Rahav G, Lee C, Dubberke ER, Kumar PN, Yacyshyn B, Kao D, Eves K, Ellison MC, Hanson ME, Guris D, Dorr MB. 2018. Bezlotoxumab for prevention of recurrent C. difficile infection in patients at increased risk for recurrence. Clin Infect Dis 67:649–656. doi: 10.1093/cid/ciy171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merck Sharp & Dohme Corp. 2016. Zinplava (bezlotoxumab) prescribing information. Merck Sharp & Dohme Corp., Kenilworth, NJ: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/761046s000lbl.pdf. [Google Scholar]
- 14.European Medicines Agency (EMA). 2016. Zinplava assessment report. European Medicines Agency, London, United Kingdom: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004136/WC500222643.pdf. [Google Scholar]
- 15.Dostalek M, Gardner I, Gurbaxani BM, Rose RH, Chetty M. 2013. Pharmacokinetics, pharmacodynamics and physiologically-based pharmacokinetic modelling of monoclonal antibodies. Clin Pharmacokinet 52:83–124. doi: 10.1007/s40262-012-0027-4. [DOI] [PubMed] [Google Scholar]
- 16.Zhou H, Mascelli MA. 2011. Mechanisms of monoclonal antibody-drug interactions. Annu Rev Pharmacol Toxicol 51:359–372. doi: 10.1146/annurev-pharmtox-010510-100510. [DOI] [PubMed] [Google Scholar]
- 17.Charlson ME, Pompei P, Ales KL, MacKenzie CR. 1987. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis 40:373–383. doi: 10.1016/0021-9681(87)90171-8. [DOI] [PubMed] [Google Scholar]
- 18.Dirks NL, Meibohm B. 2010. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet 49:633–659. doi: 10.2165/11535960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 19.Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. 2010. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet 49:493–507. doi: 10.2165/11531280-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 20.Bonate PL, Steimer J-L. 2006. Pharmacokinetic-pharmacodynamic modeling and simulation. Springer, New York, NY. [Google Scholar]
- 21.Meibohm B, Laer S, Panetta JC, Barrett JS. 2005. Population pharmacokinetic studies in pediatrics: issues in design and analysis. AAPS J 7:E475–E487. doi: 10.1208/aapsj070248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiba K, Yoshitsugu H, Kyosaka Y, Iida S, Yoneyama K, Tanigawa T, Fukushima T, Hiraoka M. 2014. A comprehensive review of the pharmacokinetics of approved therapeutic monoclonal antibodies in Japan: are Japanese phase I studies still needed? J Clin Pharmacol 54:483–494. doi: 10.1002/jcph.231. [DOI] [PubMed] [Google Scholar]
- 23.Cao Y, Balthasar JP, Jusko WJ. 2013. Second-generation minimal physiologically-based pharmacokinetic model for monoclonal antibodies. J Pharmacokinet Pharmacodyn 40:597–607. doi: 10.1007/s10928-013-9332-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim J, Hayton WL, Robinson JM, Anderson CL. 2007. Kinetics of FcRn-mediated recycling of IgG and albumin in human: pathophysiology and therapeutic implications using a simplified mechanism-based model. Clin Immunol 122:146–155. doi: 10.1016/j.clim.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ober RJ, Martinez C, Vaccaro C, Zhou J, Ward ES. 2004. Visualizing the site and dynamics of IgG salvage by the MHC class I-related receptor, FcRn. J Immunol 172:2021–2029. doi: 10.4049/jimmunol.172.4.2021. [DOI] [PubMed] [Google Scholar]
- 26.Roopenian DC, Akilesh S. 2007. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 27.Nicholson JP, Wolmarans MR, Park GR. 2000. The role of albumin in critical illness. Br J Anaesth 85:599–610. doi: 10.1093/bja/85.4.599. [DOI] [PubMed] [Google Scholar]
- 28.Goldwasser P, Feldman J. 1997. Association of serum albumin and mortality risk. J Clin Epidemiol 50:693–703. doi: 10.1016/S0895-4356(97)00015-2. [DOI] [PubMed] [Google Scholar]
- 29.Salive ME, Cornoni-Huntley J, Phillips CL, Guralnik JM, Cohen HJ, Ostfeld AM, Wallace RB. 1992. Serum albumin in older persons: relationship with age and health status. J Clin Epidemiol 45:213–221. doi: 10.1016/0895-4356(92)90081-W. [DOI] [PubMed] [Google Scholar]
- 30.Lowy I, Molrine DC, Leav BA, Blair BM, Baxter R, Gerding DN, Nichol G, Thomas WD Jr, Leney M, Sloan S, Hay CA, Ambrosino DM. 2010. Treatment with monoclonal antibodies against Clostridium difficile toxins. N Engl J Med 362:197–205. doi: 10.1056/NEJMoa0907635. [DOI] [PubMed] [Google Scholar]
- 31.Garey KW, Sethi S, Yadav Y, DuPont HL. 2008. Meta-analysis to assess risk factors for recurrent Clostridium difficile infection. J Hosp Infect 70:298–304. doi: 10.1016/j.jhin.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 32.Johnson S. 2009. Recurrent Clostridium difficile infection: a review of risk factors, treatments, and outcomes. J Infect 58:403–410. doi: 10.1016/j.jinf.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 33.Zar FA, Bakkanagari SR, Moorthi KM, Davis MB. 2007. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-associated diarrhea, stratified by disease severity. Clin Infect Dis 45:302–307. doi: 10.1086/519265. [DOI] [PubMed] [Google Scholar]