

Abstract
Background
Glycogen synthase kinase-3β (GSK3β) is a key regulator of cellular homeostasis. In neurons, GSK3β contributes to the control of neuronal transmission and plasticity, but its role in epilepsy remains to be defined.
Methods
Biochemical and electrophysiological methods were used to assess the role of GSK3β in regulating neuronal transmission and epileptogenesis. GSK3β activity was increased genetically in GSK3β[S9A] mice. Its effects on neuronal transmission and epileptogenesis induced by kainic acid were assessed by field potential recordings in mice brain slices and video electroencephalography in vivo. The ion channel expression was measured in brain samples from mice and followed by analysis in samples from patients with temporal lobe epilepsy or focal cortical dysplasia in correlation to GSK3β phosphorylation.
Findings
Higher GSK3β activity decreased the progression of kainic acid induced epileptogenesis. At the biochemical level, higher GSK3β activity increased the expression of hyperpolarization-activated cyclic nucleotide-gated (HCN) channel 4 under basal conditions and in the epileptic mouse brain and decreased phosphorylation of the glutamate α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor subunit GluA1 at Serine 831 under basal conditions. Moreover, we found a significant correlation between higher inhibitory GSK3β phosphorylation at Serine 9 and higher activating GluA1 phosphorylation at Serine 845 in brain samples from epileptic patients.
Interpretation
Our data imply GSK3β activity in the protection of neuronal networks from hyper-activation in response to epileptogenic stimuli and indicate that the anti-epileptogenic function of GSK3β involves modulation of HCN4 level and the synaptic AMPA receptors pool.
Keywords: Glycogen synthase kinases-3, GSK3, Epilepsy, AMPA receptors, GluA1 phosphorylation
Research in context.
Evidence before this study
Glycogen synthase kinase-3 α (GSK3α) and GSK3β are key homeostatic kinases that regulate many cellular processes including neuronal synaptic plasticity. Aberrant neuronal plasticity often occurs in neuropathologies, e.g. epilepsy. However, the relationship between GSK3 and epileptogenesis is not clear and available data are contradictory. Based on our previous finding that mice deficient in neuronal GSK3β are characterized by poor survival in response to the proconvulsive drug, we hypothesized that GSK3β controls neuronal excitability and epileptogenesis.
Added value of this study
In this study, we provided evidence supporting our hypothesis. We showed that mice that overexpressed a constitutively active form of GSK3β in the brain developed fewer spontaneous seizures following status epilepticus. As for a potential mechanism we indicated changes in expression of HCN4 channel and activating phosphorylation of AMPA receptor subunit GluA1 which should result in decreased excitability of neuronal networks. Finally, in brain samples from epileptic patients, we found the correlation that higher activating GluA1 phosphorylation was accompanied by higher inhibitory GSK3β phosphorylation.
Implications of all the available evidence
Our study brings an important, clinically relevant observation that under certain circumstances GSK3β acts to slow down epileptogenesis. This data are also of vital importance due to the recent proposal of GSK3 inhibitors use for epilepsy treatment.
Alt-text: Unlabelled Box
1. Introduction
Epilepsy results from abnormal excessive and synchronous activity of brain neuronal networks. Temporal lobe epilepsy (TLE) is one of the most common epilepsies in adults, involving pathological alterations in the hippocampal formation [1]. Structural abnormalities of the cerebral cortex (e.g., focal cortical dysplasia [FCD]) are increasingly recognized as important causes of severe, often intractable seizures. Epileptogenesis is the process that precedes the occurrence of epilepsy but continues also after the diagnosis and includes epilepsy progression [2,3]. During this process several cellular and structural changes occur in response to the primary cause, including head trauma, status epilepticus, infections, and genetics. In humans, this latent period can take months or even years.
Exogenously triggered epileptogenesis can be mimicked in animals by electrical or chemical brain stimulation using chemoconvulsants (e.g., kainic acid [KA]), which leads to an initial insult that triggers epileptogenesis [4]. These models are useful for identifying signaling pathways that potentially contribute to the development or manifestation of epilepsy. For example, phosphorylation of the glutamate GluA1 subunit of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) at Serine 831 (S831) and S845, increased the sensitivity to KA-induced seizures in mice [5]. Less is known, however, about the mechanisms of the resistance to epileptogenesis.
Glycogen synthase kinase-3α (GSK3α) and GSK3β are major homeostatic kinases that regulate a plethora of cellular processes [6,7] including neuronal transmission and synaptic as well as structural plasticity [[8], [9], [10], [11], [12]]. At the molecular level, GSK3β contributes to synaptic transmission, both pre- and postsynaptically, regulating the expression and trafficking of ion channels [[13], [14], [15]]. Little is known about the involvement of GSK3 kinases in epileptogenesis, while reported data are contradictory. For example, GSK3β phosphorylation at Serine 9 (S9), which inversely correlates with GSK3β activity, was reported to be increased [16] or decreased [17] in brain tissue that was resected from patients with TLE [16,17] and FCDIIa and FCDIIb patients [17]. Studies in various animal models also did not elucidate a clear relationship between GSK3 activity, status epilepticus and epileptogenesis [[18], [19], [20], [21]] but GSK3β is considered to contribute to the development of epilepsy.
Considering the multilevel impact of GSK3 on different forms of synaptic plasticity and the profound effects of pentylenetetrazol on the survival of mice deficient in neuronal GSK3β (GSK3β[n−/−]) [11,22], we hypothesized that GSK3β controls neuronal responses to proconvulsive drugs and potentially impacts epileptogenesis. Thus, the present study was designed to test this hypothesis in the context of neuronal excitability and epileptogenesis. We showed that there was no significant difference in KA induced epileptiform discharges in acute hippocampal slices from mice that overexpressed a constitutively active form of GSK3β (GSK3β[S9A]) in the brain. Consequently, there was no difference, compared to wildtype littermates, in status epilepticus induced in these mice by an intrahippocampal injection of KA. However, we provided evidence that GSK3β attenuated epileptogenesis that was triggered by such KA treatment. GSK3β[S9A] mice also expressed higher levels of hyperpolarization-activated cyclic nucleotide-gated channel 4 (HCN4) and exhibited a decrease in the phosphorylation of the AMPAR GluA1 subunit at S831. In brain tissue from TLE and FCDII and III patients, the correlation was demonstrated between GSK3β phosphorylation at S9 and GluA1 phosphorylation at S845.
2. Materials and methods
2.1. Antibodies and reagents for pharmacological treatments
The antibodies that were used for this study are listed in Table 1. IRDye-conjugated secondary antibodies (IRDye 680LT donkey anti-rabbit antibody, catalog no. 926–68,023; IRDye 800CW donkey anti-mouse antibody, catalog no. 926–32,212) were obtained from LI-COR Biosciences (Lincoln, NE, USA). Secondary antibodies were used in dilutions that were recommended by the manufacturers. The following reagents were used for the pharmacological treatments: dimethylsulfoxide (Sigma-Aldrich, St. Louis, MO, USA) and KA (Tocris Bioscience, Bristol, UK).
Table 1.
Primary antibodies.
| Antigen | Manufacturer | Catalog no. (RRID) |
Host | Dilution (Western blot) |
|---|---|---|---|---|
| GluA1 | Santa Cruz Biotechnology (Dallas, TX, USA) | sc-55,509 (AB_629532) |
Mouse | 1:100 |
| GluA1 | Millipore (Billerica, MA, USA) | AB1504 (AB_2113602) |
Rabbit | 1:500 |
| P - GluA1 (Ser845) | Millipore (Billerica, MA, USA) | 04–1073 (AB_1977219) |
Rabbit | 1:500 |
| P - GluA1 (Ser831) | Santa Cruz Biotechnology (Dallas, TX, USA) | sc-16,313 (AB_653241) |
Goat | 1:100 |
| GluA2 | Millipore (Billerica, MA, USA) | MAB397 (AB_2113875) |
Mouse | 1:500 |
| GluN1 | Millipore (Billerica, MA, USA) | MAB363 (AB_94946) |
Mouse | 1:700 |
| EAAT2 | Cell Signaling Technology (Danvers, MA, USA) | 3838 (AB_2190743) |
Rabbit | 1:1000 |
| ErbB4 | Santa Cruz Biotechnology (Dallas, TX, USA) | sc-283 (AB_2231308) |
Rabbit | 1:400 |
| TrkB | Cell Signaling Technology (Danvers, MA, USA) | 4603 (AB_2155125) |
Rabbit | 1:1000 |
| HCN4 | Alomone Labs (Jerusalem, Israel) | APC-052 (AB_2039906) |
Rabbit | 1:200 |
| GAD67 | Millipore (Billerica, MA, USA) | AB5862 | Rabbit | 1:1000 |
| GSK3α/β | Thermo Fisher Scientific (Rockford, IL, USA) | 44–610 (AB_2533693) |
Mouse | 1:5000 |
| P-S21/P-S9 GSK3α/β | Cell Signaling Technology (Danvers, MA, USA) | 9331 (AB_329830) |
Rabbit | 1:1000 |
| α-tubulin | Sigma-Aldrich (St. Louis, MO, USA) | T5168 (AB_477579) |
Mouse | 1:5000 |
2.2. Animals
All experiments described below were performed in accordance with the European Communities Council Directive of September 22nd, 2010 and Polish Act on the Protection of Animals Used for Scientific or Educational Purposes of January 15th 2015.
GSK3β[S9A] transgenic mice have been described and characterized extensively [[23], [24], [25]]. These mice carry a constitutively active form of GSK3β, with a mutation of S9 to A9, with the transgene under control of the mouse Thy-1 gene promoter for neuron-specific expression. Heterozygous GSK3β[S9A] mice were maintained on the FVB/N genetic background and compared to wildtype littermates in the same genetic background.
2.3. Electrophysiology in acute slices
All of the experiments were performed on 150 hippocampal slices that were obtained from 13 mice (bodyweight 20–22 g). Seventy-one and 79 slices were obtained from six wildtype and seven GSK3β[S9A] mice, respectively. Each animal was anesthetized with halothane and decapitated. The brain was rapidly removed and placed in cold (3–5 °C) and oxygenated (95% O2 + 5% CO2) artificial cerebrospinal fluid (aCSF) that contained 121 mM NaCl, 5 mM KCl, 2.5 mM CaCl2, 1.25 mM KH2PO4, 1.3 mM MgSO4, 26 mM NaHCO3, and 10 mM glucose. aCSF was prepared fresh before each experiment using prefiltered and deionized water. Transverse hippocampal slices (500 μm) were obtained from both hippocampi using a tissue slicer (Stoelting, Wood Dale, IL, USA). The hippocampal slices were preincubated in oxygenated aCSF at ~20 °C for 45 min after dissection. After this time, the slices were transferred to a gas-liquid interface recording chamber and maintained on nylon mesh where they were continuously perfused with oxygenated and prewarmed (35 °C) aCSF at a low flow rate (1 ml/min) for 45 min. Epileptiform discharges were evoked in hippocampal slices that were obtained from each group of animals (wildtype vs. GSK3β[S9A]) by the application of KA at 0.05 and 0.5 μM. Recordings of local field potentials were performed using glass recording electrodes (3–5 MΩ, Kwik-Fil capillaries, WP Instruments, Sarasota, FL, USA). All of the recordings were performed from the CA3c region of the hippocampus, which is known to be a principal generator of field oscillations with the highest amplitude [26]. The electrodes were positioned using a motorized micropositioner (IVM-1000, Scientifica, Uckfield, UK). In all experiments, field potentials were recorded relative to ground. The signals were filtered (0.001–0.3 kHz, band pass) and amplified (1000 ×) using a P-511 preamplifier (Grass-Astromed, West Warwick, RI, USA). Micro-encephalographic (EEG) activity was displayed on a digital storage oscilloscope (Tektronix TDS 3014, Beaverton, OR, USA) and stored on a computer hard drive using a CED-1401 data acquisition interface (Cambridge Electronic Design, Cambridge, UK). The computer analysis was performed in 5 min fragments of EEG activity that was recorded 15–60 min after KA administration. Offline spectral analysis (FFT) of those fragments was performed using Spike 2.7 software (Cambridge Electronic Design, Cambridge, UK). The detailed analysis of the frequency of epileptiform discharges covered three 20 s fragments (technical repetitions) starting in the 80, 150, 220 s of the recording, respectively, that were selected from each 5 min recording.
2.4. Surgery and video-electroencephalography
Surgery, KA injections, and video-EEG were performed according to a protocol that was approved by the 1st Ethical Committee in Warsaw, Poland (decision no. 390/2017), which is in compliance with the European Community Council Directive (2010/63/EU). For KA injections and electrode implantation, the animals were deeply anesthetized with a mixture of domitor and ketamine (0.5 g/kg and 0.8 g/kg body weight, respectively, intraperitoneal). The mice were injected with 70 nl of a 20 mM solution of KA in 0.9% NaCl at a flow rate of 50 nl/min in the left CA1 field of the hippocampus as described previously [27]. During surgery, the bipolar hippocampal electrode (Bilaney Consultants, Düsseldorf, Germany) was implanted in the injected hippocampus. Cortical recording electrodes (Bilaney Consultants) were placed bilaterally in the skull over the frontal cortex, and two electrodes (one as a reference electrode and one as a ground electrode) were placed over the cerebellum. After surgery, the mice were injected subcutaneously with antisedan (1 g/kg body weight) and connected immediately to a digital acquisition system (TWin Clinical Software for EEG, Grass Technologies. West Warwick, RI, USA). The video-EEG activity of freely behaving animals was monitored for 21 days in an isolated room. The occurrence of seizures was evaluated by visual inspection of the EEG and video recordings by trained observers. The severity of status epilepticus was analyzed during the first 24 h after the intrahippocampal injection of KA. Spontaneous seizures were defined as seizures that appeared as early as 24 h after KA administration. Seizures severity was estimated based on the modified Racine's scale [28]. Both male and female mice were used for the recordings (spontaneous seizures: n = 7 wildtype mice, n = 7 GSK3β[S9A] mice, ≥ 3 months old). All of the experiments were performed by experimenters who were blinded to the group assignment.
2.5. Human tissue samples
The human brain tissue specimens were obtained during routine surgical procedures that were related to epilepsy in a group of drug-resistant epilepsy patients. Samples from six adult patients with temporal lobe epilepsy with hippocampal sclerosis [HS ILAE type 1] [29,30] were provided by the Department of (Neuro)pathology, Amsterdam University Medical Centers, the Netherlands. The tissue was obtained and used in accordance with the Declaration of Helsinki and the AMC Research Code provided by the Medical Ethics Committee. Six samples from pediatric patients diagnosed with FCD type II and III [31] were from the Department of Neurology and Epileptology and the Department of Pathology, The Children's Memorial Health Institute, Warsaw. The study was approved by the local Ethics Board in Warsaw (approval no. 223/KBE/2015). The clinical data are summarized in Table 2.
Table 2.
Clinical characteristics of TLE-HS and FCD patients.
| TLE-HS patients | |||||||
|---|---|---|---|---|---|---|---|
| Case | Pathology* | Age | Gender | Age of onset (years) | Seizure type | Number of seizures/month | AEDs |
| P1 | TLE-HS type 1 | 25 | Male | 6 | FIAS | 5 | LEV, TPM |
| P2 | TLE-HS type 1 | 29 | Female | 13 | FAS/ FBTCS |
32 | LTG, TPM |
| P3 | TLE-HS type 1 | 57 | Female | 47 | FIAS | 1 | CNP |
| P4 | TLE-HS type 2 | 42 | Female | 4 | FIAS/ FBTCS |
1 | PHT, OXC |
| P5 | TLE-HS type 1 | 37 | Female | 20 | FIAS | 2 | CBZ |
| P6 | TLE-HS type 1 | 29 | Male | 15 | FIAS | 36 | CBZ, CLB |
| FCD patients | |||||||
| P1 | Hippocampus (adjacent to FCD2A) |
7 | Female | 5 | FIAS | 60 | LEV |
| FCD2A Temporal cortex | |||||||
| P2 | Hippocampus (adjacent to FCD3A) |
16 | Male | 10 | FIAS | 30 | CBZ, VPA |
| FCD3A Temporal cortex | |||||||
| P3 | Hippocampus (adjacent to FCD2B) |
3 | Female | 3 months | FBTCS | 60 | VGB, VPA, LEV |
| FCD2B Temporal cortex | |||||||
| P4 | Hippocampus (adjacent to FCD2B) |
8 | Female | 3 | FIAS | 10 | LEV, CLB, LTG, VPA |
| FCD2B Temporal cortex | |||||||
| P5 | Hippocampus (adjacent to FCD2A) |
16 | Female | 9 | FIAS | 10 | CBZ, VPA |
| FCD2A Temporal cortex | |||||||
| P6 | Hippocampus (adjacent to FCD2A) |
9 | Male | 4 | FIAS | 36 | VPA, LEV |
| FCD2A Frontal cortex | |||||||
FIAS, focal impaired awareness seizure; FAS, focal aware seizures; FBTCS, focal-to-bilateral tonic-clonic seizure; LEV, levetiracetam; TPM, topiramate; LTG, lamotrigine; CNP, clonazepam; PHT, phenytoin; OXC, oxcarbazepine; CBZ, carbamazepine; CLB, clobazam VPA, valproic acid; VGB, vigabatrin. *Based on Blumcke et al. [[29], [30], [31]].
2.6. Western blots
Quantitative western blot analyses were performed with use of LI-COR Odyssey Imaging System as described previously [22]. Because the samples were derived from different brain structures, what could result in different basal levels of protein expression, before the correlation analysis, the average value of the given protein expression in all samples from each structure (i.e., TLE, FCD and hippocampi adjacent to FCD) has been normalized to 1. It allowed the analysis of the proportions of the protein expression levels in the samples, regardless potential differences in the basal protein expression levels between structures.
2.7. Statistical analysis
For western blot analysis, each animal and each patient's tissue sample represent one biological repetition. For the analysis of the electrophysiological data, each animal represents one biological repetition. The statistical analyses were performed using Prism software (GraphPad, San Diego, CA, USA). The data were analyzed using the unpaired t-test, two-way ANOVA with Bonferroni correction, or Pearson product-moment and Spearman's rank correlation coefficients. The tests that were used and the number of samples that were analyzed are indicated in the respective figure legends.
3. Results
3.1. Chronically increased neuronal GSK3β activity alters expression of HNC4 and phosphorylation of GluA1
Previous research did not provide clear answer if GSK3β is involved in epileptogenesis. At the same time, it was shown in neuronal cultures, that GSK3β impacts expression or phosphorylation of proteins involved in synaptic transmission [14,15]. Thus, we first investigated biochemically the expression of selected proteins in the hippocampus of GSK3β[S9A] mice by western blotting with fluorescently labeled secondary antibodies (LI-COR Odyssey Imaging System). We selected proteins that are implicated in epilepsy and control synaptic transmission: ion channels, glutamate transporters, enzymes of GABA synthesis, trophic factor receptors, and various membrane glycoproteins and receptors. The expression of the majority of the tested proteins, i.e. GluA1, AMPA receptor subunit 2 (GluA2), N-methyl-d-aspartate receptor subunit NR1 (GluN1), excitatory amino acid transporter 2 (EAAT2), receptor tyrosine-protein kinase ErbB4 and tropomyosin receptor kinase B (TrkB) did not differ significantly between both genotypes (Fig. 1a, b). The exceptions were HCN4, which belongs to the HCN family, known to regulate neuronal excitability [32], and glutamate decarboxylase 1 (GAD67), a key enzyme in GABA synthesis. Their expression was increased and decreased, respectively, in the hippocampus of GSK3β[S9A] mice compared to wildtype littermates.
Fig. 1.

GSK3β regulates GluA1 phosphorylation and HCN4 expression in the mice brain. Hippocampi from wildtype (n = 6) and GSK3β[S9A] (n = 5) mice were isolated and lysed. (a) Western blot was used to determine the levels of GluA1, P-GluA1 (S845), P-GluA1 (S831), GluA2, GluN1, EEAT2, ErbB4, TrkB, HCN4, and GAD67. Tubulin is shown as a loading control. (b) Quantitative Western blot analysis results. Error bars indicate SEM. *p < .05, **p < .01, ***p < .001 (two-way ANOVA significant interaction effect, p = 0.0001, ***; two-way ANOVA with Bonferroni correction).
Previous research suggests that both the overall expression and the posttranscriptional modifications of GluA1 should be verified. For example, GluA1 phosphorylation at S831 and S845 is considered activating in animal models of seizures [5]. Therefore, we compared the levels of phosphorylated GluA1 at S831 (P-GluA1[S831]) and S845 (P-GluA1[S845]) in GSK3β[S9A] and wildtype mouse brains. The level of P-GluA1(S831) was significantly lower in GSK3β[S9A] mice, whereas P-GluA1 (S845) levels were comparable in both genotypes (Fig. 1a, b). These results indicate that the chronic increase in GSK3β activity altered the expression and properties of selected ion channels, known to control neuronal excitability.
3.2. GSK3β[S9A] does not impact the frequency of KA-induced epileptiform discharges in acute hippocampal slices
Based on our previous observations and available literature, we hypothesized that GSK3β increases the threshold of epileptiform discharges in response to proconvulsants. We performed a frequency analysis of epileptiform discharges that were generated in acute slices of the hippocampal formation from control and GSK3β[S9A] mice 15–60 min after KA administration (0.05 and 0.5 μM). The frequency of induced discharges is a measure of the severity of epileptiform activity [33,34].
When perfused with 0.05 μM KA, 24 of 36 (66%) of acute hippocampal slices from wildtype mice responded with epileptiform discharges. When perfused with the higher dose of KA (0.5 μM), 28 of 35 (80%) hippocampal slices from wildtype mice generated epileptiform discharges. Bath perfusion of hippocampal slices from GSK3β[S9A] mice with 0.05 and 0.5 μM KA, induced epileptiform discharges in, respectively, 20 of 41 (48%) and 23 of 38 (60%) in slices from GSK3β[S9A] mice (p > .1 for 0.05 μM KA and p = .08 for 0.5 μM KA, Fisher's exact test).
In the presence of KA, at 0.05 and 0.5 μM, hippocampal slice preparations from wildtype and GSK3β[S9A] mice responded with epileptiform discharges within 15–20 min (Fig. 2a). When perfused with 0.05 μM KA, the 24 hippocampal slices from 6 wildtype mice responded with epileptiform discharges that were characterized by a mean frequency of 0.25 ± 0.04 Hz. Similarly, the mean frequency of epileptiform discharges that were generated in the 20 hippocampal slices from 7 GSK3β[S9A] mice that were perfused with the same dose of KA was 0.22 ± 0.03, (p > .1; Fig. 2b). The mean frequency of 0.5 μM KA-induced epileptiform discharges was 0.71 ± 0.03 Hz in 28 hippocampal slices from 6 wildtype mice. Likewise, the mean frequency of 0.5 μM KA-induced epileptiform discharges in 23 hippocampal slices from the 5 GSK3β[S9A] mice was 0.62 ± 0.05 Hz, p = .072; (Fig. 2b). Based on these results, we concluded that a chronic increase in GSK3β activity did not significantly impact epileptiform activity in hippocampal slices acutely induced by KA.
Fig. 2.
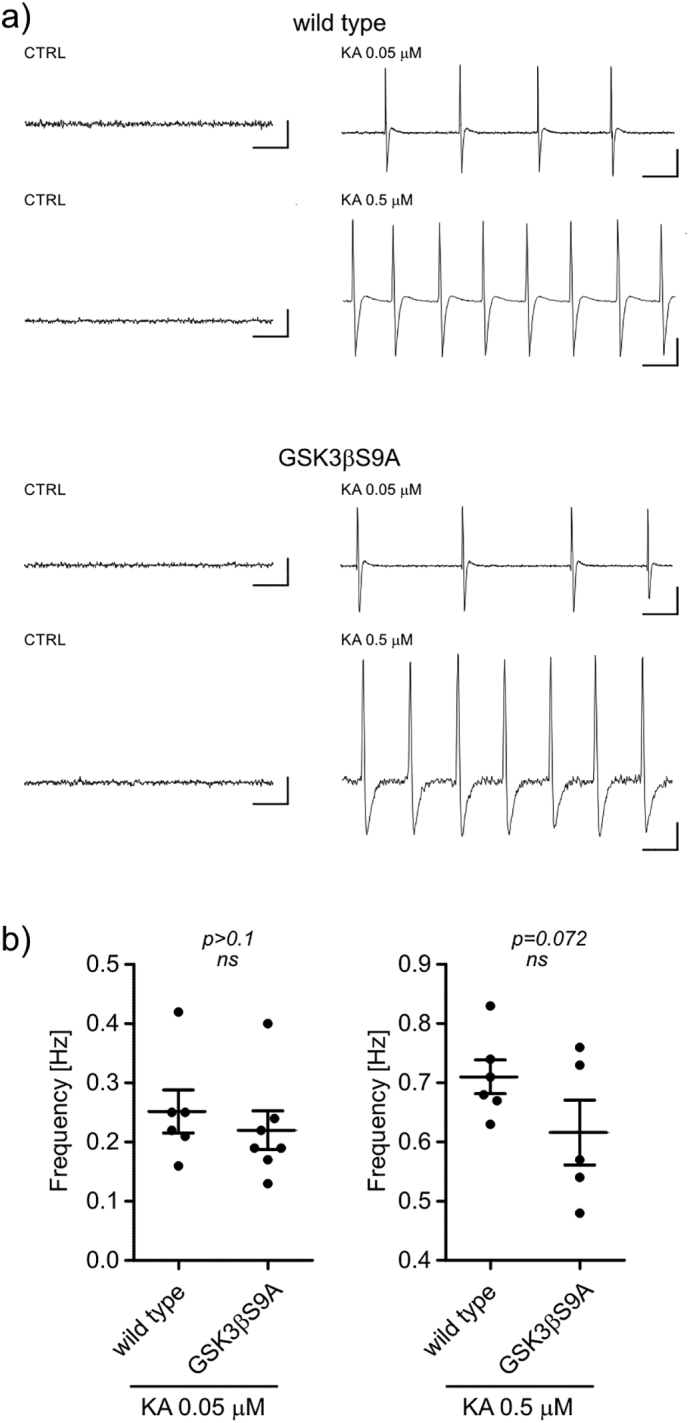
Active GSK3β does not alter epileptiform activity in acute hippocampal slices. (a) Analogue examples of epileptiform activity that was recorded 30 min after kainic acid (KA) administration in the CA3c region of hippocampal slices from wildtype and GSK3β[S9A] mice. Epileptiform discharges were induced in hippocampal slices from each group of animals by the application of 0.05 μM KA (wildtype, n = 6; GSK3β[S9A], n = 7) and 0.5 μM KA (wildtype, n = 6; GSK3β[S9A], n = 5). Calibration: 1 s and 200 μV. (b) Quantification of the effect of bath application of KA on the frequency of epileptiform field activity in hippocampal formation slices from wildtype and GSK3β[S9A] mice. Error bars indicate SEM. (one tailed t-test).
3.3. GSK3 inhibition and GluA1 phosphorylation in temporal lobe epilepsy and focal cortical dysplasia
Based on the observed changes in the levels of proteins involved in epilepsy and synaptic transmission in GSK3β[S9A] mice in comparison to wild type mice we wanted to verify if GSK3β activity have a similar effect in the human brain. To validate our data from the preclinical models we used clinical material, i.e. epileptic tissue of various origin, namely hippocampal brain samples from patients with TLE (Table 2, Fig. 3a, c) and tissue from patients with FCD II and III and the hippocampus adjacent to dysplasia (Table 2, Fig. 3b, c). The age at surgery in the adult TLE group ranged from 25 to 57 years (mean: 36.5 years; median: 33 years), and the age at seizure onset ranged from 6 to 47 years (mean: 17.5 years). The female-to-male ratio was 4/2. The predominant seizure types were focal seizures with impaired awareness (five cases) followed by focal-to-bilateral tonic-clonic seizures (two cases) and focal aware seizures (one case). In two cases, polymorphic seizures were observed. Four patients required at least two antiepileptic drugs, whereas two patients remained on monotherapy. Hippocampal histopathology indicated HS type 1 in five cases and HS type 2 in one case. In the pediatric FCD group, the age range at surgery was 7–16 years (mean: 9.8 years, median: 8.5 years), and the mean age at seizure onset was 5.2 years (range: 3 months-10 years). The female-to-male ratio was 4/2. The predominant seizure types in this group were focal seizures with impaired awareness (five cases), followed by focal-to-bilateral tonic-clonic seizures (one case). All but one patient required polytherapy with at least two antiepileptic drugs. The histopathological assessment revealed FCD2A in three cases, FCD2B in two cases and FCD3A in one case. Hippocampal slices that were obtained from areas adjacent to FCD exhibited changes that were secondary to the long-lasting epileptogenic process.
Fig. 3.

GSK3 inhibitory phosphorylation correlates with GluA1 (S845) phosphorylation in temporal lobe epilepsy and focal cortical dysplasia samples. (a,b) Western blot was used to determine the levels P-GluA1 (S845), P-GluA1 (S831), GluA1, P-GSK3α (S21), P-GSK3β (S9), GSK3α and GSK3β in samples from patients with temporal lobe epilepsy with hippocampal sclerosis (TLE-HS) and in clinical malformations of cortical development (FCD and hippocampi adjacent to FCD). Tubulin is shown as a loading control. (c) Correlation analysis of P-GluA1 (S831)/GluA1 or P-GluA1 (S845)/GluA1 and P-GSK3β (S9)/GSK3β or P-GSK3α (S21)/GSK3α. The data were analyzed as Pearson and Spearman correlation coefficients (r).
In the clinical material described above, we analyzed the correlation between the levels of total and phosphorylated GSK3α, phosphorylated GSK3β, and P-GluA1 (S845 and S831). We applied a correlative analysis of the same samples instead of comparing patients' tissue to postmortem control samples because we found the poor preservation of protein phosphorylation in the latter ones (data not shown), likely resulting from delayed autopsy tissue processing. Ratios of P-GSK3β/GSK3β and P-GluA1(S845)/GluA1 but not P-GSK3β/GSK3β and P-GluA1(S831)/GluA1 were positively correlated (Fig. 3c). In the case of P-GSK3α/GSK3α no significant correlation was observed. These data confirmed that the lower activities of GSK3β were accompanied by higher GluA1 phosphorylation at S845, in human brain samples pathologically changed due to epilepsy. These data derived from clinical material are also consistent with the data obtained from the preclinical models with regard to the correlation between GSK3 activity and GluA1 properties. As we were not able to make the same analysis in the control samples we cannot assume that the correlation is the same or different in not epileptogenic tissue.
3.4. GSK3β[S9A] decreased the frequency of KA-induced spontaneous seizures in vivo
We next investigated whether the chronic increase in the activity of GSK3β restrains KA-induced status epilepticus and subsequent epileptogenesis. GSK3β[S9A] mice and their wildtype littermates were injected intra-hippocampally with KA and the epileptiform activity was monitored by video-EEG for 21 consecutive days. The analysis of recordings from the first 24 h post-KA did not reveal significant differences between genotypes in the number, the latency, the duration, or the behavioral score of KA-induced seizures (Fig. 4). No significant differences in the latency to the first spontaneous seizures (Fig. 5b), average seizure duration, or seizure severity (Fig. 5c) were observed. In contrast, from day 14 post-KA onwards, the average number of seizures per day and the cumulative number of seizures was significantly lower in the GSK3β[S9A] mice relative to wildtype mice (Fig. 5c, d). Considering all examined period, the average number of seizures per day (Fig.5e) was significantly different between the two genotypes (two-way ANOVA, genotype effect p < .0001).
Fig. 4.
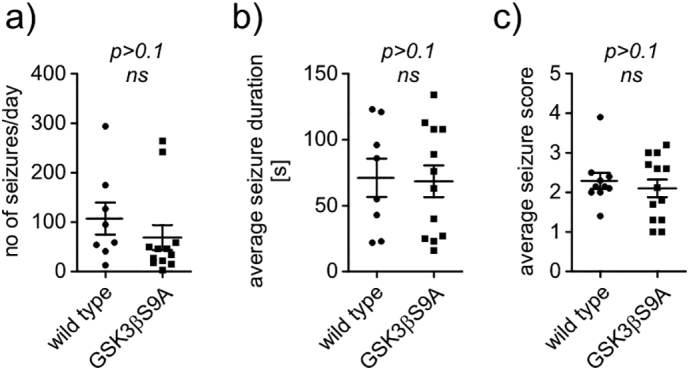
Active GSK3β does not impact KA-induced seizures in vivo in the first 24 h after administration. Quantification of the (a) average number of seizures per day, (b) average seizure duration, and (c) average seizure score in first the 24 h post-KA in wildtype (n = 7) and GSK3β[S9A] (n = 7) mice (unpaired t-test).
Fig. 5.

Active GSK3β decreases rate of KA-induced spontaneous seizures in vivo. (a) EEG recordings from wildtype and GSK3β[S9A] mice during spontaneous seizures (Racine scale: 3). (b) Latency to first spontaneous seizures. (c) Average number of spontaneous seizures, average seizure duration, and average seizure score at the indicated times in wildtype (n = 7) and GSK3β[S9A] (n = 7) mice. Error bars indicate SEM. *p < .05 (unpaired t-test). (d) Cumulative number of seizures until 21 days post-KA in wildtype (n = 7) and GSK3β[S9A] (n = 7) mice. Error bars indicate SEM. *p < .05, **p < .01, ***p < .001 (two-way ANOVA significant interaction effect, p = .0059, **; two-way ANOVA with Bonferroni correction). (e) Fluctuation of average number of daily seizures during the study (two-way ANOVA, genotype effect p < .0001).
After 21 days of seizure monitoring, we biochemically analyzed protein extracts from hippocampi of wildtype and GSK3β[S9A] mice with established epilepsy for the levels of HCN4, GAD67, P-GluA1(S831), and P-GluA1(S845). HCN4 expression was higher in GSK3β[S9A] mice compared to wildtype control mice (Fig. 6). GAD67 and P-GluA1(S831)/GluA1 levels were not different between genotypes after KA (Fig. 6), unlike in non-treated animals (Fig. 1). On the other hand we observed tendency towards lower P-GluA1(S845)/GluA1 ratios in GSK3β[S9A] mice compared to control WT mice (Fig. 6). Collectively, these results imply that GSK3β mitigates KA-induced epileptogenesis, possibly acting on HCN4 and GluA1.
Fig. 6.

GSK3β regulates GluA1 phosphorylation and HCN4 expression in the brain in established epilepsy. Hippocampi from wildtype (n = 7) and GSK3β[S9A] (n = 8) mice 21 days after the KA injection were isolated and lysed. (a) Western blot was used to determine the levels of GluA1, P-GluA1 (S845), P-GluA1 (S831), GAD67 and HCN4. Tubulin is shown as a loading control. (b) Quantitative Western blot analysis results. Error bars indicate SEM. ***p < .001 (two-way ANOVA significant interaction effect, p = .0001, ***; two-way ANOVA with Bonferroni correction).
4. Discussion
In the present study, we report that GSK3β[S9A] mice developed less severe epilepsy upon intrahippocampal KA administration. This observation correlated with increased HCN4 expression and decreased activation by phosphorylation of the GluA1 subunit of AMPAR. We further observed that the phosphorylation of GluA1 at S845 in the brain tissue that was obtained from TLE and FCD patients correlated strongly with GSK3β inactivation, revealed by its phosphorylation at S9. These results collectively support the overall working hypothesis that GSK3β protects neurons from deleterious neural network overactivity. Because this interpretation conflicts with current perceptions of the role of GSK3β in seizure development, two important issues need to be discussed: (i) the function of GSK3β in triggering of, or in response to status epilepticus and subsequent epileptogenesis and (ii) the molecular mechanisms by which GSK3β acts and performs its functions during these processes.
4.1. Involvement of GSK3β in status epilepticus and epileptogenesis
Accumulating data link GSK3 activity with status epilepticus and the progression of epilepsy, although with varying conclusions. Some researchers reported increased inhibitory phosphorylation of GSK3β at S9 and of GSK3α at S21 in response to status epilepticus, as well as in brain tissue from TLE and FCD patients. However, other studies reported completely opposite findings [[16], [17], [18], [19], [20], [21]]. Regardless of the changes in GSK3 activity that were observed in those studies, all authors agreed and acknowledged that the activity of this essential kinase can be pathogenic. Reductions of GSK3 activity in this context were explained by parallel compensatory changes that protected the nerve cells. Conversely, increased GSK3 activity could shutdown prosurvival signaling, beside or including Tau hyperphosphorylation, and regulation of cytoskeleton dynamics [[16], [17], [18],20,35]. The first two processes could trigger, acute or chronic neurodegeneration, respectively, whereas the latter process could entice aberrant axonal sprouting. Nonetheless, evidence of a causal link between changes in GSK3 activity and the pathogenesis of epilepsy, including neurodegeneration and aberrant plasticity, is practically nonexistent.
In the present study, the GSK3β[S9A] mice developed fewer spontaneous seizures following KA-induced status epilepticus. Our findings demonstrate that sustained GSK3β activity is beneficial for mitigating the course of epileptogenesis.
We previously reported that KA evoked significantly less apoptosis in hippocampal organotypic slices derived from GSK3β[S9A] mice compared to wildtype mice [22]. We attributed this effect to an increase in prosurvival signaling in the brain, involving the mammalian target of rapamycin complex 2-Akt pathway [22]. Research on Lafora disease indirectly suggested that GSK3 activity acts protectively by preventing the formation of Lafora bodies that accumulate improper glycogen molecules and thereby lead to neuronal pathology [36]. The present study provides additional evidence, namely the control of neuronal excitability, to these beneficial effects, in which GSK3β activity protects neural networks from the effects of status epilepticus and subsequent epilepsy. When this conclusion is confirmed independently, the therapeutic use of GSK3 inhibitors in epilepsy [17] should be carefully revised.
4.2. GSK3β controls neuronal excitability
Neuron-specific genetic deletion of GSK3β led to rapid and severe mortality upon pentylenetetrazol-induced status epilepticus [22]. This suggested a direct involvement of GSK3β in the regulation of neuronal excitability already under basal conditions. But the present study demonstrated that frequency of KA-induced epileptiform activity was not significantly lower in acute hippocampal slices from GSK3β[S9A] mice that express higher neuronal baseline GSK3β activity. Moreover, we did not observe less severe status epilepticus induced by KA in GSK3β[S9A] mice. Yet, these mice developed a less severe epilepsy phenotype following KA-induced status epilepticus and the biochemical analysis revealed changes potentially underlying this observation. Constitutively active GSK3β in the brain decreased the activating phosphorylation of GluA1 at Ser831 and increased HCN4 expression. We further present the correlation between increased GSK3β phosphorylation at S9 with GluA1 phosphorylation at S845 in tissue from TLE and FCD patients. These data, however, do not resolve yet the issue whether these and other ion channel-related changes functionally link GSK3β to epilepsy.
AMPARs are responsible for fast synaptic transmission in response to glutamate. AMPAR subunits can be phosphorylated at different residues, including GluA1 phosphorylation at S831 and S845. These phosphorylations are believed to activate the receptor as the result in increased channel conductance and peak-open probability, respectively [[37], [38], [39]]. Moreover, phosphorylation of S845 is considered the biochemical equivalent of the presence of GluA1 at the plasma membrane [40,41]. Increased GluA1 phosphorylation was previously observed in hypoxic seizures and subsequently correlated with greater sensitivity to KA-induced seizures in mice [5]. Other studies reported higher GluA1 expression in epilepsy models [42,43]. Our present results are fully consistent with these findings. A reasonable speculation is that GSK3 alleviates the detrimental effects of KA on neuronal activity during epileptogenesis by decreasing the pool of potentially more active GluA1 at the cell surface. Thus, the decrease in GSK3 activity that was observed in brain tissue samples from TLE and FCD patients [17] can be regarded as the manifestation of a safety mechanism, i.e. shutdown, rather than a compensatory tissue response.
The physiological role of HCN4 in neurons is poorly defined. One postulated function of HCN channels is to modulate the amplitude of postsynaptic potentials and downregulate postsynaptic excitatory responses [32]. Mutations in the genes that encode HCN2 and HCN4 were discovered both in patients who suffered from epilepsy and in animal models of epilepsy [44]. Mice that lacked Hcn1 exhibited enhanced neuronal excitability that led to increased seizure susceptibility [45,46]. Although even less is known about HCN4, a recent report showed it to be expressed in the hippocampus, mainly in CA1 and dentate gyrus fast spiking interneurons [47]. One possibility is that HCN4, in these hippocampal interneurons, which discharge rapidly, becomes activated by hyperpolarization and thereby stimulate them to secrete GABA. This, in turn, inhibits their target pyramidal neurons and reduces the overall level of excitation of hippocampal networks. The increased expression of HCN4 in the GSK3β[S9A] mice demonstrated in the present study, is proposed to reduce the probability of paroxysmal discharges. Consequently, both the downregulated synaptic expression of GluA1, as well as the upregulated expression of HCN4, are complementary in supporting our hypothesis that GSK3β activity, within a certain range, prevents excessive neuronal excitation following status epilepticus and could act antiepileptically. However, we cannot exclude indirect, homeostatic effects of prolonged activation of GSK3β on other signaling pathways not mentioned above. One example, could be a potential change in GSK3α activity as this enzyme is gaining increasing recognition for its role in regulation of neuronal plasticity (both, synaptic and structural) [10,48].
Acknowledgments
Acknowledgements
We thank M. Firkowska and A. Zielinska for technical assistance. Research was supported by the Polish National Science Centre (no. 2011/01/N/NZ3/05409 to MU; no. 2017/26/D/NZ4/00159 to PK-G, no. 2015/17/B/NZ3/03734 to TJ) and FP7 European Union grants (no. 223276, “NeuroGSK3” to FvL and JJ; no. 229676, “HealthProt” to JJ; no. 602102, “Epitarget” to EV and EA). This study was also co-financed by European Union funds from the European Social Fund (MU). The work of MU was partially financed by IUVENTUS IP2012 037872. The work of MU, KKotulska, SJ, EA and JJ was partly supported by 7FP grant “EPISTOP” (grant agreement no. 602391), and the Polish Ministerial funds for science (years 2014-2019) for the implementation of international co-financed projects. The work of AG was financed by the Polish National Science Centre (grant no. 2011/01/B/NZ3/05397). KKotulska, SJ and JJ were partly financed by the Polish National Center for Research and Development (grant EPIMARKER, no. STRATEGMED3/306306/4/2017). MU was a recipient of a L'Oreal-UNESCO for Women and Science Fellowship in Poland and EMBO Short Term Fellowship. JJ and MU were also recipients of a Foundation for Polish Science “Mistrz” Professorial Subsidy and Fellowship, respectively.
Founders had no role in the analysis of the data nor in the decision to publish the data.
Author contribution
MU, KK, FvL and JJ designed the experiments. MU, PK-G, TK, BC, KN, BP, KK, AG, HD, BL, and TJ performed the experiments and analyzed the data. WG, KS, SJ, KKotulska, EV, and EA contributed to the selection of patients, patient material, clinical data and the data analysis. MU, TK, JK, KS, KK, FvL, EV, EA, KKotulska, SJ and JJ wrote the manuscript.
Potential conflicts of interest
SJ and KKotulska received Advisory Board honoraria from Novartis Oncology. The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Contributor Information
Malgorzata Urbanska, Email: murbanska@iimcb.gov.pl.
Jacek Jaworski, Email: jaworski@iimcb.gov.pl.
References
- 1.Fisher R.S., Cross J.H., French J.A., Higurashi N., Hirsch E., Jansen F.E. Operational classification of seizure types by the International League against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):522–530. doi: 10.1111/epi.13670. [DOI] [PubMed] [Google Scholar]
- 2.Pitkänen A., Engel J. Past and present definitions of epileptogenesis and its biomarkers. Neurotherapeutics. 2014 Apr;11(2):231–241. doi: 10.1007/s13311-014-0257-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pitkänen A., Lukasiuk K., Dudek F.E., Staley K.J. Epileptogenesis. Cold Spring Harb Perspect Med. 2015 Sep;18:5(10). doi: 10.1101/cshperspect.a022822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becker A.J. Review: Animal models of acquired epilepsy: insights into mechanisms of human epileptogenesis. Neuropathol Appl Neurobiol. 2018 Feb;44(1):112–129. doi: 10.1111/nan.12451. [DOI] [PubMed] [Google Scholar]
- 5.Rakhade S.N., Fitzgerald E.F., Klein P.M., Zhou C., Sun H., Huganir R.L. Glutamate receptor 1 phosphorylation at serine 831 and 845 modulates seizure susceptibility and hippocampal hyperexcitability after early life seizures. J Neurosci. 2012 Dec 5;32(49):17800–17812. doi: 10.1523/JNEUROSCI.6121-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doble B.W., Woodgett J.R. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003 Apr 1;116(Pt 7):1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaidanovich-Beilin O., Woodgett J.R. GSK-3: Functional Insights from Cell Biology and Animal Models. Frontiers in Molecular Neuroscience [Internet] 2011;4 doi: 10.3389/fnmol.2011.00040. Available from:. [cited 2014 Feb 12] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bradley C.A., Peineau S., Taghibiglou C., Nicolas C.S., Whitcomb D.J., Bortolotto Z.A. A pivotal role of GSK-3 in synaptic plasticity. Frontiers in Molecular Neuroscience [Internet] 2012;5 doi: 10.3389/fnmol.2012.00013. Available from:. [cited 2014 Jan 31] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peineau S., Bradley C., Taghibiglou C., Doherty A., Bortolotto Z.A., Wang Y.T. The role of GSK-3 in synaptic plasticity. Br J Pharmacol. 2008 Mar;153(Suppl. 1):S428–S437. doi: 10.1038/bjp.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cymerman I.A., Gozdz A., Urbanska M., Milek J., Dziembowska M., Jaworski J. Structural Plasticity of Dendritic Spines Requires GSK3α and GSK3β. PLoS ONE. 2015;10(7) doi: 10.1371/journal.pone.0134018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kondratiuk I., Łęski S., Urbańska M., Biecek P., Devijver H., Lechat B. GSK-3β and MMP-9 Cooperate in the Control of Dendritic Spine Morphology. Mol Neurobiol. 2017;54(1):200–211. doi: 10.1007/s12035-015-9625-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gozdz A., Nikolaienko O., Urbanska M., Cymerman I.A., Sitkiewicz E., Blazejczyk M. GSK3α and GSK3β phosphorylate arc and regulate its degradation. Front Mol Neurosci. 2017;10:192. doi: 10.3389/fnmol.2017.00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen P., Gu Z., Liu W., Yan Z. Glycogen synthase kinase 3 regulates N-methyl-D-aspartate receptor channel trafficking and function in cortical neurons. Mol Pharmacol. 2007 Apr 23;72(1):40–51. doi: 10.1124/mol.107.034942. [DOI] [PubMed] [Google Scholar]
- 14.Tyagarajan S.K., Ghosh H., Yévenes G.E., Nikonenko I., Ebeling C., Schwerdel C. Regulation of GABAergic synapse formation and plasticity by GSK3beta-dependent phosphorylation of gephyrin. Proc Natl Acad Sci U S A. 2011 Jan 4;108(1):379–384. doi: 10.1073/pnas.1011824108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu L.-Q., Liu D., Hu J., Cheng J., Wang S.-H., Wang Q. GSK-3 beta inhibits presynaptic vesicle exocytosis by phosphorylating P/Q-type calcium channel and interrupting SNARE complex formation. J Neurosci. 2010 Mar 10;30(10):3624–3633. doi: 10.1523/JNEUROSCI.5223-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu C., Russin J., Heck C., Kawata K., Adiga R., Yen W. Dysregulation of PINCH signaling in mesial temporal epilepsy. J Clin Neurosci. 2017 Feb;36:43–52. doi: 10.1016/j.jocn.2016.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Talos D.M., Jacobs L.M., Gourmaud S., Coto C.A., Sun H., Lim K.-C. Mechanistic target of rapamycin complex 1 and 2 in human temporal lobe epilepsy. Ann Neurol. 2018 Feb;83(2):311–327. doi: 10.1002/ana.25149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhowmik M., Khanam R., Saini N., Vohora D. Activation of AKT/GSK3β pathway by TDZD-8 attenuates kainic acid induced neurodegeneration but not seizures in mice. Neurotoxicology. 2015 Jan;46:44–52. doi: 10.1016/j.neuro.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 19.Goodenough S., Conrad S., Skutella T., Behl C. Inactivation of glycogen synthase kinase-3beta protects against kainic acid-induced neurotoxicity in vivo. Brain Res. 2004 Nov 5;1026(1):116–125. doi: 10.1016/j.brainres.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 20.Huang W.-J., Tian F.-F., Chen J.-M., Guo T.-H., Ma Y.-F., Fang J. GSK-3β may be involved in hippocampal mossy fiber sprouting in the pentylenetetrazole-kindling model. Mol Med Rep. 2013 Nov;8(5):1337–1342. doi: 10.3892/mmr.2013.1660. [DOI] [PubMed] [Google Scholar]
- 21.Lee C.-Y., Jaw T., Tseng H.-C., Chen I.-C., Liou H.-H. Lovastatin modulates glycogen synthase kinase-3β pathway and inhibits mossy fiber sprouting after pilocarpine-induced status epilepticus. PLoS ONE. 2012;7(6) doi: 10.1371/journal.pone.0038789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urbanska M., Gozdz A., Macias M., Cymerman I.A., Liszewska E., Kondratiuk I. GSK3β controls mTOR and prosurvival signaling in neurons. Mol Neurobiol. 2018 Jul;55(7):6050–6062. doi: 10.1007/s12035-017-0823-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaworski T., Dewachter I., Lechat B., Gees M., Kremer A., Demedts D. GSK-3α/β kinases and amyloid production in vivo. Nature. 2011 Dec 8;480(7376):E4–E5. doi: 10.1038/nature10615. (discussion E6) [DOI] [PubMed] [Google Scholar]
- 24.Spittaels K., Van den Haute C., Van Dorpe J., Geerts H., Mercken M., Bruynseels K. Glycogen synthase kinase-3beta phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J Biol Chem. 2000 Dec 29;275(52):41340–41349. doi: 10.1074/jbc.M006219200. [DOI] [PubMed] [Google Scholar]
- 25.Spittaels K., Van den Haute C., Van Dorpe J., Terwel D., Vandezande K., Lasrado R. Neonatal neuronal overexpression of glycogen synthase kinase-3β reduces brain size in transgenic mice. Neuroscience. 2002;113(4):797–808. doi: 10.1016/s0306-4522(02)00236-1. Wrzesie. [DOI] [PubMed] [Google Scholar]
- 26.Kowalczyk T., Gołebiewski H., Konopacki J. Is the dentate gyrus an independent generator of in vitro recorded theta rhythm? Brain Res Bull. 2009 Sep 28;80(3):139–146. doi: 10.1016/j.brainresbull.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 27.Kuzniewska B., Nader K., Dabrowski M., Kaczmarek L., Kalita K. Adult Deletion of SRF increases Epileptogenesis and Decreases Activity-Induced Gene Expression. Mol Neurobiol. 2016 Apr;53(3):1478–1493. doi: 10.1007/s12035-014-9089-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Racine R.J. Modification of seizure activity by electrical stimulation: I. after-discharge threshold. Electroencephalogr Clin Neurophysiol. 1972 Mar 1;32(3):269–279. doi: 10.1016/0013-4694(72)90176-9. [DOI] [PubMed] [Google Scholar]
- 29.Blumcke I., Cross J.H., Spreafico R. The international consensus classification for hippocampal sclerosis: an important step towards accurate prognosis. Lancet Neurol. 2013 Sep;12(9):844–846. doi: 10.1016/S1474-4422(13)70175-3. [DOI] [PubMed] [Google Scholar]
- 30.Blümcke I., Thom M., Aronica E., Armstrong D.D., Bartolomei F., Bernasconi A. International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a Task Force report from the ILAE Commission on Diagnostic Methods. Epilepsia. 2013 Jul;54(7):1315–1329. doi: 10.1111/epi.12220. [DOI] [PubMed] [Google Scholar]
- 31.Blümcke I., Thom M., Aronica E., Armstrong D.D., Vinters H.V., Palmini A. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia. 2011 Jan;52(1):158–174. doi: 10.1111/j.1528-1167.2010.02777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kase D., Imoto K. The Role of HCN Channels on Membrane Excitability in the nervous System. J Signal Transduct. 2012;2012:619747. doi: 10.1155/2012/619747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bostanci M.O., Bağirici F. Anticonvulsive effects of carbenoxolone on penicillin-induced epileptiform activity: an in vivo study. Neuropharmacology. 2007 Feb;52(2):362–367. doi: 10.1016/j.neuropharm.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 34.Gajda Z., Szupera Z., Blazsó G., Szente M. Quinine, a blocker of neuronal cx36 channels, suppresses seizure activity in rat neocortex in vivo. Epilepsia. 2005 Oct;46(10):1581–1591. doi: 10.1111/j.1528-1167.2005.00254.x. [DOI] [PubMed] [Google Scholar]
- 35.Liu X., Ou S., Yin M., Xu T., Wang T., Liu Y. N-methyl-D-aspartate receptors mediate epilepsy-induced axonal impairment and tau phosphorylation via activating glycogen synthase kinase-3β and cyclin-dependent kinase 5. Discov Med. 2017;23(127):221–234. [PubMed] [Google Scholar]
- 36.Lohi H., Ianzano L., Zhao X.-C., Chan E.M., Turnbull J., Scherer S.W. Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy. Hum Mol Genet. 2005 Sep 15;14(18):2727–2736. doi: 10.1093/hmg/ddi306. [DOI] [PubMed] [Google Scholar]
- 37.Selvakumar B., Jenkins M.A., Hussain N.K., Huganir R.L., Traynelis S.F., Snyder S.H. S-nitrosylation of AMPA receptor GluA1 regulates phosphorylation, single-channel conductance, and endocytosis. Proc Natl Acad Sci U S A. 2013 Jan 15;110(3):1077–1082. doi: 10.1073/pnas.1221295110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roche K.W., O'Brien R.J., Mammen A.L., Bernhardt J., Huganir R.L. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996 Jun;16(6):1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 39.Banke T.G., Bowie D., Lee H., Huganir R.L., Schousboe A., Traynelis S.F. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci. 2000 Jan 1;20(1):89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Esteban J.A., Shi S.-H., Wilson C., Nuriya M., Huganir R.L., Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003 Feb;6(2):136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- 41.Oh M.C., Derkach V.A., Guire E.S., Soderling T.R. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006 Jan 13;281(2):752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- 42.Kennard J.T.T., Barmanray R., Sampurno S., Ozturk E., Reid C.A., Paradiso L. Stargazin and AMPA receptor membrane expression is increased in the somatosensory cortex of Genetic Absence Epilepsy Rats from Strasbourg. Neurobiol Dis. 2011 Apr;42(1):48–54. doi: 10.1016/j.nbd.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 43.Szczurowska E., Mareš P. NMDA and AMPA receptors: development and status epilepticus. Physiol Res. 2013;62(Suppl. 1):S21–S38. doi: 10.33549/physiolres.932662. [DOI] [PubMed] [Google Scholar]
- 44.Tu E., Waterhouse L., Duflou J., Bagnall R.D., Semsarian C. Genetic analysis of hyperpolarization-activated cyclic nucleotide-gated cation channels in sudden unexpected death in epilepsy cases. Brain Pathol. 2011 Nov;21(6):692–698. doi: 10.1111/j.1750-3639.2011.00500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saito Y., Inoue T., Zhu G., Kimura N., Okada M., Nishimura M. Hyperpolarization-activated cyclic nucleotide gated channels: a potential molecular link between epileptic seizures and Aβ generation in Alzheimer's disease. Mol Neurodegener. 2012 Oct 3;7:50. doi: 10.1186/1750-1326-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang Z., Walker M.C., Shah M.M. Loss of dendritic HCN1 subunits enhances cortical excitability and epileptogenesis. J Neurosci. 2009 Sep 2;29(35):10979–10988. doi: 10.1523/JNEUROSCI.1531-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hughes D.I., Boyle K.A., Kinnon C.M., Bilsland C., Quayle J.A., Callister R.J. HCN4 subunit expression in fast-spiking interneurons of the rat spinal cord and hippocampus. Neuroscience. 2013 May 1;237:7–18. doi: 10.1016/j.neuroscience.2013.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shahab L., Plattner F., Irvine E.E., Cummings D.M., Edwards F.A. Dynamic range of GSK3α not GSK3β is essential for bidirectional synaptic plasticity at hippocampal CA3-CA1 synapses. Hippocampus. 2014 Dec;24(12):1413–1416. doi: 10.1002/hipo.22362. [DOI] [PMC free article] [PubMed] [Google Scholar]