

Abstract
Key points
Lipin1 is critical for skeletal muscle development.
Lipin1 regulates MyoD and myocyte‐specific enhancer factor 2C (MEF2c) expression via the protein kinase C (PKC)/histone deacetylase 5‐mediated pathway.
Inhibition of PKCμ activity suppresses myoblast differentiation by inhibiting MyoD and MEF2c expression.
Abstract
Our previous characterization of global lipin1‐deficient (fld) mice demonstrated that lipin1 played a novel role in skeletal muscle (SM) regeneration. The present study using cell type‐specific Myf5‐cre;Lipin1fl/fl conditional knockout mice (Lipin1Myf5cKO) shows that lipin1 is a major determinant of SM development. Lipin1 deficiency induced reduced muscle mass and myopathy. Our results from lipin1‐deficient myoblasts suggested that lipin1 regulates myoblast differentiation via the protein kinase Cμ (PKCμ)/histone deacetylase 5 (HDAC5)/myocyte‐specific enhancer factor 2C (MEF2c):MyoD‐mediated pathway. Lipin1 deficiency leads to the suppression of PKC isoform activities, as well as inhibition of the downstream target of PKCμ, class II deacetylase HDAC5 nuclear export, and, consequently, inhibition of MEF2c and MyoD expression in the SM of lipin1Myf5cKO mice. Restoration of diacylglycerol‐mediated signalling in lipin1 deficient myoblasts by phorbol 12‐myristate 13‐acetate transiently activated PKC and HDAC5, and upregulated MEF2c expression. Our findings provide insights into the signalling circuitry that regulates SM development, and have important implications for developing intervention aimed at treating muscular dystrophy.
Keywords: Skeletal muscle development, myoblast differentiation, Myf5‐expressing progenitors
Key points
Lipin1 is critical for skeletal muscle development.
Lipin1 regulates MyoD and myocyte‐specific enhancer factor 2C (MEF2c) expression via the protein kinase C (PKC)/histone deacetylase 5‐mediated pathway.
Inhibition of PKCμ activity suppresses myoblast differentiation by inhibiting MyoD and MEF2c expression.
Introduction
Lipin1 catalyses the penultimate step in triglyceride synthesis and acts as a nuclear transcriptional coactivator (Finck et al. 2006; Han et al. 2006; Kim et al. 2013). Studies of lipin1 have revealed multiple roles: its lipid‐storing function in adipose tissue, its role in Schwann cell and macrophage biology and its role in hepatic lipid metabolism (Rehnmark et al. 1998; Finck et al. 2006; Nadra et al. 2008; Reue & Dwyer, 2009; Ren et al. 2010; Nadra et al. 2012; Hu et al. 2013; Kim et al. 2013; Mitra et al. 2013; Schweitzer et al. 2015; Wang et al. 2016; Vozenilek et al. 2018), as well as new roles in autophagy and mitophagy in other recent studies (Zhang et al. 2014; Alshudukhi et al. 2018; Schweitzer et al. 2018). There are three mammalian lipin genes, lipin1, lipin2 and lipin3, with distinct but partially overlapping expression pattern (Donkor et al. 2007), although lipin1 is the predominant lipin isoform in skeletal muscle (SM), accounting for almost all phosphatidic acid phosphatase activity in SM (Han et al. 2006; Reue & Dwyer, 2009). Clinical studies have identified a population carrying a homozygous or compound heterozygous LPIN1 gene mutation, which could be the result of a premature stop codon insertion, a large intragenic deletion, frameshift mutation and single amino acid replacements, comprising the major cause of rhabdomyolysis (Zeharia et al. 2008; Michot et al. 2010; Michot et al. 2012). Our earlier work in global lipin1‐deficient (fld) mice demonstrated that lipin1 plays a major role in SM regeneration (Jiang et al. 2015). In particular, fld mice exhibited smaller regenerated muscle fibre cross‐sectional areas in response to injury, accompanied by reduced MyoD expression compared to wild‐type (WT) mice. A key challenge in these studies, however, is that lipin1‐deficient fld mice have systemic metabolic abnormalities and develop severe neuropathy and locomotor defects that complicate or preclude a detailed analysis of SM function (Rehnmark et al. 1998). Thus, for the present study, we generated Lipin1Myf5cKO mice to specifically deplete lipin1 expression in Myf5‐expressing progenitors to further investigate the role of lipin1 in myogenic determination and differentiation.
SM development involves a highly ordered cascade of events comprising myogenic lineage commitment, myoblast proliferation and terminal differentiation (Buckingham, 2001; Bentzinger et al. 2012). These steps are tightly controlled by transcription factor families, including the myogenic basic helix‐loop‐helix (bHLH) proteins and the myocyte enhancer factor 2 (MEF2). Members of the myogenic bHLH proteins, MyoD and Myf5, are required for myoblast specification and differentiation, whereas myogenin and MRF4 play key roles in myoblast fusion (Hernandez‐Hernandez et al. 2017). Myoblast differentiation requires the co‐operative activation of members of the MyoD families and MEF2 (Dodou et al. 2003), which need to be dissociated from class II HDACs to release the repression function (McKinsey et al. 2000; Lu et al. 2000b); however, this regulatory mechanism is still not fully understood.
In the present study, we employed our newly generated Lipin1Myf5cKO mice to investigate the role of lipin1 in myogenic differentiation. The results obtained suggest that lipin1 deficiency results in reduced SM mass. Lipin1 is a major determinant of skeletal myogenesis by the promotion of MEF2c and MyoD‐regulated muscle differentiation. We evaluated the expression of the myogenic transcription factors and proteins involved in SM differentiation, and identified a lipin1‐associated regulatory pathway that was further confirmed in lipin1‐depleted myoblasts. Our experiments uncover the key role of lipin1 in a regulatory circuit that facilitates gene programme transition from proliferation to differentiation.
Methods
Animals
Lipin1Myf5cKO mice were generated by crossing Lipin1fl/fl mice (Nadra et al. 2008) with Myf5‐Cre mice (Stock No: 007893; Jackson Laboratories, Bar Harbor, ME, USA). Both Lipin1fl/fl and Myf5‐Cre mice were in a C57/B6 background. Experiments were performed on 2–4‐months‐old mice. These mice had free access to drinking water and regular chow, unless otherwise noted. All animal experiments were performed in accordance with the relevant guidelines and regulations approved by the Animal Care and Use Committee of Wright State University (#1058).
Cell culture and differentiation of C2C12 myoblasts
Mouse C2C12 myoblasts were cultured in high‐glucose Dulbecco's modified Eagle's medium (DMEM) (#10569‐010; Gibco, Gaithersburg, MD, USA) supplemented with 10% (v/v) fetal bovine serum (#16000‐044; Gibco) and 1% penicillin–streptomycin (#15140‐122; Gibco) under humidified air containing 5% CO2 at 37°C. Myotube formation was induced at confluence by replacing growth medium with differentiation medium consisting of DMEM supplemented with 2% horse medium (#16050‐122; Gibco) and 1% penicillin–streptomycin.
Adenoviruses‐mediated short hairpin RNA (shRNA) knockdown
Adenoviruses expressing shRNA to knockdown lipin1 (shLpin1) and control shLacZ were generous gifts from Dr Thurl Harris (University of Virginia, Charlottesville, VA, USA). For knockdown of lipin1 expression, C2C12 myoblasts were infected with shLpin1 and shLacZ control in growth medium containing 8 g mL−1 polybrene (#107689; Sigma, St Louis, MO, USA) for 24 h. The medium was then replaced with DMEM supplemented with 10% fetal bovine serum. The myoblasts were incubated for an additional 24 h until they were confluent. Differentiation was then initiated by switching the medium to DMEM differentiation medium.
Immunofluorescence, microscopy and image processing
Cells were fixed with 4% formaldehyde and permeabilized in PBS containing 0.1% Triton X‐100 for 20 min followed by blocking in PBS containing 1% BSA (#BP9704; Thermo Fisher Scientific, Waltham, MA, USA) for 1 h at room temperature. The cells were then incubated with the Mf20 monoclonal antibody (#AB_2147781; dilution 1:10; Developmental Studies Hybridoma Bank, Iowa City, IA, USA) to detect MHC‐expressing cells overnight at 4°C and subsequently with an Alexa Fluor 488‐ or Alexa Fluor 555‐conjugated secondary antibody (#A‐21411; dilution 1:500, Thermo Fisher Scientific) for 1 h at room temperature. Nuclei of cells were detected by staining with 4′,6‐diamidino‐2‐phenylindole (DAPI) (#D1306; 2.5 μg mL−1; Thermo Fisher Scientific) for 10 min. Images were obtained using an inverted microscope (IX70; Olympus, Tokyo, Japan) equipped with a DFC7000T camera (Leica Microsystems, Wetzlar, Germany). The fusion index was calculated as the ratio of nuclei in MyHC‐positive myotubes to the total number of nuclei in the field for five random fields.
Cos7 cells were seeded onto 13 mm round glass coverslips. Cells were transfected with HDAC5‐GFP (#32211; Addgene, Cambridge, MA, USA) with/without lipin1‐HA, lipin1 D712A‐HA mutant or PKCu‐HA (#10808; Addgene) plasmids. After 24 h, cells were fixed in 4% paraformaldehyde. The cells were then permeabilized with 0.1% Triton X‐100 (#28314; Thermo Fisher Scientific) made with PBS for 10 min at room temperature. After washes, the cells were blocked with 1% BSA, incubated with rabbit anti‐HA primary antibody (#Ab9110; dilution 1:1000; Abcam, Cambridge, MA, USA) using 1% BSA blocking reagent overnight at 4˚C. The next day, the cells were incubated with secondary antibody, goat anti‐rabbit 555 (#A21428; dilution 1:1000; Thermo Fisher Scientific) with 1% BSA blocking reagent. Nuclei were then labeled with DAPI. Co‐localization of HDAC5 and lipin1, lipin1D712A or PKCu was examined.
Quantitative real‐time PCR
Total RNA from C2C12 myoblasts and muscle tissues was extracted using TRIzol reagent (#15596018; Thermo Fisher Scientific). The concentration and quality of RNA were assessed with a NanoDrop 2000 (Thermo Fisher Scientific). One microgram of total RNA was used for reverse transcription with the High‐Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). The quantitative real‐time PCR reaction was performed in a QuantStudio Real‐Time PCR System (Thermo Fisher Scientific) using SYBR Green Real‐time PCR Master Mix (#A25742; Thermo Fisher Scientific). The sequence of primers is provided in Table 1. The C t (2−ΔΔCt) method was used to analyse the relative gene expression data.
Table 1.
Primer sequence used for RT‐PCR
| Primer | Forward 5'‐ to 3' | Reverse 5'‐ to 3' |
|---|---|---|
| Lipin1 | CCTTCTATGCTGCTTTTGGGAACC | GTGATCGACCACTTCGCAGAGC |
| Myf‐5 | TGTATCCCCTCACCAGAGGAT | GGCTGTAATAGTTCTCCACCTGTT |
| MyoD | GCAGGCTCTGCTGCGCGACC | TGCAGTCGATCTCTCAAAGCACC |
| Mef2c | TCTGTCTGGCTTCAACACTG | TGGTGGTACGGTCTCTAGGA |
| Gapdh | CCAATGTGTCCGTCGTGGATCT | GTTGAAGTCGCAGGAGACAACC |
Western blotting
Cells and muscle tissues were lysed in RIPA buffer containing 10 mm Tris‐HCL (pH 7.4), 30 mm NaCl, 1 mm EDTA and 1% Nonidet P‐40, supplemented with proteinase inhibitors and phosphatase inhibitors before use. Protein concentration was determined for each sample and equal amounts of proteins were used, boiled at 95°C for 5 min in 1 × SDS sample buffer and separated by 7.5–15% SDS‐PAGE. Thereafter, proteins were transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA) using a Mini Trans‐Blot Cell System (Bio‐Rad, Hercules, CA, USA). The membrane was blocked with 1% casein buffer for 1 h, and incubated with the primary antibodies overnight at 4°C. After probing with secondary antibodies for 1h at 25°C, protein bands were detected by using Amersham Imager 600 (GE Healthcare Life Sciences, Little Chalfont, UK). β‐actin (#A1978; dilution 1:5000; Sigma) or GAPDH (Ab181602; dilution 1:20000; Abcam) antibodies were used as a loading control. The densitometry values were normalized by the corresponding loading control densitometry values obtained from the same sample. Antibodies used include lipin1 (#14906; dilution 1:1000; Cell Signaling Technology, Beverly, MA, USA), phospho‐PKCμ (#2054; dilution 1:1000; Cell Signaling Technology), MyoD (#Ab16148; dilution 1:1000; Abcam), MEF2c (#5030; dilution 1:1000; Cell Signaling Technology), phospho‐PKCα (#sc‐377565 dilution 1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), phospho‐PKCθ isoforms (#sc‐271922; dilution 1:1000; Santa Cruz Biotechnology) and Pax7 (#AB_528428; dilution 1:1000; Developmental Studies Hybridoma Bank) and (#Ab34360; dilution 1:1000; Abcam), along with goat anti‐mouse IgG‐HRP (#w402B; dilution 1:5000; Promega, Madison, WI, USA) and goat anti‐rabbit IgG‐HRP (#w401B; dilution 1:5000; Promega).
Statistical analysis
Unless noted otherwise, data are provided as the mean ± SD number (n) of independent experiments. Statistical significance was calculated using a two‐tailed Student's t test. P < 0.05 was considered statistically significant.
Results
Depletion of lipin1 in Myf5‐expressing precursors causes suppression of SM development
To investigate the role of lipin1 in SM development, we generated Lipin1Myf5cKO mice to specifically deplete lipin1 expression in Myf5‐expressing progenitors. Myf5 is the first muscle regulatory factor (i.e. Myf5, MyoD, myogenin and Mrf4) expressed during embryonic development (Ott et al. 1991; Buckingham, 1992; Bentzinger et al. 2012). Loss of lipin1 in Myf5‐expressing progenitors did not induce neurological phenotypes such as hind‐limb clasping reflexes, tremors or an unsteady gait, as observed in global lipin1‐deficient fld mice shortly after 10 days of age. However, compared to control mice, selectively deleting lipin1 in Myf5‐expressing precursors caused the inhibition of SM development, characterized by a 24% reduction in muscle mass of both TA and GAS muscle in Lipin1Myf5cKO mice (Fig. 1 A–C). Haematoxylin and eosin staining exhibited a reduction in mean fibre cross‐sectional area in both TA and GAS muscle of Lipin1Myf5cKO mice compared to WT mice (Fig. 1 D–E). Consistent with recent reports in skeletal muscle‐specific lipin1 deficient (MCK promoter) (Schweitzer et al. 2018) and fld (Zhang et al. 2014) mice, SM of Lipin1Myf5cKO mice also displayed a large number of centrally nucleated fibres compared to control mice, suggesting that lipin1 deficiency results in myopathy. The centrally positioned nuclei are associated with excessive degeneration/regeneration processes similar to those reported in patients with lipin1 deficiency (Zeharia et al. 2008; Michot et al. 2012). However, we did not observe any obvious body weight difference in these knockout mice. These mice also have altered fat deposition and metabolism (Fig. 1 A) and increased intramuscular lipid accumulation (data not shown). We are currently exploring the impact of lipin1 deficiency on fat‐associated phenotypes in a separate project.
Figure 1. Lipin1 deficiency in Myf5‐expressing cells results in reduced SM mass.
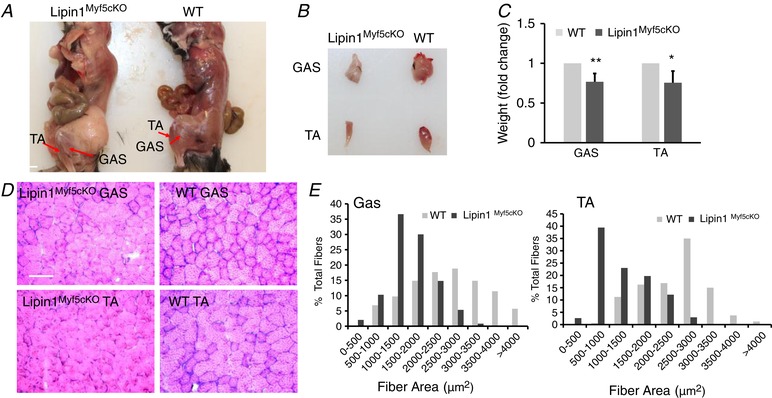
Appearance of TA and GAS muscles in WT and Lipin1Myf5cKO mice at 3 months of age (TA and GAS are indicated by arrows) (A). Isolated TA and GAS muscle (B) and the muscle weight (C) in WT and Lipin1Myf5cKO mice. (WT vs. Lipin1Myf5cKO, * P < 0.05; ** P < 0.01 Student's t test). Histology examination (D) and quantification of reduced myofibre area (E) of GAS and TA muscle from WT and Lipin1Myf5cKO mice (scale bar = 100 μm).
We next examined the expression levels of lipin1 in the TA and GAS muscles of Lipin1Myf5cKO mice. As shown in Fig. 2 A, lipin1 was expressed in the control WT mice but depleted in Lipin1Myf5cKO mice. We also measured the protein expression levels of Myf5, MyoD and MEF2c in GAS and TA muscles of Lipin1Myf5cKO and control mice. Although lipin1 was depleted in Myf5‐expression progenitor cells, the expression level of Myf5 was not affected. Interestingly, the expression levels of MyoD and MEF2c were reduced by ∼48% and 60%, respectively, in GAS and TA muscle of Lipin1Myf5cKO mice compared to the control group (Fig. 2 B). We also measured mRNA expression levels, which were consistently decreased, whereas the mRNA expression levels of Myf5 were not (Fig. 2 C).
Figure 2. Lipin1 depletion in Myf5‐expressing progenitor cells inhibits MEF2 and MyoD expression but does not alter Myf5 expression.
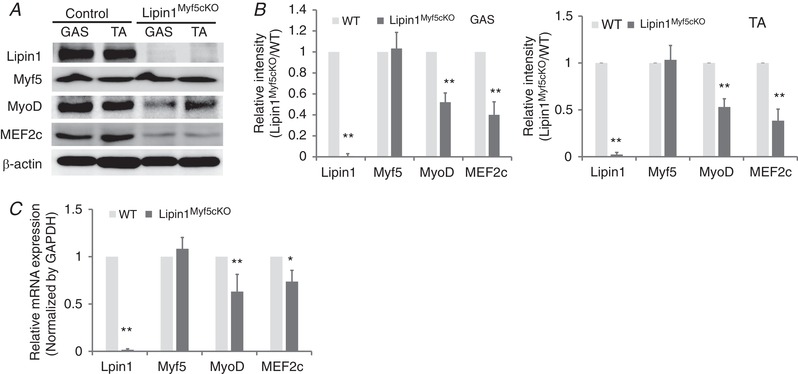
Western blotting (A), densitometry graphs (B) and mRNA expression (C) of lipin1, myogenic regulatory factors (Myf5 and MyoD) and of myogenic enhancer factor (MEF2c) in GAS and TA muscles of 3‐month‐old WT and Lipin1Myf5cKO mice. Unless otherwise indicated, data are from one representative (n = 3) of at least three independent experiments (* P < 0.05; ** P < 0.01 Student's t test).
Lipin1 deficiency diminishes PKC activation
Lipin1 catalyses diacylglycerol (DAG) biosynthesis (Han et al. 2006). Overexpression of lipin1 increases cellular DAG, whereas a deficiency contributes to decreased DAG (Ren et al. 2010). DAG can recruit and activate a variety of downstream effector molecules by phosphorylation, including PKCμ (renamed as protein kinase D; PKD) and other PKC isoforms, including classical (PKCα, PKCβI, PKCβII and PKCγ) and novel (PKCδ, PKCε, PKCη and PKCθ) isoforms (Brose et al. 2004; Toker, 2005; Gallegos & Newton, 2008; Schmitz‐Peiffer & Biden, 2008; Newton, 2009; Leonard & Hurley, 2011). PKC activation has been suggested to be involved in MyoD expression, although the detailed mechanisms of its action are unknown (Brunelli et al. 2007). To investigate whether any particular isoform was specifically implicated in the signalling mechanisms by which lipin1 deficiency induces its myogenesis defects, we assessed the activation of conventional PKCα and novel θ isoforms, as well as PKCμ and PKA, in the GAS and TA muscle of WT vs. Lipin1Myf5cKO mice. The activation of PKC isoforms was measured by performing western blotting using antibodies directed against the phosphorylated proteins. As shown in Fig. 3, both phosphorylated PKCμ at Ser744/748 decreased by more than 70%, and PKCα at Ser657 reduced by 63%, in GAS and TA muscle, suggesting an inhibition of PKCμ and PKCα activation in the SM of Lipin1Myf5cKO mice. Phosphorylated novel PKCθ isoform was also reduced by 40% in the TA muscle of Lipin1Myf5cKO mice compared to WT mice. By contrast, total levels of PKA and PKC did not exhibit significant differences.
Figure 3. Lipin1 deficiency diminishes PKC activation.
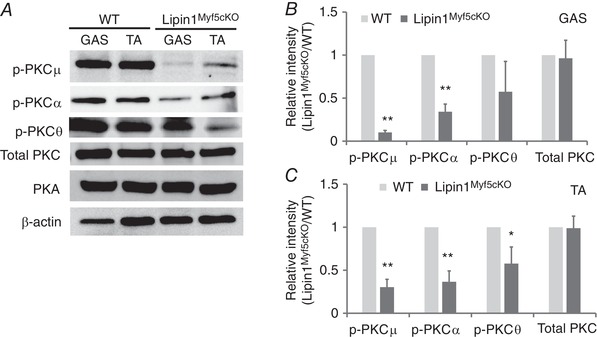
Representative immunoblots (A) and densitometry graphs of lipin1 and different protein kinase expression and phosphorylation in GAS (B) and TA (C) muscles in WT and Lipin1Myf5cKO mice at 3 months. Unless otherwise indicated, data are from one representative (n = 3) of at least three independent experiments. (* P < 0.05; ** P < 0.01 Student's t test).
Lipin1 is required for myoblast differentiation in cell culture
To clarify the role of lipin1 in the myoblast differentiation process, we knocked down lipin1 expression in C2C12 cells using adenovirus‐driven shRNA against the Lpin1 gene and evaluated the effect of lipin1 deficiency on myoblast differentiation at days 0, 4 and 6 after differentiation treatment. At day 6 post‐differentiation treatment, we fixed the differentiated cells and examined myotube formation using Mf20 immunostaining to detect myosin heavy chain‐positive myotubes. We found that lipin1 deficiency in C2C12 myoblasts induced a significantly reduced myoblast differentiation and myotube formation compared to shLacZ‐treated cells (Fig. 4 A). Consistently, the lipin1 shRNA‐treated myotubes had a reduced fusion index, which is calculated as the percentage of nuclei contained in myosin‐positive myotubes. This index was reduced from 49% to 30% in shRNA‐treated myotubes compared to the control (Fig. 4 B). This is consistent with a previous study in which we revealed that lipin1 is required for myoblast differentiation using lipin1 depleted myoblasts and primary myoblasts (Jiang et al. 2015).
Figure 4. Lipin1 is required for myoblast differentiation in cell culture.
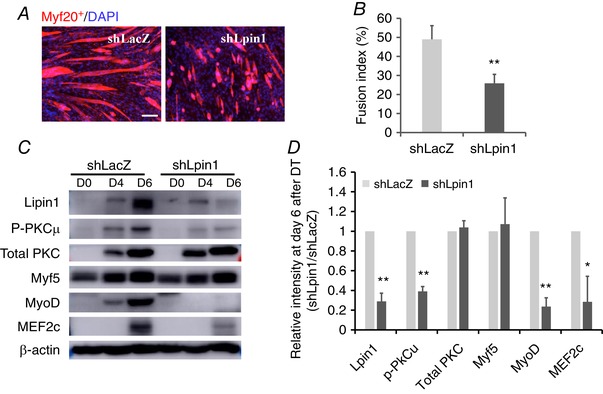
C2C12 myoblasts were infected with adenovirus to knockdown lipin1 (shLpin1) or control shLacZ before differentiation. A, cells were fixed and stained for Mf20+ and DAPI (scale bars = 50 μm). B, fusion index was determined by counting the percentage of nuclei within Mf20+ myotubes over total nuclei within five randomly selected fields per sample. Protein expression levels (C) and densitometric analysis (D) of PKC, myogenic differentiation markers and MEF2c in differentiated myoblasts. * P < 0.05; ** P < 0.01.
We also examined the expression levels of myogenic regulatory proteins after differentiation treatment (Fig. 4 C–D). The expression level of Myf5 was not altered, although MyoD and MEF2c were dramatically reduced at day 6 after differentiation treatment. PKCμ activation indicated by phosphorylated PKCμ levels was reduced by 60% at day 6. In addition, the expression levels of the myogenic transcriptional factor, MyoD, were also decreased by 75% at day 6 after differentiation treatment. These results are consistent with our findings obtained in vivo and confirm that lipin1 is essential for effective differentiation during the early‐stage myogenesis.
Inhibition of PKCμ activity suppresses myoblast differentiation by inhibiting MyoD and MEF2c expression in vitro
Next, we determined whether PKCμ activity is required for MyoD and MEF2 expression during myogenesis. C2C12 cells were differentiated for 6 days by fetal bovine serum withdrawal in the presence of CID755673 to selectively inhibit PKCμ activity. As shown in Fig. 5 A, treatment with CID755673 reduced the degree of C2C12 differentiation, as indicated by visualizing MHC‐positive myotubes detected by Mf20 immunostaining (Fig. 5 A). The fusion index, which is calculated by the percentage of nuclei inside the myotube, was decreased from 50% to 31% after CID755673 treatment (Fig. 5 B). To assess whether myogenesis was regulated by the activation of PKC, myoblasts were shifted from a growth medium to a differentiation medium (DMEM containing 2% horse serum) and differentiation was examined at days 0, 4 and 6. The phosphorylation of PKCu, MyoD and MEF2c expression in differentiated myoblasts was reduced by 70%, 75% and 63%, respectively, at day 6 after differentiation treatment (Fig. 5 C–D).
Figure 5. Inhibition of PKCμ activity suppresses myoblast differentiation by inhibiting MyoD and MEF2c expression in vitro .
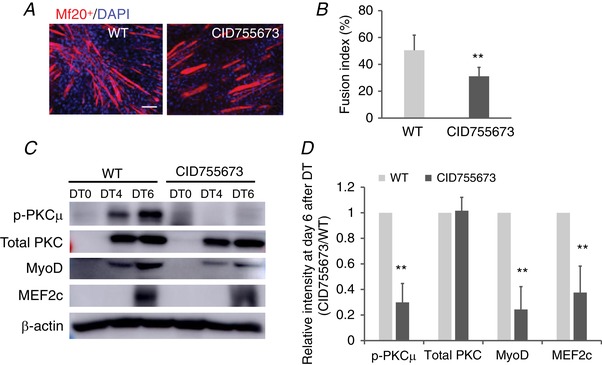
C2C12 myoblasts were treated with PKCμ inhibitor, CID 755673, for 6 days, and subjected to myoblast differentiation. Six days after differentiation, cells were fixed and stained with Mf20 and DAPI (scale bars = 50 μm) (A) and the fusion index was calculated (B). C, cell lysate was harvested, and protein expression was measured by western blot. D, protein expression levels in (C) were quantified by densitometric analysis and normalized to β‐actin. ** P < 0.01.
Lipin1 deficiency decreases MEF2 and MyoD by inhibiting the nuclear export of HDAC5
MyoD and MEF2c have been reported to interact with each other via their DNA‐binding and dimerization motifs to synergistically activate transcription and myogenic programing (Kaushal et al. 1994; Molkentin et al. 1995; Molkentin et al. 1996; Black et al. 1998). The MEF2c protein also interacts with the class II deacetylase, HDAC5, resulting in the repression of MEF2c‐dependent genes (Lu et al. 2000a). The release of MEF2c from repression by HDAC5 is controlled by the nuclear export of HDAC5, which can be regulated by CaMK (Ginnan et al. 2012), protein kinase A (Ha et al. 2010) and salt inducible kinase (Takemori et al. 2009). Because MyoD–MEF2c interaction and activation require the nuclear export of Class II HDAC to release the suppression function, we also examined the protein expression of HDAC5. As shown in Fig. 6 A, we did not observe any obvious differences in the expression levels of total HDAC5, although the phosphorylated HDAC5 was substantially suppressed in the SM of Lipin1Myf5cKO mice. To further confirm our observation in tissue samples, we measured HDAC5 and phosphorylated HDAC5 expression levels in CID755673 treated differentiated C2C12 as shown in Fig. 5. As a result of a low transfection efficiency in myoblasts, we used Cos7 cells for this experiment. We consider that the overexpression of the indicated plasmids in C2C12 myoblasts should have a similar effect in Cos7 cells. PKCμ suppression consistently did not alter protein expression levels of the total HDAC5, although it inhibited its phosphorylated form, suggesting that PKCμ activation is required during myogenesis to translocate HDAC5 from the nucleus to release its transcriptional repressor function.
Figure 6. Lipin1 regulates HDAC5 nuclear export by activation of PKCμ.
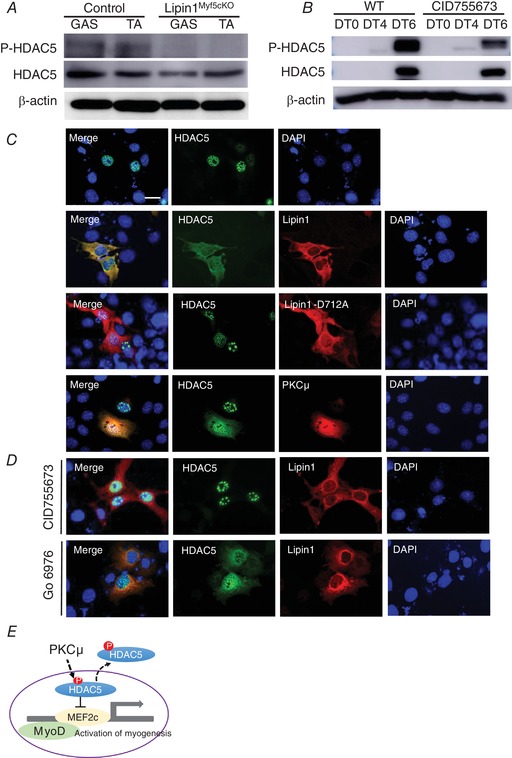
A, western blot of HDAC5 and phosphorylated HDAC5 (Ser259) in the GAS and TA muscle of WT and Lipin1Myf5cKO mice. B, C2C12 myoblasts were treated with CID 755673 for 6 days and subjected to myoblast differentiation. Six days after differentiation, cell lysate was harvested, the protein expression levels of HDAC5 and phosphorylated HDAC5 were measured by western blotting. C, Cos7 cells were transfected with either HDAC5‐GFP or along with lipin1‐HA, catalytic mutant lipin1‐D712A‐HA or PKCμ‐HA. Cells were fixed 24 h after transfection, and subcellular localization of lipin1 and HDAC5 were examined by anti‐HA (red), anti‐GFP (green) antibodies. Nuclei were detected by DAPI (blue). Co‐localization of HDAC5 and lipin1 or PKCμ was determined. Scale bars = 50 μm. D, cells co‐transfected with HDAC5‐GFP and lipin1‐HA were treated with either selective PKCμ inhibitor, CID755673 or a pan‐PKC inhibitor against for PKCα, PKCβ and PKCγ isoforms, Go 6976. After treatment for 6 h, the subcellular localization of lipin1 and HDAC5 was evaluated by immunostaining using anti‐HA and anti‐GFP antibodies. E, a model for regulating HDAC5 nuclear export.
We next examined the subcellular localization of HDAC5 and found that overexpressed HDAC5 was almost exclusively localized in the nucleus (Fig. 6 C). Interestingly, co‐overexpression of lipin1 and HDAC5 induced HDAC5 nuclear export. HDAC5 nuclear export was dependent on lipin1 catalytic activity because the catalytic inactive mutant, D712A, lost its capability to induce HDAC5 nuclear translocation. Co‐transfection of PKCμ with HDAC5 also induced HDAC5 nuclear export. Selective PKCμ inhibitor, CID755673, inhibited HDAC5 nuclear export in the presence of lipin1 (Fig 6 D). Interestingly, inhibition of Ca2+‐dependent isoforms of PKC (PKCα, PKCβ and PKCγ) by Go 6976 (Guh et al. 1998) failed to inhibit HDAC5 nuclear export in the presence of lipin1, suggesting that lipin1 regulates HDAC5 nuclear export via the activation of PKCμ. These results suggest that PKCμ‐mediated phosphorylation of class IIa HDACs is required to release the suppression function of MEF2 transcription to activate genes that govern muscle differentiation and growth (Fig 6 E). Lipin1 deficiency failed to induce HDAC5 nuclear export that releases the inhibition function of HDAC5 to MEF2c via PKCμ activity.
Restoring DAG‐mediated signalling response by phorbol 12‐myristate 13‐acetate (PMA) treatment transiently activated PKCμ/HDAC5 and upregulated MEF2c expression
PMA, an analogue of DAG, activates PKC by mimicking the activating ligand DAG (Way et al. 2000). To assess whether restoration of DAG‐mediated signalling could alter HDAC5 subcellular localization, we treated cells transfected with HDAC5 with 10 μm PMA for 30 min, and then evaluated the HDAC5 subcellular localization (Fig. 7 A). We found that PMA treatment drives HDAC5 subcellular translocation from the nucleus to cytoplasm.
Figure 7. PMA treatment activates PKC/HDAC5 and promotes the upregulation of MEF2c.
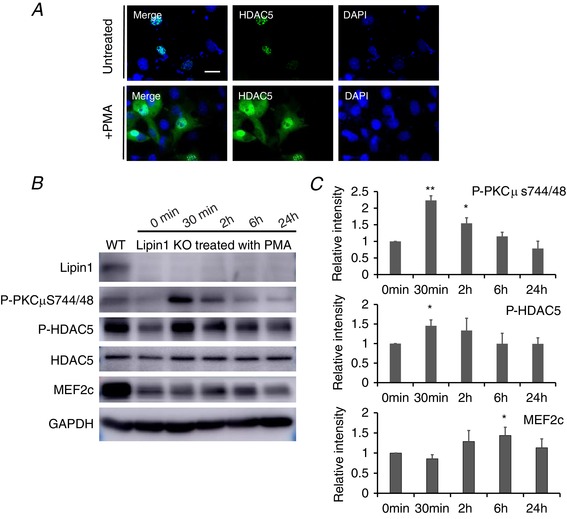
A, Cos7 cells transfected with HDAC5 treated with/without PKC agonist, PMA, for 30 min. Subcellular localization of HDAC5 was examined by immunostaining (scale bars = 50 μm). B, lipin1 deficient C2C12 myoblasts were subjected to myogenic differentiation for 6 days followed by PMA treatment for 0 min, 30 min, 2 h, 6 h and 24 h. The activation of PKCμ, HDAC5 and the expression of MEF2c were measured by western blotting at differentiate time points followed by PMA treatment. C, densitometry analysis of western blots. Data are shown as the fold change in the absolute fluorescence intensity at various time points after PMA treatment compared to zero time point (0 min). Data were from three independent experiments. * P < 0.05.
To evaluate whether restoring DAG‐mediated PKC activation could rescue myoblast differentiation defect induced by lipin1‐deficiency, lipin1 deficient myoblasts were subjected to myogenic differentiation for 6 days. Cells were then treated with 10 μm of PMA for 0 min, 30 min, 2 h, 6 h and 24 h. After treatment, the activation of PKCμ and HDAC5 and the expression level of MEF2c were assessed (Fig. 7 B and C). We found that PKCμ and HDAC5 were activated after 30 min of PMA treatment. However, the activation of PKCμ and HDAC5 was terminated immediately after 30 min of treatment, as indicated by downregulation of phospho‐PKCμ and phosphor‐HDAC5 and MEF2c, and it returned to baseline levels 24 h later. Moreover, the expression of MEF2c was upregulated at 2 and 6 h of PMA treatment, most probably as a result of translational delay, returning to basal levels by 24 h.
Discussion
The present study suggests that lipin1 plays a critical role in SM development (Fig. 8). Lipin1 deficiency resulted in reduced SM mass by suppressing the activity of PKCμ, which is required for regulating HDAC5 nuclear‐cytoplasmic shuttling. HDAC5 phosphorylation‐induced cytoplasm translocation may release the suppression function of MEF2c and activate MEF2c and MyoD expression, resulting in myoblast differentiation in vitro.
Figure 8. Lipin1 in SM differentiation.
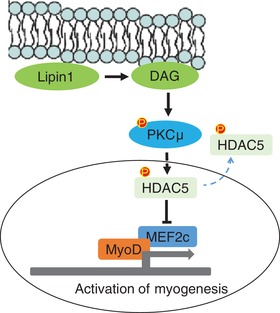
Lipin1 is suggested to play a critical role in SM development
In the present study, we found that the absence of lipin1 resulted in inhibited myoblast differentiation in vitro. The observed reduction was associated with the inhibition of phosphorylation/activation of PKCμ. PKCμ selective inhibitor treatment suppressed MyoD and MEF2c expression in vitro (Fig. 5) and inhibited lipin1‐ mediated HDAC5 nucleo‐cytoplasmic translocation (Fig. 6), suggesting that lipin1 regulates myogenesis mainly by activation of PKCμ. Our results are consistent with a previous study showing that PKCμ (PKD2) depletion by shRNAs inhibits C2C12 myoblast differentiation in vitro, and the ectopic expression of PKCμ increases myogenic differentiation (Kleger et al. 2011). We cannot rule out that other PKC isoforms, including PKCα and θ, may also play an important role in myogenesis. Capiati et al. (1999) reported that PKCα promote chick myoblast differentiation. Additional studies using isoform specific inhibitors or their specific shRNA are needed to comprehensively evaluate the role of PKC in myogenesis.
MyoD is a master regulator of skeletal myogenesis. Overexpression of MyoD in mouse embryonic fibroblasts converts cells into SM (Yao et al. 2013) and MyoD deficiency suppresses myoblast differentiation potential (Sabourin et al. 1999). The activation of MyoD is dependent on its association with members of the MEF2 family of transcription factors. SM‐specific deletion of MEF2c isoform in mice results in disorganized myofibres and perinatal lethality demonstrating a critical role of MEF2 proteins in SM development (Potthoff et al. 2007). By contrast, neither MEF2a, nor MEF2d isoform is required for normal SM development in vivo. The combinatorial activity of MyoD and MEF2c transcription factors promotes the expression of myogenic genes during the transition from undifferentiated myoblasts to differentiated myotubes (Lu et al. 2000b). Our results suggest that a lipin1 deficiency induces decreased MEF2c and MyoD expression, and also that their respective expression impacts upon each other. Although previous work suggests that MyoD and MEF2c interact during skeletal myogenesis throughout the DNA binding domains (Tapscott, 2005; Potthoff & Olson, 2007), further research is needed to understand how they participate in a regulatory loop by inducing and maintaining the expression of each other during skeletal myogenesis.
We found that lipin1 regulates MEF2c expression by the manipulation of HDAC5 nuclear export. Lipin1 regulates phosphorylated‐associated HDAC5 nucleocytoplasmic trafficking depending on its catalytic activity because the lipin1 inactive mutant D712A lost its capacity to mediate HDAC5 nuclear export. In the present study, treatment with the PKC agonist, PMA, also promoted HDAC5 nuclear export. Treatment with specific PKCμ inhibitor, CID755673, blocked HDAC5 nuclear export in the presence of lipin1, whereas treatment with a pan‐PKC inhibitor against for PKCα, PKCβ and PKCγ isoforms failed to block HDAC5 nuclear export induced by lipin1. Based on our findings, we propose that lipin1 regulates HDAC5 nucleocytoplasmic trafficking via DAG/PKCμ. In the nucleus, HDAC5 binds to MEF2c via the HDAC‐interaction domain and represses its activity (Lemercier et al. 2000). During myogenesis, MEF2c repression is relieved in response to phosphorylation‐induced translocation of HDAC5 to the cytoplasm. Therefore, lipin1 deficiency increases nuclear‐localized HDAC5 and represses MEF2 expression. HDAC7 has also been reported to affect MEF2c transcriptional function (Dressel et al. 2001). Further studies will be conducted to examine the role of lipin1 on HDAC7 expression. A previous study also suggested that lipin1 could directly interact with MEF2c (Liu & Gerace, 2009). It is also possible that, in addition to activating MEF2c via the DAG/HDAC5 mediated pathway, lipin1 may also be able to activate MEF2c via its transcriptional regulation. The nuclear transcription regulation of MEF2c by lipin1 requires further investigation.
We also explored whether restoration of DAG by treating cells with PMA could rescue the myoblast differentiation defect in lipin1 deficient cells. We found that PMA treatment can activate PKCμ and HDAC5, in turn upregulating MEF2c expression. However, PKCμ and HDAC5 activation by PMA was rapid and transient, showing a robust activation at 30 min and returning to baseline by 24 h. PKCμ activation requires membrane recruitment by DAG and other PKC isoforms for its catalytic activity. PKCμ has to be phosphorylated by novel PKC isoforms within its activation loop at Ser744 and Ser748 to promote its activity (Brandlin et al. 2002; Yuan et al. 2002). The phosphorylated and activated PKCμ dissociates from the plasma membrane, translocates to the cytosol, and subsequently enters into the nucleus to activate HDAC5. The activation of PKCμ has been suggested to be sustained for a few hours once it leaves the membrane and is dependent on its phosphorylation status (Matthews et al. 2000). An ubiquitin E3 ligase, RINCK, has been reported to be able to bind the C1 domain of PKC and lead to the ubiquitination and degradation of PKCs (Chen et al. 2007). However, the detailed mechanisms that govern the termination of the cellular action of PKCμ, as well as how PKCμ is regulated spatially and temporally in biological systems, are largely unknown and remain to be determined.
In summary, we identified an essential role of lipin1 in regulating SM development via the activation of PKC, which regulates MyoD and MEF2c expression by manipulating HDAC5 nuclear export. Because patients with a lipin1 deficiency are associated with severe rhabdomyolysis and muscle atrophy in type 2 muscle fibres, the regulatory circuitry identified in the present study provides an insight into how the dysfunction of lipin1 causes muscle rhabdomyolysis.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
HR designed the experiments. RC provided Lipin1fl/fl mice. AJ, DH and AA performed the research. AJ, DH, AA and HR analysed the data. HR wrote the manuscript. All authors read and approved the final version of the manuscript submitted for publication.
Funding
This project was supported by startup funds from Wright State University to HR, the NIH Center of Biomedical Research Excellence on Obesity and Cardiovascular Diseases (P20 GM103527‐06), and a Beginning Grant‐in‐Aid (11BGIA7710059), as well as a Scientist Development Grant (12SDG12050697) from the American Heart Association to HR.
Acknowledgements
We sincerely thank Dr Thurl Harris (University of Virginia) for the generous gift of lipin1 adenoviruses (shLpin1) and control shLacZ. C2C12 myoblasts were generously provided by Dr Marcia Abbott at Chapman University (Orange, CA, USA). We thank Drs Reuben Shaw (Salk Institute for Biological Studies, La Jolla, CA, USA) and Alex Toker (Harvard Medical School, Boston, MA, USA) for their kind provision of plasmids HDAC5‐GFP and PKCu‐HA.
Biography
Abdulrahman Jama is a first‐year PhD student in the Department of Biochemistry & Molecular Biology at Wright State University. He has been working on identifying the molecular mechanisms underlying myopathy induced by lipin1 deficiency.

Edited by: Michael Hogan and Troy Hornberger
References
- Alshudukhi AA, Zhu J, Huang D, Jama A, Smith JD, Wang QJ, Esser KA & Ren H (2018). Lipin‐1 regulates Bnip3‐mediated mitophagy in glycolytic muscle. FASEB J, fj201800374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzinger CF, Wang YX & Rudnicki MA (2012). Building muscle: molecular regulation of myogenesis. Cold Spring Harb Perspect Biol 4, a008342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BL, Molkentin JD & Olson EN (1998). Multiple roles for the MyoD basic region in transmission of transcriptional activation signals and interaction with MEF2. Mol Cell Biol 18, 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandlin I, Hubner S, Eiseler T, Martinez‐Moya M, Horschinek A, Hausser A, Link G, Rupp S, Storz P, Pfizenmaier K & Johannes FJ (2002). Protein kinase C (PKC)eta‐mediated PKC mu activation modulates ERK and JNK signal pathways. J Biol Chem 277, 6490–6496. [DOI] [PubMed] [Google Scholar]
- Brose N, Betz A & Wegmeyer H (2004). Divergent and convergent signaling by the diacylglycerol second messenger pathway in mammals. Curr Opin Neurobiol 14, 328–340. [DOI] [PubMed] [Google Scholar]
- Brunelli S, Relaix F, Baesso S, Buckingham M & Cossu G (2007). Beta catenin‐independent activation of MyoD in presomitic mesoderm requires PKC and depends on Pax3 transcriptional activity. Dev Biol 304, 604–614. [DOI] [PubMed] [Google Scholar]
- Buckingham M (1992). Making muscle in mammals. Trends Genet 8, 144–148. [DOI] [PubMed] [Google Scholar]
- Buckingham M (2001). Skeletal muscle formation in vertebrates. Curr Opin Genet Dev 11, 440–448. [DOI] [PubMed] [Google Scholar]
- Capiati DA, Téllez‐Iñón MT & Boland RL (1999). Participation of protein kinase C alpha in 1,25‐dihydroxy‐vitamin D3 regulation of chick myoblastproliferation and differentiation. Mol Cell Endocrinol 153, 39–45. [DOI] [PubMed] [Google Scholar]
- Chen D, Gould C, Garza R, Gao T, Hampton RY & Newton AC (2007). Amplitude control of protein kinase C by RINCK, a novel E3 ubiquitin ligase. J Biol Chem 282, 33776–33787. [DOI] [PubMed] [Google Scholar]
- Dodou E, Xu SM & Black BL (2003). mef2c is activated directly by myogenic basic helix‐loop‐helix proteins during skeletal muscle development in vivo. Mech Dev 120, 1021–1032. [DOI] [PubMed] [Google Scholar]
- Donkor J, Sariahmetoglu M, Dewald J, Brindley DN & Reue K (2007). Three mammalian lipins act as phosphatidate phosphatases with distinct tissue expression patterns. J Biol Chem 282, 3450–3457. [DOI] [PubMed] [Google Scholar]
- Dressel U, Bailey PJ, Wang SC, Downes M, Evans RM & Muscat GE (2001). A dynamic role for HDAC7 in MEF2‐mediated muscle differentiation. J Biol Chem 276, 17007–17013. [DOI] [PubMed] [Google Scholar]
- Finck BN, Gropler MC, Chen Z, Leone TC, Croce MA, Harris TE, Lawrence JC, Jr. & Kelly DP (2006). Lipin 1 is an inducible amplifier of the hepatic PGC‐1alpha/PPARalpha regulatory pathway. Cell Metab 4, 199–210. [DOI] [PubMed] [Google Scholar]
- Gallegos LL & Newton AC (2008). Spatiotemporal dynamics of lipid signaling: protein kinase C as a paradigm. IUBMB Life 60, 782–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginnan R, Sun LY, Schwarz JJ & Singer HA (2012). MEF2 is regulated by CaMKIIdelta2 and a HDAC4‐HDAC5 heterodimer in vascular smooth muscle cells. Biochem J 444, 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guh JH, Chueh SC, Hwang TL, Chen J & Teng CM (1998). Cell proliferation in human prostatic smooth muscle cells involves the modulation of protein kinase C isozymes. Eur J Pharmacol 359, 281–284. [DOI] [PubMed] [Google Scholar]
- Ha CH, Kim JY, Zhao J, Wang W, Jhun BS, Wong C & Jin ZG (2010). PKA phosphorylates histone deacetylase 5 and prevents its nuclear export, leading to the inhibition of gene transcription and cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A 107, 15467–15472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han GS, Wu WI & Carman GM (2006). The Saccharomyces cerevisiae lipin homolog is a Mg2+‐dependent phosphatidate phosphatase enzyme. J Biol Chem 281, 9210–9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez‐Hernandez JM, Garcia‐Gonzalez EG, Brun CE & Rudnicki MA (2017). The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin Cell Dev Biol 72, 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Yin H, Mitra MS, Liang X, Ajmo JM, Nadra K, Chrast R, Finck BN & You M (2013). Hepatic‐specific lipin‐1 deficiency exacerbates experimental alcohol‐induced steatohepatitis in mice. Hepatology 58, 1953–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Zhu J, Zhuang X, Zhang X, Luo T, Esser KA & Ren H (2015). Lipin1 regulates skeletal muscle differentiation through extracellular signal‐regulated kinase (ERK) activation and cyclin D complex‐regulated cell cycle withdrawal. J Biol Chem 290, 23646–23655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal S, Schneider JW, Nadal‐Ginard B & Mahdavi V (1994). Activation of the myogenic lineage by MEF2A, a factor that induces and cooperates with MyoD. Science 266, 1236–1240. [DOI] [PubMed] [Google Scholar]
- Kim HE, Bae E, Jeong DY, Kim MJ, Jin WJ, Park SW, Han GS, Carman GM, Koh E & Kim KS (2013). Lipin1 regulates PPARgamma transcriptional activity. Biochem J 453, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleger A, Loebnitz C, Pusapati GV, Armacki M, Muller M, Tumpel S, Illing A, Hartmann D, Brunner C, Liebau S, Rudolph KL, Adler G & Seufferlein T (2011). Protein kinase D2 is an essential regulator of murine myoblast differentiation. PLoS ONE 6, e14599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemercier C, Verdel A, Galloo B, Curtet S, Brocard MP & Khochbin S (2000). mHDA1/HDAC5 histone deacetylase interacts with and represses MEF2A transcriptional activity. J Biol Chem 275, 15594–15599. [DOI] [PubMed] [Google Scholar]
- Leonard TA & Hurley JH (2011). Regulation of protein kinases by lipids. Curr Opin Struct Biol 21, 785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GH & Gerace L (2009). Sumoylation regulates nuclear localization of lipin‐1alpha in neuronal cells. PLoS ONE 4, e7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, McKinsey TA, Nicol RL & Olson EN (2000a). Signal‐dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci U S A 97, 4070–4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, McKinsey TA, Zhang CL & Olson EN (2000b). Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell 6, 233–244. [DOI] [PubMed] [Google Scholar]
- Matthews SA, Iglesias T, Rozengurt E & Cantrell D (2000). Spatial and temporal regulation of protein kinase D (PKD). EMBO J 19, 2935–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL & Olson EN (2000). Activation of the myocyte enhancer factor‐2 transcription factor by calcium/calmodulin‐dependent protein kinase‐stimulated binding of 14‐3‐3 to histone deacetylase 5. Proc Natl Acad Sci U S A 97, 14400–14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michot C, Hubert L, Brivet M, De Meirleir L, Valayannopoulos V, Muller‐Felber W, Venkateswaran R, Ogier H, Desguerre I, Altuzarra C, Thompson E, Smitka M, Huebner A, Husson M, Horvath R, Chinnery P, Vaz FM, Munnich A, Elpeleg O, Delahodde A, de Keyzer Y & de Lonlay P (2010). LPIN1 gene mutations: a major cause of severe rhabdomyolysis in early childhood. Hum Mutat 31, E1564–E1573. [DOI] [PubMed] [Google Scholar]
- Michot C, Hubert L, Romero NB, Gouda A, Mamoune A, Mathew S, Kirk E, Viollet L, Rahman S, Bekri S, Peters H, McGill J, Glamuzina E, Farrar M, von der Hagen M, Alexander IE, Kirmse B, Barth M, Laforet P, Benlian P, Munnich A, JeanPierre M, Elpeleg O, Pines O, Delahodde A, de Keyzer Y & de Lonlay P (2012). Study of LPIN1, LPIN2 and LPIN3 in rhabdomyolysis and exercise‐induced myalgia. J Inherit Metab Dis 35, 1119–1128. [DOI] [PubMed] [Google Scholar]
- Mitra MS, Chen Z, Ren H, Harris TE, Chambers KT, Hall AM, Nadra K, Klein S, Chrast R, Su X, Morris AJ & Finck BN (2013). Mice with an adipocyte‐specific lipin 1 separation‐of‐function allele reveal unexpected roles for phosphatidic acid in metabolic regulation. Proc Natl Acad Sci U S A 110, 642–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD, Black BL, Martin JF & Olson EN (1995). Cooperative activation of muscle gene expression by MEF2 and myogenic bHLH proteins. Cell 83, 1125–1136. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Firulli AB, Black BL, Martin JF, Hustad CM, Copeland N, Jenkins N, Lyons G & Olson EN (1996). MEF2B is a potent transactivator expressed in early myogenic lineages. Mol Cell Biol 16, 3814–3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadra K, de Preux Charles AS, Medard JJ, Hendriks WT, Han GS, Gres S, Carman GM, Saulnier‐Blache JS, Verheijen MH & Chrast R (2008). Phosphatidic acid mediates demyelination in Lpin1 mutant mice. Genes Dev 22, 1647–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadra K, Medard JJ, Mul JD, Han GS, Gres S, Pende M, Metzger D, Chambon P, Cuppen E, Saulnier‐Blache JS, Carman GM, Desvergne B & Chrast R (2012). Cell autonomous lipin 1 function is essential for development and maintenance of white and brown adipose tissue. Mol Cell Biol 32, 4794–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton AC (2009). Lipid activation of protein kinases. J Lipid Res 50 (Suppl), S266–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott MO, Bober E, Lyons G, Arnold H & Buckingham M (1991). Early expression of the myogenic regulatory gene, myf‐5, in precursor cells of skeletal muscle in the mouse embryo. Development 111, 1097–1107. [DOI] [PubMed] [Google Scholar]
- Potthoff MJ, Arnold MA, McAnally J, Richardson JA, Bassel‐Duby R & Olson EN (2007). Regulation of skeletal muscle sarcomere integrity and postnatal muscle function by Mef2c. Mol Cell Biol 27, 8143–8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potthoff MJ & Olson EN (2007). MEF2: a central regulator of diverse developmental programs. Development 134, 4131–4140. [DOI] [PubMed] [Google Scholar]
- Rehnmark S, Giometti CS, Slavin BG, Doolittle MH & Reue K (1998). The fatty liver dystrophy mutant mouse: microvesicular steatosis associated with altered expression levels of peroxisome proliferator‐regulated proteins. J Lipid Res 39, 2209–2217. [PubMed] [Google Scholar]
- Ren H, Federico L, Huang H, Sunkara M, Drennan T, Frohman MA, Smyth SS & Morris AJ (2010). A phosphatidic acid binding/nuclear localization motif determines lipin1 function in lipid metabolism and adipogenesis. Mol Biol Cell 21, 3171–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reue K & Dwyer JR (2009). Lipin proteins and metabolic homeostasis. J Lipid Res 50 (Suppl), S109‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabourin LA, Girgis‐Gabardo A, Seale P, Asakura A & Rudnicki MA (1999). Reduced differentiation potential of primary MyoD‐/‐ myogenic cells derived from adult skeletal muscle. J Cell Biol 144, 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz‐Peiffer C & Biden TJ (2008). Protein kinase C function in muscle, liver, and beta‐cells and its therapeutic implications for type 2 diabetes. Diabetes 57, 1774–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer GG, Chen Z, Gan C, McCommis KS, Soufi N, Chrast R, Mitra MS, Yang K, Gross RW & Finck BN (2015). Liver‐specific loss of lipin‐1‐mediated phosphatidic acid phosphatase activity does not mitigate intrahepatic TG accumulation in mice. J Lipid Res 56, 848–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer GG, Collier SL, Chen Z, McCommis KS, Pittman SK, Yoshino J, Matkovich SJ, Hsu FF, Chrast R, Eaton JM, Harris TE, Weihl CC & Finck BN (2018). Loss of lipin 1‐mediated phosphatidic acid phosphohydrolase activity in muscle leads to skeletal myopathy in mice. FASEB J, fj201800361R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemori H, Katoh Hashimoto Y, Nakae J, Olson EN & Okamoto M (2009). Inactivation of HDAC5 by SIK1 in AICAR‐treated C2C12 myoblasts. Endocr J 56, 121–130. [DOI] [PubMed] [Google Scholar]
- Tapscott SJ (2005). The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development 132, 2685–2695. [DOI] [PubMed] [Google Scholar]
- Toker A (2005). The biology and biochemistry of diacylglycerol signalling. Meeting on molecular advances in diacylglycerol signalling. EMBO Rep 6, 310–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vozenilek AE, Navratil AR, Green JM, Coleman DT, Blackburn CMR, Finney AC, Pearson BH, Chrast R, Finck BN, Klein RL, Orr AW & Woolard MD (2018). Macrophage‐associated lipin‐1 enzymatic activity contributes to modified low‐density lipoprotein‐induced proinflammatory signaling and atherosclerosis. Arterioscler Thromb Vasc Biol 38, 324–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Kim C, Jogasuria A, Han Y, Hu X, Wu J, Shen H, Chrast R, Finck BN & You M (2016). Myeloid cell‐specific lipin‐1 deficiency stimulates endocrine adiponectin‐FGF15 axis and ameliorates ethanol‐induced liver injury in mice. Sci Rep 6, 34117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way KJ, Chou E & King GL (2000). Identification of PKC‐isoform‐specific biological actions using pharmacological approaches. Trends Pharmacol Sci 21, 181–187. [DOI] [PubMed] [Google Scholar]
- Yao Z, Fong AP, Cao Y, Ruzzo WL, Gentleman RC & Tapscott SJ (2013). Comparison of endogenous and overexpressed MyoD shows enhanced binding of physiologically bound sites. Skelet Muscle 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JZ, Bae D, Cantrell D, Nel AE & Rozengurt E (2002). Protein kinase D is a downstream target of protein kinase C theta. Biochem Bioph Res Co 291, 444–452. [DOI] [PubMed] [Google Scholar]
- Zeharia A, Shaag A, Houtkooper RH, Hindi T, de Lonlay P, Erez G, Hubert L, Saada A, de Keyzer Y, Eshel G, Vaz FM, Pines O & Elpeleg O (2008). Mutations in LPIN1 cause recurrent acute myoglobinuria in childhood. Am J Hum Genet 83, 489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Verity MA & Reue K (2014). Lipin‐1 regulates autophagy clearance and intersects with statin drug effects in skeletal muscle. Cell Metab 20, 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]