

Abstract
Key points
Cardiac alternans refers to a beat‐to‐beat alternation in contraction, action potential (AP) morphology and Ca2+ transient (CaT) amplitude, and represents a risk factor for cardiac arrhythmia, including atrial fibrillation.
We developed strategies to pharmacologically manipulate the AP waveform with the goal to reduce or eliminate the occurrence of CaT and contraction alternans in atrial tissue.
With combined patch‐clamp and intracellular Ca2+ measurements we investigated the effect of specific ion channel inhibitors and activators on alternans. In single rabbit atrial myocytes, suppression of Ca2+‐activated Cl− channels eliminated AP duration alternans, but prolonged the AP and failed to eliminate CaT alternans. In contrast, activation of K+ currents (IKs and IKr) shortened the AP and eliminated both AP duration and CaT alternans.
As demonstrated also at the whole heart level, activation of K+ conductances represents a promising strategy to suppress alternans, and thus reducing a risk factor for atrial fibrillation.
Abstract
At the cellular level alternans is observed as beat‐to‐beat alternations in contraction, action potential (AP) morphology and magnitude of the Ca2+ transient (CaT). Alternans is a well‐established risk factor for cardiac arrhythmia, including atrial fibrillation. This study investigates whether pharmacological manipulation of AP morphology is a viable strategy to reduce the risk of arrhythmogenic CaT alternans. Pacing‐induced AP and CaT alternans were studied in rabbit atrial myocytes using combined Ca2+ imaging and electrophysiological measurements. Increased AP duration (APD) and beat‐to‐beat alternations in AP morphology lowered the pacing frequency threshold and increased the degree of CaT alternans. Inhibition of Ca2+‐activated Cl− channels reduced beat‐to‐beat AP alternations, but prolonged APD and failed to suppress CaT alternans. In contrast, AP shortening induced by activators of two K+ channels (ML277 for Kv7.1 and NS1643 for Kv11.1) abolished both APD and CaT alternans in field‐stimulated and current‐clamped myocytes. K+ channel activators had no effect on the degree of Ca2+ alternans in AP voltage‐clamped cells, confirming that suppression of Ca2+ alternans was caused by the changes in AP morphology. Finally, activation of Kv11.1 channel significantly attenuated or even abolished atrial T‐wave alternans in isolated Langendorff perfused hearts. In summary, AP shortening suppressed or completely eliminated both CaT and APD alternans in single atrial myocytes and atrial T‐wave alternans at the whole heart level. Therefore, we suggest that AP shortening is a potential intervention to avert development of alternans with important ramifications for arrhythmia prevention and therapy.
Keywords: action potential duration, alternans, arrhythmia, atria, calcium, excitation‐contraction coupling, K+ current
Key points
Cardiac alternans refers to a beat‐to‐beat alternation in contraction, action potential (AP) morphology and Ca2+ transient (CaT) amplitude, and represents a risk factor for cardiac arrhythmia, including atrial fibrillation.
We developed strategies to pharmacologically manipulate the AP waveform with the goal to reduce or eliminate the occurrence of CaT and contraction alternans in atrial tissue.
With combined patch‐clamp and intracellular Ca2+ measurements we investigated the effect of specific ion channel inhibitors and activators on alternans. In single rabbit atrial myocytes, suppression of Ca2+‐activated Cl− channels eliminated AP duration alternans, but prolonged the AP and failed to eliminate CaT alternans. In contrast, activation of K+ currents (IKs and IKr) shortened the AP and eliminated both AP duration and CaT alternans.
As demonstrated also at the whole heart level, activation of K+ conductances represents a promising strategy to suppress alternans, and thus reducing a risk factor for atrial fibrillation.
Introduction
At the cellular level cardiac alternans is observed as periodic beat‐to‐beat variation in contraction amplitude, action potential (AP) morphology and cytosolic Ca2+ transient (CaT) amplitude at constant stimulation frequency. Atrial alternans is closely linked to the development of atrial arrhythmias such as atrial fibrillation and flutter (Narayan et al. 2002; Hiromoto et al. 2005; Jousset et al. 2012; Kanaporis & Blatter, 2017a ). Atrial fibrillation (AF), the most prevalent cardiac arrhythmia, is associated with an increased risk of stroke, cardiomyopathies and heart failure, and accounts for significant morbidity and mortality (Chinitz et al. 2013; Chugh et al. 2014). Therapeutic strategies for AF include ablation procedures and pharmacological approaches. Pharmacological treatment of AF involves rate‐controlling drugs, such as beta‐blockers, calcium channel inhibitors or cardiac glycosides (e.g. digoxin), or rhythm‐controlling compounds that suppress sodium or potassium channels. However, the efficacy of current pharmacological therapies for treatment and prevention of AF is limited and inconsistent, as reflected in high AF recurrence rates (Morillo et al. 2014; Geng et al. 2017). In accordance with ongoing clinical efforts to find new, more personalized and less invasive therapeutic approaches, this study explores the role of AP morphology for the development and severity of atrial alternans, and based on these findings, tests the hypothesis that targeted pharmacological modification of AP morphology is a potential therapeutic tool for prevention of arrhythmia.
It is well established that development of AP duration (APD) and CaT alternans is strongly coupled (Pruvot et al. 2004; Kanaporis & Blatter, 2015). The relationship between cytosolic Ca2+ concentration ([Ca2+]i) and membrane potential (V m) plays a key role in the generation of alternans. Changes in [Ca2+]i shape AP waveforms by modulating the activity of Ca2+‐regulated ion conductances. On the other hand, AP morphology affects intracellular Ca2+ handling by regulating Ca2+ entry through L‐type Ca2+ channels and extrusion through Na+/Ca2+ exchange. Although it is still debated whether disturbances of V m or [Ca2+]i regulation are the primary cause of alternans (Edwards & Blatter, 2014; Kanaporis & Blatter, 2017a ), growing evidence indicates that alternans is initiated by disturbances in intracellular Ca2+ handling (Chudin et al. 1999; Diaz et al. 2004; Goldhaber et al. 2005; Shkryl et al. 2012; Kanaporis & Blatter, 2015). However, recently we have demonstrated that AP morphology and its beat‐to‐beat alternation have strong effects on the development and degree of alternans in atrial myocytes (Kanaporis & Blatter, 2017b ) by regulating diastolic sarcoplasmic reticulum (SR) Ca2+ load and affecting the kinetics of L‐type Ca2+ currents. These findings raise the possibility that targeted pharmacological manipulation of AP morphology will allow to establish desirable AP waveforms that carry a low risk of sustaining APD, CaT and contraction alternans, thus opening a window towards novel strategies for arrhythmia prevention and therapy. The hypothesis of AP modulation for alternans prevention and rescue was tested in this study. In summary, we demonstrate that APD prolongation through inhibition of Ca2+‐activated Cl− channels (CaCCs) attenuated APD but not CaT alternans, whereas APD shortening through activation of K+ currents suppressed or completely eliminated both CaT and APD alternans in single atrial myocytes and atrial T‐wave alternans at the whole heart level.
Methods
Ethical approval
All procedures and protocols were approved by the Institutional Animal Care and Use Committee of Rush University Chicago, and comply with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and UK regulations on animal experimentation (Grundy, 2015).
Myocyte isolation
Atrial myocytes were isolated from male New Zealand White rabbits (2.5–2.9 kg; 69 rabbits; Charles River Laboratories, Wilmington, MA, USA). Rabbits were anaesthetized with an intravenous injection of sodium pentobarbital (100 mg kg−1) and heparin (1000 IU kg−1). The depth of anaesthesia was evaluated by foot pinch or checking corneal reflexes to ensure a deep anaesthesia and that the animal did not experience any pain. Hearts were excised, mounted on a Langendorff apparatus and retrogradely perfused via the aorta. After an initial 5–10 min of perfusion with oxygenated Ca2+‐free Tyrode solution (mmol l−1: 140 NaCl, 4 KCl, 10 d‐glucose, 5 Hepes, 1 MgCl2, 1000 IU l−1 heparin; pH 7.4 with NaOH), the heart was perfused with minimal essential medium Eagle (MEM) solution containing 20 μmol l−1 Ca2+ and 22.5 μg ml−1 Liberase TH (Roche Diagnostic Corporation, Indianapolis, IN, USA) for ∼20 min at 37 ˚C. The left atrium was dissected from the heart and minced, filtered and washed in MEM solution containing 50 μmol l−1 Ca2+ and 10 mg ml−1 bovine serum albumin. Isolated cells were washed and kept in MEM solution with 50 μmol l−1 Ca2+ at room temperature (20–24°C) and were used within 1–8 h after isolation.
Patch clamp experiments
The external Tyrode solution was composed of (mmol l−1): 135 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 Hepes and 10 d‐glucose; pH 7.4 with NaOH. All chemicals and reagents were from Sigma‐Aldrich (St. Louis, MO, USA), unless otherwise stated. Patch clamp pipettes (1.5–3 MΩ filled with internal solution) were pulled from borosilicate glass capillaries (WPI, Sarasota, FL, USA) with a P‐97 horizontal puller (Sutter Instruments, Novato, CA, USA). For all voltage‐ and current‐clamp experiments with the exception of the K+ current measurements (see below) pipettes were filled with internal solution containing (mmol l−1): 130 K+ glutamate, 10 NaCl, 10 KCl, 0.33 MgCl2, 4 MgATP and 10 Hepes with pH adjusted to 7.2 with KOH. For simultaneous [Ca2+]i measurements, 100 μmol l−1 Fluo‐4 pentapotassium salt or Indo‐1 pentapotassium salt (both from Thermo Fisher Scientific, Waltham MA, USA) was added to the internal solution. The internal solution was filtered through 0.22‐μm pore filters. Electrophysiological signals were recorded from single cardiac myocytes in the whole‐cell ruptured patch clamp configuration using an Axopatch 200A patch clamp amplifier, the Axon Digidata 1440A interface and pCLAMP 10.3 software (Molecular Devices, Sunnyvale, CA, USA). Current and AP recordings were low‐pass filtered at 5 kHz and digitized at 10 kHz. All patch clamp experiments were performed at room temperature (20–24°C).
For AP measurements the whole‐cell ‘fast’ current clamp mode of the Axopatch 200A was used and APs were evoked by 4 ms stimulation pulses with a magnitude ∼1.5 times higher than AP activation threshold. V m measurements were corrected for a junction potential error of −10 mV. During APD alternans subsequent APs of alternating duration were termed odd and even APs. For APD and APD alternans analysis an average of at least eight consecutive odd and eight consecutive even APs was used.
For AP clamp experiments voltage commands in the form of atrial APs were generated from averages of APs recorded from three individual cells paced at 1.3 Hz and exhibiting CaT alternans (Kanaporis & Blatter, 2017b ). Two voltage commands were generated: APCaT_Large representing APs recorded during large CaTs, and APCaT_Small, the AP waveform observed during small CaTs (Fig. 1). These two distinct AP waveforms were used to generate the following pacing protocols: (1) same‐shape APCaT_Large protocol consisting of 24 consecutive APCaT_Large waveforms; (2) same‐shape APCaT_Small protocol consisting of 24 consecutive APCaT_Small waveforms; and (3) alternans AP protocol consisting of 12 consecutive APCaT_Large–APCaT_Small pairs. Stimulation frequency was modified by changing diastolic intervals between voltage commands. For statistical analysis of CaT properties and CaT alternans only the last six CaTs elicited by the pacing protocol were analysed to ensure that recordings were done under steady‐state conditions.
Figure 1. Action potential waveforms used as voltage commands for voltage clamp experiments.
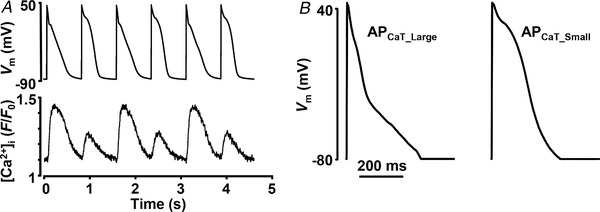
A, simultaneous recordings of APD and CaT alternans from current‐clamped single atrial myocyte. B, voltage commands used to pace cells during AP voltage clamp experiments representing AP waveforms observed during large (APCaT_Large) and small (APCaT_Small) alternans CaTs. These AP waveforms were used in all AP voltage clamp experiments in this study.
K+ currents were recorded using the whole cell configuration of the patch clamp technique in voltage clamp mode (Lu et al. 2001). The external solution contained (mmol l−1): 149 N‐methyl‐d‐glucamine, 5 MgCl2, 0.9 CaCl2 and 5 Hepes; pH 7.35 with HCl. Patch pipettes were filled with an internal solution composed of (mmol l−1): 120 K+ aspartate, 20 KCl, 5 Hepes, 10 EGTA, 5 MgATP, 5 Na+ creatine kinase phosphate and 0.65 CaCl2; pH 7.2 with KOH. L‐type Ca2+ channels were blocked by 30 μmol l−1 nisoldipine. For K+ current measurements, myocytes were voltage clamped and held at a holding V m of −60 mV. K+ currents were elicited with 3 s voltage steps ranging from −70 mV to +50 mV (10 mV intervals). Upon completion of each step command, the command potential was returned to −60 mV for 1.8 s. K+ currents were quantified as the difference current at the holding V m at the beginning of the stimulation protocol and at the end of the 3 s stimulation pulse. Tail currents were measured as the difference current at the holding V m at the beginning of the stimulation protocol and the peak of the tail current observed after V m was returned to the holding potential following the stimulation step. Currents were normalized to cell capacitance. No attempt was made to isolate specific K+ currents, i.e. measurements were performed in the absence of any K+ channel blockers. The rationale behind such an approach was to avoid unspecific action of the blockers as well as keeping conditions comparable to those during AP recordings.
Cytosolic [Ca2+] measurements
[Ca2+]i measurements
Fluo‐4 fluorescence was excited at 485 nm with a xenon arc lamp and [Ca2+]i‐dependent Fluo‐4 signals were collected at 515 nm using a photomultiplier tube. Background‐subtracted fluorescence emission signals (F) were normalized to resting fluorescence (F 0) recorded under steady‐state conditions at the beginning of an experiment, and changes of [Ca2+]i are presented as changes of F/F 0. Indo‐1 fluorescence was excited at 357 nm (xenon arc lamp) and emitted cellular fluorescence was recorded simultaneously at 405 nm (F 405) and 485 nm (F 485) with photomultiplier tubes. F 405 and F 485 signals were background subtracted and changes of [Ca2+]i are expressed as changes of the ratio F 405/F 485. Data recording and digitization were achieved using the Axon Digidata 1440A interface and pCLAMP 10.3 software. Fluorescence signals were low‐pass filtered at 50 Hz.
Field stimulation
Atrial myocytes were loaded with 5 μmol l−1 Indo‐1/AM or 5 μmol l−1 Fluo‐4/AM in the presence of 0.05% Pluronic F‐127 for 20–30 min at room temperature, and then washed for 20 min in Tyrode solution to allow for de‐esterification of the dye. CaTs were triggered by electrical field stimulation with a pair of platinum electrodes. The electrical stimulus was set at a voltage ∼50% greater than threshold to induce myocyte SR Ca2+ release. During the course of experiments cells were continuously superfused (∼1 ml min−1) with Tyrode solution.
Patch clamp
[Ca2+]i was monitored simultaneously with APs (in current clamp mode) or membrane currents (AP voltage clamp mode). For [Ca2+]i measurements cells were loaded with fluorescent probes Fluo‐4 pentapotassium salt (100 μmol l−1) or Indo‐1 pentapotassium salt (100 μmol l−1) via the patch pipette.
CaT alternans
CaT alternans was induced by incrementally increasing the pacing frequency until stable alternans was observed (typical range where stable CaTs were observed was 1.6–2.5 Hz). The degree of Ca2+ alternans was quantified as the alternans ratio (AR). AR = 1 − [Ca2+]i,Small/[Ca2+]i,Large, where [Ca2+]i,Large and [Ca2+]i,Small are the amplitudes of the large and small CaTs of a pair of alternating CaTs. By this definition AR values fall between 0 and 1, where AR = 0 indicates no Ca2+ alternans and AR = 1 indicates a situation where SR Ca2+ release is completely abolished on every other beat. CaTs were considered alternating when the beat‐to‐beat difference in CaT amplitude exceeded 10% (AR > 0.1) (Kanaporis & Blatter, 2015). The amplitude of a CaT was measured as the difference in F 405/F 485 or F/F 0 measured immediately before the stimulation pulse and at the peak of the CaT.
Langendorff perfused heart
Rabbits were anaesthetized with an intravenous injection of sodium pentobarbital (100 mg kg−1) and heparin (1000 IU kg−1). Hearts were excised and mounted on a whole heart Langendorff perfusion system (Harvard Apparatus, Holliston, MA, USA) after the aorta was cannulated. The heart was perfused with modified Krebs–Henseleit solution composed of (mmol l−1): 119 NaCl, 4 KCl, 1.2 KH2PO4, 25 NaHCO3, 10 d‐glucose, 2 sodium pyruvate, 1.8 CaCl2, 2 MgSO4, oxygenated with a 95% O2/5% CO2 gas mix and pH adjusted to 7.4 with NaOH at 37°C. Atrial electrograms were recorded from two electrodes placed on the surface of the left atrium, while electrical stimulation was applied to the right atria. To induce atrial T‐wave alternans, stimulation frequency was gradually increased until stable alternans was observed, typically in the range of 6.5–8 Hz. Temperature was maintained at 37°C.
Data analysis and presentation
Results are presented as individual observations or as mean ± SEM, and n represents the number of individual cells. Statistical significance was evaluated using unpaired and paired Student's t‐test and differences were considered significant at P < 0.05.
Results
AP morphology determines degree of CaT alternans
To determine the effect of AP morphology on the development of CaT alternans, voltage‐clamped atrial myocytes were stimulated at various frequencies with the following pacing protocols: (1) the same‐shape APCaT_Large protocol; (2) the same‐shape APCaT_Small protocol; and (3) the alternans AP protocol. AP waveforms used for voltage command were generated from prerecorded APs from current‐clamped atrial myocytes exhibiting CaT and APD alternans (Fig. 1). The degree of CaT alternans was quantified by the AR. Figure 2 A demonstrates a typical course of alternans development during the same‐shape and alternans AP clamp protocols (all traces were recorded from the same atrial myocyte). Compared to the same‐shape APCaT_Large protocol, the same‐shape APCaT_Small protocol (i.e. AP waveforms with prolonged APD) elicited CaT alternans with larger AR at any given frequency (Fig. 2 A,C). The alternans AP clamp protocols not only further increased the degree of CaT alternans, but also lowered the pacing threshold for the induction of CaT alternans (Fig. 2 A,C). AR values observed in individual cells paced with all three stimulation protocols at a cycle length (CL) of 540 ms are shown in Fig. 2 B. These results clearly demonstrate that AP morphology plays an important role in modulating intracellular Ca2+ release. AP morphology affects the threshold frequency for CaT alternans induction and determines the degree of CaT alternans, where beat‐to‐beat alternation in AP waveform caused the highest degree of severity of CaT alternans.
Figure 2. Severity of CaT alternans is determined by AP morphology.
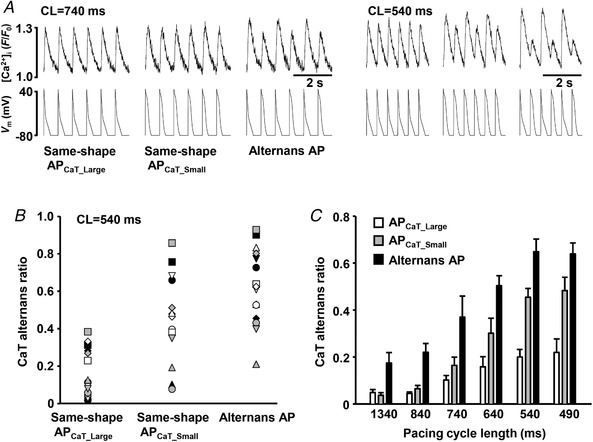
A, CaTs elicited with the same‐shape APCaT_Large, the same‐shape APCaT_Small and the alternans AP clamp protocols recorded from the same atrial myocyte paced at cycle lengths (CL) of 740 ms (left) and 540 ms (right). B, CaT alternans ratios recorded from 15 individual cells (10 rabbits) stimulated with the same‐shape APCaT_Large, the same‐shape APCaT_Small and the alternans AP clamp protocols at CL = 540 ms. Individual symbols represent individual cells. C, pacing with the same‐shape APCaT_Small waveforms (grey bars) enhances the degree of CaT alternans compared to the APCaT_Large protocol (white bars). CaT alternans ratio (AR) is further increased during the alternans AP voltage clamp protocol (black bars). n for individual data sets varies from 7 to 34.
Inhibition of Ca2+‐activated Cl− channels does not prevent CaT alternans
The results shown in Fig. 2 demonstrate that alternation in AP morphology in voltage‐clamped cells leads to an increased degree of CaT alternans. Inspired by these results we tested whether the opposite holds true, i.e. whether suppression of beat‐to‐beat variation in AP morphology would reduce the severity of CaT alternans. Previously we have demonstrated that in rabbit atrial myocytes CaCCs are a major contributor to the beat‐to‐beat APD alternation (Kanaporis & Blatter, 2016). Thus, we expected that inhibition of these channels would diminish APD alternans and consequently lessen the degree of CaT alternans. However, inhibition of CaCCs with 4,4‐diisothiocyanatostilbene‐2,2′‐disulfonic acid disodium salt hydrate (DIDS; 500 μmol l−1) in field‐stimulated atrial myocytes failed to abolish CaT alternans (Fig. 3 Aa) and on average had no effect on the AR (Fig. 3 Ac). Previously, we have demonstrated that inhibition of CaCCs indeed suppressed APD alternans (Kanaporis & Blatter, 2016), but also leads to an overall stable APD prolongation [Fig. 3 B and our previous observations (Kanaporis & Blatter, 2016))] While the former is expected to translate into a suppression of CaT alternans, a prolonged APD is likely to promote CaT alternans (Fig. 2). Thus, inhibition of CaCCs has two opposite effects on APs which results in inconsistent effects on CaT alternans, where indeed increases, decreases or no change in AR were observed (Fig. 3 Ab).
Figure 3. Suppression of Ca2+‐activated chloride channels does not prevent CaT alternans.
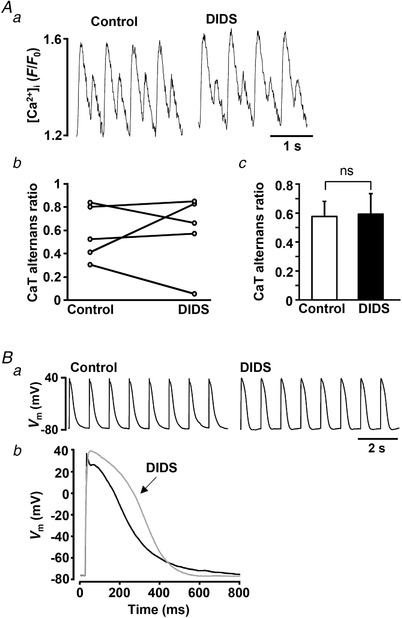
Aa, CaT alternans recorded in control and in the presence of Cl− channel blocker DIDS (500 μmol l−1) in a field stimulated atrial myocyte; A b, CaT alternans ratios (ARs) in control and after the application of DIDS in five individual field‐stimulated cells; Ac, mean CaT ARs in control and in the presence of DIDS (paired t test, P = 0.89, n = 5, 3 rabbits). B, CaCC inhibition with DIDS leads to APD prolongation. Ba, raw traces of APs recorded in control and after application of DIDS. Bb, overlay of APs recorded in control (black) and after the application of DIDS (grey).
Activation of K+ currents shortens AP duration
Data obtained in AP‐clamp experiments (Fig. 2) suggest strongly that APD shortening might be a viable approach to prevent the development of CaT alternans. To test this hypothesis APD was pharmacologically shortened by the application of K+ channel agonists. For this purpose, we used the compounds NS1643 (20 μmol l−1), a selective agonist of Kv11.1 channels (rapid delayed rectifier K+ current, I Kr), and ML277 (0.5 μmol l−1), an activator of Kv7.1 (I Ks, slow delayed rectifying K+ current) channels. In voltage clamp experiments (Fig. 4) we confirmed that both compounds increased K+ currents in rabbit atrial myocytes. Typical traces of K+ currents before and after the application of the agonists are shown in Fig. 4 Aa for NS1643 and Fig. 4 Ba for ML277. ML277 significantly increased K+ current across all test voltages ≥−40 mV (Fig. 4 Bb). The effects of NS1643 were more complex and voltage‐dependent. At test voltages >0 mV there was no significant effect of the drug, whereas at voltages <0 mV (i.e. in the voltage range crucial for repolarization of the atrial AP and APD) the drug increased the current (Fig. 4 Ab). In contrast, an increase of tail currents across the entire voltage range was observed for both drugs (Fig. 4 Ac,Bc). The increase in tail current was statistically significant at voltages ≥−30 mV for ML277 and ≥−20 mV for NS1643.
Figure 4. K+ channel agonists increase K+ current in atrial myocytes.
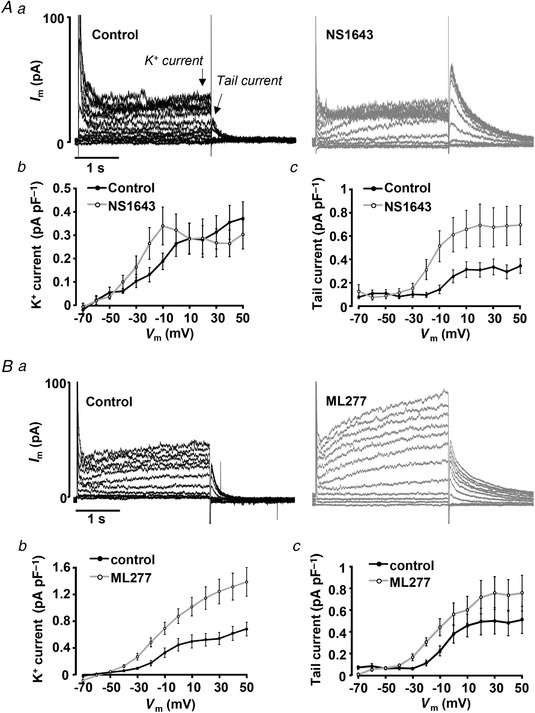
Aa, K+ current traces elicited by voltage steps (−70 to +50 mV) from V H = −60 mV before and after application of Kv11.1 agonist NS1643 (20 μmol l−1). Arrows indicate where the amplitudes of K+ and tail currents were measured. I–V curves of K+ currents (Ab) and tail currents (Ac) recorded in control and in the presence of NS1643 (n = 5, 4 rabbits). B, representative current traces (Ba) elicited by the same protocol as in A before and after application of Kv7.1 agonist ML277 (0.5 μmol l−1). I–V curves of K+ currents (Bb) and tail currents (Bc) recorded in control and after application of ML277 (n = 8, 3 rabbits).
Next, we tested the effect of the K+ channel agonists on APD in current clamped atrial myocytes paced at 1 Hz with no CaT alternans. Typical traces of APs recorded before and after the application of K+ agonists are shown in Fig. 5. Both NS1643 (Fig. 5 A) and ML277 (Fig. 5 B) resulted in APD shortening. APD at 30% repolarization (APD30) decreased by 25 ± 9% in the presence of NS1643 (paired t‐test, P = 0.17, n = 9, 7 rabbits) and by 37 ± 8% in ML277 (P = 0.010, n = 10, 6 rabbits). Application of NS1643 resulted in statistically significant shortening of APD at 50% repolarization (APD50) by 47 ± 8% (P = 0.019) while at 90% repolarization (APD90) APD was decreased by 36 ± 8% (P = 0.0012). Similarly, ML277 shortened APD50 and APD90 by 32 ± 8% (P = 0.012) and 26 ± 8% (P = 0.013), respectively.
Figure 5. K+ channel agonists shorten AP duration (APD).
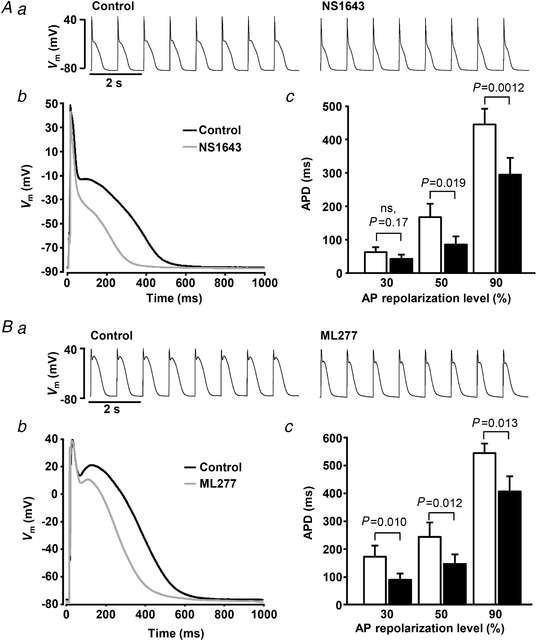
Aa, traces of APs recorded in control and after the application of NS1643 (20 μmol l−1); Ab, overlay of APs recorded in control (black) and after the application of NS1643 (grey); Ac, mean APD at 30, 50 and 90% repolarization level before and after application of NS1643 (n = 9, 7 rabbits). Ba, traces of APs recorded in control and after the application of ML2773 (0.5 μmol l−1); Bb, overlay of APs recorded in control and after the application of ML277; Bc, mean APD at 30, 50 and 90% repolarization level before and after the application of ML277 (n = 10, 6 rabbits).
APD shortening suppresses development and severity of CaT alternans
To investigate the effect of APD shortening on the development of CaT alternans, both K+ channel agonists were applied to field‐stimulated single atrial myocytes. In this series of experiments the stimulation rate was gradually increased (typically to 1.6–2.5 Hz) until stable atrial CaT alternans was observed. Stable alternans was recorded for ∼1 min (mean 53 ± 10 s; range 20–185 s; n = 18) before cells were exposed to NS1643 (20 μmol l−1) or ML277 (0.5 μmol l−1) and changes in CaT AR were monitored. APD shortening induced by NS1643 or ML277 significantly reduced CaT AR (Fig. 6) and essentially abolished CaT alternans. The maximal effect on CaT AR was reached 37 ± 7 and 79 ± 10 s after application of NS1643 and ML277, respectively. In the presence of NS1643, CaT AR decreased from 0.58 ± 0.05 in control to 0.09 ± 0.01 (paired t‐test, P = 0.000004, n = 11, 4 rabbits), while ML277 reduced CaT AR from 0.60 ± 0.06 to 0.09 ± 0.03 (n = 10, P = 0.00004, 6 rabbits). To exclude the possibility that the observed decrease in CaT AR was due to time‐dependent but drug‐independent changes of CaT alternans, we performed control experiments in which atrial myocytes (n = 11) exhibiting CaT alternans were paced at a constant rate for 10 min. The data shown in Fig. 7 A demonstrate that CaT AR remained stable during this prolonged period of constant stimulation under control conditions, after which application of ML277 led to a rapid AR decrease (Fig. 7 B).
Figure 6. AP shortening reduces degree of CaT alternans in field‐stimulated atrial myocytes.
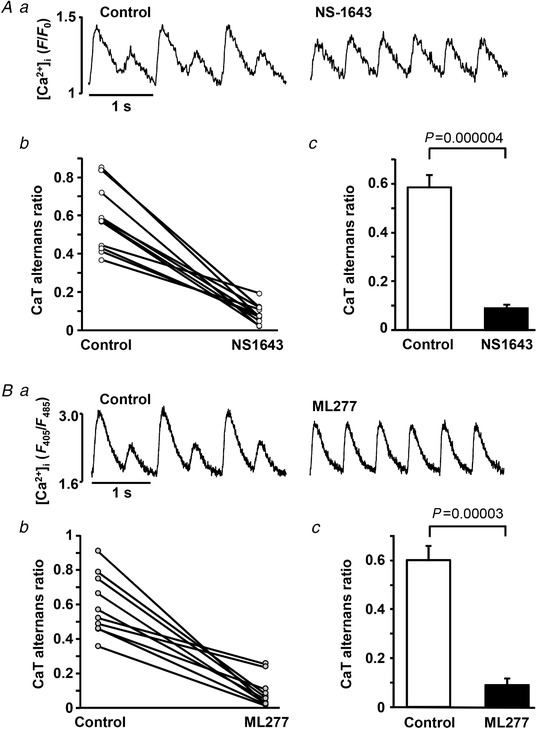
Aa, CaTs recorded before and after application of Kv11.1 channel agonist NS1643 in field‐stimulated atrial myocytes. Ab, changes of CaT alternans ratio (AR) in control and in the presence of NS1643 from 11 individual cells. Ac, mean CaT AR in control and after application of NS1643 (n = 11, 4 rabbits). Ba, CaTs recorded before and after application of Kv7.1 channel agonist ML277 in field stimulated atrial myocytes. Bb, changes in CaT AR from 10 individual cells. Bc, mean CaT AR in control and after application of ML277 (n = 10, 6 rabbits).
Figure 7. Degree of CaT alternans remains stable during prolonged pacing.
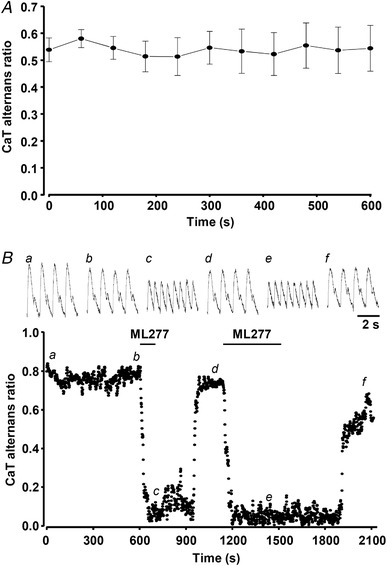
A, mean CaT alternans ratio (AR) over time in field‐stimulated atrial myocytes (n = 11, 4 rabbits) continuously paced for 10 min. B, application of ML277 (0.5 μmol l−1), following 10 min recording of stable CaT alternans, leads to fast reduction in CaT AR in field‐stimulated atrial myocytes. Recovery of CaT AR is observed during the washout of ML277. Top panel: representative CaT traces observed at the times indicated by the corresponding letter on the main graph.
In the next series of experiments, we simultaneously monitored CaT and APD alternans dynamics in current clamped atrial myocytes. Representative traces of V m and [Ca2+]i before and after the application of NS1643 or ML277 are shown in Fig. 8. In the presence of both K+ agonists the APD became shorter and the degree of CaT decreased significantly. Not only CaT alternans, but also APD alternans was abolished. Figure 8 Ad and Fig. 8 Bd show the ratios of APD recorded on odd beats to APD of even beats at 30, 50 and 90% AP repolarization. When CaT alternans was observed under control conditions, APs concurring with large CaTs were arbitrarily defined as odd beats. Figure 8 Ad and Fig. 8 Bd demonstrate that in the presence of K+ channel agonists the APDOdd/APDEven ratio approached 1, i.e. beat‐to‐beat APD alternans was no longer observed.
Figure 8. AP shortening suppresses CaT and APD alternans in current‐clamped atrial myocytes.
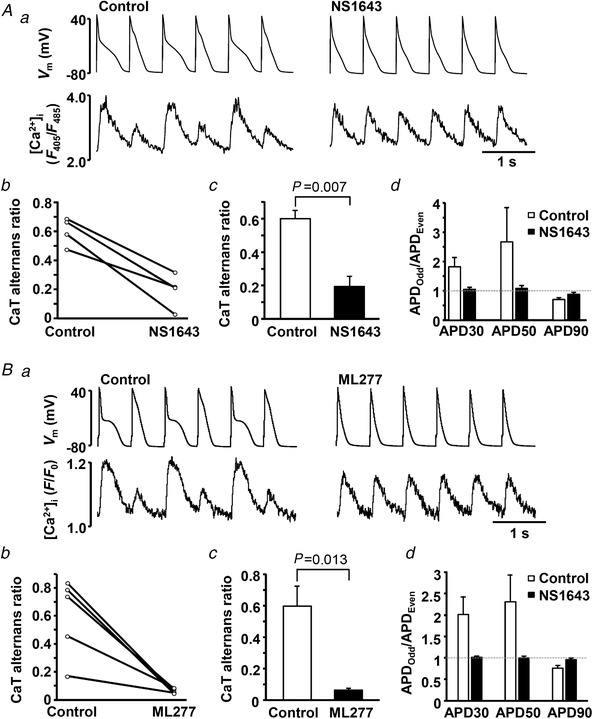
A and B, effect of K+ channel agonists NS1643 (A) and ML277 (B) on CaT alternans in current‐clamped atrial myocytes. a, action potential and [Ca2+]i traces observed before and after application of NS1643 (n = 4, 3 rabbits) or ML277 (n = 5, 5 rabbits). b, CaT alternans ratio in individual cells in control and after the application of K+ channel agonists. c, mean CaT alternans ratio in control and in the presence of K+ channel agonists. d, in addition to CaT alternans, APD alternans also was abolished in the presence of NS1643 and ML277. Ratios of APDs of odd and even beats at 30, 50 and 90% of AP repolarization are shown. In the presence of K+ channel agonists the ratio approaches 1 (marked by dashed line), indicating elimination of APD alternans.
To further verify that CaT alternans suppression was caused specifically by shortening of the AP and not by other potential effects of the K+ channel agonists, we tested NS1643 and ML277 in AP voltage‐clamped cells. In this case the voltage applied to the cell is determined by the stimulation protocol and is not modified pharmacologically. The data show that under voltage‐clamp conditions the two K+ channel agonists had no significant effect on the development of CaT alternans (Fig. 9). During all three AP voltage‐clamp protocols ML227 and NS1634 had no effect on the ARs. These experiments indeed confirmed that the effect of the K+ channel agonists is due to APD shortening.
Figure 9. K+ channel agonists have no effect on CaT alternans under voltage clamp conditions.
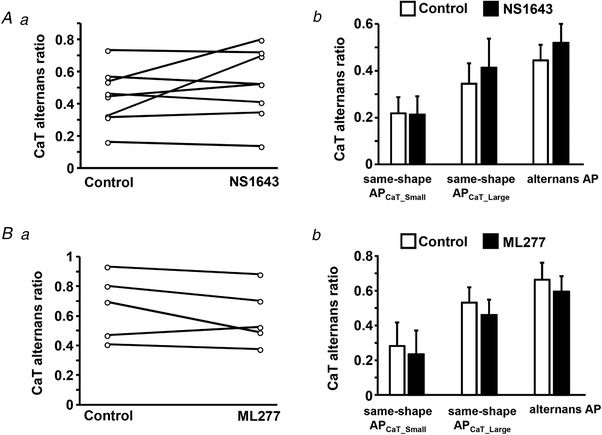
A and B, application of NS1643 (n = 8, 7 rabbits) (A) and ML277 (n = 5, 5 rabbits) (B) has no effect on the degree of CaT alternans in AP voltage‐clamped atrial myocytes, demonstrating that the effect of these compounds is caused by shortening of APD. a, CaT alternans ratio (AR) in individual atrial myocytes before and after application of agonists of K+ channels observed during the alternans AP stimulation protocol. b, mean CaT ARs in the absence and presence of the K+ agonists during the same‐shape APCaT_Small, the same‐shape APCaT_Large and the alternans AP protocol. In all groups mean CaT ARs in control and in the presence of agonists are not statistically different.
Activation of K+ channels suppresses alternans at the whole heart level
Finally, we tested if activation of K+ channels is effective in attenuating APD alternans at the organ level. Atrial T‐wave (Ta) alternans was monitored in Langendorf‐perfused isolated rabbit hearts. Electrograms were recorded from two electrodes placed on the left atria, while high rate stimulation was applied to the right atria. To induce Ta‐wave alternans, the stimulation frequency was gradually increased until stable Ta‐wave alternans was observed (in the range 6.5–8 Hz at 37°C). Electrogram sequences recorded in control and after the application of NS1643 (20 μmol l−1) via the perfusate are shown in Fig. 10. NS1643 was tested in four hearts. In two hearts NS1643 completely abolished Ta‐wave alternans whereas in the other two a significant reduction of the degree of Ta‐wave alternans was observed. In summary, the data show that the protective effect against atrial alternans of K+ channel agonists and presumably APD shortening observed in single cells could be verified at the whole organ level.
Figure 10. NS1643 abolishes atrial T‐wave (Ta) alternans in Langendorff‐perfused hearts.
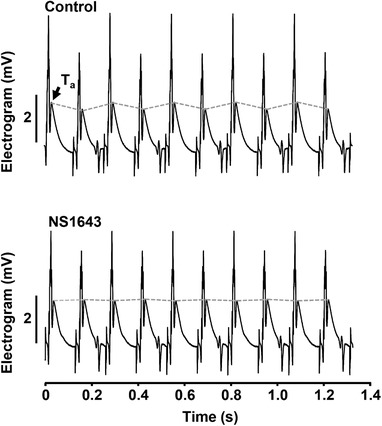
Atrial electrograms recorded from the same Langendorf‐perfused isolated rabbit heart in control and after the application of NS1643 (20 μmol l−1). In the presence of NS1643 beat‐to‐beat alternation of the Ta‐wave is eliminated.
Discussion
In this study we investigated the effect of AP morphology on the development of CaT alternans in rabbit atrial myocytes, and whether CaT alternans can be controlled by targeted modulation of the shape of the AP. The main findings are as follows: (1) AP clamp experiments applying various APCaT_Large and APCaT_Small command voltage protocols revealed that AP morphology determined the alternans pacing frequency threshold and degree of CaT alternans (Fig. 2); (2) Previously we have demonstrated that suppression of CaCCs greatly reduced beat‐to‐beat variation in APD (Kanaporis & Blatter, 2016). However, suppression of CaCCs failed to reduce the degree of CaT alternans (Fig. 3) because the concomitant APD prolongation favours the occurrence of CaT alternans (see also Fig. 2); (3) APD shortening by pharmacological activation of I Ks or I Kr efficiently suppressed CaT and APD alternans in single atrial myocytes (Figs 6 and 8); (4) Application of a K+ channel agonist significantly attenuated or even abolished Ta alternans at the whole heart level (Fig. 10).
Mechanisms of atrial alternans and the role of AP morphology
The coincidence of APD and CaT alternans is well documented (Fig. 1) (Pruvot et al. 2004; Kanaporis & Blatter, 2015) and it is generally agreed that the bi‐directional coupling between [Ca2+]i and V m plays a key role in the development of alternans. V m directly affects the activity of Ca2+ handling mechanisms that are voltage‐dependent, while [Ca2+]i dynamics modulate V m through Ca2+‐dependent ion currents and transporters. Due to the complex nature of the bi‐directional coupling between V m and [Ca2+]i, the experimental distinction between effects of Ca2+ and V m is difficult and therefore the mechanisms underlying alternans remain incompletely understood, especially in atrial tissue. Computational (Nolasco & Dahlen, 1968; Watanabe & Koller, 2002; Tolkacheva et al. 2004; Jordan & Christini, 2006, 2007) and several experimental studies (Koller et al. 2005; Tolkacheva et al. 2006) have suggested that self‐sustaining beat‐to‐beat oscillations of APD can occur if the slope of diastolic APD restitution is steep enough, and that the role of V m as a causative factor of alternans becomes more prominent as pacing rates increase (Jordan & Christini, 2007; Bayer et al. 2010). However, contrary to these results, other experimental studies found a poor relationship between experimentally determined APD restitution kinetics and inducibility of alternans (Saitoh et al. 1988; Banville & Gray, 2002; Kalb et al. 2004; Pruvot et al. 2004; Wu & Patwardhan, 2006). In addition to the APD restitution hypothesis, there is strong evidence that alternans can be initiated by disturbances of intracellular Ca2+ signalling (Chudin et al. 1999; Diaz et al. 2004; Goldhaber et al. 2005; Shkryl et al. 2012; Kanaporis & Blatter, 2015). Major support for this notion stems from the demonstration that Ca2+ alternans can be elicited in voltage‐clamped ventricular (Chudin et al. 1999) and atrial (Kanaporis & Blatter, 2015) myocytes where V m is kept constant on every beat, i.e. in the absence of APD alternans. In addition, beat‐to‐beat alternation in AP morphology was abolished when intracellular Ca2+ release was blocked (Kanaporis & Blatter, 2015), suggesting that electrical alternans is a consequence rather than the cause of cardiac alternans. A popular candidate for the underlying mechanism for CaT alternans is beat‐to‐beat variation in SR Ca2+ concentration ([Ca2+]SR) (Diaz et al. 2004; Eisner et al. 2006; Nivala & Qu, 2012; Kanaporis & Blatter, 2017b), although there is also direct experimental evidence that CaT alternans can be observed without alternans in diastolic [Ca2+]SR (Huser et al. 2000; Picht et al. 2006). As an alternative to the SR Ca2+ load hypothesis, beat‐to‐beat differences in the refractoriness of SR Ca2+ release was suggested (Kornyeyev et al. 2012; Shkryl et al. 2012; Lugo et al. 2014; Wang et al. 2014).
Irrespective of whether V m or [Ca2+]i plays the leading role in the initiation of alternans, here and in our previous study (Kanaporis & Blatter, 2017b ) we demonstrate that AP morphology and beat‐to‐beat variation of APD are pivotal for development and sustainability of atrial alternans. We generated AP waveforms that emulate experimentally recorded V m changes during CaT alternans and used these AP waveforms in AP voltage‐clamp experiments. When cells were paced with these AP waveforms it was observed that: (1) CaT alternans was induced at lower pacing rate and degree of alternans was higher if there is APD variation on a beat‐to‐beat basis (AP alternans voltage clamp; Fig. 2); and (2) when the same‐shape AP clamp protocols were applied, the AP waveform with prolonged APD (APCaT_Small) induced more pronounced CaT alternans compared to the APCaT_Large protocol (Fig. 2). While several mechanisms could be envisaged as to how this might occur, in a recent study we demonstrated two distinct mechanisms that involved end‐diastolic SR Ca2+ content and the kinetics of L‐type Ca2+ current (Kanaporis & Blatter, 2017b ). The AP waveform observed during large Ca2+ release results in a larger peak L‐type Ca2+ current that provides a more efficient trigger for intracellular Ca2+ release. The APCaT_Large waveform also leads to lower [Ca2+]SR available for the subsequent beat. Consequently, both mechanisms result in enhanced Ca2+ alternans (Kanaporis & Blatter, 2017b ). A direct effect of AP morphology on atrial Ca2+ release and contraction has been observed previously, and occurred through prolongation of atrial APD after pharmacological inhibition of K+ currents, which in turn enhanced Ca2+ entry via reverse mode Na+/Ca2+ exchange (Schotten et al. 2007).
Atrial alternans and atrial fibrillation
The majority of clinical data that relate cardiac alternans and arrhythmias have so far been obtained in ventricles. The beat‐to‐beat alternation of ventricular AP repolarization, reflected in the ECG as T‐wave alternans (TWA), has proven to be a valuable prognostic tool for ventricular arrhythmia risk stratification (Merchant et al. 2013). The clinical use of atrial APD alternans as a diagnostic and prognostic tool, however, is limited by the fact that the atrial repolarization signal is masked by the ventricular QRS complex in conventional ECG recordings. Nevertheless, several experimental (Jousset et al. 2012; Monigatti‐Tenkorang et al. 2014; Verrier et al. 2016) and clinical studies (Narayan et al. 2002, 2011a, b ; Hiromoto et al. 2005; Lalani et al. 2013), using monophasic AP electrodes to monitor atrial repolarization alternans in vivo, have provided convincing evidence that atrial APD alternans may lead directly to AF or its transition from atrial flutter. In addition, it was suggested that, similarly to ventricular TWA, Ta‐wave alternans can be suitable to predict vulnerability to AF (Narayan et al. 2011a; Lalani et al. 2013). For example, it was demonstrated that atrial APD alternans preceded AF episodes (Narayan et al. 2002, 2011a; Monigatti‐Tenkorang et al. 2014). Moreover, in patients with persistent AF Ta‐wave alternans is induced at relatively low pacing rates (Narayan et al. 2011a). Also, atrial APD alternans was more severe and more prevalent in patients with persistent AF than in subjects with paroxysmal AF (Lalani et al. 2013), while alternans was not observed in the control group (Narayan et al. 2011a; Lalani et al. 2013). The putative mechanisms connecting APD alternans to arrhythmias involve the development of spatially discordant alternans (Wilson & Rosenbaum, 2007; Gelzer et al. 2008; Wilson et al. 2009; Hsieh et al. 2016). Similarly to findings in ventricle, discordant alternans was also observed in the atria and has been directly linked to the development of AF (Hiromoto et al. 2005; Jousset et al. 2012; Verrier et al. 2016).
AP shortening as a tool for arrhythmia prevention
Anti‐arrhythmic drug therapy and catheter ablation are the most common approaches for AF treatment. However, at present both approaches are far from optimal, particularly in patients with persistent forms of AF as the rate of arrhythmia recurrence and hospitalizations remains high (Morillo et al. 2014). Furthermore, meta‐data analysis suggests that the currently available anti‐arrhythmic drug therapy is inferior to catheter ablation (Morillo et al. 2014; Geng et al. 2017; Chen et al. 2018). Therefore, to develop a more effective drug therapy for AF, a better mechanistic understanding of atrial arrhythmogenesis and the mechanisms of action of different anti‐arrhythmic agents is paramount. Great variability in the treatment outcomes also implies that the causes and mechanisms leading to AF are heterogeneous and therefore need to be addressed in a more personalized fashion.
Our study demonstrates that AP morphology plays an important role in the development and sustainability of atrial alternans. We have determined that both prolongation of APD and beat‐to‐beat alternation in APD increase the severity of CaT alternans (Fig. 2). Furthermore, we investigated whether pharmacological modulations of AP morphology and dynamics can be used to reduce or eliminate CaT alternans as data obtained in voltage‐clamped myocytes have suggested. CaCCs are a major contributor to Ca2+‐induced APD alternans in rabbit atria (Kanaporis & Blatter, 2016). Therefore, we blocked CaCCs to reduce alternation in APD. While inhibition of CaCCs indeed greatly reduced APD alternans, it failed to rescue or significantly reduce the degree of CaT alternans (Fig. 3). We speculate that CaCC inhibition fails to abolish CaT alternans due to substantial APD prolongation (Fig. 3 B) which by itself increases the propensity of CaT alternans. While attenuation of APD alternans alone is expected to diminish the risk of arrhythmogenicity and can be beneficial for arrhythmia prevention, the fact that CaT alternans precipitates into mechanical alternans constitutes a major concern. As known for the ventricle, mechanical alternans in failing hearts is associated with increased mortality (Kim et al. 2014). The role of atrial contractility is often underestimated. Active atrial contraction is crucial for ventricular filling during ventricular diastole and is referred to as atrial booster function or ‘atrial kick’ (Rahimtoola et al. 1975; Nicod et al. 1986; Blume et al. 2011; Trafford et al. 2013; Hoit, 2014; Mehrzad et al. 2014). This booster function contributes 20–40% of the end‐diastolic filling, and becomes a critical factor for cardiac output, especially at high heart rates and under pathological conditions involving the atrioventricular valves. The significance of the atrial kick under pathological conditions can be illustrated by a striking, albeit unusual case where the left ventricular filling and resulting cardiac output were reported to be exclusively dependent on the atrial kick (Neuman et al. 2011). Given the importance of atrial contraction for cardiac output, mechanical alternans of the atria is expected to have profound effects on overall cardiac performance. Thus, from the perspective of cardiac performance it is equally important to prevent and treat electrical (arrhythmia risk) and mechanical (impaired haemodynamics and cardiac output) alternans. Consequently, a desirable strategy of alternans prevention or treatment would entail both, interventions that lower the propensity of APD as well as mechanical and CaT alternans.
Based on the data shown in Fig. 2, we hypothesized that APD shortening could reduce AR and ameliorate CaT alternans. While there is a wide range of pharmacological agents known to modulate APD, in this study we aimed at interventions that should have minor effects on Ca2+ and Na+ currents and therefore minimally affect excitability, excitation–contraction coupling and Ca2+ release. We focused on the repolarization phase of the atrial AP that is largely governed by K+ conductances and used the K+ channel agonists NS1643 and ML277 to selectively increase I Kr and I Ks (Fig. 4) and shorten APD (Fig. 5). Application of K+ channel agonists efficiently abolished or attenuated CaT alternans in both field‐stimulated (Fig. 6) and current‐clamped atrial myocytes (Fig. 8). Experiments on current‐clamped atrial myocytes also revealed that NS1643 and ML277 not only abolished CaT alternans, but also eliminated APD alternans (Fig. 8), and thus showed the targeted therapeutic effect as discussed above. In addition, application of NS1643 eliminated or reduced high‐rate pacing‐induced atrial repolarization alternans at the whole heart level (Fig. 10). Because both K+ channel agonists, albeit targeting different K+ channels, essentially produced the same results, it was concluded that the key effect here is shortening of the AP, and that – in a more general sense – interventions aimed at ADP shortening represent potentially an efficient tool to limit or prevent CaT alternans, concomitant with the elimination of APD alternans. Furthermore, in AP voltage‐clamp experiments (Fig. 9) where APD is fixed, NS1643 and ML277 had no effect on the CaT AR, clearly demonstrating that suppression of CaT alternans was due to the shortening of AP.
APD shortening might strike as a counterintuitive strategy for AF prevention because of the notion that APD shortening carries its own increased arrhythmogenic risk. However, the mechanisms leading to AF are complex and AF risk increases significantly under various pathological conditions, such as heart failure or Brugada syndrome (Francis & Antzelevitch, 2008). Interestingly, increased AF risk was found in connection with both APD shortening and prolongation (Nielsen et al. 2013). Based on our results, we suggest that APD shortening might be a viable anti‐arrhythmic strategy in some cohorts of patients, particularly in those exhibiting prolonged APD. The link between AF and increased APD durations is well documented. Atrial arrhythmias in the form of polymorphic atrial tachycardia and AF have been observed regularly in patients with genetically established long QT syndrome (LQTS) (Kirchhof et al. 2003; Johnson et al. 2008; Zellerhoff et al. 2009; Guerrier et al. 2013). A relationship between a prolonged QT interval and an increased risk of AF was demonstrated in various patient cohorts as well as in general population groups (Kirchhof et al. 2003; Mandyam et al. 2013; Nielsen et al. 2013; Hoshino et al. 2015; O'Neal et al. 2015). Furthermore, the risk of AF correlated with the degree of APD prolongation (Nielsen et al. 2013; Hoshino et al. 2015). Although the atrial and ventricular myocardium differ in their endowment with ion channels (see below), both tissues still show important commonalities, a fact that suggests that many patients with LQTS are also likely to exhibit delayed atrial repolarization (Kirchhof et al. 2003). Moreover, management of AF in LQTS patients is challenging because Class Ia and Class III anti‐arrhythmics commonly used for AF treatment would result in additional AP prolongation. Nonetheless, a case of AF prevention in a patient with type 1 LQTS by late Na+ current blocker mexiletine has been reported (El Yaman et al. 2008), although it remains unclear if AF suppression was achieved due to mexiletine‐induced APD shortening or due to other mechanisms related to the blockage of late Na+ current.
While K+ agonists are not currently used for the management of cardiac arrhythmias, they have been used to modify AP morphology and reduce arrhythmias in several experimental studies. Activation of I Ks (Ma et al. 2015) or I Kr (Zhang et al. 2012) in cardiomyocytes derived from induced pluripotent stem cells isolated from patients with LQTS was shown to restore (at least partially) AP parameters to control levels. Several studies have attempted to use K+ agonists to shorten APs in intact hearts of LQTS animal models. For example, activation of I Ks was demonstrated to shorten APD and suppress early afterdepolarizations in rabbit hearts with acquired LQTS2 and left ventricular hypertrophy (Xu et al. 2002). The I Kr agonist NS1643 was shown to successfully shorten the QT interval and suppress ventricular arrhythmias in rabbits with LQTS induced by 3 weeks of atrioventricular block with ventricular bradypacing, and in rabbits where APD was prolonged by dofetilide‐induced I Kr inhibition (Diness et al. 2008). In another study ICA‐105574 (an I Kr agonist), but not NS1643, completely prevented ventricular arrhythmias in Langendorff‐perfused guinea pig hearts caused by I Kr and I Ks inhibition, although both ICA‐105574 and NS1643 could reverse the drug‐induced prolongation of APD in ventricular myocytes (Meng et al. 2013). All aforementioned studies have focused on ventricular arrhythmias and, to the best of our knowledge, AP shortening as a therapeutic strategy for atrial arrhythmias has not yet been tested. Atrial and ventricular cells are endowed with unique, tissue‐specific sets of ion channels leading to distinctive AP morphologies, but also to tissue‐specific options for ion‐channel modulation and AP manipulation. For example, CaCCs are larger in the atrium than in the ventricle (Szigeti et al. 1998), and small‐conductance Ca2+‐activated (Tuteja et al. 2005; Hsueh et al. 2013), two‐pore domain (Limberg et al. 2011), acetylcholine‐activated (Bingen et al. 2013) and ultra‐rapid rectifier (Ravens & Wettwer, 2011) K+ channels are predominantly expressed in the atria. In addition, mutations in Kv7.1 (Chen et al. 2003; Bartos et al. 2011) and Kv11.1 (Andreasen et al. 2013) have been associated with familial AF. This opens an opportunity to develop atria‐specific modulators of AP morphology and thus tissue‐specific anti‐arrhythmic strategies.
While we report here a potential use of I K activation as an anti‐arrhythmic strategy in the atrium, our study also has some limitations. All experiments were performed at room temperature (20–24°C). We (Huser et al. 2000) and others (summarized in Euler, 1999) have shown that the pacing frequency threshold for the occurrence of alternans decreases with lower temperatures. By conducting experiments at room temperature, alternans could be studied in isolated myocytes with pacing protocols that were significantly less stressful, allowing for prolonged stimulation protocols and extended cell viability over time. Furthermore, kinetic limitations of fluorescent Ca2+ indicator dyes make reliable recording of high‐frequency CaTs difficult, and in the case of CaT alternans the small‐amplitude CaT typically starts to fuse with the declining phase of the preceding large‐amplitude transient and thus becomes difficult to detect and to be quantified. However, Ca2+ handling mechanisms and pathways are temperature‐dependent, particularly ion channels showing faster kinetics at physiological temperatures. Thus, AP morphology during alternans might be affected differently at higher temperature. Nonetheless, previous studies found that electrophysiological properties of the myocardium that are potentially relevant for the development of alternans are not significantly different whether experiments were performed at 30 or 37°C (Myles et al. 2011), consistent with a study on alternans by Pastore et al. (1999) that found no mechanistic differences between 27 and 37°C.
The use of pharmacological tools to modulate ion channel activity raises the question of specificity of the drugs used. DIDS, for example, is not especially selective for CaCCs. DIDS is a widely used inhibitor of cellular anion conductances and was demonstrated to suppress various Cl− channels (Duan & Nattel, 1994), the Cl−/HCO3 − exchanger (Helbig et al. 1988) and voltage‐dependent anion channel in mitochondria (Skonieczna et al. 2017). The K+ channel agonists ML277 and NS1643 are not very widely used and therefore data on their specificity are scarce. To date no major unspecific action of these drugs has been reported in the heart. ML277 has a more than 100‐fold higher selectivity for Kv7.1 over Kv7.2 or Kv7.4 (Yu et al. 2010). A more pronounced effect of NS1643 on the Kv11.1b isoform than on the Kv11a isoform is known (Larsen et al. 2010). NS1643 also activates other members of the hERG channel family, including Kv11.2 (Elmedyb et al. 2007) and Kv11.3 (Bilet & Bauer, 2012), and also affects a large‐conductance Ca2+‐activated K+ channel (Wu et al. 2008). However, the aim of our study was not to determine the effect of activation of a particular channel, rather K+ activators were used as a tool to shorten the AP, i.e. to achieve the desired AP morphology that was predicted to prevent the development of CaT alternans. Therefore, in our settings a high specificity of the compounds, while desirable, was not essential.
The perfused whole heart data shown in Fig. 10 reveal indications of a second degree 2:1 AV block, an occurrence that was observed in several hearts tested at high pacing rates. However, this pattern showed no obvious correlation with the degree of Ta‐wave alternans and was not affected by the application of NS1643, i.e. the AV block remained unchanged while NS1643 abolished or reduced Ta‐wave alternans. Therefore, it is highly unlikely that these 2:1 AV block‐like patterns contribute in any way to the development of atrial alternans in our experimental context.
Additional information
Competing interests
None.
Author contributions
Conception and design of the work: G.K and L.A.B. Collection, analysis and interpretation of data and drafting the article: G.K, Z.M.K and L.A.B. All authors have approved the final version of the manuscript.
Funding
This work was supported by National Institutes of Health grants HL057832, HL132871 and HL134781 to L.A.B. G.K. was supported by American Heart Association grant 16GRNT30130011.
Biography
Giedrius Kanaporis received his PhD in Biophysics from Kaunas University of Medicine, Lithuania. He worked as a postdoctoral research fellow at Stony Brook University in the USA and Kaunas University of Medicine in Lithuania. Currently he is an Assistant Professor at Rush University in Chicago, USA. His research interests are cell‐to‐cell communication, gap junctions, calcium signalling, electrophysiology of the heart, pathophysiological cardiac remodelling and mechanisms of cardiac arrhythmias.

Edited by: Peying Fong & Robert Harvey
References
- Andreasen L, Nielsen JB, Christophersen IE, Holst AG, Sajadieh A, Tveit A, Haunso S, Svendsen JH, Schmitt N & Olesen MS (2013). Genetic modifier of the QTc interval associated with early‐onset atrial fibrillation. Can J Cardiol 29, 1234–1240. [DOI] [PubMed] [Google Scholar]
- Banville I & Gray RA (2002). Effect of action potential duration and conduction velocity restitution and their spatial dispersion on alternans and the stability of arrhythmias. J Cardiovasc Electrophysiol 13, 1141–1149. [DOI] [PubMed] [Google Scholar]
- Bartos DC, Duchatelet S, Burgess DE, Klug D, Denjoy I, Peat R, Lupoglazoff JM, Fressart V, Berthet M, Ackerman MJ, January CT, Guicheney P & Delisle BP (2011). R231C mutation in KCNQ1 causes long QT syndrome type 1 and familial atrial fibrillation. Heart Rhythm 8, 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer JD, Narayan SM, Lalani GG & Trayanova NA (2010). Rate‐dependent action potential alternans in human heart failure implicates abnormal intracellular calcium handling. Heart Rhythm 7, 1093–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilet A & Bauer CK (2012). Effects of the small molecule HERG activator NS1643 on Kv11.3 channels. PLoS One 7, e50886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingen BO, Neshati Z, Askar SF, Kazbanov IV, Ypey DL, Panfilov AV, Schalij MJ, de Vries AA & Pijnappels DA (2013). Atrium‐specific Kir3.x determines inducibility, dynamics, and termination of fibrillation by regulating restitution‐driven alternans. Circulation 128, 2732–2744. [DOI] [PubMed] [Google Scholar]
- Blume GG, McLeod CJ, Barnes ME, Seward JB, Pellikka PA, Bastiansen PM & Tsang TS (2011). Left atrial function: physiology, assessment, and clinical implications. Eur J Echocardiogr 12, 421–430. [DOI] [PubMed] [Google Scholar]
- Chen C, Zhou X, Zhu M, Chen S, Chen J, Cai H, Dai J, Xu X & Mao W (2018). Catheter ablation versus medical therapy for patients with persistent atrial fibrillation: a systematic review and meta‐analysis of evidence from randomized controlled trials. J Interv Card Electrophysiol 52, 9–18. [DOI] [PubMed] [Google Scholar]
- Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, Jin HW, Sun H, Su XY, Zhuang QN, Yang YQ, Li YB, Liu Y, Xu HJ, Li XF, Ma N, Mou CP, Chen Z, Barhanin J & Huang W (2003). KCNQ1 gain‐of‐function mutation in familial atrial fibrillation. Science 299, 251–254. [DOI] [PubMed] [Google Scholar]
- Chinitz JS, Vaishnava P, Narayan RL & Fuster V (2013). Atrial fibrillation through the years: contemporary evaluation and management. Circulation 127, 408–416. [DOI] [PubMed] [Google Scholar]
- Chudin E, Goldhaber J, Garfinkel A, Weiss J & Kogan B (1999). Intracellular Ca2+ dynamics and the stability of ventricular tachycardia. Biophys J 77, 2930–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugh SS, Havmoeller R, Narayanan K, Singh D, Rienstra M, Benjamin EJ, Gillum RF, Kim YH, McAnulty JH, Jr. , Zheng ZJ, Forouzanfar MH, Naghavi M, Mensah GA, Ezzati M & Murray CJ (2014). Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation 129, 837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz ME, O'Neill SC & Eisner DA (2004). Sarcoplasmic reticulum calcium content fluctuation is the key to cardiac alternans. Circ Res 94, 650–656. [DOI] [PubMed] [Google Scholar]
- Diness TG, Yeh YH, Qi XY, Chartier D, Tsuji Y, Hansen RS, Olesen SP, Grunnet M & Nattel S (2008). Antiarrhythmic properties of a rapid delayed‐rectifier current activator in rabbit models of acquired long QT syndrome. Cardiovasc Res 79, 61–69. [DOI] [PubMed] [Google Scholar]
- Duan D & Nattel S (1994). Properties of single outwardly rectifying Cl− channels in heart. Circ Res 75, 789–795. [DOI] [PubMed] [Google Scholar]
- Edwards JN & Blatter LA (2014). Cardiac alternans and intracellular calcium cycling. Clin Exp Pharmacol Physiol 41, 524–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner DA, Li Y & O'Neill SC (2006). Alternans of intracellular calcium: mechanism and significance. Heart Rhythm 3, 743–745. [DOI] [PubMed] [Google Scholar]
- El Yaman M, Perry J, Makielski JC & Ackerman MJ (2008). Suppression of atrial fibrillation with mexiletine pharmacotherapy in a young woman with type 1 long QT syndrome. Heart Rhythm 5, 472–474. [DOI] [PubMed] [Google Scholar]
- Elmedyb P, Olesen SP & Grunnet M (2007). Activation of ERG2 potassium channels by the diphenylurea NS1643. Neuropharmacology 53, 283–294. [DOI] [PubMed] [Google Scholar]
- Euler DE (1999). Cardiac alternans: mechanisms and pathophysiological significance. Cardiovasc Res 42, 583–590. [DOI] [PubMed] [Google Scholar]
- Francis J & Antzelevitch C (2008). Atrial fibrillation and Brugada syndrome. J Am Coll Cardiol 51, 1149–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelzer AR, Koller ML, Otani NF, Fox JJ, Enyeart MW, Hooker GJ, Riccio ML, Bartoli CR & Gilmour RF, Jr (2008). Dynamic mechanism for initiation of ventricular fibrillation in vivo. Circulation 118, 1123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng J, Zhang Y, Wang Y, Cao L, Song J, Wang B, Song W, Li J & Xu W (2017). Catheter ablation versus rate control in patients with atrial fibrillation and heart failure: a multicenter study. Medicine (Baltimore) 96, e9179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldhaber JI, Xie LH, Duong T, Motter C, Khuu K & Weiss JN (2005). Action potential duration restitution and alternans in rabbit ventricular myocytes: the key role of intracellular calcium cycling. Circ Res 96, 459–466. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrier K, Czosek RJ, Spar DS & Anderson J (2013). Long QT genetics manifesting as atrial fibrillation. Heart Rhythm 10, 1351–1353. [DOI] [PubMed] [Google Scholar]
- Helbig H, Korbmacher C, Kuhner D, Berweck S & Wiederholt M (1988). Characterization of Cl−/HCO3 − exchange in cultured bovine pigmented ciliary epithelium. Exp Eye Res 47, 515–523. [DOI] [PubMed] [Google Scholar]
- Hiromoto K, Shimizu H, Furukawa Y, Kanemori T, Mine T, Masuyama T & Ohyanagi M (2005). Discordant repolarization alternans‐induced atrial fibrillation is suppressed by verapamil. Circ J 69, 1368–1373. [DOI] [PubMed] [Google Scholar]
- Hoit BD (2014). Left atrial size and function: role in prognosis. J Am Coll Cardiol 63, 493–505. [DOI] [PubMed] [Google Scholar]
- Hoshino T, Nagao T, Shiga T, Maruyama K, Toi S, Mizuno S, Ishizuka K, Shimizu S, Uchiyama S & Kitagawa K (2015). Prolonged QTc interval predicts poststroke paroxysmal atrial fibrillation. Stroke 46, 71–76. [DOI] [PubMed] [Google Scholar]
- Hsieh YC, Lin JC, Hung CY, Li CH, Lin SF, Yeh HI, Huang JL, Lo CP, Haugan K, Larsen BD & Wu TJ (2016). Gap junction modifier rotigaptide decreases the susceptibility to ventricular arrhythmia by enhancing conduction velocity and suppressing discordant alternans during therapeutic hypothermia in isolated rabbit hearts. Heart Rhythm 13, 251–261. [DOI] [PubMed] [Google Scholar]
- Hsueh CH, Chang PC, Hsieh YC, Reher T, Chen PS & Lin SF (2013). Proarrhythmic effect of blocking the small conductance calcium activated potassium channel in isolated canine left atrium. Heart Rhythm 10, 891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huser J, Wang YG, Sheehan KA, Cifuentes F, Lipsius SL & Blatter LA (2000). Functional coupling between glycolysis and excitation‐contraction coupling underlies alternans in cat heart cells. J Physiol 524, 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JN, Tester DJ, Perry J, Salisbury BA, Reed CR & Ackerman MJ (2008). Prevalence of early‐onset atrial fibrillation in congenital long QT syndrome. Heart Rhythm 5, 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan PN & Christini DJ (2006). Action potential morphology influences intracellular calcium handling stability and the occurrence of alternans. Biophys J 90, 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan PN & Christini DJ (2007). Characterizing the contribution of voltage‐ and calcium‐dependent coupling to action potential stability: implications for repolarization alternans. Am J Physiol Heart Circ Physiol 293, H2109–2118. [DOI] [PubMed] [Google Scholar]
- Jousset F, Tenkorang J, Vesin JM, Pascale P, Ruchat P, Rollin AG, Fromer M, Narayan SM & Pruvot E (2012). Kinetics of atrial repolarization alternans in a free‐behaving ovine model. J Cardiovasc Electrophysiol 23, 1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalb SS, Dobrovolny HM, Tolkacheva EG, Idriss SF, Krassowska W & Gauthier DJ (2004). The restitution portrait: a new method for investigating rate‐dependent restitution. J Cardiovasc Electrophysiol 15, 698–709. [DOI] [PubMed] [Google Scholar]
- Kanaporis G & Blatter LA (2015). The mechanisms of calcium cycling and action potential dynamics in cardiac alternans. Circ Res 116, 846–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaporis G & Blatter LA (2016). Calcium‐activated chloride current determines action potential morphology during calcium alternans in atrial myocytes. J Physiol 594, 699–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaporis G & Blatter LA (2017a). Alternans in atria: mechanisms and clinical relevance. Medicina (Kaunas) 53, 139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaporis G & Blatter LA (2017b). Membrane potential determines calcium alternans through modulation of SR Ca2+ load and L‐type Ca2+ current. J Mol Cell Cardiol 105, 49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim R, Cingolani O, Wittstein I, McLean R, Han L, Cheng K, Robinson E, Brinker J, Schulman SS, Berger RD, Henrikson CA & Tereshchenko LG (2014). Mechanical alternans is associated with mortality in acute hospitalized heart failure: prospective mechanical alternans study (MAS). Circ Arrhythm Electrophysiol 7, 259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhof P, Eckardt L, Franz MR, Monnig G, Loh P, Wedekind H, Schulze‐Bahr E, Breithardt G & Haverkamp W (2003). Prolonged atrial action potential durations and polymorphic atrial tachyarrhythmias in patients with long QT syndrome. J Cardiovasc Electrophysiol 14, 1027–1033. [DOI] [PubMed] [Google Scholar]
- Koller ML, Maier SK, Gelzer AR, Bauer WR, Meesmann M & Gilmour RF, Jr (2005). Altered dynamics of action potential restitution and alternans in humans with structural heart disease. Circulation 112, 1542–1548. [DOI] [PubMed] [Google Scholar]
- Kornyeyev D, Petrosky AD, Zepeda B, Ferreiro M, Knollmann B & Escobar AL (2012). Calsequestrin 2 deletion shortens the refractoriness of Ca2+ release and reduces rate‐dependent Ca2+‐alternans in intact mouse hearts. J Mol Cell Cardiol 52, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalani GG, Schricker AA, Clopton P, Krummen DE & Narayan SM (2013). Frequency analysis of atrial action potential alternans: a sensitive clinical index of individual propensity to atrial fibrillation. Circ Arrhythm Electrophysiol 6, 859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen AP, Bentzen BH & Grunnet M (2010). Differential effects of Kv11.1 activators on Kv11.1a, Kv11.1b and Kv11.1a/Kv11.1b channels. Br J Pharmacol 161, 614–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limberg SH, Netter MF, Rolfes C, Rinne S, Schlichthorl G, Zuzarte M, Vassiliou T, Moosdorf R, Wulf H, Daut J, Sachse FB & Decher N (2011). TASK‐1 channels may modulate action potential duration of human atrial cardiomyocytes. Cell Physiol Biochem 28, 613–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Kamiya K, Opthof T, Yasui K & Kodama I (2001). Density and kinetics of IKr and IKs in guinea pig and rabbit ventricular myocytes explain different efficacy of IKs blockade at high heart rate in guinea pig and rabbit: implications for arrhythmogenesis in humans. Circulation 104, 951–956. [DOI] [PubMed] [Google Scholar]
- Lugo CA, Cantalapiedra IR, Penaranda A, Hove‐Madsen L & Echebarria B (2014). Are SR Ca content fluctuations or SR refractoriness the key to atrial cardiac alternans?: insights from a human atrial model. Am J Physiol Heart Circ Physiol 306, H1540–1552. [DOI] [PubMed] [Google Scholar]
- Ma D, Wei H, Lu J, Huang D, Liu Z, Loh LJ, Islam O, Liew R, Shim W & Cook SA (2015). Characterization of a novel KCNQ1 mutation for type 1 long QT syndrome and assessment of the therapeutic potential of a novel IKs activator using patient‐specific induced pluripotent stem cell‐derived cardiomyocytes. Stem Cell Res Ther 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandyam MC, Soliman EZ, Alonso A, Dewland TA, Heckbert SR, Vittinghoff E, Cummings SR, Ellinor PT, Chaitman BR, Stocke K, Applegate WB, Arking DE, Butler J, Loehr LR, Magnani JW, Murphy RA, Satterfield S, Newman AB & Marcus GM (2013). The QT interval and risk of incident atrial fibrillation. Heart Rhythm 10, 1562–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrzad R, Rajab M & Spodick DH (2014). The three integrated phases of left atrial macrophysiology and their interactions. Int J Mol Sci 15, 15146–15160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J, Shi C, Li L, Du Y & Xu Y (2013). Compound ICA‐105574 prevents arrhythmias induced by cardiac delayed repolarization. Eur J Pharmacol 718, 87–97. [DOI] [PubMed] [Google Scholar]
- Merchant FM, Sayadi O, Moazzami K, Puppala D & Armoundas AA (2013). T‐wave alternans as an arrhythmic risk stratifier: state of the art. Curr Cardiol Rep 15, 398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monigatti‐Tenkorang J, Jousset F, Pascale P, Vesin JM, Ruchat P, Fromer M, Narayan SM & Pruvot E (2014). Intermittent atrial tachycardia promotes repolarization alternans and conduction slowing during rapid rates, and increases susceptibility to atrial fibrillation in a free‐behaving sheep model. J Cardiovasc Electrophysiol 25, 418–427. [DOI] [PubMed] [Google Scholar]
- Morillo CA, Verma A, Connolly SJ, Kuck KH, Nair GM, Champagne J, Sterns LD, Beresh H, Healey JS & Natale A (2014). Radiofrequency ablation vs antiarrhythmic drugs as first‐line treatment of paroxysmal atrial fibrillation (RAAFT‐2): a randomized trial. JAMA 311, 692–700. [DOI] [PubMed] [Google Scholar]
- Myles RC, Burton FL, Cobbe SM & Smith GL (2011). Alternans of action potential duration and amplitude in rabbits with left ventricular dysfunction following myocardial infarction. J Mol Cell Cardiol 50, 510–521. [DOI] [PubMed] [Google Scholar]
- Narayan SM, Bode F, Karasik PL & Franz MR (2002). Alternans of atrial action potentials during atrial flutter as a precursor to atrial fibrillation. Circulation 106, 1968–1973. [DOI] [PubMed] [Google Scholar]
- Narayan SM, Franz MR, Clopton P, Pruvot EJ & Krummen DE (2011a). Repolarization alternans reveals vulnerability to human atrial fibrillation. Circulation 123, 2922–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan SM, Wright M, Derval N, Jadidi A, Forclaz A, Nault I, Miyazaki S, Sacher F, Bordachar P, Clementy J, Jais P, Haissaguerre M & Hocini M (2011b). Classifying fractionated electrograms in human atrial fibrillation using monophasic action potentials and activation mapping: evidence for localized drivers, rate acceleration, and nonlocal signal etiologies. Heart Rhythm 8, 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman Y, Pereg D & Mosseri M (2011). Living on an atrial kick‐an unusual case of a stuck mitral valve. Eur Heart J 32, 3106. [DOI] [PubMed] [Google Scholar]
- Nicod P, Hillis LD, Winniford MD & Firth BG (1986). Importance of the "atrial kick" in determining the effective mitral valve orifice area in mitral stenosis. Am J Cardiol 57, 403–407. [DOI] [PubMed] [Google Scholar]
- Nielsen JB, Graff C, Pietersen A, Lind B, Struijk JJ, Olesen MS, Haunso S, Gerds TA, Svendsen JH, Kober L & Holst AG (2013). J‐shaped association between QTc interval duration and the risk of atrial fibrillation: results from the Copenhagen ECG study. J Am Coll Cardiol 61, 2557–2564. [DOI] [PubMed] [Google Scholar]
- Nivala M & Qu Z (2012). Calcium alternans in a couplon network model of ventricular myocytes: role of sarcoplasmic reticulum load. Am J Physiol Heart Circ Physiol 303, H341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolasco JB & Dahlen RW (1968). A graphic method for the study of alternation in cardiac action potentials. J Appl Physiol 25, 191–196. [DOI] [PubMed] [Google Scholar]
- O'Neal WT, Efird JT, Kamel H, Nazarian S, Alonso A, Heckbert SR, Longstreth WT, Jr & Soliman EZ (2015). The association of the QT interval with atrial fibrillation and stroke: the Multi‐Ethnic Study of Atherosclerosis. Clin Res Cardiol 104, 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore JM, Girouard SD, Laurita KR, Akar FG & Rosenbaum DS (1999). Mechanism linking T‐wave alternans to the genesis of cardiac fibrillation. Circulation 99, 1385–1394. [DOI] [PubMed] [Google Scholar]
- Picht E, DeSantiago J, Blatter LA & Bers DM (2006). Cardiac alternans do not rely on diastolic sarcoplasmic reticulum calcium content fluctuations. Circ Res 99, 740–748. [DOI] [PubMed] [Google Scholar]
- Pruvot EJ, Katra RP, Rosenbaum DS & Laurita KR (2004). Role of calcium cycling versus restitution in the mechanism of repolarization alternans. Circ Res 94, 1083–1090. [DOI] [PubMed] [Google Scholar]
- Rahimtoola SH, Ehsani A, Sinno MZ, Loeb HS, Rosen KM & Gunnar RM (1975). Left atrial transport function in myocardial infarction. Importance of its booster pump function. Am J Med 59, 686–694. [DOI] [PubMed] [Google Scholar]
- Ravens U & Wettwer E (2011). Ultra‐rapid delayed rectifier channels: molecular basis and therapeutic implications. Cardiovasc Res 89, 776–785. [DOI] [PubMed] [Google Scholar]
- Saitoh H, Bailey JC & Surawicz B (1988). Alternans of action potential duration after abrupt shortening of cycle length: differences between dog Purkinje and ventricular muscle fibers. Circ Res 62, 1027–1040. [DOI] [PubMed] [Google Scholar]
- Schotten U, de Haan S, Verheule S, Harks EG, Frechen D, Bodewig E, Greiser M, Ram R, Maessen J, Kelm M, Allessie M & Van Wagoner DR (2007). Blockade of atrial‐specific K+‐currents increases atrial but not ventricular contractility by enhancing reverse mode Na+/Ca2+‐exchange. Cardiovasc Res 73, 37–47. [DOI] [PubMed] [Google Scholar]
- Shkryl VM, Maxwell JT, Domeier TL & Blatter LA (2012). Refractoriness of sarcoplasmic reticulum Ca2+ release determines Ca2+ alternans in atrial myocytes. Am J Physiol Heart Circ Physiol 302, H2310–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skonieczna M, Cieslar‐Pobuda A, Saenko Y, Foksinski M, Olinski R, Rzeszowska‐Wolny J & Wiechec E (2017). The impact of DIDS‐induced inhibition of voltage‐dependent anion channels (VDAC) on cellular response of lymphoblastoid cells to ionizing radiation. Med Chem 13, 477–483. [DOI] [PubMed] [Google Scholar]
- Szigeti G, Rusznak Z, Kovacs L & Papp Z (1998). Calcium‐activated transient membrane currents are carried mainly by chloride ions in isolated atrial, ventricular and Purkinje cells of rabbit heart. Exp Physiol 83, 137–153. [DOI] [PubMed] [Google Scholar]
- Tolkacheva EG, Anumonwo JM & Jalife J (2006). Action potential duration restitution portraits of mammalian ventricular myocytes: role of calcium current. Biophys J 91, 2735–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolkacheva EG, Romeo MM, Guerraty M & Gauthier DJ (2004). Condition for alternans and its control in a two‐dimensional mapping model of paced cardiac dynamics. Phys Rev E Stat Nonlin Soft Matter Phys 69, 031904. [DOI] [PubMed] [Google Scholar]
- Trafford AW, Clarke JD, Richards MA, Eisner DA & Dibb KM (2013). Calcium signalling microdomains and the t‐tubular system in atrial mycoytes: potential roles in cardiac disease and arrhythmias. Cardiovasc Res 98, 192–203. [DOI] [PubMed] [Google Scholar]
- Tuteja D, Xu D, Timofeyev V, Lu L, Sharma D, Zhang Z, Xu Y, Nie L, Vazquez AE, Young JN, Glatter KA & Chiamvimonvat N (2005). Differential expression of small‐conductance Ca2+‐activated K+ channels SK1, SK2, and SK3 in mouse atrial and ventricular myocytes. Am J Physiol Heart Circ Physiol 289, H2714–2723. [DOI] [PubMed] [Google Scholar]
- Verrier RL, Fuller H, Justo F, Nearing BD, Rajamani S & Belardinelli L (2016). Unmasking atrial repolarization to assess alternans, spatiotemporal heterogeneity, and susceptibility to atrial fibrillation. Heart Rhythm 13, 953–961. [DOI] [PubMed] [Google Scholar]
- Wang L, Myles RC, De Jesus NM, Ohlendorf AK, Bers DM & Ripplinger CM (2014). Optical mapping of sarcoplasmic reticulum Ca2+ in the intact heart: ryanodine receptor refractoriness during alternans and fibrillation. Circ Res 114, 1410–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe MA & Koller ML (2002). Mathematical analysis of dynamics of cardiac memory and accommodation: theory and experiment. Am J Physiol Heart Circ Physiol 282, H1534–1547. [DOI] [PubMed] [Google Scholar]
- Wilson LD, Jeyaraj D, Wan X, Hoeker GS, Said TH, Gittinger M, Laurita KR & Rosenbaum DS (2009). Heart failure enhances susceptibility to arrhythmogenic cardiac alternans. Heart Rhythm 6, 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson LD & Rosenbaum DS (2007). Mechanisms of arrythmogenic cardiac alternans. Europace 9(Suppl 6), vi77–82. [DOI] [PubMed] [Google Scholar]
- Wu R & Patwardhan A (2006). Mechanism of repolarization alternans has restitution of action potential duration dependent and independent components. J Cardiovasc Electrophysiol 17, 87–93. [DOI] [PubMed] [Google Scholar]
- Wu SN, Peng H, Chen BS, Wang YJ, Wu PY & Lin MW (2008). Potent activation of large‐conductance Ca2+‐activated K+ channels by the diphenylurea 1,3‐bis‐[2‐hydroxy‐5‐(trifluoromethyl)phenyl]urea (NS1643) in pituitary tumor (GH3) cells. Mol Pharmacol 74, 1696–1704. [DOI] [PubMed] [Google Scholar]
- Xu X, Salata JJ, Wang J, Wu Y, Yan GX, Liu T, Marinchak RA & Kowey PR (2002). Increasing IKs corrects abnormal repolarization in rabbit models of acquired LQT2 and ventricular hypertrophy. Am J Physiol Heart Circ Physiol 283, H664–670. [DOI] [PubMed] [Google Scholar]
- Yu H, Lin Z, Xu K, Huang X, Long S, Wu M, McManus OB, Le Engers J, Mattmann ME, Engers DW, Le UM, Lindsley CW, Hopkins CR & Li M (2010). Identification of a Novel, Small Molecule Activator of KCNQ1 Channels. National Center for Biotechnology Information (US), Bethesda, MD: http://www.ncbi.nlm.nih.gov/books/NBK143558/ [PubMed] [Google Scholar]
- Zellerhoff S, Pistulli R, Monnig G, Hinterseer M, Beckmann BM, Kobe J, Steinbeck G, Kaab S, Haverkamp W, Fabritz L, Gradaus R, Breithardt G, Schulze‐Bahr E, Bocker D & Kirchhof P (2009). Atrial arrhythmias in long‐QT syndrome under daily life conditions: a nested case control study. J Cardiovasc Electrophysiol 20, 401–407. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zou B, Yu H, Moretti A, Wang X, Yan W, Babcock JJ, Bellin M, McManus OB, Tomaselli G, Nan F, Laugwitz KL & Li M (2012). Modulation of hERG potassium channel gating normalizes action potential duration prolonged by dysfunctional KCNQ1 potassium channel. Proc Natl Acad Sci U S A 109, 11866–11871. [DOI] [PMC free article] [PubMed] [Google Scholar]