

Abstract
Key points
Exogenous Na+/H+ exchanger 1 (NHE1) expression stimulated the collective migration of epithelial cell sheets
Stimulation with epidermal growth factor, a key morphogen, primarily increased migration of the front row of cells, whereas NHE1 increased that of submarginal cell rows, and the two stimuli were additive
Accordingly, NHE1 localized not only to the leading edges of leader cells, but also in cryptic lamellipodia in submarginal cell rows
NHE1 expression disrupted the morphology of epithelial cell sheets and three‐dimensional cysts
Abstract
Collective cell migration plays essential roles in embryonic development, in normal epithelial repair processes, and in many diseases including cancer. The Na+/H+ exchanger 1 (NHE1, SLC9A1) is an important regulator of motility in many cells and has been widely studied for its roles in cancer, although its possible role in collective migration of normal epithelial cells has remained unresolved. In the present study, we show that NHE1 expression in MDCK‐II kidney epithelial cells accelerated collective cell migration. NHE1 localized to the leading edges of leader cells, as well as to cryptic lamellipodia in submarginal cell rows. Epidermal growth factor, a kidney morphogen, increased displacement of the front row of collectively migrating cells and reduced the number of migration fingers. NHE1 expression increased the number of migration fingers and increased displacement of submarginal cell rows, resulting in additive effects of NHE1 and epidermal growth factor. Finally, NHE1 expression resulted in disorganized development of MDCK‐II cell cysts. Thus, NHE1 contributes to collective migration and epithelial morphogenesis, suggesting roles for the transporter in embryonic and early postnatal development.
Keywords: MDCK cells, NHE1, Na+/H+ exchanger, collective cell migration, metastasis, EGF
Key points
Exogenous Na+/H+ exchanger 1 (NHE1) expression stimulated the collective migration of epithelial cell sheets
Stimulation with epidermal growth factor, a key morphogen, primarily increased migration of the front row of cells, whereas NHE1 increased that of submarginal cell rows, and the two stimuli were additive
Accordingly, NHE1 localized not only to the leading edges of leader cells, but also in cryptic lamellipodia in submarginal cell rows
NHE1 expression disrupted the morphology of epithelial cell sheets and three‐dimensional cysts
Introduction
Collective cell migration, which refers to cells migrating together in sheets, streams or other multicellular arrangements, is pivotal for physiological processes such as embryonic development and wound healing. In addition, collective cell migration plays central roles in diseases such as cancer and fibrosis (Haeger et al. 2015; Mayor & Etienne‐Manneville, 2016; Friedl & Mayor, 2017; Khalil et al. 2017). The Na+/H+ exchanger 1 (NHE1, SLC9A1) is a ubiquitously expressed, acid‐extruding transporter with essential roles in cell and tissue acid‐base homeostasis (Orlowski & Grinstein, 2004; Boedtkjer et al. 2012). NHE1 plays important roles in the regulation of cell motility (Schwab et al. 2012), in a manner dependent both on its ion transport activity and on its interaction with the actin cytoskeleton via ezrin/radixin/moesin (Denker et al. 2000). In single migrating cells, NHE1 localizes to leading edge lamellipodia and is an important driver of motility (Lagana et al. 2000; Schneider et al. 2009; Martin et al. 2011). Its activity contributes to an increased intracellular pH (pHi) in the front of the migrating cell (Martin et al. 2011), which favours polarity and motility by pH‐dependent regulation of Cdc42 (Frantz et al. 2007), cofilin (Frantz et al. 2008), cortactin (Magalhaes et al. 2011) and focal adhesion kinase (Choi et al. 2013). The concomitant acidification of the tumor microenvironment further drives NHE1‐dependent cell motility, as well as invasiveness in cancer cells (Counillon et al. 2016; Stock & Pedersen, 2017). Furthermore, NHE1‐mediated Na+ uptake in the leading edge favours aquaporin‐mediated water flux, facilitating leading edge protrusion (Schwab et al. 2012; Stroka et al. 2014; Jensen et al. 2016). Accordingly, knockdown or inhibition of NHE1 decreases cancer cell motility and invasiveness, and reduces metastasis in in vivo models (Klein et al. 2000; Chiang et al. 2008; Chang et al. 2014; Cardone et al. 2015).
Interesting recent work indicates that NHE1 regulates epithelial development and homeostasis. Thus, NHE1 was assigned a role downstream of transforming growth factor alpha stimulated branching morphogenesis of mammary organoids (Jenkins et al. 2012). Similarly, human cancer cells with high NHE1 expression formed less regular spheroids with weaker cell–cell adhesions (Hofschröer et al. 2017). In a Drosophila model, NHE1 overexpression led to cell proliferation and tissue expansion, and overexpressed NHE1 co‐operated with Ras to reduce cell–cell coordination (Grillo‐Hill et al. 2015). Finally, movement of clusters of head and neck squamous cell carcinoma cells was decreased upon NHE1 knockdown (Kaminota et al. 2017). Expression of NHE1 is developmentally regulated; NHE1 levels are particularly high in murine kidney at postnatal week 2, which is the time when normal kidney function is established, compared to both prenatal and adult stages (Rieder & Fliegel, 2002). Collective migration is essential both for kidney morphogenesis during development (Vasilyev et al. 2009) and kidney repair after acute injury (Palmyre et al. 2014). Notably, epidermal growth factor (EGF), which generally stimulates NHE1 activity (Maly et al. 2002; Coaxum et al. 2009), is an important morphogen and growth factor in the kidneys (Zeng et al. 2009).
Accordingly, we investigated whether NHE1 upregulation and EGF stimulation regulate the collective migration of kidney epithelial cells. We used 3D MDCK‐II cysts to evaluate the effects of NHE1 expression on normal kidney tubule formation. We demonstrate that expression of NHE1 in MDCK‐II kidney epithelial cells increased collective migration and altered the organization of migrating cell sheets. NHE1 localized to the leading edge of migrating cells both in the front and in cryptic lamellipodia several cell rows further back in the sheet. Cell tracking showed that NHE1 expression in particular drove the increased motility of cells behind the leading edge. Stimulation with EGF also increased collective motility, although this was manifested in the front of the collectively migrating sheets. NHE1 overexpression and EGF stimulation had accumulative effects on collective migration. Finally, NHE1 overexpression was sufficient to elicit structural disorganization of MDCK cysts. The results of the present study demonstrate, for the first time, a role for NHE1 in collective migration of normal mammalian epithelial cells. This is important for understanding how NHE1 contributes to tissue homeostasis and development, as well as disease states where NHE1 is overexpressed, such as early cancer development.
Methods
Cell culture
Wild‐type (WT) Madin–Darby canine kidney (MDCK II/G) cells (Gaush et al. 1966; Louvard, 1980) were obtained from Professor W. James Nelson (Stanford University, CA, USA). MDCK cells expressing GFP‐tagged NHE1 (NHE1‐MDCK) are stable with a low but homogeneous NHE1 expression and have been described previously (Pedraz‐Cuesta et al. 2016). Because NHE1‐GFP was overexpressed at low levels, which could not be obtained for green fluorescent protein (GFP) or enhanced GFP (EGFP), and because EGFP overexpression in MDCK did not appear to affect cellular polarity and migration (data not shown), WT MDCK cells were employed as the control cell line.
Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 1 g L–1 glucose (31600‐083; Gibco, Gaithersburg, MD, USA), supplemented with 1 g L–1 NaHCO3, 10% (v/v) fetal bovine serum (FBS) (Gibco) and a cocktail of 0.5 U mL–1 penicillin (Sigma, St Louis, MO, USA), 0.5 g mL–1 streptomycin (Gibco) and 1 mg mL–1 kanamycin (Gibco) (PSK). MDCK II/G were routinely grown in low bicarbonate to avoid the formation of intracellular vacuoles (Yeaman et al. 2004). Cells were maintained at 37°C in a humidified atmosphere with 5% CO2. Cells were cultured at up to 80% confluency and passaged every 1–4 days. For transfections, cells were seeded at 50% confluency and transfected using Lipofectamine 2000 (Life Technologies, Grand Island, NY, USA) in accordance with the manufacturer's instructions. The constructs encoding tubulin‐RFP and paxilin‐RFP were a generous gift from W. James Nelson (Stanford University). The construct encoding Ankyrin‐G‐mCherry was generously provided by Camilla S. Jensen, University of Copenhagen, Denmark. The dynamin2‐RFP construct was generously provided by Ching Hwa‐Sung (Weill Cornell Medical College, New York, NY, USA).
Drug treatment and acid loading
Cells were stimulated with 100 ng mL–1 EGF (E9644; Sigma). EGF was used from a stock solution at 100 μg mL–1 in 10 mm acetic acid with 0.1% BSA, which was also used as the control solvent. For drug treatments, cells were treated with 0.2 μm jasplakinolide (ALX‐350‐27‐C050; Enzo Diagnostics, Farmingdale, NY, USA), 50 μm blebbistatin (B0560; Sigma), 1 μg mL–1 cytochalasin D, 10 mm methyl‐β‐cyclodextrin (MBCD) (4555; Sigma), 10 μm 5‐(N‐ethyl‐N‐isopropyl)amiloride (EIPA) (E3111; Life Technologies) or 80 μm dynasore (9754; Sigma) for 10 min. MBCD was dissolved in H2O and used at a dilution of 1:100. All other drugs were dissolved in dimethyl sulphoxide (DMSO) and used 1:1000. All drug treatments were performed for 10 min. Shorter and longer times were also tested.
To image clusters in acid loaded cells, cells were washed in Ringer solution (in mm: 135 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES and 10 glucose) and then acid loaded for 5 min (in mm: 20 NH4Cl, 115 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES and 10 glucose). NHE1‐mediated acid release was prevented in Na+‐free Ringer solution for 5 min (in mm: 135 N‐methyl‐d‐glucamine, 5 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES and 10 glucose) before release in Ringer solution.
Migration assays
Trypsinized WT MDCK and NHE1‐MDCK cells were resuspended at 106 mL–1 in Ca2+ free DMEM (D9800‐10; US Biological, Salem, MA, USA) supplemented with 1 g L–1 NaHCO3 and PSK and with or without 10% dialysed FBS. Next, 1000 μL of cell suspension was seeded in each well of a collagen‐coated 24‐well plate. After 1 h of incubation, 1000 μL of Ca2+ containing medium was added to allow the formation of adherens junctions. The Ca2+ medium was based on Ca2+ free DMEM with PSK and with or without 10% dialysed FBS, as well as the addition of final concentrations of 1.8 mm CaCl2. Cells were incubated an additional 1 h before initiating migration by scratching the cell layer with a rubber scraper. After washing, DMEM low w/o phenol red (11880‐028; Gibco) supplemented with PSK, 4 mm l‐glutamine and 0–10% FBS was added to the cell layers. For EGF stimulation, cells were stimulated with 100 ng mL–1 EGF or a control solvent as indicated above.
For wound quantifications, 2 μg mL–1 Hoechst 33342 (H3570; Life Technologies) was added at the same time as Ca2+ containing medium. Plates with migrating cells were imaged after scratching (0 h). Cells were then allowed to migrate in a CO2 incubator. After 16 h, the cells were fixed and visualized again. Three experiments were performed, and two to four measurements were obtained from each condition in each experiment.
For live tracking, the cells were allowed to migrate in a CO2 incubator. After 16 h, the cells were loaded for 15 min with 2 μg mL–1 Hoechst 33342. The migrating cells were then imaged in DMEM low w/o phenol red supplemented with PSK, 4 mm l‐glutamine and 5% FBS using time‐lapse microscopy for 5 h with a 5 min frame rate at 10× magnification.
Migration was only quantified from sheets and wounds that did not close because the cells accelerated when in close proximity to the opposing sheet.
Staining
Cells were fixed 10 min in 4% paraformaldehyde. For observations of microtubules, cells were fixed for 10 min in ice‐cold methanol. For staining, cells were permeabilized for 5–10 min in 0.1% Triton X‐100 and 3% BSA in PBS. For immunofluorescence staining, the samples were stained in primary antibodies (dilution 1:1000, mouse anti‐α‐tubulin; sc‐32293, Santa Cruz Biotechnology, Santa Cruz, CA, USA; dilution 1:500, rabbit anti‐ezrin, 3142, Cell Signaling Technology, Beverly, MA, USA) overnight at 4ᵒC before three washes in PBS and labelling in secondary antibodies (Alexa Fluor anti‐mouse‐647 and Alexa Fluor anti‐rabbit‐594) for 1 h at room temperature. For labelling of nuclei and actin, the cells were labelled with 2 μg mL–1 Hoechst 33342 and 4 ng mL–1 phalloidin‐rhodamine (P1951; Sigma) diluted in 3% BSA in PBS for 20–60 min. The samples were finally washed three times in PBS and kept in PBS. Antibodies and dyes were diluted in 3% BSA in PBS.
Cysts
Cysts were grown in eight‐chamber Lab‐Tek slides with Geltrex (A14132‐02; Gibco). Next, 2500 cells were seeded in the presence of 10 ng mL–1 EGF. After 8 days with medium changes every 2–3 days, the cysts were fixed and stained. Fixation was performed for 30 min in 4% PFA followed by 3 × 10 min washes in 1 m glycine in PBS. The cysts were then permeabilized for 5 min in 0.5% Triton X‐100 in PBS before blocking 1.5 h in 5% BSA in PBS. Staining with Hoechst (dilution 1:400) and phalloidin‐647 (dilution 1:400) was performed for 1 h in PBS with 5% BSA. Finally, the cysts were washed three times in PBS and kept in PBS.
Microscopy
Imaging of migration assays was performed using a Ti Eclipse inverted microscope (Nikon, Tokyo, Japan) equipped with a motorized stage, Plan Fluor 4× (NA 0.13) PhL and CFI Plan Fluor DL 10× (NA 0.30) Ph1 objectives, and the TI‐ND6‐PFS3 Perfect Focus System controlled with NIS‐Elements software (Nikon). Images were captured with a Zyla 5.5 sCMOS camera (Andor Technology Ltd, Belfast, UK). The epifluorescence light source was a pE‐300 white LED unit (CoolLED) with appropriate filter sets.
Imaging of NHE1 clusters was performed on a Nikon Ti Eclipse inverted total internal reflection fluorescence (TIRF) microscope equipped with a motorized stage, a CFI Apochromat TIRF 100× NA 1.49 objective and the TI‐ND6‐PFS3 Perfect Focus System controlled with NIS‐Elements software (Nikon). Images were captured with an Andor iXon3 EMCCD camera or a Zyla 5.5 sCMOS camera (Andor). The epifluorescence light source was a pE‐300white LED unit (CoolLED, Andover, UK), which was paired with hard coated TIRF HQ filter sets.
Cysts were imaged on a LSM 700 confocal microscope system (Carl Zeiss, Oberkochen, Germany) with a 100x objective, as well as 405/420‐800 nm and 555/300‐630 nm lasers handled with ZEN software (Carl Zeiss).
Image analysis of migration assays
Image analysis was primarily carried out using NIS Elements software (Nikon) or FIJI/ImageJ (NIH, Bethesda, MD, USA). NIS Elements was used for quantification of migration assays. Briefly, the contrast was enhanced before blurring the signal with a 15 × 15 average kernel. The gap in the cell layer was then detected by thresholding and manual evaluation. For measurements of structural index, the same binaries were used to measure the length of the migrating cell front. The numbers from the same position at 0 and 16 h were divided to obtain a measure of structural index. The Automated Spot Detection function was used to detect all nuclei based on the Hoechst signal. The number of cell nuclei within the region that cells had moved into at 16 h was used to calculate the average cell area. Finally, the measurement ‘Nearest Neighbor’ in NIS Elements software was employed on the detected nuclei.
For time‐lapse acquisitions, cells were tracked based on Hoechst labelling of nuclei. Nuclei were detected and tracked frame‐to‐frame using the FIJI‐plugin Trackmate (Tinevez et al. 2017). Tracks at different positions were manually identified and extracted to Excel (Microsoft Corp., Redmond, WA, USA) or MATLAB (MathWorks Inc., Natick, MA, USA).
Quantification of proliferation rate
For quantification of the proliferation rate, 50,000 cells of either WT MDCK or NHE1‐MDCK were seeded into collagen‐coated wells of a six‐well plate. The cells were allowed to grow at 37°C and 5% CO2 for 2, 24, 48 or 72 h. At these time points, the cells were fixed and stained with Hoechst. Twelve random positions were imaged using a 10x objective on a widefield microscope for each sample. Automated Spot Detection was used to identify and count cells in the images with NIS Elements software.
Intracellular pH measurements
Measurements of pHi were carried out essentially as described previously (Lauritzen et al. 2010; Pedraz‐Cuesta et al. 2016). Cells were loaded with 1.6 μm BCECF‐AM (B1150; Thermo Fisher Scientific, Wlatham, MA, USA) by directly adding from a 1:1000 DMSO stock into the cell culture medium and incubated for 30 min. To calibrate to pH values, the cellular BCECF fluorescence ratio was measured in KCl Ringer solution (in mm: 1 MgSO4, 1 K2HPO4, 156 KCl, 1 CaCl2, 3.3 Mops, 3.3 Tes and 3.3 Hepes) with 10 μm nigericin (N7143; Sigma) at pH 7.0, 7.4, 7.8 and 6.7. There were no differences in fluorescence emission ratios at a given pHi for WT and NHE1‐MDCK cells.
For experiments, the dish was continuously perfused with warm HCO3 – buffered Ringer solution (in mm: 25 NaHCO3, 1 CaCl2, 1 Na2HPO4, 118 NaCl, 1 MgSO4, 4 KCl, 3.3 MOPS, 3.3 TES and 3.3 HEPES, pH 7.4) saturated with 5% CO2. For measurements of H+ transport capacity, the cells were initially imaged for 5 min and then the perfusion was switched to HCO3 –‐Ringer buffer with 20 mm NH4Cl to acid load the cells using the NH4Cl prepulse method. After 5 min, the perfusion was switched back to HCO3 –‐Ringer buffer to release H+. The steady‐state pHi was measured during the initial 5 min of perfusion with HCO3 –‐Ringer buffer.
Imaging was performed on a Eclipse Ti (Nikon) with a 40× objective and a Coolsnap camera (Photometrics, Huntington Beach, CA, USA). Excitation light at 440 nm and 485 nm was obtained with a DeltaRAM X monochromator (Horoba, Kyoto, Japan) and perfusion was enabled with a perfusion system (ALA Scientific Instruments, Farmingdale, NY, USA), as well as a Warner pump (Warner Pump Malaysia, Petaling Jaya, Malaysia).
Proton fluxes were calculated based on the initial recovery after acid release multiplied by the buffering capacity. The buffer capacity was calculated using: βi = ([NH4 +]i,loaded – [NH4 +]i,release)/(pHi,loaded – pHi,release), where [NH4 +]I = [NH4Cl] (10pHo‐pK)/[(1 + 10pHo‐pK)(10pHi‐pK)] and pK(NH4 +) = 8.9 (Lauritzen et al. 2010).
Statistical analysis
Unless otherwise stated, all of the reported experiments were performed at least three times. Data analysis and statistical analyses were handled in Excel (Microsoft Corp.) or MATLAB (MathWorks Inc.). For most experiments, one‐way, two‐way, or three‐way ANOVA and Tukey's post hoc test were used. Some experiments were evaluated using Student's t test. P < 0.05 was considered statistically significant. Equal variance and normal distribution were confirmed by visualization of raw data.
Data availability
Data are available from the authors upon request.
Results
NHE1‐GFP localized to the leading edge of collectively migrating cells
MDCK cells, comprising a widely‐used system of normal epithelial cells with respect to investigating cell polarity, the polarized trafficking of proteins and transport proteins, and as a model of collective cell migration (Poujade et al. 2007; Marlar et al. 2014), were employed to study the effect of NHE1 on collective cell movement. To allow visual inspection of NHE1 localization and direct comparison between individual experiments, a cell line stably expressing NHE1 with a C‐terminal GFP tag (referred to as NHE1‐MDCK) was used (Pedraz‐Cuesta et al. 2016) (Fig. 1 A). In this cell line, the expression level of NHE1‐GFP is generally homogeneous between individual cells and over time.
Figure 1. Overexpressed NHE1 in MDCK cells localizes as clusters and is recruited to the leading edge of collectively migrating cells.
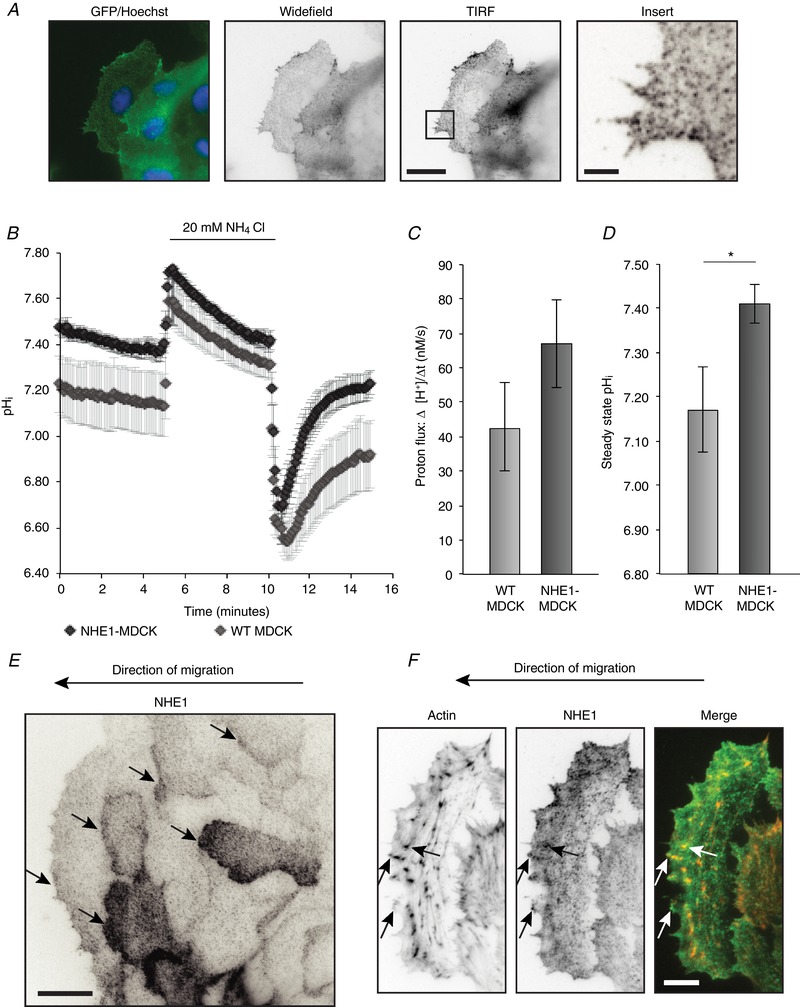
A, NHE1 tagged with GFP stably expressed in MDCK cells (NHE1‐MDCK) was imaged using wide‐field fluorescence imaging and TIRF microscopy. Left: the overlay is from widefield fluorescence microscopy of NHE1 (green) and Hoechst (blue). Other images show an inverted contrast of the NHE1‐GFP signal. Scale bars: large images = 20 μm; insert = 3 μm. B–D, overexpressed NHE1 increased steady state pHi, as well as the capacity of MDCK cells to transport H+. B, cells were loaded with BCECF‐AM to ratiometrically image pHi in HCO3 − buffered Ringer solution. The cells were acid loaded in NH4Cl in buffer for 5 min before releasing in Ringer buffer; n = 7 for NHE1‐MDCK and n = 8 for WT MDCK, each with analysis of 20–40 cells. C, flux of H+ after acid release and corrected for buffer capacity. There was no statistically significant difference between NHE1‐MDCK and WT MDCK cells. D, steady‐state pHi was measured within the initial 5 min of imaging. The graph represents nine and 10 individual experiments on NHE1‐MDCK and WT MDCK cells, respectively, each with analysis of 20–40 cells. Statistics were performed using Student's t test; P < 0.05 was considered statistically significant. Error bars represent the SEM. E, during collective migration, NHE1‐MDCK clusters localized to the leading edge of migrating cells several rows behind the wound. Arrows indicate leading edges of migrating cells. Scale bar = 20 μm. F, example of a migrating cell, stained with phalloidin‐rhodamine to visualize actin. NHE1 clusters partially localized to actin in the lamellipodium and filopodia. Arrows indicate examples of NHE1 clusters localizing to actin. Scale bar = 10 μm. Fluorescence images (A, E and F) are representative of at least five individual experiments. [Color figure can be viewed at wileyonlinelibrary.com]
To confirm that the GFP‐tagged NHE1 was functional, the H+ transport capacity of the cells was measured. Using BCECF‐AM, the cellular pHi was measured during recovery from an NH4Cl pulse and subsequently corrected for buffer capacity. Recovery from the acid load was not significantly increased in NHE1‐MDCK cells compared to WT MDCK cells (Fig. 1 B–C). By contrast, the steady‐state pHi was significantly higher in NHE1‐MDCK cells compared to WT MDCK cells (7.41 in NHE1‐MDCK vs. 7.17 in WT MDCK), indicating that, in these cells, the overexpressed NHE1 affected cellular pHi regulation (Fig. 1 D).
In single migrating cancer cells, NHE1 drives cell movement by localizing to the leading edge (Martin et al. 2011). In sheets of collectively migrating NHE1‐MDCK cells, GFP‐tagged NHE1 localized to the leading edge (Fig. 1 E) and some NHE1‐GFP localized at actin puncta (Fig. 1 F). Importantly, NHE1‐GFP was not only recruited to the leading edge of the leader cells, but also to cryptic lamellipodia of cells further behind in the migrating cell sheet (Fig. 1 E, arrows) (Farooqui & Fenteany, 2005). This leading edge localization in the sheet suggests that NHE1 may contribute to the migration of both leader cells and cells further back in the cell sheet. Interestingly, we also observed that NHE1‐GFP localized in a punctuate pattern resembling clusters (Fig. 1 A and E). The punctuate localization pattern of NHE1‐GFP was particularly evident using TIRF microscopy in non‐migrating, subconfluent cells (Fig. 1 A, insert) as well as migrating cells (Fig. 1 E–F). A punctuate NHE1 localization has previously been observed in antibody labelling of endogenous NHE1 (Stock et al. 2008; Schneider et al. 2009; Ludwig et al. 2013) but, to our knowledge, it has never been studied using TIRF imaging. Moreover, puncta or clusters of NHE1‐GFP were motile in the TIRF plane of subconfluent cells (see Supporting information, Movie S1). The clusters were not disrupted by a number of actin‐ and membrane‐disrupting drugs or the NHE1‐inhibitor EIPA. Moreover, they did not co‐localize to tested cytoskeletal proteins, as shown by representative line scans of the green (NHE1‐GFP) and red (cytoskeletal proteins) fluorescence intensities (Fig. 2). Interestingly, colocalization of NHE1‐GFP clusters with actin was more pronounced when NHE1 was activated by an acid load (Fig. 3, arrows).
Figure 2. Localization of NHE‐GFP clusters.
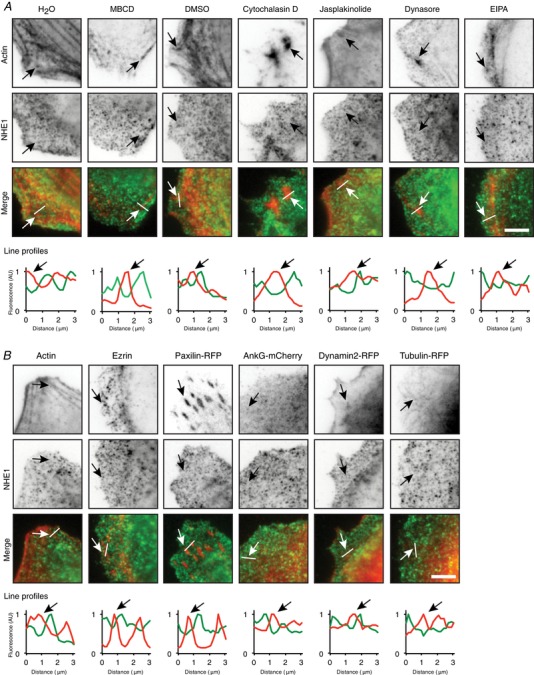
A, subconfluent MDCK cells stably expressing NHE1 with a GFP tag (NHE1‐MDCK) were treated for 10 min with MBCD (methyl‐β‐cyclodextrin, cholesterol depletion), cytochalasin D (actin depolymerization), jasplakinolide (actin stabilization), dynasore (endocytosis inhibition) or EIPA (NHE1 inhibition). Cells were then fixed and stained with phalloidin‐rhodamine to visualize actin. H2O (1:100) is control for MBCD and DMSO (1:1000) is control for the other treatments. B, NHE1‐MDCK were either transiently transfected with ankyrin G‐mCherry, paxilin‐RFP, dynamin2‐RFP or tubulin‐RFP, or stained with phalloidin‐rhodamine to label actin or an anti‐ezrin antibody. The cells were imaged with TIRF microscopy of green (NHE1 fused to GFP) and red fluorescence (stainings and transient transfections). In (A) and (B), inverted contrast images are shown at the top, and merged images with actin in red and NHE1 in green are shown below. Line profiles show the relative intensities of red and green fluorescence along the indicated lines. Arrows indicate the same positions of localization of the stained or transfected proteins in the images and on the line profiles. The cells are subconfluent and not migrating. AU, arbitrary units. Scale bar = 5 μm. The images are representative of three individual experiments. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 3. NHE1‐GFP clusters localized to actin in acid loaded cells.
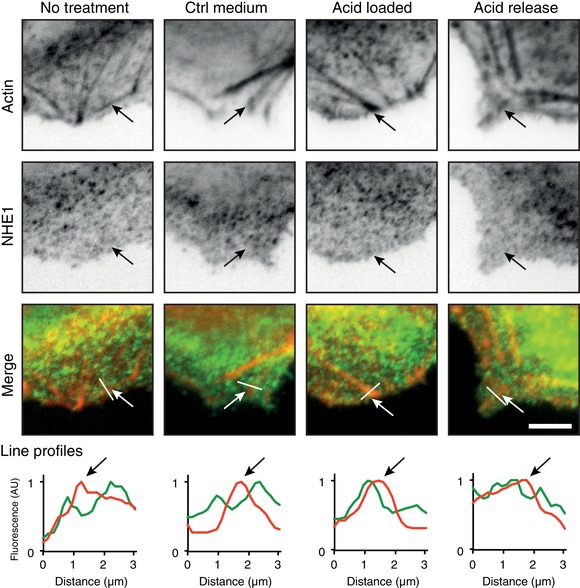
Subconfluent NHE1‐MDCK cells were acid loaded by a 20 mm NH4Cl prepulse in Ringer solution followed by NH4Cl removal to stimulate NHE1 activity. Cells were fixed and stained with phalloidin‐rhodamine to label actin and subsequently imaged using TIRF microscopy. Line profiles show the relative intensities of red and green fluorescence along the indicated lines. Arrows indicate positions of actin in the images and on the line profiles. AU, arbitrary units. Scale bar = 5 μm. The images are representative of three individual experiments. [Color figure can be viewed at wileyonlinelibrary.com]
Together, these results show that NHE1 expression in MDCK cells contributes to elevated pHi. NHE1 localizes in a punctate, clustered pattern in the leading edge of the leader cells and in cryptic lamellipodia of cells further back in the collectively migrating sheets.
NHE1 expression increases collective cell migration
Because NHE1 localizes to the leading edge and in cryptic lamellipodia further back, we hypothesized that NHE1 could affect the collective migration of MDCK‐II cells. Because overexpressed NHE1 increases the proliferation rate in some cell types, we first tested whether this is the case in MDCK cells. This was not observed (Fig. 4 A) and therefore migration could be directly quantified from 16 h migration experiments. WT MDCK and NHE1‐MDCK cells were seeded as instant confluent monolayers shortly prior to scratching to avoid monolayer peeling (Benjamin et al. 2010; Pedersen et al. 2017). Cells were serum starved during seeding and then allowed to migrate for 16 h in the presence of varying serum concentrations (0–10%) (Fig. 4 B). When the cells were fully deprived of serum, migration was decreased in both cell types. Across all serum conditions, NHE1‐MDCK cells exhibited significantly increased migration compared to WT cells (Fig. 4 C). We next investigated whether collective movement was further accelerated by stimulation with EGF, a known stimulator of NHE1 activity (Maly et al. 2002; Coaxum et al. 2009). Migrating cells were treated with EGF or a control medium. Collective migration of WT MDCK cells and NHE1‐MDCK cells significantly increased in the presence of EGF (Fig. 4 D). The maximal migration distance was observed when treating with EGF in combination with 5% serum.
Figure 4. Overexpression of NHE1 increased the migration of MDCK cells.
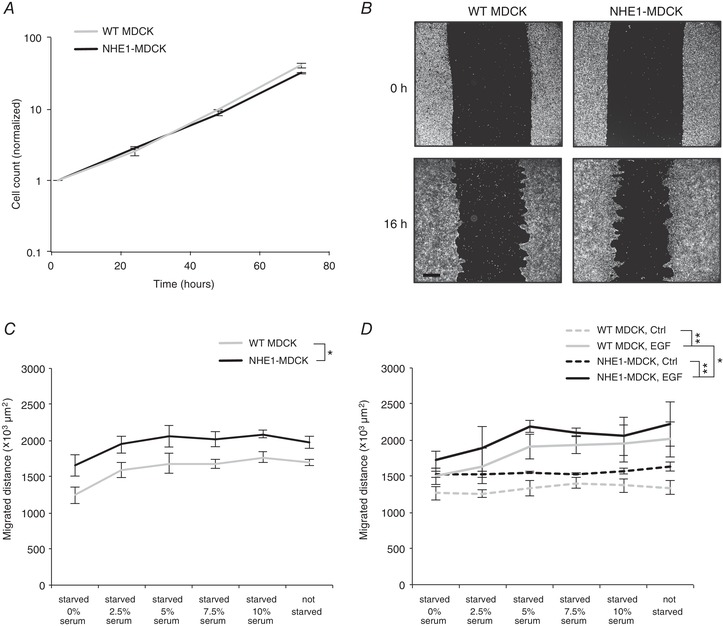
A, overexpression of NHE1 in MDCK cells did not increase the proliferation rate. Equal numbers of NHE1‐MDCK and WT MDCK were seeded on collagen and fixed after 2, 24, 48 or 72 h. The cells were then fixed and counted. The graph represents two individual experiments. B–D, NHE1‐MDCK and WT MDCK cells were seeded in Ca2+ free medium for 1 h to pack cell layers, then Ca2+ was added to allow formation of adherens junctions. This was performed without serum; thus, the cells were starved for 2 h. Afterwards, migration was induced by scratching the cell layer and adding migration medium with indicated concentrations of serum. Images of identical positions at 0 h and 16 h were acquired using Hoechst labelling. B, examples of WT MDCK and NHE1‐MDCK cell layers at 0 h and 16 h of migration. Scale bar = 500 μm. C, across all serum concentrations, NHE1‐MDCK cells migrated significantly more than WT MDCK cells. Moreover, the same statistical test showed that the migration speed was lower for 0% serum than 5%, 7.5% and 10% serum. D, migrating cells were treated with EGF or control medium. EGF significantly increased the migration of both NHE1‐MDCK and WT MDCK cells. EGF‐treated NHE1‐MDCK cells migrated significantly more compared to EGF‐treated WT MDCK cells. Statistical tests in (B) to (C): two‐way and three‐way ANOVA. * P < 0.05, ** P < 0.001 (n = 3). Error bars represent the SEM.
In conclusion, overexpression of NHE1 increased collective MDCK cell migration, in a manner potentiated by treatment with EGF and serum.
NHE1 alters cell–cell organization in epithelial sheets
At the front of collectively migrating MDCK cells, migration fingers form spontaneously. They consist of cells organized as groups that explore the gap and drive collective cell migration (Poujade et al. 2007). Each group or finger of cells is guided by a leader cell that has a characteristic large, fanlike morphology and is generally considered to guide the follower cells (Poujade et al. 2007). Because NHE1 increased collective cell migration as shown by the previous experiment, this could be manifested by the altered formation and morphology of migration fingers in the cell collectives. Notably, migrating NHE1‐MDCK cells formed large, irregular migration fingers compared to WT MDCK cells (Fig. 5 A). Individual NHE1‐MDCK cells appeared less regular in the sheet compared to WT MDCK cells. To quantify the formation of migration fingers, we used a structural index, calculated as the ratio between the length of the wound edge at 16 h and 0 h (Fig. 5 B). Without EGF treatment (no treatment, NT; treatment with control medium, Ctrl), NHE1‐MDCK cells exhibited a significantly higher structural index compared to WT MDCK cells (Fig. 5 C), confirming that these cells had more migration fingers and/or larger migration fingers. Upon EGF treatment, the cell sheets of both WT MDCK and in particular NHE1‐MDCK cells appeared less organized with fewer migration fingers (Fig. 5 A); this was manifested in a decrease of the structural index for NHE1‐MDCK cells (Fig. 5 C). Further analysis demonstrated that NHE1‐MDCK cells were larger and flatter in morphology than WT MDCK cells (Fig. 5 D). Moreover, for both cell types, treatment with EGF also increased the average area covered by each cell and thus spacing. A similar trend was observed when measuring the Nearest Neighbor Distance (NND) as an alternative measure of cell spacing (Fig. 5 E). These data suggested that overexpression of NHE1 affected the cellular organization of cell sheets. Cell–cell junctions contribute to cellular organization; however, no differences were observed in the expression of adherens junction proteins β‐catenin and E‐cadherin in WT and NHE1‐MDCK cells (data not shown).
Figure 5. Migrating NHE1‐MDCK cells formed less organized migration fingers.
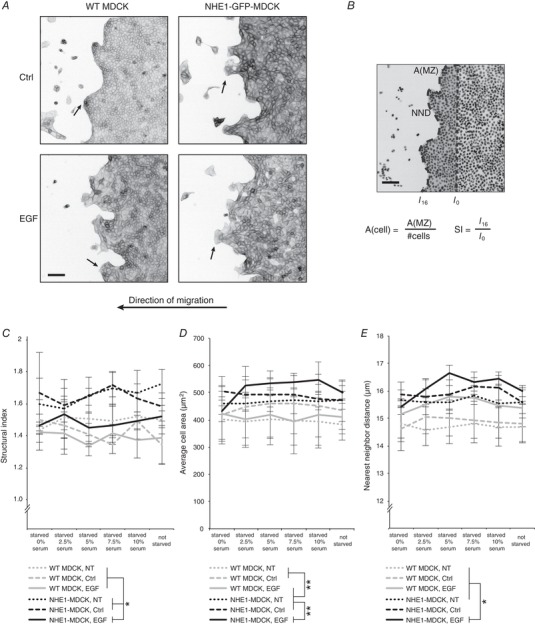
A, collectively migrating cells were treated with indicated serum concentrations, as well as EGF, a control solvent (Ctrl) or no treatment (NT). The cells were fixed after 16 h and stained with phalloidin‐rhodamine to visualize actin. Image examples are from cells treated with 5% serum. Arrows indicate examples of migration fingers. Actin is shown as an inverted contrast image. B, quantification of cell sheet morphology was based on Hoechst labelling of the cell sheets (shown as inverted contrast). NIS Elements software was employed to measure the length of the wound edge in images from 0 h and 16 h. Structural index (SI) was obtained as a ratio between these numbers, reflecting the level of migration finger formation. Automated spot detection was used to identify all Hoechst‐stained nuclei. The average cell size was then calculated by dividing the migration zone (MZ) by the number of cells. The Nearest Neighbor Distance (NND) could be directly measured in the software. Scale bars = 100 μm. C, treatment with EGF did not change the structural index significantly on WT MDCK cells. NHE1‐MDCK cells had a significantly higher structural index than WT MDCK cells. However, treatment with EGF decreased the structural index to the same level as WT MDCK cells. D and E, relative cell area and NND are both measures of the spacing between cell nuclei. Both measures showed that NHE1‐MDCK cells were more spaced than WT MDCK cells. Moreover, treatment with EGF increased the spacing significantly. All data were analysed using three‐way ANOVA and Tukey's post hoc test. * P < 0.01, ** P < 0.001 (n = 3). Error bars represent the SEM.
We therefore next investigated how NHE1 affected cell polarization in 3D and the behaviour of cell collectives, using a 3D cyst forming assay (Debnath et al. 2003). We found that, in 3D cultures, WT MDCK cells formed regular cysts with a central lumen and characteristic apical actin bands. By contrast, NHE1‐MDCK cysts were disorganized, lacked a central lumen and did not form apical actin bands (Fig. 6). Because there was no difference in the proliferation level of the two cell lines, as well as no difference in apoptosis levels in the cysts (data not shown), these differences may be attributed to differential regulation of organization of cell collectives upon overexpression of NHE1.
Figure 6. Polarity disruption in MDCK cysts overexpressing NHE1.
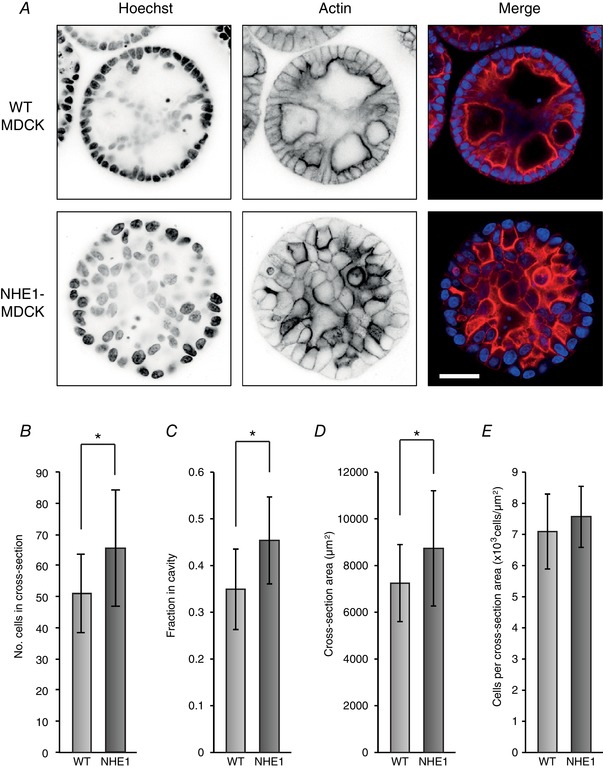
NHE1‐MDCK cells and WT MDCK cells were grown as cysts for 8 days. The cysts were then fixed and stained with Hoechst to visualize cell nuclei, as well as phalloidin to visualize actin. A, in cysts of WT MDCK cells, most cells were organized as a sphere with apical actin strands and cavities, although with some cells within the cyst. NHE1‐MDCK cysts were less organized with no or few cavities and no clear apical actin bands. B–E, cysts of NHE1‐MDCK cells contained more cells (B) and were larger (D), and more cells were localized in the cavity (C). However, there were similar numbers of cells relative to the size (E); n = 30 cysts (WT MDCK) and n = 28 cysts (NHE1‐MDCK) were included. * P < 0.05 using Student's t test. [Color figure can be viewed at wileyonlinelibrary.com]
Together, these experiments demonstrate that NHE1 expression increases the average area covered by each migrating cell and thus the cell spacing. Overexpressed NHE1 also created more and irregular migration fingers, detected as an increase in structural index. Treatment with EGF further increased the average area covered by each cell but decreased the formation of migration fingers.
Time‐lapse detection of migrating NHE1‐MDCK cells
Given that NHE1 and EGF treatment had differential effects on the morphology of the cell sheets, we next investigated whether the behaviour of individual cells within the monolayer was also differentially affected.
To assess this, we performed live cell imaging of collectively migrating cells. We used Hoechst labelling of cell nuclei and, subsequently, automated tracking to identify and follow all cells during collective movement (Fig. 7 A; see Supporting information, Movies S2 and S3). First, migration fingers and leader cells were identified, and leader cell tracks were extracted and the average displacement over time was calculated (Fig. 7 B). Analysis demonstrated that there was no effect of NHE1 on leader cell movement, whereas EGF treatment led to significantly decreased leader cell displacement (Fig. 7 C). To evaluate how persistent individual cells were in their direction of movement (directional persistence index), the total displacement relative to the total track length was calculated (Fig. 7 D). Neither NHE1, nor EGF treatment affected the directional persistence of these cells. Thus, the potentiated collective migration of MDCK cells by NHE1 and EGF was not reflected in the movement of leader cells.
Figure 7. Live collective migration showed that increased collective movement was not leader cell‐driven.
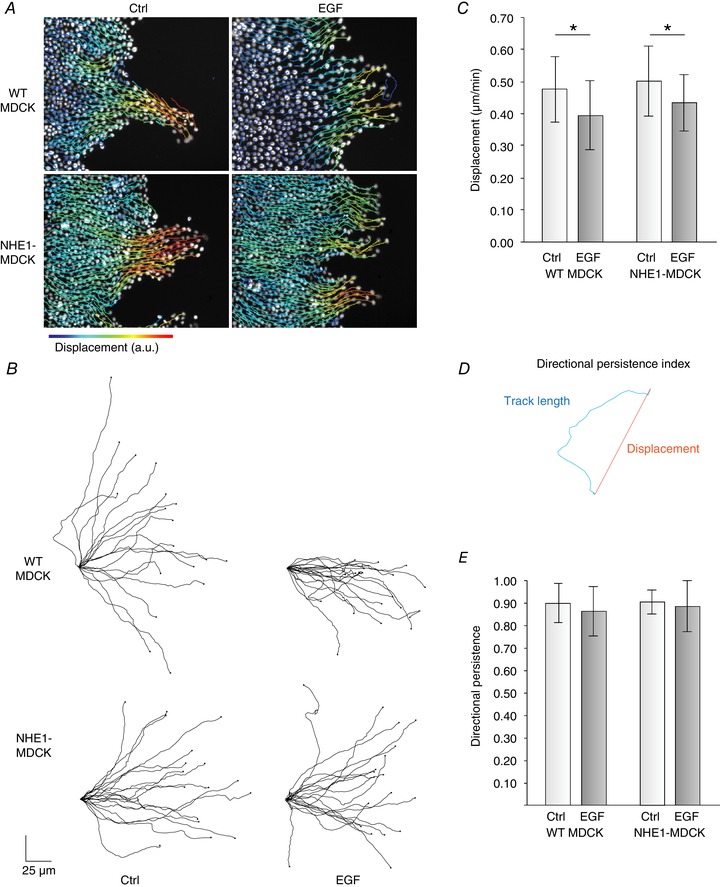
WT MDCK and NHE1‐MDCK cells were allowed to migrate in a wound healing assay overnight in the presence of EGF or control (Ctrl) medium. The cells were then loaded with Hoechst and imaged using fluorescence microscopy at 37°C and 5% CO2. A, individual cells were tracked using the Trackmate plugin in FIJI. The complete movies are provided in the Supporting information (Movie S3). B, leader cells were identified and tracks were extracted. C, net displacement of leader cells was not different between WT MDCK cells and MDCK cells overexpressing NHE1. By contrast, EGF treatment significantly decreased leader cell displacement. D, directional persistence was evaluated using an index, where the total track length was divided by the displacement. E, there was no effect of EGF treatment and NHE1 overexpression on the directionality of the leader cells. * P < 0.005 using two‐way ANOVA. Error bars represent the SD. Three individual experiments were performed and 18 (WT MDCK, Ctrl), 18 (WT MDCK, EGF), 18 (NHE1‐MDCK, Ctrl), and 19 (NHE1‐MDCK, EGF) leader cells were included. [Color figure can be viewed at wileyonlinelibrary.com]
Because both NHE1 overexpression and EGF treatment were associated with altered organization of the cell sheets, we investigated how the movement of other cells in the sheet was affected. We identified cells that were in the front of the monolayers but were neither leader cells, nor part of defined migration fingers (Fig. 8 A). Treatment with EGF significantly increased the displacement and the directional persistence of non‐leader cells in the sheet front (Fig. 8 B and C), whereas there was no difference between WT MDCK cells and NHE1‐MDCK cells (Fig. 8 B and C). Thus, upon treatment with EGF, MDCK monolayers formed fewer migration fingers, and cells outside the fingers were more motile.
Figure 8. Overexpressed NHE1 and EGF increased sheet movement differently.
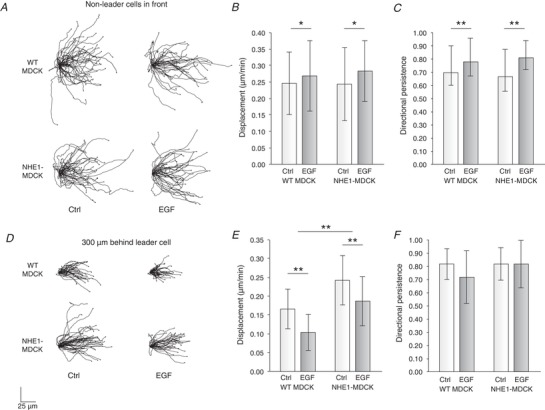
A, cells located in the front of the cell sheet, which were not leader cells, were identified. In these cells, the displacement (B) and directional persistence (C) were significantly increased by EGF treatment, whereas there was no difference between WT MDCK and NHE1‐MDCK cells. From three experiments, 47 (WT MDCK, Ctrl), 41 (WT MDCK, EGF), 38 (NHE1‐MDCK, Ctrl) and 39 (NHE1‐MDCK, EGF) cells were used. D, cells located 300 μm behind the leader cells were identified. E, in these cells, the overexpressed NHE1 contributed to increased overall displacement, whereas it was decreased by EGF treatment. F, there was no effect of overexpressed NHE1 or EGF on directional persistence. From three experiments, 35 (WT MDCK, Ctrl), 37 (WT MDCK, EGF), 42 (NHE1‐MDCK, Ctrl) and 35 (NHE1‐MDCK, EGF) cells were used. Error bars represent the SD. * P < 0.05 and ** P < 0.001 using two‐way ANOVA.
We next identified and extracted track information from cells located in the cell sheets, 300 μm behind the leader cells (Fig. 8 C). Here, the cellular displacement was significantly higher for NHE1‐MDCK cells compared to WT MDCK cells, whereas EGF treatment decreased the cellular displacement (Fig. 8 D), with no effect on directional persistence (Fig. 8 E).
Thus, EGF increased displacement of non‐leader cells in the front of the sheet, and NHE1 increased overall motility throughout the cell sheet behind the leader cells.
Discussion
Collective migration is a widespread principle during epithelial morphogenesis in embryonic development and wound healing, as well as being common in pathologies including cancer (Jenkins et al. 2012; Grillo‐Hill et al. 2015; Hofschröer et al. 2017). NHE1 is a major driver of single cell migration in many cell types (Lagana et al. 2000; Schneider et al. 2008; Martin et al. 2011), has been assigned a role in mammary epithelial morphogenesis (Jenkins et al. 2012) and is developmentally regulated in the kidney (Rieder & Fliegel, 2002). A role of NHE1 in collective migration of head and neck squamous cell carcinoma cells was recently suggested, albeit in cell clusters and without detailed analysis (Kaminota et al. 2017). We therefore hypothesized that NHE1 upregulation may stimulate collective epithelial cell migration and, in this way, contribute to kidney epithelial morphogenesis and other processes involving epithelial collective cell migration.
The key findings of the present study are that NHE1 localized both to leading edge lamellipodia and to cryptic lamellipodia in submarginal MDCK cell sheets. Increased NHE1 expression was observed to increase collective migration speed not by affecting the front row of cells but, instead, by increasing displacement of the submarginal rows. These observations are important for the understanding of NHE1 in the context of embryonic development and epithelial organization, as well as in cancer. NHE1 is already known to affect the behaviour of individual cells, although the present study highlights the importance of understanding the role of NHE1 in cell collectives.
The localization of NHE1 in the leading edge of single migrating cells is well established (Klein et al. 2000; Schneider et al. 2009; Martin et al. 2011). In the present study, we now show that NHE1 also localizes in cryptic lamellipodia of multiple rows of cells behind the wound edge, which is in line with this observation and suggests that NHE1 is an important and general driver of movement of all cells in the collective. The mechanisms through which polarization in collectively migrating sheets occur are complex, in part cell‐type specific, and involve both mechanical forces and signalling by numerous intracellular signals (Haeger et al. 2015). Our prior work identified Akt and ERK signalling as being important for leading edge NHE1 localization in single migrating fibroblasts (Clement et al. 2013) and future studies should assess whether this is also the case in collectively migrating epithelial cells.
By using TIRF microscopy and GFP‐tagging to obtain high‐resolution images of basal NHE1 localization, we could identify it as clearly localized in clusters or puncta, which co‐localized with actin in acid loaded or migrating cells but not in resting cells. This suggests that NHE1–actin interactions are dynamically regulated, perhaps via the binding of NHE1 to the ezrin/radixin/moesin complex (Denker et al. 2000). Also consistent with a dynamic interaction of NHE1 with cytoskeletal elements, NHE1 localized to membrane ruffles in T47D cells but did not colocalize with focal adhesions in these cells (Pedraz‐Cuesta et al. 2016). In migrating melanoma cells, NHE1 localization was increased at focal adhesions in the leading edge but not in the trailing part (Ludwig et al. 2013). NHE1 is known to function as a dimer in the membrane (Fafournoux et al. 1994; Counillon et al. 2016). We do not know the nature of the NHE1 clusters, although dynamic regulation in the plasma membrane in response to physiological stimuli (Marlar et al. 2014; Koffman et al. 2015; Arnspang et al. 2016) and higher‐order assemblies of transporters is a known phenomenon (Yang et al. 1996; Subach et al. 2009; Rossi et al. 2012; Gao et al. 2017) and has also been proposed for the brush‐border NHE (presumably NHE3).
What is the mechanism underlying the observed effects of NHE1 on collective cell migration? A role for NHE1 in cell–cell adhesion and epithelial organization has recently been proposed (Grillo‐Hill et al. 2015; Hofschröer et al. 2017) and would be consistent our findings. NHE1 expression increased steady‐state pHi in MDCK cells; hence, a role for NHE1‐induced alkalinization is probable, given multiple previous studies showing important roles for pHi in regulation of cytoskeletal dynamics and cell motility (Frantz et al. 2008; Schwab et al. 2012; Webb et al. 2016) In addition, the effects of NHE1 may involve scaffolding effects related to the physical interaction of NHE1 with cytoskeletal elements. Although not addressed further in the present study, a dual role of NHE1 as a pH regulator and physical cytoskeletal scaffold has been demonstrated in a previous study showing that a transport‐dead NHE1 mutant retains partial effect on single cell motility (Denker & Barber, 2002). Although the precise roles of cryptic lamellipodia in generating traction forces to move the collective is debated, it is clear that, similar to the leading edge lamellipodia, they interact with the substratum and can contribute to these forces (Haeger et al. 2015). Thus, the roles of NHE1 in polarization and adhesion control in the lamellipodia of submarginal cell rows are probably similar to those in the front edge of the sheet. It is possible that NHE1 may additionally contribute to epithelial sheet organization and cell–cell interactions in the moving sheet. In support of such a model, NHE1‐MDCK cells were less organized, had increased distances between nuclei and formed more migration fingers. NHE1‐expressing cells also formed dys‐organized cysts with poor formation of apical actin strands and cavity formation, in excellent agreement with recent reports of NHE1‐mediated morphological disorganization in melanoma cells (Hofschröer et al. 2017) and in Drosophila (Grillo‐Hill et al. 2015).
EGF receptor family signalling plays central roles in kidney development and physiology (Zeng et al. 2009) and contributes to pathological conditions such as renal fibrosis (Zeng et al. 2009; Zhuang & Liu, 2014), which can ultimately lead to chronic kidney failure. Upon treatment with EGF, MDCK cells were less confined to migration fingers, and cells in the front of the sheets migrated more independently. This is consistent with reports suggesting that EGF‐stimulated cells have a greater probability of adopting leader‐cell morphologies and features of epithelial‐to‐mesenchymal transformation (Lo et al. 2007; Khalil & Friedl, 2010). In many cell types, NHE1 is activated by EGF (Maly et al. 2002; Coaxum et al. 2009) and NHE1‐dependent cancer cell migration has been reported to be accelerated by EGF (Chiang et al. 2008; Cardone et al. 2015). Importantly, however, in the present study, we show that, although EGF and NHE1 expression both stimulated collective cell migration, they did so via separate mechanisms, with NHE1 primarily increasing displacement of cells in submarginal rows. This observation indicates that regulation and roles of NHE1 in collective and single cell migration, although sharing several characteristics, are not identical.
Conclusions
The present study shows that NHE1 localizes not only to the front of collectively migrating kidney epithelial cells, but also to cryptic lamellipodia of submarginal cell rows, where it was found in distinct membraneous clusters. The present study identifies NHE1 as an important overall driver of collective migration, acting via increased collective movement by increasing the speed of follower cells. EGF stimulation also increased collective migration but by stimulating the motility of cells at the wound edge. Our results have relevance for the role of NHE1 in development and morphogenesis of normal epithelial cells, as well as for pathological conditions characterized by increased collective migration.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
LNN and SFP conceived and designed the project. LNN, SFP and MP supervised the project. HHJ, GAP and JJM carried out the experiments. HHJ analysed the data. HHJ wrote the manuscript with inputs and comments from LNN and SFP. pHi measurements were performed at the Department of Biology, Section for Cell Biology and Physiology, University of Copenhagen, Denmark. Cyst culturing was performed at Randall Division of Cell and Molecular Biophysics, King's College London, UK. All other experiments were performed at the Department of Clinical Medicine and Department of Molecular Biology and Genetics, Aarhus University, Denmark. All authors have seen, commented and approved the manuscript submitted for publication. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All who qualify for authorship are included as authors, and all authors listed had qualified contributions. We thank Katrine Franklin Mark for excellent technical assistance and Signe H. Kramer for help with real time imaging of pHi.
Funding
This work was supported by a Lundbeck Junior Group Leader Fellowship to LNN from the Lundbeck Foundation, by the Graduate School of Science and Technology (HHJ) and by a Novo Nordisk Foundation grant to SFP (NNF16OC0023194). The Nikon microscope was funded by the Lundbeck Foundation, the Carlsberg Foundation and MEMBRANES (Aarhus University, Denmark).
Supporting information
Movie S1. NHE1 clusters moved fast in the TIRF zone. Crop of a single non‐migrating NHE1‐MDCK cell imaged using TIRF microscopy of GFP fused to NHE1. The movie was acquired at 10 fps and is shown at the same speed in the time‐lapse presentation. The movie is shown as inverted contrast.
Movie S2. Time‐lapse imaging of collectively migrating cells. WT MDCK and NHE1‐MDCK cells were treated with EGF or control medium and loaded with Hoechst. The cells were imaged every 5 min during collective cell migration. Scale bar = 300 μm.
Movie S3. Live tracking of collectively migrating cells. WT MDCK and NHE1‐MDCK cells were treated with EGF or control medium and loaded with Hoechst. The cells were imaged during collective cell migration. Individual cells were tracked using the FIJI‐plugin TrackMate. The tracks are colour‐coded according to displacement. The same example is shown in Fig. 7A. Scale bar = 100 μm.
Biography
Helene H. Jensen is a molecular cell biologist with a strong interest in membrane transport proteins and their involvement in disease. She obtained her PhD degree under the supervision of Associate Professor Lene N. Nejsum, at Aarhus University, Denmark. The research group investigates why and how ion and aquaporin water transporters contribute to epithelial motility and can accelerate cancer development and metastases. The present study expands our understanding of membrane transporters being important (and in some cases surprising) players in these processes. Further studies may thus explore their role in cell‐cell interactions, three‐dimensional cell complexes, and their potential as drug targets or biomarkers. Helene H. Jensen is currently working as a post doctoral fellow at Aalborg University, Denmark, under the supervision of Professor Michael Toft Overgaard. Here, she utilizes her expertise in cell biology and fluorescence microscopy to investigate how mutations in the Ca2+‐sensor calmodulin affect Ca2+ transport and cellular health.

Edited by: Kim Barrett & Ruth Murrell‐Lagnado
Linked articles: This article is highlighted in a Perspectives article by Roger. To read this article, visit https://doi.org/10.1113/JP277472.
References
- Arnspang E, Login F, Koffman J, Sengupta P & Nejsum L (2016). AQP2 plasma membrane diffusion is altered by the degree of AQP2‐S256 phosphorylation. Int J Mol Sci 17, 1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin JM, Kwiatkowski AV., Yang C, Korobova F, Pokutta S, Svitkina T, Weis WI & Nelson WJ (2010). AlphaE‐catenin regulates actin dynamics independently of cadherin‐mediated cell‐cell adhesion. J Cell Biol 189, 339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boedtkjer E, Bunch L & Pedersen SF (2012). Physiology, pharmacology and pathophysiology of the pH regulatory transport proteins NHE1 and NBCn1: similarities, differences, and implications for cancer therapy. Curr Pharm Des 18, 1345–1371. [DOI] [PubMed] [Google Scholar]
- Cardone RA, Greco MR, Zeeberg K, Zaccagnino A, Saccomano M, Bellizzi A, Bruns P, Menga M, Pilarsky C, Schwab A, Alves F, Kalthoff H, Casavola V & Reshkin SJ (2015). A novel NHE1‐centered signaling cassette drives epidermal growth factor receptor‐dependent pancreatic tumor metastasis and is a target for combination therapy. Neoplasia 17, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang G, Wang J, Zhang H, Zhang Y, Wang C, Xu H, Zhang H, Lin Y, Ma L, Li Q & Pang T (2014). CD44 targets Na+/H– exchanger 1 to mediate MDA‐MB‐231 cells’ metastasis via the regulation of ERK1/2. Br J Cancer 110, 916–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang Y, Chou C‐Y, Hsu K‐F, Huang Y‐F & Shen M‐R (2008). EGF upregulates Na+/H+ exchanger NHE1 by post‐translational regulation that is important for cervical cancer cell invasiveness. J Cell Physiol 214, 810–819. [DOI] [PubMed] [Google Scholar]
- Choi C H, Webb BA, Chimenti MS, Jacobson MP & Barber DL (2013). pH sensing by FAK‐His58 regulates focal adhesion remodeling. J Cell Biol 202, 849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement DL, Mally S, Stock C, Lethan M, Satir P, Schwab A, Pedersen SF & Christensen ST (2013). PDGFR signaling in the primary cilium regulates NHE1‐dependent fibroblast migration via coordinated differential activity of MEK1/2‐ERK1/2‐p90RSK and AKT signaling pathways. J Cell Sci 126, 953–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coaxum SD, Garnovskaya MN, Gooz M, Baldys A & Raymond JR (2009). Epidermal growth factor activates Na+/H+ exchanger in podocytes through a mechanism that involves Janus kinase and calmodulin. Biochim Biophys Acta – Mol Cell Res 1793, 1174–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counillon L, Bouret Y, Marchiq I & Pouysségur J (2016). Na+/H+ antiporter (NHE1) and lactate/H+ symporters (MCTs) in pH homeostasis and cancer metabolism. Biochim Biophys Acta – Mol Cell Res 1863, 2465–2480. [DOI] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK & Brugge JS (2003). Morphogenesis and oncogenesis of MCF‐10A mammary epithelial acini grown in three‐dimensional basement membrane cultures. Methods 30, 256–268. [DOI] [PubMed] [Google Scholar]
- Denker SP & Barber DL (2002). Cell migration requires both ion translocation and cytoskeletal anchoring by the Na‐H exchanger NHE1. J Cell Biol 159, 1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denker SP, Huang DC, Orlowski J, Furthmayr H & Barber DL (2000). Direct binding of the Na‐H exchanger NHE1 to ERM proteins regulates the cortical cytoskeleton and cell shape independently of H+ translocation. Mol Cell 6, 1425–1436. [DOI] [PubMed] [Google Scholar]
- Fafournoux P, Noël J & Pouysségur J (1994). Evidence that Na+/H+ exchanger isoforms NHE1 and NHE3 exist as stable dimers in membranes with a high degree of specificity for homodimers. J Biol Chem 269, 2589–2596. [PubMed] [Google Scholar]
- Farooqui R & Fenteany G (2005). Multiple rows of cells behind an epithelial wound edge extend cryptic lamellipodia to collectively drive cell‐sheet movement. J Cell Sci 118, 51–63. [DOI] [PubMed] [Google Scholar]
- Frantz C, Barreiro G, Dominguez L, Chen X, Eddy R, Condeelis J, Kelly MJS, Jacobson MP & Barber DL (2008). Cofilin is a pH sensor for actin free barbed end formation: role of phosphoinositide binding. J Cell Biol 183, 865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz C, Karydis A, Nalbant P, Hahn KM & Barber DL (2007). Positive feedback between Cdc42 activity and H+ efflux by the Na‐H exchanger NHE1 for polarity of migrating cells. J Cell Biol 179, 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P & Mayor R (2017). Tuning collective cell migration by cell‐cell junction regulation. Cold Spring Harb Perspect Biol 9, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Chen J, Gao J, Wang H & Xiong W (2017). Super‐resolution microscopy reveals the insulin‐resistance‐regulated reorganization of GLUT4 on plasma membranes. J Cell Sci 130, 396–405. [DOI] [PubMed] [Google Scholar]
- Gaush CR, Hard WL & Smith TF (1966). Characterization of an established line of canine kidney cells (MDCK). Proc Soc Exp Biol Med 122, 931–935. [DOI] [PubMed] [Google Scholar]
- Grillo‐Hill BK, Choi C, Jimenez‐Vidal M & Barber DL (2015). Increased H+ efflux is sufficient to induce dysplasia and necessary for viability with oncogene expression. Elife 2015, 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeger A, Wolf K, Zegers MM & Friedl P (2015). Collective cell migration: guidance principles and hierarchies. Trends Cell Biol 25, 556–566. [DOI] [PubMed] [Google Scholar]
- Hofschröer V, Koch KA, Ludwig FT, Friedl P, Oberleithner H, Stock C & Schwab A (2017). Extracellular protonation modulates cell‐cell interaction mechanics and tissue invasion in human melanoma cells. Sci Rep 7, 42369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins EC, Debnath S, Gundry S, Gundry S, Uyar U & Fata JE (2012). Intracellular pH regulation by Na+/H+ exchanger‐1 (NHE1) is required for growth factor‐induced mammary branching morphogenesis. Dev Biol 365, 71–81. [DOI] [PubMed] [Google Scholar]
- Jensen HH, Login FH, Koffman JS, Kwon T‐H & Nejsum LN (2016). The role of aquaporin‐5 in cancer cell migration: a potential active participant. Int J Biochem Cell Biol 79, 271–276. [DOI] [PubMed] [Google Scholar]
- Kaminota T, Yano H, Shiota K, Nomura N, Yaguchi H, Kirino Y, Ohara K, Tetsumura I, Sanada T, Ugumori T, Tanaka J & Hato N (2017). Elevated Na+ /H+ exchanger‐1 expression enhances the metastatic collective migration of head and neck squamous cell carcinoma cells. Biochem Biophys Res Commun 486, 101–107. [DOI] [PubMed] [Google Scholar]
- Khalil AA & Friedl P (2010). Determinants of leader cells in collective cell migration. Integr Biol 2, 568. [DOI] [PubMed] [Google Scholar]
- Khalil AA, Ilina O, Gritsenko PG, Bult P, Span PN & Friedl P (2017). Collective invasion in ductal and lobular breast cancer associates with distant metastasis. Clin Exp Metastasis 34, 421–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein M, Seeger P, Schuricht B, Alper SL & Schwab A (2000). Polarization of Na+/H+ and Cl–/HCO3 – exchangers in migrating renal epithelial cells. J Gen Physiol 115, 599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffman JS, Arnspang EC, Marlar S & Nejsum LN (2015). Koffman JS, Arnspang EC, Marlar S & Nejsum LN. Opposing effects of cAMP and T259 phosphorylation on plasma membrane diffusion of the water channel aquaporin‐5 in Madin–Darby canine kidney cells. 1. Opposing effects of cAMP and T259 phosphorylation on plasma. PLoS ONE 10, e0133324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagana A, Vadnais J, Le P, Nguyen T, Laprade R, Nabi I & Noel J (2000). Regulation of the formation of tumor cell pseudopodia by the Na(+)/H(+) exchanger NHE1. J Cell Sci 113, 3649–3662. [DOI] [PubMed] [Google Scholar]
- Lauritzen G, Jensen F, Boedtkjer E, Dybboe R, Aalkjaer C, Nylandsted J & Pedersen SF (2010). NBCn1 and NHE1 expression and activity in ΔNErbB2 receptor‐expressing MCF‐7 breast cancer cells: contributions to pHi regulation and chemotherapy resistance. Exp Cell Res 316, 2538–2553. [DOI] [PubMed] [Google Scholar]
- Lo H‐W, Hsu S‐C, Xia W, Cao X, Shih J‐Y, Wei Y, Abbruzzese JL, Hortobagyi GN & Hung M‐C (2007). Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial‐mesenchymal transition in cancer cells via up‐regulation of TWIST gene expression. Cancer Res 67, 9066–9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louvard D (1980). Apical membrane aminopeptidase appears at site of cell‐cell contact in cultured kidney epithelial cells. Proc Natl Acad Sci U S A 77, 4132–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig FT, Schwab A & Stock C (2013). The Na+/H+‐exchanger (NHE1) generates pH nanodomains at focal adhesions. J Cell Physiol 228, 1351–1358. [DOI] [PubMed] [Google Scholar]
- Magalhaes MAO, Larson DR, Mader CC, Bravo‐Cordero JJ, Gil‐Henn H, Oser M, Chen X, Koleske AJ & Condeelis J (2011). Cortactin phosphorylation regulates cell invasion through a pH‐dependent pathway. J Cell Biol 195, 903–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maly K, Strese K, Kampfer S, Ueberall F, Baier G, Ghaffari‐Tabrizi N, Grunicke HH & Leitges M (2002). Critical role of protein kinase C α and calcium in growth factor induced activation of the Na+/H+ exchanger NHE1. FEBS Lett 521, 205–210. [DOI] [PubMed] [Google Scholar]
- Marlar S, Arnspang EC, Koffman JS, Locke EM, Christensen BM & Nejsum LN (2014). Elevated cAMP increases aquaporin‐3 plasma membrane diffusion. Am J Physiol Cell Physiol 306, C598–C606. [DOI] [PubMed] [Google Scholar]
- Martin C, Pedersen SF, Schwab A & Stock C (2011). Intracellular pH gradients in migrating cells. Am J Physiol Cell Physiol 300, C490–C495. [DOI] [PubMed] [Google Scholar]
- Mayor R & Etienne‐Manneville S (2016). The front and rear of collective cell migration. Nat Rev Mol Cell Biol 17, 97–109. [DOI] [PubMed] [Google Scholar]
- Orlowski J & Grinstein S (2004). Diversity of the mammalian sodium/proton exchanger SLC9 gene family. Pflugers Arch Eur J Physiol 447, 549–565. [DOI] [PubMed] [Google Scholar]
- Palmyre A, Lee J, Ryklin G, Camarata T, Selig MK, Duchemin AL, Nowak P, Arnaout MA, Drummond IA & Vasilyev A (2014). Collective epithelial migration drives kidney repair after acute injury. PLoS ONE 9, e101304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen GA, Jensen HH, Schelde A‐SB, Toft C, Pedersen HN, Ulrichsen M, Login FH, Amieva MR & Nejsum LN (2017). The basolateral vesicle sorting machinery and basolateral proteins are recruited to the site of enteropathogenic E. coli microcolony growth at the apical membrane ed. Valenti G. PLoS ONE 12, e0179122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedraz‐Cuesta E, Fredsted J, Jensen HH, Bornebusch A, Nejsum LN, Kragelund BB & Pedersen SF (2016). Prolactin signaling stimulates invasion via Na(+)/H(+) exchanger NHE1 in T47D human breast cancer cells. Mol Endocrinol 30, 693–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poujade M, Grasland‐Mongrain E, Hertzog A, Jouanneau J, Chavrier P, Ladoux B, Buguin A & Silberzan P (2007). Collective migration of an epithelial monolayer in response to a model wound. Proc Natl Acad Sci U S A 104, 15988–15993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CV & Fliegel L (2002). Developmental regulation of Na+/H+ exchanger expression in fetal and neonatal mice. Am J Physiol Circ Physiol 283, H273–H283. [DOI] [PubMed] [Google Scholar]
- Rossi A, Moritz TJ, Ratelade J & Verkman AS (2012). Super‐resolution imaging of aquaporin‐4 orthogonal arrays of particles in cell membranes. J Cell Sci 125, 4405–4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L, Klausen TK, Stock C, Mally S, Christensen ST, Pedersen SF, Hoffmann EK & Schwab A (2008). H‐ras transformation sensitizes volume‐activated anion channels and increases migratory activity of NIH3T3 fibroblasts. Pflugers Arch 455, 1055–1062. [DOI] [PubMed] [Google Scholar]
- Schneider L, Stock CM, Dieterich P, Jensen BH, Pedersen LB, Satir P, Schwab A, Christensen ST & Pedersen SF (2009). The Na+/H+ exchanger NHE1 is required for directional migration stimulated via PDGFR‐α in the primary cilium. J Cell Biol 185, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab A, Fabian A, Hanley PJ & Stock C (2012). Role of ion channels and transporters in cell migration. Physiol Rev 92, 1865–1913. [DOI] [PubMed] [Google Scholar]
- Stock C, Mueller M, Kraehling H, Mally S, Noël J, Eder C & Schwab A (2008). pH nanoenvironment at the surface of single melanoma cells. Cell Physiol Biochem 20, 679–686. [DOI] [PubMed] [Google Scholar]
- Stock C & Pedersen SF (2017). Roles of pH and the Na+/H+ exchanger NHE1 in cancer: from cell biology and animal models to an emerging translational perspective? Semin Cancer Biol 43, 5–16. [DOI] [PubMed] [Google Scholar]
- Stroka KM, Jiang H, Chen SH, Tong Z, Wirtz D, Sun SX & Konstantopoulos K (2014). Water permeation drives tumor cell migration in confined microenvironments. Cell 157, 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subach FV, Patterson GH, Manley S, Gillette JM, Lippincott‐Schwartz J & Verkhusha V. V (2009). Photoactivatable mCherry for high‐resolution two‐color fluorescence microscopy. Nat Methods 6, 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez JY, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL & Eliceiri KW (2017). TrackMate: an open and extensible platform for single‐particle tracking. Methods 115, 80–90. [DOI] [PubMed] [Google Scholar]
- Vasilyev A, Liu Y, Mudumana S, Mangos S, Lam P‐Y, Majumdar A, Zhao J, Poon K‐L, Kondrychyn I, Korzh V & Drummond IA (2009). Collective cell migration drives morphogenesis of the kidney nephron. PLoS Biol 7, e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb BA, White KA, Grillo‐Hill BK, Schönichen A, Choi C & Barber DL (2016). A histidine cluster in the cytoplasmic domain of the Na‐H exchanger NHE1 confers pH‐sensitive phospholipid binding and regulates transporter activity. J Biol Chem 291, 24096–24104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Brown D & Verkman AS (1996). The mercurial insensitive water channel (AQP‐4) forms orthogonal arrays in stably transfected Chinese hamster ovary cells. J Biol Chem 271, 4577–4580. [PubMed] [Google Scholar]
- Yeaman C, Grindstaff KK & Nelson WJ (2004). Mechanism of recruiting Sec6/8 (exocyst) complex to the apical junctional complex during polarization of epithelial cells. J Cell Sci 117, 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng F, Singh AB & Harris RC (2009). The role of the EGF family of ligands and receptors in renal development, physiology and pathophysiology. Exp Cell Res 315, 602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang S & Liu N (2014). EGFR signaling in renal fibrosis. Kidney Int Suppl 4, 70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. NHE1 clusters moved fast in the TIRF zone. Crop of a single non‐migrating NHE1‐MDCK cell imaged using TIRF microscopy of GFP fused to NHE1. The movie was acquired at 10 fps and is shown at the same speed in the time‐lapse presentation. The movie is shown as inverted contrast.
Movie S2. Time‐lapse imaging of collectively migrating cells. WT MDCK and NHE1‐MDCK cells were treated with EGF or control medium and loaded with Hoechst. The cells were imaged every 5 min during collective cell migration. Scale bar = 300 μm.
Movie S3. Live tracking of collectively migrating cells. WT MDCK and NHE1‐MDCK cells were treated with EGF or control medium and loaded with Hoechst. The cells were imaged during collective cell migration. Individual cells were tracked using the FIJI‐plugin TrackMate. The tracks are colour‐coded according to displacement. The same example is shown in Fig. 7A. Scale bar = 100 μm.
Data Availability Statement
Data are available from the authors upon request.