

Abstract
Key points
Respiratory muscle weakness is a major feature of Duchenne muscular dystrophy (DMD), yet little is known about the neural control of the respiratory muscles in DMD and animal models of dystrophic disease.
Substantial diaphragm muscle weakness is apparent in young (8‐week‐old) mdx mice, although ventilatory capacity in response to maximum chemostimulation in conscious mice is preserved.
Peak volume‐ and flow‐related measures during chemoactivation are equivalent in anaesthetized, vagotomized wild‐type and mdx mice.
Diaphragm and T3 external intercostal electromyogram activities are lower during protracted sustained airway occlusion in mdx compared to wild‐type mice. Yet, peak inspiratory pressure generation is remarkably well preserved.
Despite profound diaphragm weakness and lower muscle activation during maximum non‐ventilatory efforts, inspiratory pressure‐generating capacity is preserved in young adult mdx mice, revealing compensation in support of respiratory system performance that is adequate, at least early in dystrophic disease.
Abstract
Diaphragm dysfunction is recognized in the mdx mouse model of muscular dystrophy; however, there is a paucity of information concerning the neural control of dystrophic respiratory muscles. In young adult (8 weeks of age) male wild‐type and mdx mice, we assessed ventilatory capacity, neural activation of the diaphragm and external intercostal (EIC) muscles and inspiratory pressure‐generating capacity during ventilatory and non‐ventilatory behaviours. We hypothesized that respiratory muscle weakness is associated with impaired peak inspiratory pressure‐generating capacity in mdx mice. Ventilatory responsiveness to hypercapnic hypoxia was determined in conscious mice by whole‐body plethysmography. Diaphragm isometric and isotonic contractile properties were determined ex vivo. In anaesthetized mice, thoracic oesophageal pressure, and diaphragm and EIC electromyogram (EMG) activities were recorded during baseline conditions and sustained tracheal occlusion for 30–40s. Despite substantial diaphragm weakness, mdx mice retain the capacity to enhance ventilation during hypercapnic hypoxia. Peak volume‐ and flow‐related measures were also maintained in anaesthetized, vagotomized mdx mice. Peak inspiratory pressure was remarkably well preserved during chemoactivated breathing, augmented breaths and maximal sustained efforts during airway obstruction in mdx mice. Diaphragm and EIC EMG activities were lower during airway obstruction in mdx compared to wild‐type mice. We conclude that ventilatory capacity is preserved in young mdx mice. Despite profound respiratory muscle weakness and lower diaphragm and EIC EMG activities during high demand in mdx mice, peak inspiratory pressure is preserved, revealing adequate compensation in support of respiratory system performance, at least early in dystrophic disease. We suggest that a progressive loss of compensation during advancing disease, combined with diaphragm dysfunction, underpins the development of respiratory system morbidity in dystrophic diseases.
Keywords: DMD, mdx, breathing, oesophageal pressure, EMG, diaphragm, intercostal
Key points
Respiratory muscle weakness is a major feature of Duchenne muscular dystrophy (DMD), yet little is known about the neural control of the respiratory muscles in DMD and animal models of dystrophic disease.
Substantial diaphragm muscle weakness is apparent in young (8‐week‐old) mdx mice, although ventilatory capacity in response to maximum chemostimulation in conscious mice is preserved.
Peak volume‐ and flow‐related measures during chemoactivation are equivalent in anaesthetized, vagotomized wild‐type and mdx mice.
Diaphragm and T3 external intercostal electromyogram activities are lower during protracted sustained airway occlusion in mdx compared to wild‐type mice. Yet, peak inspiratory pressure generation is remarkably well preserved.
Despite profound diaphragm weakness and lower muscle activation during maximum non‐ventilatory efforts, inspiratory pressure‐generating capacity is preserved in young adult mdx mice, revealing compensation in support of respiratory system performance that is adequate, at least early in dystrophic disease.
Introduction
Dystrophin is a structural protein expressed in muscle and the central nervous system (Lidov, 1996; Muntoni et al. 2003). Duchenne muscular dystrophy (DMD) is a severe neuromuscular disease caused by dystrophin deficiency (Hoffman et al. 1987; Ervasti, 2007). Respiratory muscle dysfunction is described in DMD patients (De Bruin et al. 1997; Beck et al. 2006) with deleterious consequences for respiratory system performance (Mayer et al. 2015). Loss of ambulation and cardiorespiratory failure are cardinal features of DMD (Yiu & Kornberg, 2008). Life expectancy for DMD boys is severely curtailed and there is currently no cure for the devastating disease.
Substantial diaphragm muscle weakness is reported in patients and animal models of DMD (Stedman et al. 1991; Khirani et al. 2014). However, there remains an incomplete understanding of the neural control of breathing in DMD and dystrophic animal models, where further deficits or compensation may arise with implications for respiratory performance (Burns et al. 2017c). A thorough understanding of the consequences of dystrophin deficiency for the control of breathing is essential with respect to the consideration of disease progression and therapeutic strategies that aim to combat respiratory insufficiency in human dystrophinopathies.
Dystrophin is ordinarily expressed in neurons (Lidov, 1996). In the mdx mouse model of DMD, studies indicate reduced number of corticospinal neurons (Sbriccoli et al. 1995), damage to motor pathways (Carretta et al. 2001), loss of neuronal projections (Pinto et al. 2008), GABAergic dysfunction (Sekiguchi et al. 2009; Vaillend & Chaussenot, 2017) and cognitive deficits (Chaussenot et al. 2015). It is unclear whether dystrophin deficiency affects the nervous system control of breathing. A comprehensive understanding of the motor control of the dystrophin deficient respiratory musculature is lacking. It is essential to determine whether dystrophin deficiency is deleterious to the motor control of breathing and respiratory‐related behaviours, or whether there is intrinsic compensation within the neural control circuits, facilitating performance. Recently, we described evidence of enhanced motor drive to the mdx diaphragm during chemoactivation of breathing (Burns et al. 2017c), suggesting intrinsic compensation in the neural control of ventilation in dystrophin deficient mice. Our studies have revealed a retained capacity to increase ventilation in response to modest chemostimulation in young mdx mice (Burns et al. 2017c). However, the extent to which dystrophin deficiency affects inspiratory pressure‐generating capacity across a range of ventilatory and non‐ventilatory behaviours (Mantilla et al. 2010; Seven et al. 2014) is unclear, although this is important to establish. Clearly, aberrant neural control of breathing could serve to exacerbate respiratory morbidity in DMD, compounding mechanical constraints arising from respiratory muscle dysfunction.
The principal objective of the present study was to perform an assessment of respiratory system performance during ventilatory and non‐ventilatory behaviours in the mdx mouse model of DMD. We assessed ventilatory capacity during hypercapnic hypoxic breathing in conscious mice. In anaesthetized mice, we determined inspiratory pressure‐generating capacity and diaphragm and external intercostal (EIC) electromyogram (EMG) activities during ventilatory (chemoactivation and augmented breaths) and non‐ventilatory (airway obstruction) behaviours. We hypothesized that respiratory muscle weakness is associated with impaired peak inspiratory pressure‐generating capacity in mdx mice.
Methods
Ethical approval
Procedures on live animals were performed under licence in accordance with Irish and European law following approval by University College Cork animal research ethics committee (AEEC no. 2013/035). Experiments were carried out in accordance with guidelines laid down by University College Cork Animal Welfare Body, and conform to the principles and regulations described by Grundy (2015).
Experimental animals
Male wild‐type (C57BL/10ScSnJ; n = 28) and mdx (C57BL/10ScSn‐Dmdmdx/J; n = 26) mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and studied at 8 weeks of age. Animals were housed in individually ventilated cages in our institution's animal housing facility. All animals were housed in temperature‐ and humidity‐controlled rooms, operating under a 12:12 h light/dark cycle with food and water available ad libitum.
Whole‐body plethysmography
Whole‐body plethysmography was used to assess respiratory flow in unrestrained, unanaesthetized mice. Mice were introduced into plethysmograph chambers (Model PLY4211; volume 600 mL; Buxco Research Systems, Wilmington, NC, USA) and allowed 60–90 min to acclimate to the chamber environment. Following exploration and grooming behaviours, mice settled and were studied during quiet rest. Recordings were typically performed contemporaneously in a wild‐type and mdx mouse using a pair of plethysmograph chambers.
Experimental protocol
Following acclimation and a settling period, a 20 min baseline recording was performed in normoxia. This was followed by a graded hypercapnic challenge where animals were challenged with increasing levels of inspired carbon dioxide: = 0.02, 0.04 and 0.06 ( = 0.21) consecutively for 5 min each. This was immediately followed by maximal chemoreceptor stimulation with hypercapnic hypoxia ( = 0.10/ = 0.06) for 5 min, to examine ventilatory capacity in wild‐type (n = 9) and mdx (n = 7) mice. Mice were subsequently killed by urethane overdose.
Data analysis
Ventilation during hypercapnia and hypercapnic hypoxia was determined during steady‐state conditions in the fifth minute of exposure and compared with the preceding baseline normoxic period. Tidal volume (V T), minute ventilation () and mean inspiratory flow (V T Ti −1) were normalized for body mass (g).
Diaphragm EMG and oesophageal pressure recordings
Anaesthesia was induced with 5% isoflurane in 60% O2 (balance N2) followed by urethane (1.7 g kg−1 i.p.). Wild‐type (n = 9) and mdx (n = 9) mice were then placed in the supine position, gradually weaned from the isoflurane and the body temperature was maintained at 37°C via a rectal probe and thermostatically‐controlled heating blanket (Harvard Apparatus, Holliston, MA, USA). Supplemental anaesthetic was administered if necessary to maintain an adequate surgical plane of anaesthesia, which was by assessment of pedal withdrawal reflex to noxious pinch. A pulse oximeter clip (MouseOxTM; Starr Life Sciences Corporation, Oakmount, PA, USA) was placed on a shaved thigh of each mouse for the measurement of peripheral capillary O2 saturation (). A mid‐cervical tracheotomy was performed. All animals were maintained with a bias flow of supplemental O2 ( = 0.60) under baseline conditions. End‐tidal carbon dioxide (ETCO2) was measured using a MicroCapStar (CWE, Ardmore, PA, USA). To estimate the intra‐pleural sub‐atmospheric pressure generated by the respiratory musculature during inspiration, we measured oesophageal pressure using a pressure‐tip catheter (Mikro‐Tip; Millar Inc., Houston, TX, USA), which was positioned in the thoracic oesophagus through the mouth. During inspiratory activity, oesophageal recordings displayed phasic sub‐atmospheric pressure swings. Concentric needle electrodes (26 G; Natus Manufacturing Ltd, Gort, Ireland) were inserted into the costal diaphragm for the continuous measurement of diaphragm EMG activity, which was amplified (× 5000), filtered (500 Hz low cut‐off to 5000 Hz high cut‐off) and integrated (50 ms time constant; Neurolog system; Digitimer Ltd, Welwyn Garden City, UK). All signals were passed through an analogue‐to‐digital converter (Powerlab r8/30; ADInstruments, Colorado Springs, CO, USA) and were acquired using LabChart 7 (ADInstruments).
Experimental protocol
Following instrumentation, animals were allowed at least 10 min to stabilize before baseline parameters were measured for a period of 10 min. Next, animals were challenged with hypercapnic hypoxia ( = 0.15 and = 0.05; 1 min) to examine the effects of chemostimulation on diaphragm EMG activity and oesophageal inspiratory pressure generation. Following the experimental protocol, mice were killed by decapitation. Diaphragm muscle was excised for ex vivo functional analysis.
Data analysis
The amplitudes of integrated inspiratory diaphragm EMG activity and peak inspiratory sub‐atmospheric oesophageal pressure were analysed and averaged under steady‐state basal conditions and averaged for the final 15 breaths (maximal response) of the hypercapnic hypoxia challenge. Oesophageal pressure is reported in absolute units (cmH2O). EMG data are reported in arbitrary units (A.U.). In one wild‐type and three mdx mice, responses to chemostimulation were characterized by tachypnoea and a reduction in EMG amplitude. Because our aim was to examine the magnitude of the increase in EMG amplitude (motor recruitment) of the diaphragm during maximal chemoreceptor stimulation with hypercapnic hypoxia, we determined a priori that trials characterized by frequency‐only responses to gas challenge in wild‐type and mdx mice would be excluded from group analysis, consistent with our approach in a recent study (Burns et al. 2017c). Spontaneous augmented breaths (sighs) observed during baseline conditions were analysed and compared with the average of the preceding five breaths. Augmented breaths were classified as breaths with more than double the baseline EMG amplitude (Mantilla et al. 2011; Seven et al. 2014). For two wild‐type mice, no breath met this criterion during the recording period.
Diaphragm and EIC EMG and oesophageal pressure recordings
In separate studies, wild‐type (n = 10) and mdx (n = 10) mice were anaesthetized and tracheotomized as described above. Oesophageal pressure and costal diaphragm EMG activity were recorded. In addition, a concentric needle electrode was inserted in the third intercostal space (T3) for the measurement of EIC EMG, which was amplified (× 5000), filtered (500 Hz low cut‐off to 5000 Hz high cut‐off) and integrated (50 ms time constant; Neurolog system, Digitimer Ltd). All signals were passed through an analogue‐to‐digital converter and were acquired using LabChart 7 (ADInstruments).
Experimental protocol
Following instrumentation, animals were allowed at least 10 min to stabilize before baseline parameters were measured for a period of 10 min. Next, a pneumotachometer was connected to the tracheal cannula to record respiratory airflow for a period of 1–2 min. The pneumotachometer and ETCO2 were disconnected and following a baseline period, animals were challenged with sustained tracheal occlusion for 30–40s until a plateau was observed in the inspiratory pressure recordings during sustained maximum non‐ventilatory efforts. Following recovery, animals were instrumented for the measurement of ETCO2 and tracheal airflow and the vagi were sectioned bilaterally at the cervical level. Respiratory parameters were recorded for 10 min under steady‐state conditions following vagotomy. Next, animals were challenged with hypercapnic hypoxia ( = 0.15 and = 0.06; 3 min) to examine the effects of chemostimulation on diaphragm and EIC EMG activities and tracheal airflow. Following the experimental protocol, mice were killed by decapitation.
Data analysis
The amplitudes of integrated inspiratory diaphragm and EIC EMG activities and peak inspiratory sub‐atmospheric oesophageal pressure were analysed and averaged under steady‐state basal conditions and averaged for the five successive maximal sustained efforts (maximal response) of the airway occlusion challenge. During baseline breathing and chemoactivation, V T was derived from tracheal airflow measurements. Spontaneous peak breaths observed during chemoactivation in vagotomized mice, notably greater in amplitude than neighbouring breaths, were analysed and compared with the preceding baseline period to provide a measure of maximum ventilatory effort. Spontaneous peak breaths during hypercapnic hypoxia were observed in five wild‐type and five mdx mice and were analysed as a measure of peak ventilatory performance.
Ex vivo diaphragm muscle function
Diaphragm muscle was excised with a rib and central tendon attached. Muscle bundles with longitudinally arranged muscle fibres were prepared for functional assessment and suspended vertically between two platinum plate electrodes. The rib was attached to an immobile hook and the central tendon was attached to a dual‐mode force transducer (Aurora Scientific Inc., Aurora, ON, Canada) with non‐elastic string. Diaphragm muscle preparations from wild‐type (n = 8) and mdx (n = 9) mice were studied in a water‐jacketed tissue bath at 35°C containing Krebs solution (in mm: 120 NaCl, 5 KCl, 2.5 Ca2+, 1.2 MgSO4, 1.2 NaH2PO4, 25 NaHCO3 and 11.5 glucose) and d‐tubocurarine (25 μm) and were continuously aerated with carbogen ( = 0.95 and = 0.05). Muscle optimum length (L o) was determined by adjusting the position of the force transducer, in turn adjusting the length of the muscle preparations, using a micro‐positioner between intermittent twitch contractions (Burns & O'Halloran, 2016; Burns et al. 2017b). L o was determined as the muscle length which revealed maximal isometric twitch force in response to single isometric twitch stimulation (supramaximal stimulation, 1 ms in duration). Preparations remained at L o for the duration of the protocol.
Experimental protocol
A single isometric twitch contraction was measured. Peak isometric twitch force (P t), contraction time (CT) and half‐relaxation time (½ RT) were determined. Peak isometric force at 100 Hz (P o) was determined. The force–frequency relationship was examined by stimulating the muscle sequentially at 10, 20, 40, 60, 80, 100, 120, 140 and 160 Hz (300 ms train duration). Contractions were interspersed by an interval of 1 min. Next, an isotonic contraction was elicited in preparations at 0% load to examine maximum unloaded muscle shortening and velocity of shortening (Burns & O'Halloran, 2016; Burns et al. 2017b).
Data analysis
Muscle force was normalized for muscle cross‐sectional area (CSA) and expressed as specific force (N cm−2). The CSA of each muscle bundle was determined by dividing muscle mass (g) by the product of muscle L o (cm) and muscle density (assumed to be 1.06 g cm−3). CT and ½ RT were measured as indices of isometric twitch kinetics. Total muscle shortening was determined as the maximum distance shortened during contraction. Total muscle shortening (S max) was determined in absolute units (cm) and was normalized to L o and expressed as L L o −1. Shortening velocity was determined as the distance shortened during the initial 30 ms of shortening (Lewis et al. 2015; 2016). Shortening velocity (V max) was determined in absolute units (cm.s−1) and was normalized to L o and expressed as L o s−1.
Statistical analysis
Values are expressed as the mean ± SD or as box and whisker plots (median, 25–75th percentile and scatter plot). Data were compared statistically using Prism, version 6.0 (Graphpad Software Inc., San Diego, CA, USA). Data were tested for normal distribution and equal variances. Data sets that were normally distributed and of equivalent variance were compared statistically using an unpaired two‐tailed Student's t test. Welch's correction was applied in the case of unequal variance. Data that were not normally distributed were compared using Mann–Whitney non‐parametric tests. Data for the diaphragm muscle force–frequency relationship, ventilatory responsiveness to chemostimulation in conscious mice, oesophageal pressure and EMG activities during obstruction, and flow, volume and EMG measures during peak breaths in vagotomized mice during chemoactivation were compared statistically by repeated measures two‐way analysis of variance (ANOVA) with a Bonferroni post hoc test. P < 0.05 was considered statistically significant.
Results
Diaphragm muscle contractile function ex vivo
Figure 1 shows representative original recordings for wild‐type (black) and mdx (grey) diaphragm twitch (Fig. 1 A) and tetanic (Fig. 1 B) contractions, force–frequency relationship (Fig. 1 C) and maximal unloaded shortening (Fig. 1 D). Twitch kinetics (CT and ½ RT) and isotonic contractile parameters (S max and V max) are shown in Table 1. Twitch CT was significantly increased in mdx compared to wild‐type diaphragm (P = 0.027; unpaired Student's t test). Twitch ½ RT and isotonic contractile parameters were not significantly different between wild‐type and mdx diaphragm preparations, with the exception of absolute shortening and absolute velocity of shortening, which were significantly less in mdx compared to wild‐type mice (Table 1). Diaphragm twitch (P = 0.0241; unpaired Student's t test) (Fig. 1 E, P t) and tetanic force (P < 0.0001; unpaired Student's t test with Welch's correction) (Fig. 1 E, P o) were both significantly depressed in mdx compared to wild‐type muscle. For the force–frequency relationship, diaphragm specific force was significantly lower in mdx compared to wild‐type preparations (P = 0.0003, genotype; repeated measures two‐way ANOVA) (Fig. 1 F). Post hoc analysis confirmed significant force depression in mdx across a broad range of stimulation frequencies (60–160 Hz).
Figure 1. Ex vivo wild‐type and mdx diaphragm muscle contractile function.
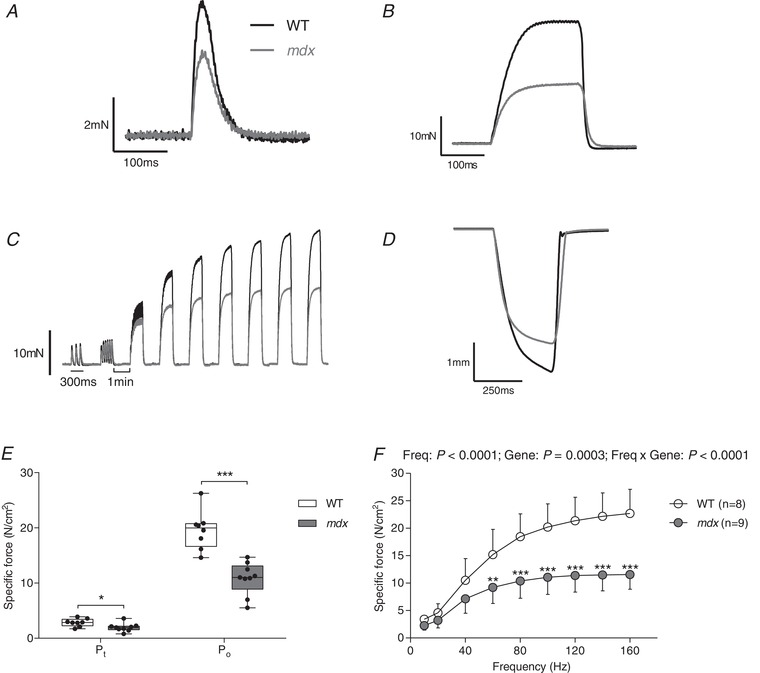
A–D, original traces of ex vivo diaphragm muscle twitch contraction (A), tetanic contraction (B), force–frequency relationship (C) and maximum unloaded shortening (D) for wild‐type (WT) (black) and mdx (grey) preparations. E, group data for diaphragm muscle twitch (P t) and tetanic (P o) force in WT (n = 8) and mdx (n = 9) mice. Tetanic force was measured following stimulation at 100 Hz ex vivo. Values are expressed as box and whisker plots (median, 25–75th percentile and scatter plot). Data were statistically compared by unpaired Student's t tests. * P < 0.05 and *** P < 0.001 compared to the corresponding WT value. F, group data (mean ± SD) for diaphragm muscle force–frequency relationship ex vivo in WT (open) and mdx (grey) preparations. Data were statistically compared by repeated measures two‐way ANOVA (frequency × gene) followed by a Bonferroni post hoc test. ** P < 0.01 and *** P < 0.001 compared to the corresponding WT value.
Table 1.
Ex vivo wild‐type and mdx diaphragm muscle contractile kinetics
| Wild‐type (n = 8) | mdx (n = 9) | P (Student's t test) | |
|---|---|---|---|
| CT (ms) | 17.3 ± 2.3 | 20.3 ± 2.6 | 0.0270 |
| ½ RT (ms) | 19.6 ± 6.9 | 23.2 ± 4.3 | 0.2177 |
| S max (cm) | 0.36 ± 0.07 | 0.26 ± 0.1 | 0.0386 |
| S max (L L o −1) | 0.36 ± 0.07 | 0.30 ± 0.13 | 0.3252 |
| V max (cm s−1) | 4.1 ± 1.1 | 2.8 ± 1.3 | 0.0420 |
| V max (L o s−1) | 4.1 ± 1.1 | 3.3 ± 1.9 | 0.3373 |
| L o (cm) | 1.0 ± 0.1 | 0.9 ± 0.2 | 0.1024 |
CT, contraction time; ½ RT, half‐relaxation time; S max, peak shortening; V max, peak shortening velocity; L o, optimum length. Data are shown as the mean ± SD and were statistically compared using two‐tailed unpaired Student's t tests.
Ventilatory responsiveness to hypercapnic hypoxia in conscious mice
Figure 2 A shows representative respiratory flow traces for wild‐type and mdx mice during exposure to baseline air ( = 0.21) and hypercapnic hypoxia ( = 0.10 and = 0.06). Chemostimulation with graded hypercapnia and hypercapnic hypoxia resulted in a significant increase in f R (P < 0.0001, gas; repeated measures two‐way ANOVA) (Fig. 2 B), V T (P < 0.0001) (Fig. 2 C), (P < 0.0001) (Fig. 2 D) and V T Ti −1 (P < 0.0001) (Fig. 2 E) for both wild‐type and mdx mice, with no difference between the two groups.
Figure 2. Ventilation in response to chemostimulation in conscious wild‐type and mdx mice.
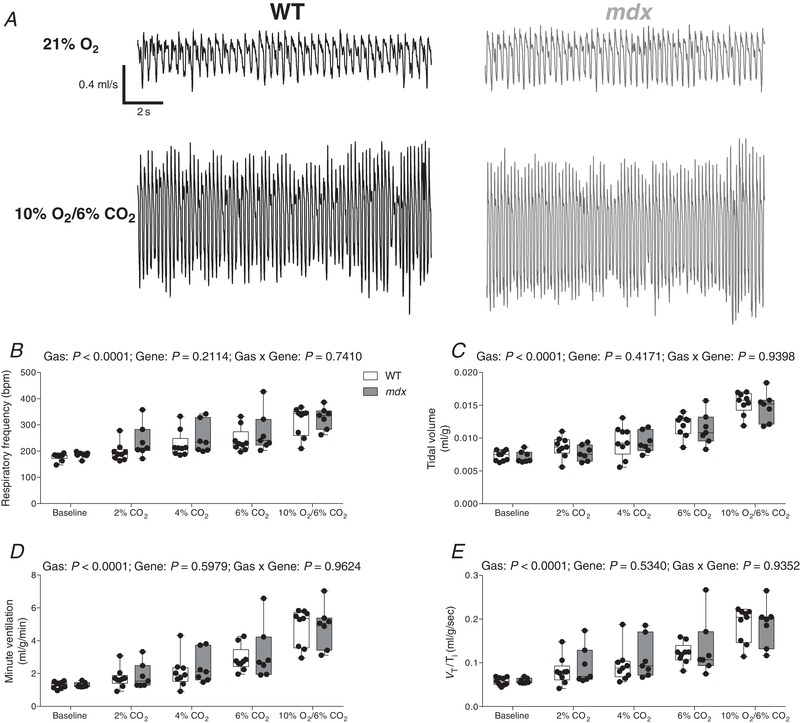
A, representative respiratory flow traces during normoxia (21% O2) and hypercapnic hypoxia (10% O2/6% CO2) in a wild‐type (WT) (black) and mdx (grey) mouse; inspiration downwards. B–E, group data for respiratory frequency (B), tidal volume (C), minute ventilation (D) and mean inspiratory flow (E) (V T/T i) during graded hypercapnia (2%, 4% and 6% CO2) and hypercapnic hypoxia (10% O2/6% CO2). Values are expressed as box and whisker plots (median, 25–75th percentile and scatter plot). Data were statistically compared by repeated measures two‐way ANOVA.
Oesophageal pressure and diaphragm EMG activity in anaesthetized mice
Representative original recordings of oesophageal pressure and diaphragm EMG during baseline conditions ( = 0.60) and during a representative spontaneous augmented breath (shaded) are shown in Figure 3 A. Table 2 shows baseline respiratory measurements in wild‐type and mdx mice. Respiratory frequency was significantly higher in mdx compared to wild‐type mice (P < 0.0001; unpaired Student's t test) (Table 2). There was no significant difference in and ETCO2 between wild‐type and mdx mice (Table 2).
Figure 3. Oesophageal pressure and diaphragm EMG activity during ventilatory behaviours in anaesthetized wild‐type and mdx mice.
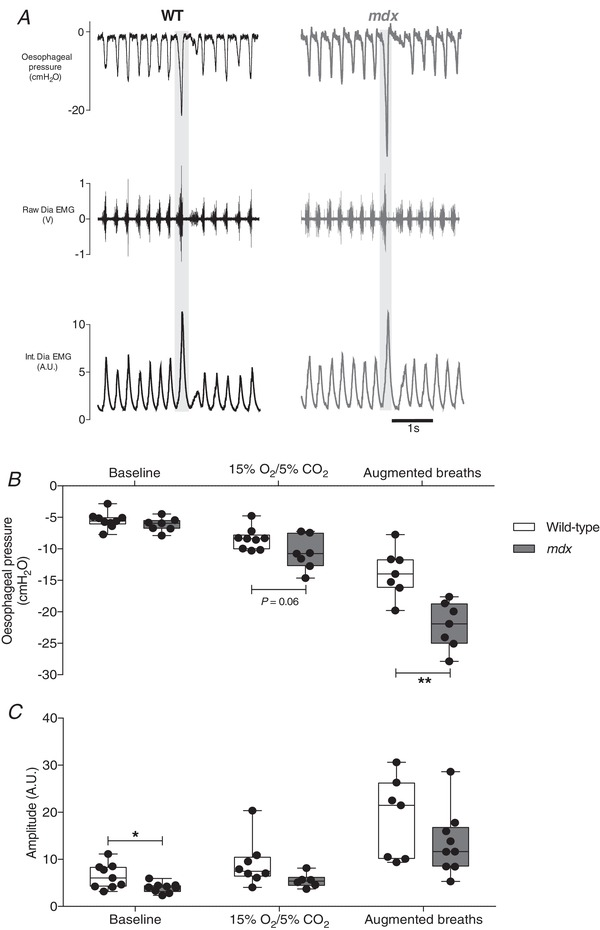
A, representative traces of oesophageal pressure and diaphragm (Dia) muscle raw and integrated (Int.) EMG activity in a wild‐type (WT) (black) and mdx (grey) mouse during baseline (60% inspired O2) and during an augmented breath (shaded). B and C, group data for oesophageal pressure (B) and diaphragm integrated EMG activity (C) during baseline, chemostimulation (15% O2/5% CO2) and augmented breaths for WT (n = 7–9) and mdx (n = 6–9) mice. Values are expressed as box and whisker plots (median, 25–75th percentile and scatter plot). Data were statistically compared by two‐tailed unpaired Student's t tests. * P = 0.025 and ** P = 0.0013 compared to the corresponding WT values.
Table 2.
Baseline respiratory parameters in anaesthetized wild‐type and mdx mice
| Wild‐type (n = 9) | mdx (n = 9) | P (Student's t test) | |
|---|---|---|---|
| f R (breaths min−1) | 195 ± 11 | 230 ± 11 | < 0.0001 |
| (%) | 97.2 ± 1.9 | 96.5 ± 2.0 | 0.4069 |
| ETCO2 (mmHg) | 32.9 ± 3.3 | 30.3 ± 3.0 | 0.0966 |
| Body mass (g) | 24.9 ± 1.8 | 25.0 ± 2.3 | 0.9110 |
f R, respiratory frequency; , peripheral capillary oxygen saturation; ETCO2, end‐tidal carbon dioxide. Data are shown as the mean ± SD and were statistically compared using two‐tailed unpaired Student's t tests.
Peak inspiratory oesophageal pressure during baseline recordings was equivalent between wild‐type and mdx mice (P = 0.2887; unpaired Student's t test) (Fig. 3 B). Baseline diaphragm EMG activity was significantly lower in mdx (P = 0.025; unpaired Student's t test with Welch's correction) (Fig. 3 C) compared to wild‐type mice. The peak inspiratory pressure generated during chemoactivation ( = 0.15 and = 0.05) was equivalent in wild‐type and mdx mice, with a trend towards greater sub‐atmospheric pressure generation in mdx mice (P = 0.0563; unpaired Student's t test) (Fig. 3 B). Diaphragm EMG activity during chemoactivation was not different between wild‐type and mdx mice (P = 0.0853; unpaired Student's t test with Welch's correction) (Fig. 3 C). Peak inspiratory oesophageal pressure during augmented breaths was greater in mdx compared to wild‐type mice (P = 0.0013; unpaired Student's t test) (Fig. 3 B). Diaphragm EMG during augmented breaths was equivalent between wild‐type and mdx mice (P = 0.2031; unpaired Student's t test) (Fig. 3 C).
Oesophageal pressure and diaphragm and EIC EMGs during airway obstruction in anaesthetized mice
Representative original recordings of oesophageal pressure, diaphragm and EIC EMG activities during baseline conditions ( = 0.60) and peak sustained inspiratory efforts during protracted airway obstruction are shown in Figure 4 A and B. Obstruction significantly increased sub‐atmospheric pressure generation in wild‐type (P < 0.0001; repeated measures two‐way ANOVA with Bonferroni) (Fig. 5 A) and mdx mice (P < 0.0001) (Fig. 5 A), with no significant difference between the two groups (P = 0.1931, gene; repeated measures two‐way ANOVA) (Fig. 5 A). Diaphragm EMG activity was significantly increased during airway obstruction compared to baseline in wild‐type (P < 0.0001; repeated measures two‐way ANOVA with Bonferroni) (Fig. 5 B) and mdx mice (P < 0.001). However, diaphragm EMG activity was lower in mdx (P = 0.0479, gene; repeated measures two‐way ANOVA) (Fig. 5 B) and mdx mice had significantly lower diaphragm EMG activity compared to wild‐type during airway obstruction (P < 0.05; repeated measures two‐way ANOVA with Bonferroni) (Fig. 5 B). Airway obstruction significantly increased EIC EMG activity in wild‐type (P < 0.0001; repeated measures two‐way ANOVA with Bonferroni) (Fig. 5 C) but not mdx mice (P > 0.05). EIC EMG activity was lower in mdx compared to wild‐type (P = 0.0003, gene; repeated measures two‐way ANOVA) (Fig. 5 C). Post hoc analysis revealed that EIC EMG activity was significantly lower during airway obstruction in mdx compared to wild‐type mice (P < 0.0001; repeated measures two‐way ANOVA with Bonferroni) (Fig. 5 C). In light of these findings, we extended our analyses to determine inspiratory duration and respiratory EMG activities expressed as the area under the curve (AUC) of the integrated EMG signals before and during airway obstruction (Table 3). During baseline recordings, inspiratory duration was shorter in mdx compared with wild‐type mice (P < 0.05; repeated measures two‐way ANOVA with Bonferroni). Inspiratory duration was significantly longer during obstructed airway efforts, both in wild‐type and mdx mice, with no genotype difference. Consistent with data for respiratory EMG amplitude during airway obstruction, both diaphragm (P < 0.05) and EIC (P < 0.001) EMG AUC activities were significantly lower in mdx compared with wild‐type mice (Table 3).
Figure 4. Oesophageal pressure and diaphragm and EIC EMG activities in anaesthetized wild‐type and mdx mice: original recordings.
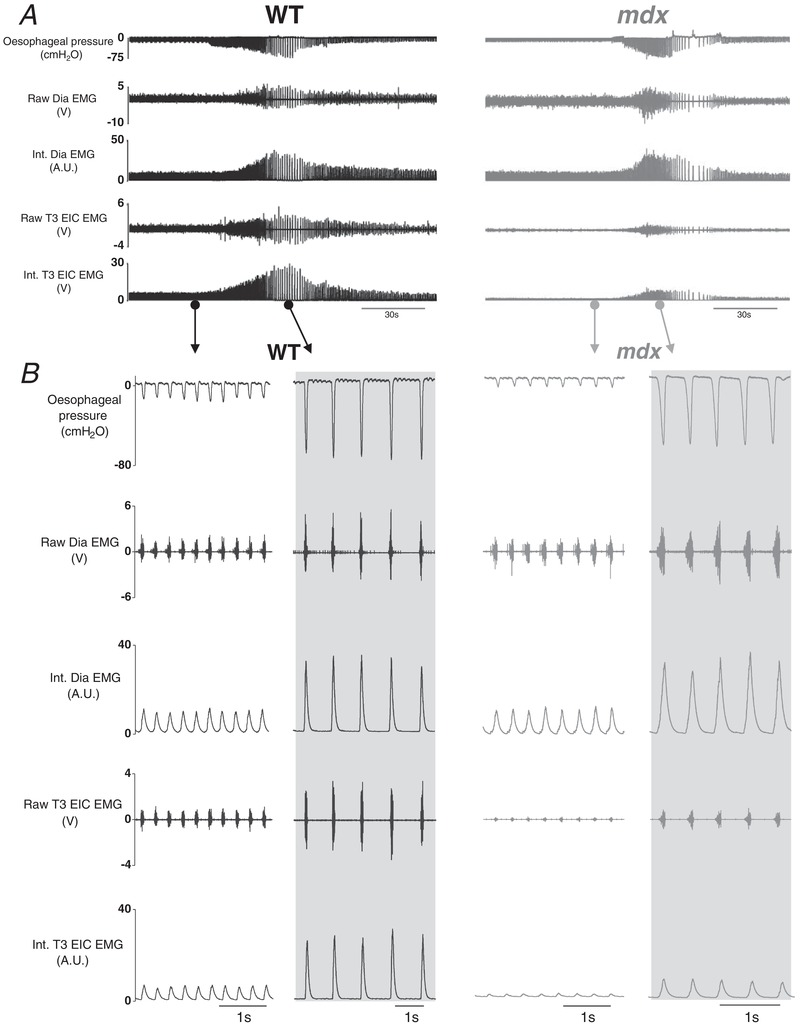
A, representative recordings of oesophageal pressure and diaphragm (Dia) and T3 external intercostal (T3 EIC) raw and integrated (Int.) electromyogram (EMG) activity in a wild‐type (WT) (black) and mdx (grey) mouse during baseline ( = 0.60) and protracted tracheal occlusion (∼30–40 s). B, representative traces of oesophageal pressure and Dia and T3 EIC raw and Int. EMG activity in a WT and mdx mouse during baseline and peak inspiratory efforts (shaded) during a protracted tracheal occlusion.
Figure 5. Oesophageal pressure and diaphragm and EIC EMG activities during tracheal occlusion in anaesthetized wild‐type and mdx mice: group data.
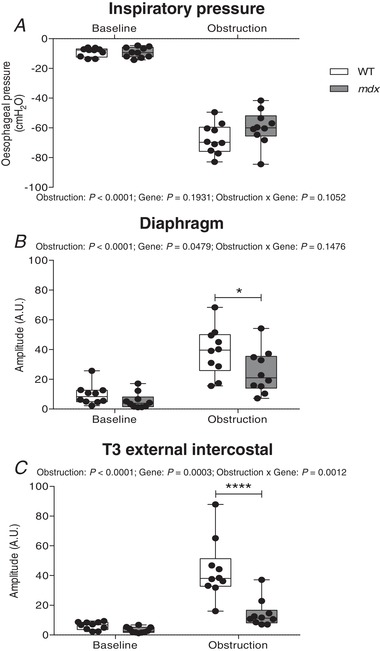
A–C, group data for oesophageal pressure (A), diaphragm EMG activity (B) and T3 external intercostal (T3 EIC) EMG activity (C) during baseline conditions and during tracheal occlusion (average of five successive peak breaths) in wild‐type (WT) (n = 10) and mdx (n = 10) mice. Values are expressed as box and whisker plots (median, 25–75th percentile and scatter plot). Data were statistically compared by repeated measures two‐way ANOVA with a Bonferroni post hoc test. * P < 0.05 and **** P < 0.0001 compared to the corresponding WT values.
Table 3.
Baseline respiratory parameters in anaesthetized wild‐type and mdx mice before and during peak sustained efforts during airway occlusion
| Wild‐type (n = 10) | mdx (n = 10) | ||||
|---|---|---|---|---|---|
| Baseline | Obstruction | Baseline | Obstruction | P (RM two‐way ANOVA) | |
| f R (breaths min−1) | 217 ± 34 | 76 ± 18 | 227 ± 41 | 109 ± 32 | Obstruction: P < 0.0001; Gene: P = 0.0802; Obstruction × Gene: P = 0.1962 |
| (%) | 95.1 ± 2.1 | 40.1 ± 9.5 | 95.8 ± 1.9 | 46.3 ± 9.1 | Obstruction: P < 0.0001; Gene: P = 0.116; Obstruction × Gene: P = 0.2132 |
| T i (ms) | 105 ± 15 | 144 ± 18 | 88 ± 14* | 135 ± 15 | Obstruction: P < 0.0001; Gene: P = 0.0441; Obstruction × Gene: P = 0.2073 |
| TTP (ms) | 58 ± 15 | 76 ± 11 | 50 ± 10 | 82 ± 13 | Obstruction: P < 0.0001; Gene: P = 0.7911; Obstruction × Gene: P = 0.0438 |
| Dia AUC (A.U.) | 101 ± 76 | 585 ± 241 | 57 ± 56 | 382 ± 242$ | Obstruction: P < 0.0001; Gene: P = 0.0582; Obstruction × Gene: P = 0.1349 |
| T3 EIC AUC (A.U.) | 65 ± 28 | 594 ± 215 | 34 ± 24 | 198 ± 131$$$ | Obstruction: P < 0.0001; Gene: P < 0.0001; Obstruction × Gene: P = 0.0001 |
f R, respiratory frequency; , peripheral capillary oxygen saturation; T i, inspiratory duration; TTP, time to peak sub‐atmospheric pressure; Dia AUC, area under the curve of diaphragm integrated EMG signal (A.U. = arbitrary units); T3 EIC AUC, area under the curve of T3 external intercostal integrated EMG signal (A.U. = arbitrary units). Data are shown as the mean ± SD and were statistically compared by repeated measures two‐way analysis of variance (RMANOVA).
* P < 0.05 compared to wild‐type baseline,
$ P < 0.05,
$$$ P < 0.001 compared to wild‐type obstruction.
Tracheal airflow and diaphragm and EIC EMG activity in anaesthetized, vagotomized mice
Table 4 shows respiratory data in anaesthetized, vagotomized wild‐type and mdx mice during baseline conditions ( = 0.60). There was no significant difference in f R, V T or between wild‐type and mdx mice. Similarly, peak inspiratory and expiratory flows and also ETCO2 and were equivalent between groups. Figure 6 A shows representative original recordings of tracheal airflow, diaphragm and EIC EMG activities in anaesthetized, vagotomized mice during baseline conditions ( = 0.60). Peak respiratory efforts during chemoactivation increased V T (P < 0.0001, peak breath; repeated measures two‐way ANOVA) (Fig. 6 B), peak inspiratory flow (P = 0.027) (Fig. 6 C) and peak expiratory flow (P = 0.016) (Fig. 6 D), with no difference between wild‐type and mdx mice. Diaphragm EMG activity increased during peak respiratory efforts (P = 0.0128, peak breath; repeated measures two‐way ANOVA) (Fig. 6 E) in wild‐type and mdx mice, with equivalent diaphragm EMG activity between groups (P = 0.4055, gene; repeated measures two‐way ANOVA) (Fig. 6 E). Diaphragm EMG recruitment (delta EMG amplitude) during peak respiratory efforts compared to baseline was equivalent between wild‐type and mdx (P = 0.9953; unpaired Student's t test) (Fig. 6 F). EIC EMG activity was significantly lower during baseline conditions in mdx mice compared to wild‐type (P < 0.05; repeated measures two‐way ANOVA with Bonferroni) (Fig. 6 G) and during peak chemoactivation (P < 0.01). EIC EMG recruitment (delta EMG amplitude) during peak ventilation compared to baseline was significantly lower in mdx compared to wild‐type mice (P = 0.0434; unpaired Student's t test) (Fig. 6 H).
Table 4.
Baseline respiratory parameters in anaesthetized, vagotomized wild‐type and mdx mice
| Wild‐type (n = 10) | mdx (n = 7) | P (Student's t test) | |
|---|---|---|---|
| f R (breaths min−1) | 64 ± 19 | 60 ± 14 | 0.6932 |
| V T (mL g−1) | 0.032 ± 0.005 | 0.032 ± 0.003 | 0.9800 |
| (mL g−1 min−1) | 2.0 ± 0.7 | 1.9 ± 0.4 | 0.6343 |
| PIF (mL g−1 s−1) | 1.03 ± 0.16 | 1.15 ± 0.13 | 0.1345 |
| PEF (mL g−1 s−1) | 1.04 ± 0.18 | 1.08 ± 0.29 | 0.7207 |
| ETCO2 (mmHg) | 53.2 ± 3.7 | 50.5 ± 6.1 | 0.2735 |
| (%) | 94.8 ± 2.5 | 94.1 ± 3.1 | 0.5821 |
| Body mass (g) | 24.8 ± 1.5 | 26.1 ± 0.8 | 0.0382 |
f R, respiratory frequency; V T, tidal volume; , minute ventilation; PIF, peak inspiratory flow; PEF, peak expiratory flow; ETCO2, end‐tidal carbon dioxide; , peripheral capillary oxygen saturation. Data are shown as the mean ± SD and were statistically compared using two‐tailed unpaired Student's t tests.
Figure 6. Tracheal airflow and diaphragm and EIC EMG activities in anaesthetized, vagotomized wild‐type and mdx mice.
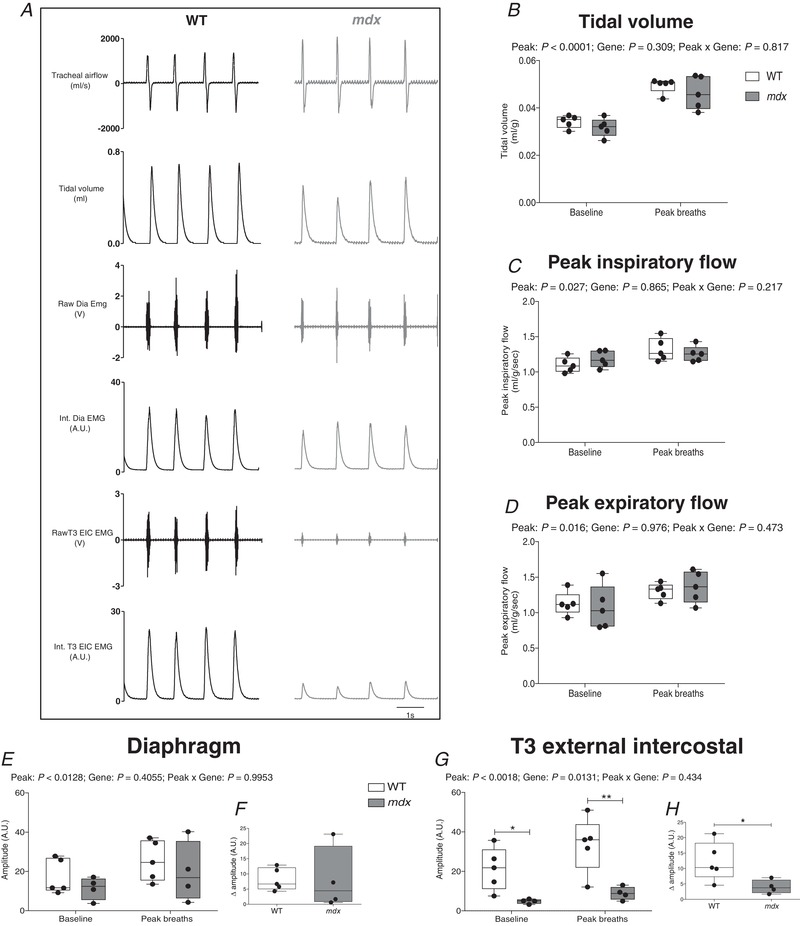
A, representative recordings of tracheal airflow, tidal volume, raw and integrated (Int.) diaphragm (Dia) and T3 external intercostal (T3 EIC) EMG activities for an anaesthetized WT mouse (black) and mdx mouse (grey) during baseline conditions ( = 0.60). B–D, group data for tidal volume (B), peak inspiratory flow (C) and peak expiratory flow (D) during baseline conditions and during spontaneous peak breaths during chemoactivation ( = 0.15/ = 0.06). E, diaphragm EMG activity during baseline and peak breaths. F, diaphragm EMG response (expressed as absolute change from baseline amplitude) during peak breaths. G, EIC EMG activity during baseline and peak breaths. H, EIC EMG activity (expressed as absolute change from baseline amplitude) during peak breaths. Values are expressed as box and whisker plots (median, 25–75th percentile and scatter plot). Data were statistically compared by repeated measures two‐way ANOVA or two‐tailed unpaired Student's t tests. * P < 0.05 and ** P < 0.01 compared to the corresponding WT values.
Discussion
The main findings of the present study are that: (i) there is substantial diaphragm muscle weakness in young adult mdx mice; (ii) ventilatory capacity in response to maximum chemostimulation is equivalent between conscious wild‐type and mdx mice; (iii) peak tidal volume and peak inspiratory and expiratory flows during maximum chemoactivation are equivalent in anaesthetized, vagotomized wild‐type and mdx mice; (iv) peak inspiratory pressure‐generating capacity is preserved in mdx mice during ventilatory (chemoactivated breathing and augmented breaths) and non‐ventilatory (sustained tracheal occlusion) behaviours; and (v) diaphragm and EIC EMG activities are lower during airway obstruction in mdx compared with wild‐type mice.
Our study confirmed substantial diaphragm muscle weakness in young adult mdx mice, consistent with our recent reports (Burns et al. 2017a,c). Diaphragm deficits in mdx mice were indicated by a significantly reduced twitch and tetanic isometric force compared to wild‐type. The force–frequency relationship revealed considerable force loss in the frequency range of 60–160 Hz, corresponding to a broad range of ventilatory and non‐ventilatory behaviours (Sieck et al. 2013; Mantilla et al. 2014). We have previously documented considerable muscle fibre damage and remodelling in dystrophic diaphragm at this age, with increased variability of muscle fibre size, centralized myonuclei, altered myosin heavy chain isoform composition and collagen deposition (Burns et al. 2017c, 2018). Force loss was accompanied by decreases in muscle shortening and velocity of shortening.
Notwithstanding the profound diaphragm weakness in mdx, we reasoned that ventilatory behaviours might be protected as a result of the large reserve capacity of the diaphragm muscle, such that maximum ventilation during chemoactivation can be achieved with <40% of peak diaphragm force (Sieck & Fournier, 1989; Mantilla et al. 2010; Medina‐Martínez et al. 2015; Greising et al. 2016). By contrast, we hypothesized that non‐ventilatory behaviours, such as airway obstruction, airway clearance, and respiratory reflexes encompassing coughing and sneezing, requiring substantially elevated respiratory muscle activation, and which are critical for the safeguarding of pulmonary function, are compromised in dystrophic disease. Dysphagia and poor airway control increase the risk of obstructive airway events during sleep, which are common in teenage DMD boys (Barbé et al. 1994; Khan & Heckmatt, 1994; Suresh et al. 2005; Sawnani et al. 2015). Peak respiratory pressures are low in DMD and are known to decline progressively with advancing disease (Khirani et al. 2014).
We assessed ventilation in response to hypercapnic and hypercapnic hypoxic challenges, which confirmed the capacity for mdx mice to enhance ventilation. Ventilation during graded hypercapnia was equivalent in mdx and wild‐type mice, revealing that CO2 chemosensitivity is intact in young adult mdx mice, which retain considerable ventilatory reserve and, despite diaphragm dysfunction, maintain a capacity to elevate to wild‐type levels through increases in rate and volume. This preserved capacity to elevate pulmonary ventilation was further revealed during exposure to hypercapnic hypoxia with peak ventilation increasing more than three‐fold in mdx mice, equivalent to responses in wild‐type mice. We acknowledge the limitations of accurate determination of tidal volume by the method of plethysmography, which may have been further limited in the present study as a result of our estimation (not continuous measurement) of body temperature.
In anaesthetized mice, we demonstrated that peak inspiratory oesophageal pressure generation was equivalent during basal breathing ( = 0.60) in mdx and wild‐type mice. Acute chemoactivation ( = 0.15 & = 0.05) increased ventilatory drive and the magnitude of the enhanced peak inspiratory oesophageal pressure was equivalent in mdx and wild‐type mice. This confirmed the lack of mechanical deficit in young adult mdx mice and hence no limitation in sub‐atmospheric pressure‐generating capacity during chemoactivation. Diaphragm EMG responses to hypercapnic hypoxia were equivalent in mdx compared to wild‐type mice. As such, even the compromised dystrophic mdx diaphragm is probably capable of supporting high ventilatory demand.
We extended this line of enquiry by way of assessment of spontaneous augmented breaths (sighs), which result in elevated diaphragm motor unit recruitment beyond that observed during chemoactivation (Mantilla et al. 2011; Seven et al. 2014), producing up to 50% of peak transdiaphragmatic pressure in mice (Greising et al. 2013; 2015; 2016). Interestingly, during augmented breaths, peak inspiratory oesophageal pressure generation was preserved and significantly greater in mdx compared to wild‐type mice, again convincingly demonstrating a lack of mechanical constraint in 8‐week‐old mdx mice despite substantial diaphragm weakness (approaching 50% force loss compared with wild‐type in the range 60–100 Hz). Of note, diaphragm EMG recruitment during spontaneous augmented breaths was equivalent (not potentiated) in mdx compared to wild‐type mice. Thus, as is the case for ventilation during chemoactivation, augmented breaths are not compromised in young adult mdx mice, insofar as enhanced inspiratory pressures can be achieved even with 50% reduction in force‐generating capacity of the diaphragm, presumably because such efforts either remain within the reserve capacity of the dystrophic diaphragm, or they are facilitated by accessory muscle recruitment, which can compensate for efforts exceeding the reserve capacity of the weakened mdx diaphragm. Of note in the present study, inspiratory pressure generation during augmented breaths was greater in mdx compared to wild‐type mice, although diaphragm EMG recruitment was equivalent between groups.
In addition, we examined tracheal airflow in anaesthetized, vagotomized mice during chemoactivation, where we characterized maximum ventilatory effort, exceeding that of volume‐related measures in conscious mice as a result of the release of vagal inhibition of breathing with superimposed chemoactivation of respiratory motor outflow. Direct measures of peak V T and peak inspiratory and expiratory flows were equivalent in vagotomized wild‐type and mdx mice, confirming and extending observations from our studies using plethysmography. Peak diaphragm EMG activity was equivalent in wild‐type and mdx mice, whereas EIC EMG was significantly lower in mdx mice. Preserved neural drive to the weakened mdx diaphragm may be wholly adequate to generate even the large volumes and respiratory flows observed in vagotomized mdx mice. In this respect, our findings are consistent with a variety of other studies revealing that transdiaphragmatic pressure‐generating capacity during high ventilatory demand (chemoactivation; augmented breaths) is within 50% of maximum capacity (Sieck & Fournier, 1989; Mantilla et al. 2010), including studies in mice (Greising et al. 2013, 2015, 2016), and these behaviours can be adequately met even in the context of motor neuron loss (Khurram et al. 2018a) or age‐related sarcopenia of the diaphragm (Khurram et al. 2018b). Although force loss is severe in mdx diaphragm, ventilatory behaviours are accommodated, at least early in dystrophic disease.
Our consideration of respiratory‐related capacity to this point was limited to peak ventilation during chemoactivation of breathing, albeit including observations in animals with removal of vagal afferent inhibition of respiratory drive. We reasoned that the preserved capacity in mdx might be limited to the ventilatory range, such that mechanical deficit might be revealed during non‐ventilatory efforts, particularly over sustained periods, which evoke near‐maximum respiratory system activation, or at least activation well beyond the reserve capacity of the weakened mdx diaphragm. To investigate this, we extended our studies to examine peak inspiratory pressure‐generating capacity in wild‐type and mdx mice during protracted sustained tracheal occlusion, which results in greater motor recruitment of respiratory muscles beyond that of augmented breaths (Mantilla et al. 2011; Greising et al. 2013; Seven et al. 2014). Previously, it has been shown that sustained tracheal occlusions lasting 15 s in anaesthetized mice result in the generation of transdiaphragmatic pressures ∼65–82% of peak pressures evoked by bilateral phrenic nerve stimulation (Greising et al. 2013, 2015, 2016), with transdiaphragmatic pressure values of ∼72 cmH20 during obstruction in young male mice, equating to ∼78% of maximum transdiaphragmatic pressure (Greising et al. 2015). In the present study, oesophageal pressure recordings were performed in anaesthetized mice with contemporaneous measurement of diaphragm and EIC EMG activity. Tracheal occlusions sustained for 30–40s generated impressive sub‐atmospheric inspiratory pressures in wild‐type and mdx mice. The sustained efforts during the final phase of airway obstruction were probably reflective of maximum or near‐maximum respiratory system performance, as indicated by progressively greater sub‐atmospheric inspiratory pressures during occlusion to a sustained nadir associated with maximal diaphragm and EIC recruitment. Indeed, sub‐atmospheric pressures were often noted to decrease further (become more ‘negative’) both in wild‐type and mdx mice, when diaphragm EMG activity reached a maximum plateau or even decreased compared to preceding efforts.
Tracheal occlusion in the present study resulted in sub‐atmospheric pressure generation that clearly exceeded the reserve capacity of the dystrophic diaphragm. Moreover, both diaphragm and EIC EMG activities (determined both as amplitude and AUC of the integrated EMG signals) were substantially lower during peak sustained efforts in mdx compared to wild‐type mice, with a relatively greater reduction of activity in EIC EMG compared to diaphragm EMG in mdx mice. Decreased EMG activity during airway obstruction could reflect impaired motor control of the diaphragm and EIC muscles, although it is probably fully explained by impaired neuromuscular transmission given the evidence in young mdx mice of an increased variability of neural transmission to the diaphragm (Personius & Sawyer, 2006) and a decreased amplitude of mdx diaphragm motor end plate potentials (Carlson & Roshek, 2001). Neuromuscular impairment during high demand behaviours that exceed the reduced reserve capacity of the mdx diaphragm would be expected to translate to a substantial inability to generate peak inspiratory pressures equivalent to wild‐type, beyond that predicted by assessment of diaphragm peak force. However, in the present study, peak sustained inspiratory pressures during airway obstruction were equivalent in wild‐type and mdx mice. We determined that inspiratory duration, which is prolonged during obstructed efforts, was equivalent during peak sustained efforts in wild‐type and mdx mice, highlighting that compensation in the capacity to generate equivalent sub‐atmospheric pressures in mdx mice does not depend upon an altered control of inspiratory timing. We therefore reason that recruitment of support muscles beyond the primary muscles of inspiration (diaphragm and EIC) contributes substantively to non‐ventilatory pressure‐generating capacity in young dystrophic mdx mice, as a result of the necessary and sufficient compensation that is capable of facilitating increased thoracic volume. It is clear from the present study that T3 EIC muscle activity is severely curtailed in mdx mice, suggesting that intercostal muscles do not provide compensation during high demand behaviours in mdx mice. Of note, ventilatory capacity is maintained in rat models of amyotrophic lateral sclerosis (ALS) at end‐stage disease despite substantial loss of phrenic motor neurons (Nichols et al. 2013; Nichols et al. 2014). Indeed, it is established that scalene and trapezius muscles enhance ventilation in ALS mice (Romer et al. 2017).
Elegant work in dystrophic dogs with the GRMD mutation showed a reduced diaphragm contribution to inspiratory tidal volume during wakefulness at rest in the lateral recumbent position (Mead et al. 2014). Moreover, there was loss of ventilatory capacity during pharmacological stimulation of breathing, although capacity is partly compensated for by expiratory muscles that serve to lower end‐expiratory chest wall volume. Mead et al. (2014) also provided an excellent perspective on a proposed redistribution of the work of breathing beyond the diaphragm to accessory and auxiliary muscles in the context of dystrophic disease. Of further interest, a strong argument is also presented in favour of viewing increased diaphragm stiffness as an adaptive benefit in muscular dystrophy (Mead et al. 2014), comprising an outcome that may defend against cranial movement of the diaphragm during subatmospheric pressure generation by accessory and support muscles, particularly during high demand behaviours. Similar changes in the two‐compartment chest wall model of breathing are reported in DMD patients (LoMauro et al. 2010), with outcomes dependent upon posture (lower diaphragm contribution to tidal volume in the supine position) but, surprisingly, these are not influenced by the presence or absence of scoliosis (LoMauro et al. 2010). It is plausible that expiratory muscle activation is increased in dystrophic disease in support of ventilatory capacity. Beyond the recognized contribution of active expiration to increased ventilatory capacity in support of inspiratory muscle work, expiratory muscle recruitment could serve to shorten expiratory duration and decrease end‐expiratory lung volume, accessing expiratory reserve volume in dystrophic animals and DMD boys that allows an increase in tidal volume (Mead et al. 2014), until compensation is progressively lost with advancing disease.
In respect of the preserved capacity of mdx mice to generate peak inspiratory pressures during airway obstruction, we suggest that auxiliary muscle recruitment in support of peak pressure‐generating capacity offers the most plausible explanation for respiratory compensation in young mdx mice, notwithstanding that the muscle group(s) pivotal to the support of respiratory system compensation during non‐ventilatory behaviours were not identified in the present study. Fibrosis in mdx (and DMD) leads to decreased chest wall compliance and increased work with respect to breathing. Although the extent of fibrosis and its expected impact on chest wall compliance was not determined in the present study, and may not be a major pathology in young mdx mice but presumaby has greater relevance at later stages of disease, we reason that compensated inspiratory pressure‐generating capacity in mdx mice does not relate to increased chest wall compliance. Indeed, compensation is probably limited by increased chest wall stiffness during progressive disease in mdx and DMD, adding further to the burden of a redistributed workload on accessory and auxiliary muscles.
The long‐term compensatory capacity of primary and secondary respiratory motor pathways and muscles in mdx (and DMD) is not known. We speculate that, in dystrophic disease, there may be serial recruitment of accessory and other support muscles that facilitate respiratory performance, although, in turn, these show progressive deterioration. Mechanical strain leads to injury in dystrophic muscle, triggering a spiral of progressive dysfunction. It is known, for example, that recruitment of the mdx diaphragm in exercise expedites diaphragm dysfunction (Capogrosso et al. 2017). As such, the early modest decline in inspiratory pressure‐generating capacity in mdx mice that we observed in the present study (∼10% loss comparing median values in Fig. 5 A) could conceivably decline further with advancing disease. We suggest that compensation does not prevail during progressive dystrophic disease, a hypothesis previously proposed by Gayraud et al. (2007). It is especially interesting to consider that peak hypercapnic ventilation is normal in 5‐month‐old mdx mice (Gayraud et al. 2007), whereas it decreased at 7 months, as well as at 10–12 months (Gosselin et al. 2003) and 16 months of age (Gayraud et al. 2007). Interestingly, diaphragm dysfunction presents early in the mdx model, with relatively modest further deterioration in force‐generating capacity with advancing disease (e.g. 50–66% force loss at 7–14 months of age) (Lynch et al. 2001; Gosselin et al. 2003; Selsby et al. 2016). We therefore suggest that a progressive decline in ventilatory performance in mdx (and perhaps DMD) potentially relates to an increased dependence upon, and a progressive loss of compensatory mechanisms, which, when combined with diaphragm dysfunction, underpin the development of respiratory morbidity. In DMD, there is a progressive decline in pulmonary function and respiratory muscle strength culminating in reduced ventilation (Smith et al. 1989; De Bruin et al. 1997; Hukins & Hillman, 2000; Khirani et al. 2014). The decline in peak respiratory pressure generation in DMD has important prognostic value (Phillips et al. 2001; Khirani et al. 2014).
In summary, the present study highlights new facets to breathing with neuromuscular disease. We demonstrate profound diaphragm muscle weakness during early disease progression in young adult mdx mice but a capacity to preserve ventilatory behaviours over a wide range. Ventilatory demand is probably adequately met by even the compromised dystrophic diaphragm, at least in early disease. We reveal the remarkable preservation of inspiratory pressure‐generating capacity during sustained airway obstruction in mdx mice, which illustrates a necessary and sufficient compensation within the respiratory control system. We hypothesize that there is an increased reliance on compensatory support, which is progressively lost in mdx (and DMD), which, when combined with devastating primary respiratory muscle (and neuromuscular junction) dysfunction, contributes to respiratory‐related morbidity, culminating in the sequential loss of non‐ventilatory and ventilatory capacity and, ultimately, leading to respiratory morbidity and failure in advanced disease. Our findings have relevance to human dystrophinopathies and therapeutic strategies for pulmonary support and rehabilitation in DMD boys.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
DPB, EFL and KDO'H were responsible for the experimental design. DPB, KHM, EFL and KDO'H were responsible for the acquisition of data. DPB, KHM and KDO'H were responsible for data analysis. DPB and KDO'H were responsible for statistical analysis. DPB and KDO'H were responsible for interpretation of data. DPB and KDO'H were responsible for drafting the original manuscript. All authors contributed to the drafting and critical revision of the manuscript for important intellectual content. All authors have approved the final version of the manuscript submitted for publication and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
DPB and KHM were supported by funding from the Department of Physiology, UCC.
Translational perspective.
Duchenne muscular dystrophy (DMD) is an X‐linked fatal neuromuscular disease. Respiratory‐related morbidity and mortality are widely recognized features of DMD, yet there is a considerable knowledge gap in respect of a comprehensive understanding of the neural control of breathing and respiratory‐related behaviours in the human dystrophinopathies. The dystrophin‐deficient mdx mouse is a widely used pre‐clinical model of DMD. In young adult mdx mice, we examined inspiratory pressure‐generating capacity across a range of ventilatory and non‐ventilatory behaviours aiming to explore inherent deficits and compensations that manifest in dystrophic disease. The present study reveals remarkable compensation in support of respiratory system performance, at least early in disease, despite evidence of profound diaphragm muscle weakness. Our data illustrate a capacity to preserve peak inspiratory pressure generation, despite respiratory muscle weakness and impaired neural recruitment of primary inspiratory muscles (diaphragm and intercostal muscles). Our findings point to a considerable reserve in the capacity of the respiratory system to perform a broad range of function, probably as a result of the recruitment of auxiliary muscles in support of peak respiratory‐related performance. Peak inspiratory pressure generation is most relevant to protective reflexes related to airway maintenance and clearance, which is particularly relevant to neuromuscular (and neurodegenerative) conditions, given that dysphagic patients are at risk of obstructive airway events during sleep, as well as aspiration pneumonia. It will be important to determine whether similar compensation presents in DMD boys and also whether the progressive loss of respiratory performance with advancing disease relates to a decline or loss of compensatory mechanisms. Clinical assessment of auxiliary muscle control of breathing during maximal pressure‐generating manoeuvres is clearly warranted (e.g. assessment of scalene, sternocleidomastoid and trapezius EMG activities and oesophageal pressure measurement during maximal sniff manoeuvres). Rehabilitative strategies focussed on auxiliary muscles that support inspiratory pressure generation may prove promising. Further delineation of the compensatory signature of the neuromuscular control system governing respiratory performance in animal models of DMD may provide insight into mechanisms that can be protected and/or further harnessed in the development of therapeutic strategies for DMD.
Acknowledgements
We are grateful to staff of the Biological Services Unit, University College Cork for help with animal care.
Biography
David P. Burns has a primary research interest in the control of breathing in muscular dystrophy, with wider interests in cardiorespiratory physiology in animal models of disease. He received the Usha Award of the American Physiological Society, presented at the Experimental Biology 2017 meeting in Chicago, and he was selected as one of three early career scientists working in Ireland to participate at the 68th Nobel Lindau meeting in Germany in 2018. This paper is the third in a series of studies published in The Journal of Physiology that have been performed as part of his doctoral training at University College Cork, Ireland.

Edited by: Scott Powers & Frank Powell
References
- Barbé F, Quera‐Salva MA, McCann C, Gajdos P, Raphael JC, de Lattre J & Agustí AG (1994). Sleep‐related respiratory disturbances in patients with Duchenne muscular dystrophy. Eur Respir J 7, 1403–1408. [DOI] [PubMed] [Google Scholar]
- Beck J, Weinberg J, Hamnegård CH, Spahija J, Olofson J, Grimby G & Sinderby C (2006). Diaphragmatic function in advanced Duchenne muscular dystrophy. Neuromuscul Disord 16, 161–167. [DOI] [PubMed] [Google Scholar]
- Burns DP, Ali I, Rieux C, Healy J, Jasionek G & O'Halloran KD (2017a). Tempol supplementation restores diaphragm force and metabolic enzyme activities in mdx mice. Antioxidants (Basel) 6, E101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns DP, Canavan L, Rowland J, O'Flaherty R, Brannock M, Drummond SE, O'Malley D, Edge D & O'Halloran KD (2018). Recovery of respiratory function in mdx mice co‐treated with neutralizing interleukin‐6 receptor antibodies and urocortin‐2. J Physiol 596, 5175–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns DP & O'Halloran KD (2016). Evidence of hypoxic tolerance in weak upper airway muscle from young mdx mice. Respir Physiol Neurobiol 226, 68–75. [DOI] [PubMed] [Google Scholar]
- Burns DP, Rowland J, Canavan L, Murphy KH, Brannock M, O'Malley D, O'Halloran KD & Edge D (2017b). Restoration of pharyngeal dilator muscle force in dystrophin‐deficient (mdx) mice following co‐treatment with neutralizing interleukin‐6 receptor antibodies and urocortin 2. Exp Physiol 102, 1177–1193. [DOI] [PubMed] [Google Scholar]
- Burns DP, Roy A, Lucking EF, McDonald FB, Gray S, Wilson RJ, Edge D & O'Halloran KD (2017c). Sensorimotor control of breathing in the mdx mouse model of Duchenne muscular dystrophy. J Physiol 595, 6653–6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capogrosso RF, Mantuano P, Cozzoli A, Sanarica F, Massari AM, Conte E, Fonzino A, Giustino A, Rolland JF, Quaranta A, De Bellis M, Camerino GM, Grange RW & De Luca A (2017). Contractile efficiency of dystrophic mdx mouse muscle: in vivo and ex vivo assessment of adaptation to exercise of functional end points. J Appl Physiol (1985) 122, 828–843. [DOI] [PubMed] [Google Scholar]
- Carlson CG & Roshek DM (2001). Adult dystrophic (mdx) endplates exhibit reduced quantal size and enhanced quantal variation. Pflugers Arch 442, 369–375. [DOI] [PubMed] [Google Scholar]
- Carretta D, Santarelli M, Vanni D, Carrai R, Sbriccoli A, Pinto F & Minciacchi D (2001). The organisation of spinal projecting brainstem neurons in an animal model of muscular dystrophy. A retrograde tracing study on mdx mutant mice. Brain Res 895, 213–222. [DOI] [PubMed] [Google Scholar]
- Chaussenot R, Edeline JM, Le Bec B, El Massioui N, Laroche S & Vaillend C (2015). Cognitive dysfunction in the dystrophin‐deficient mouse model of Duchenne muscular dystrophy: a reappraisal from sensory to executive processes. Neurobiol Learn Mem 124, 111–122. [DOI] [PubMed] [Google Scholar]
- De Bruin PF, Ueki J, Bush A, Khan Y, Watson A & Pride NB (1997). Diaphragm thickness and inspiratory strength in patients with Duchenne muscular dystrophy. Thorax 52, 472–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti JM (2007). Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta 1772, 108–117. [DOI] [PubMed] [Google Scholar]
- Gayraud J, Matecki S, Hnia K, Mornet D, Prefaut C, Mercier J, Michel A & Ramonatxo M (2007). Ventilation during air breathing and in response to hypercapnia in 5 and 16 month‐old mdx and C57 mice. J Muscle Res Cell Motil 28, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin LE, Barkley JE, Spencer MJ, McCormick KM & Farkas GA (2003). Ventilatory dysfunction in mdx mice: impact of tumor necrosis factor‐alpha deletion. Muscle Nerve 28, 336–343. [DOI] [PubMed] [Google Scholar]
- Greising SM, Mantilla CB, Medina‐Martínez JS, Stowe JM & Sieck GC (2015). Functional impact of diaphragm muscle sarcopenia in both male and female mice. Am J Physiol Lung Cell Mol Physiol 309, L46–L52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greising SM, Mantilla CB & Sieck GC (2016). Functional measurement of respiratory muscle motor behaviors using transdiaphragmatic pressure. Methods Mol Biol 1460, 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greising SM, Sieck DC, Sieck GC & Mantilla CB (2013). Novel method for transdiaphragmatic pressure measurements in mice. Respir Physiol Neurobiol 188, 56–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EP, Brown RH & Kunkel LM (1987). Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928. [DOI] [PubMed] [Google Scholar]
- Hukins CA & Hillman DR (2000). Daytime predictors of sleep hypoventilation in Duchenne muscular dystrophy. Am J Respir Crit Care Med 161, 166–170. [DOI] [PubMed] [Google Scholar]
- Khan Y & Heckmatt JZ (1994). Obstructive apnoeas in Duchenne muscular dystrophy. Thorax 49, 157–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khirani S, Ramirez A, Aubertin G, Boulé M, Chemouny C, Forin V & Fauroux B (2014). Respiratory muscle decline in Duchenne muscular dystrophy. Pediatr Pulmonol 49, 473–481. [DOI] [PubMed] [Google Scholar]
- Khurram OU, Fogarty MJ, Rana S, Vang P, Sieck GC & Mantilla CB (2018a). Diaphragm muscle function following mid‐cervical contusion injury in rats. J Appl Physiol (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurram OU, Fogarty MJ, Sarrafian TL, Bhatt A, Mantilla CB & Sieck GC (2018b). Impact of aging on diaphragm muscle function in male and female Fischer 344 rats. Physiol Rep 6, e13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis P, Sheehan D, Soares R, Coelho AV & O'Halloran KD (2016). Redox remodeling is pivotal in murine diaphragm muscle adaptation to chronic sustained hypoxia. Am J Respir Cell Mol Biol 55, 12–23. [DOI] [PubMed] [Google Scholar]
- Lewis P, Sheehan D, Soares R, Varela Coelho A & O'Halloran KD (2015). Chronic sustained hypoxia‐induced redox remodeling causes contractile dysfunction in mouse sternohyoid muscle. Front Physiol 6, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidov HG (1996). Dystrophin in the nervous system. Brain Pathol 6, 63–77. [DOI] [PubMed] [Google Scholar]
- LoMauro A, D'Angelo MG, Romei M, Motta F, Columbo D, Comi GP, Pedotti A, Marchi E, Turconi AC, Bresolin N & Aliveri A (2010). Abdominal volume contribution to tidal volume as an early indicator of respiratory impairment in Duchenne muscular dystrophy. Eur Respir J 35, 1118–1125. [DOI] [PubMed] [Google Scholar]
- Lynch GS, Hinkle RT & Faulkner JA (2001). Force and power output of diaphragm muscle strips from mdx and control mice after clenbuterol treatment. Neuromuscul Disord 11, 192–196. [DOI] [PubMed] [Google Scholar]
- Mantilla CB, Seven YB, Hurtado‐Palomino JN, Zhan WZ & Sieck GC (2011). Chronic assessment of diaphragm muscle EMG activity across motor behaviors. Respir Physiol Neurobiol 177, 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantilla CB, Seven YB & Sieck GC (2014). Convergence of pattern generator outputs on a common mechanism of diaphragm motor unit recruitment. Prog Brain Res 209, 309–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantilla CB, Seven YB, Zhan WZ & Sieck GC (2010). Diaphragm motor unit recruitment in rats. Respir Physiol Neurobiol 173, 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer OH, Finkel RS, Rummey C, Benton MJ, Glanzman AM, Flickinger J, Lindström BM & Meier T (2015). Characterization of pulmonary function in Duchenne Muscular Dystrophy. Pediatr Pulmonol 50, 487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead AF, Petrov M, Malik AS, Mitchell MA, Childers MK, Bogan JR, Seidner G, Kornegay JN & Stedman HH (2014). Diaphragm remodeling and compensatory respiratory mechanics in a canine model of Duchenne muscular dystrophy. J Appl Physiol (1985) 116, 807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina‐Martínez JS, Greising SM, Sieck GC & Mantilla CB (2015). Semi‐automated assessment of transdiaphragmatic pressure variability across motor behaviors. Respir Physiol Neurobiol 215, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntoni F, Torelli S & Ferlini A (2003). Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol 2, 731–740. [DOI] [PubMed] [Google Scholar]
- Nichols NL, Gowing G, Satriotomo I, Nashold LJ, Dale EA, Suzuki M, Avalos P, Mulcrone PL, McHugh J, Svendsen CN & Mitchell GS (2013). Intermittent hypoxia and stem cell implants preserve breathing capacity in a rodent model of amyotrophic lateral sclerosis. Am J Respir Crit Care Med 187, 535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols NL, Johnson RA, Satriotomo I & Mitchell GS (2014). Neither serotonin nor adenosine‐dependent mechanisms preserve ventilatory capacity in ALS rats. Respir Physiol Neurobiol 197, 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Personius KE & Sawyer RP (2006). Variability and failure of neurotransmission in the diaphragm of mdx mice. Neuromuscul Disord 16, 168–177. [DOI] [PubMed] [Google Scholar]
- Phillips MF, Quinlivan RC, Edwards RH & Calverley PM (2001). Changes in spirometry over time as a prognostic marker in patients with Duchenne muscular dystrophy. Am J Respir Crit Care Med 164, 2191–2194. [DOI] [PubMed] [Google Scholar]
- Pinto ML, Tokunaga HH, Souccar C, Schoorlemmer GH & de Cassia Ribeiro da Silva Lapa R (2008). Loss of neuronal projections in the dystrophin‐deficient mdx mouse is not progressive. Brain Res 1224, 127–132. [DOI] [PubMed] [Google Scholar]
- Romer SH, Seedle K, Turner SM, Li J, Baccei ML & Crone SA (2017). Accessory respiratory muscles enhance ventilation in ALS model mice and are activated by excitatory V2a neurons. Exp Neurol 287, 192–204. [DOI] [PubMed] [Google Scholar]
- Sawnani H, Thampratankul L, Szczesniak RD, Fenchel MC & Simakajornboon N (2015). Sleep disordered breathing in young boys with Duchenne muscular dystrophy. J Pediatr 166, 640–645.e641. [DOI] [PubMed] [Google Scholar]
- Sbriccoli A, Santarelli M, Carretta D, Pinto F, Granato A & Minciacchi D (1995). Architectural changes of the cortico‐spinal system in the dystrophin defective mdx mouse. Neurosci Lett 200, 53–56. [DOI] [PubMed] [Google Scholar]
- Sekiguchi M, Zushida K, Yoshida M, Maekawa M, Kamichi S, Sahara Y, Yuasa S, Takeda S & Wada K (2009). A deficit of brain dystrophin impairs specific amygdala GABAergic transmission and enhances defensive behaviour in mice. Brain 132, 124–135. [DOI] [PubMed] [Google Scholar]
- Selsby JT, Ballmann CG, Spaulding HR, Ross JW & Quindry JC (2016). Oral quercetin administration transiently protects respiratory function in dystrophin‐deficient mice. J Physiol 594, 6037–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seven YB, Mantilla CB & Sieck GC (2014). Recruitment of rat diaphragm motor units across motor behaviors with different levels of diaphragm activation. J Appl Physiol (1985) 117, 1308–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieck GC, Ferreira LF, Reid MB & Mantilla CB (2013). Mechanical properties of respiratory muscles. Compr Physiol 3, 1553–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieck GC & Fournier M (1989). Diaphragm motor unit recruitment during ventilatory and nonventilatory behaviors. J Appl Physiol (1985) 66, 2539–2545. [DOI] [PubMed] [Google Scholar]
- Smith PE, Edwards RH & Calverley PM (1989). Ventilation and breathing pattern during sleep in Duchenne muscular dystrophy. Chest 96, 1346–1351. [DOI] [PubMed] [Google Scholar]
- Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettieri RA, Petrof B, Narusawa M, Leferovich JM, Sladky JT & Kelly AM (1991). The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 352, 536–539. [DOI] [PubMed] [Google Scholar]
- Suresh S, Wales P, Dakin C, Harris MA & Cooper DG (2005). Sleep‐related breathing disorder in Duchenne muscular dystrophy: disease spectrum in the paediatric population. J Paediatr Child Health 41, 500–503. [DOI] [PubMed] [Google Scholar]
- Vaillend C & Chaussenot R (2017). Relationships linking emotional, motor, cognitive and GABAergic dysfunctions in dystrophin‐deficient mdx mice. Hum Mol Genet 26, 1041–1055. [DOI] [PubMed] [Google Scholar]
- Yiu EM & Kornberg AJ (2008). Duchenne muscular dystrophy. Neurol India 56, 236–247. [DOI] [PubMed] [Google Scholar]