

Abstract
During viral infections, cell death can be induced as a direct result of cytopathic virus replication in various cell types and tissues or as an immune response of the host to the infectious agent. This leads to an infiltration of inflammatory cells, causing subsequent tissue damage. The balance between effective elimination of the pathogen and prevention of fatal tissue damage is decisive for life. The host has developed various mechanisms to inhibit excessive immune responses.
Subject terms: Innate lymphoid cells, Infection
Glucocorticoids (GCs) are well known to inhibit the immune response. GCs are synthesized after activation of the hypothalamic–pituitary–adrenal (HPA) axis by various viral infections and systemic inflammation. Neurons in the hypothalamus express the corticotropin-releasing hormone (CRH). CRH in turn induces a signaling cascade, which ends with an activation and release of GCs in the adrenal cortex.1 GCs can act as suppressors or inducers of the immune system by binding to the glucocorticoid receptor (GR) (Fig. 1). The HPA axis is activated in response to various viral infections or systemic inflammation, and is required to restore homeostasis by limiting inflammation and tissue damage. The underlying mechanisms remained unclear so far.
Fig. 1.
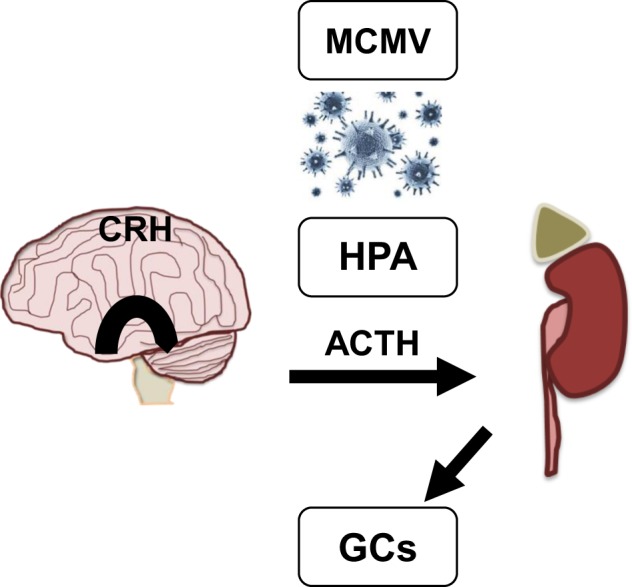
Viral induction of the HPA axis increases GC levels. The HPA axis is activated early during infection with viruses, such as human cytomegalovirus (CMV), and murine MCMV through the release of innate cytokines. Induction of the HPA leads to secretion of CRH from the hypothalamus that in turn activates the distribution of adrenocorticosterone hormone (ACTH) from the anterior pituitary. Subsequently, ACTH promotes the adrenal cortex to release GCs
In a recent issue of Nature Immunology, Quatrini et al. associate the induction of HPA axis and suppression of immunopathology to surface presentation of the immunological check point receptor PD-1.2 Interferon gamma (IFN-γ), produced by innate lymphoid cells (ILCs), is claimed to be the driving force in antiviral defense. To assess the role of GCs in natural killer (NK) cells, the authors employed mice with loxP-flanked alleles of the glucocorticoid receptor, expressing the Cre recombinase under the activating NK cell receptor NCR1 (NKp46) promoter (called GRNcr1-iCre). This leads to cell-specific knockout in NCR1+ cells, including NK cells, ILC1s, and ILC3s.3
In GRNcr1-iCre mice, NK cell function was regulated by GCs in response to murine cytomegalovirus (MCMV) infection. Neither ILC1 or ILC3 cells, nor liver NK cells from GRNcr1-iCre mice showed changes in cytokine response upon infection. Thus, regulation of IFN-γ expression by GCs is tissue and cell type specific.
Histological analysis of these spleens revealed significantly enhanced tissue damage and immunopathology in GR Ncr1-iCre mice compared with WT Ncr1-iCre mice upon infection. Interestingly, higher-IFN-γ induction does not influence viral titers, indicating that GCs inhibit rather immunopathology than mediating direct antiviral effects.
To identify the genes responsible for IFN-γ regulation in splenic NK cells, Quatrini et al. performed next-generation sequencing and pairwise comparison of the gene expression of splenic and liver NK cells. They identified Pdcd1, which encodes the immunological check point receptor PD-1, uniquely induced by GCs in splenic NK cells. Subsequently, the authors showed that endogenous GCs enabled a tissue selective PD-1 de novo expression on splenic NK cells isolated from WTNcr1-iCre mice, but not on splenic NK cells from GRNcr1-iCre mice. Moreover, PD-1 expression was not present on liver NK cells although they are responsive to GR signaling. To clarify the tissue selective expression profile of PD-1, Quatrini et al. analyzed the inflammatory cytokine expression. Profound differences between spleen and liver were detected during MCMV infection. IL-15 is exclusively induced in spleen upon MCMV infection, whereas IL-12 was highly induced in the liver and only moderately induced in spleen, indicating a different cytokine milieu in the spleen and liver. To assess the influence of the different cytokine milieus in the various organs on NK cell activation several combinations of IL-12/-15/-18 and GCs in splenic and liver NK cells were tested to potentially induce PD-1 surface expression. The presence of GCs, IL-15, and IL-18 lead to profound PD-1 expression on splenic NK cells isolated from WTNcr1-iCre mice, but not from GRNcr1-iCre mice. Additional treatment with IL-12 inhibits PD-1 expression in this context. Hence, PD-1 expression on splenic NK cells essentially depends on the cytokine environment, but the underlying molecular mechanism remains still unknown. To assess the biological importance of GR-mediated PD-1 expression, the authors perform in vivo experiments. GRNcr1-iCre mice were highly susceptible to MCMV infection, and blockage of PD-1 by antibodies in WT mice leads to death of infected mice. Genetic GR deficiency in NK cells and blocking of PD-1 leads to high IFN-γ levels. Interestingly, viral load was not affected but mice showed tissue specific pathology in the spleens. Furthermore, blockage of IFN-γ prevents spleen pathology. These data indicate that GR-mediated PD-1 expression on NK cells is responsible to inhibit augmented IFN-γ expression and prevent immunopathology (Fig. 2).
Fig. 2.

Endogenous GCs and the cytokine microenvironment decide about tissue pathology and pathogen elimination. Viral induction of the HPA leads to secretion of endogenous GCs. The cytokine microenvironment, especially the presence/absence of IL-12 and IL-15, together with the secreted GCs mediate tissue-specific PD-1 expression on splenic NK cells. In the liver, high-IL-12 levels lead to PD-1neg NK cells, whereas elevated IL-15 presence in the spleen activated a high proportion of NK cells to express PD-1 on their surface. Subsequently, PD-1 expression limits the secretion of the pro-inflammatory cytokine interferon gamma (IFN-γ), especially in the spleen, preserving tissue integrity and promoting effective pathogen elimination. In contrast, absence of this pathology preventive pathway leads to high-IFN-γ levels causing severe tissue damage and inflammation. Hence, activation of this neuroendocrine pathway sustains tissue integrity and effective pathogen elimination
The publication addresses important aspects of immune regulation during viral infections, but some aspects remain open in this story. NK cells are essential during early infection of mice with MCMV infection and produce both IFN-γ and granzyme B to (i) recruit adaptive immune cells and to (ii) mediate cytotoxicity against infected cells.4 PD-1 expression on NK cells selectively dampens IFN-γ production, but has no influence on cytotoxicity. Molecular mechanisms mediating NK cell cytotoxicity are tightly regulated by the net signal of engaged activating and inhibitory receptors; how this interaction is influenced by PD-1 expression remains unclear. Understanding how the regional effect of GC on NK cells leads to tissue-specific expression of PD-1 on splenic NK cells is still missing. Elucidation of the molecular mechanism would be helpful. PD-1 expression can be regulated in many different ways. Various mechanisms such as cis-DNA elements, transcription factors, and epigenetic components including DNA methylation and histone modification control PD-1 expression.5 The interplay between these regulators could explain the fine-tuning of PD-1 expression in different inflammatory environments and across numerous cell types. The different cytokine milieu could be a starting point in the elucidation of this mechanism. In T-cells PD-1 can be induced by the common γ chain cytokines interleukin-2 (IL-2), IL-7, IL-15, and IL-21.6 The impact of additional cytokines and analysis of downstream signaling pathways constitute a promising approach.
The therapeutic inactivation of PD-1 is a double-edged sword. While the blocking of immune checkpoint pathways, such as PD-1, by therapeutic antibodies is an effective therapy7 and highlighted by the Nobel Prize, the same blockade of PD-1 during an acute viral infection may lead to immunopathology. The therapeutic manipulation of PD-1 must be carefully planned in the context of the treatment to prevent any unwanted damage during an acute infection. Identification of the mechanisms that regulate expression in a tissue-specific manner could be the first step for therapeutic fine-tuning of PD-1 expression.
In summary, although not all questions are resolved, the finding that GCs induce PD-1 on NK cells, which limit immunopathology by inhibiting IFN-γ expression, seems to be an important insight into the mechanism by which the immune system keeps the balance between effective elimination of pathogens and the limiting/reduction of necessary inflammatory responses. The answers to the newly arisen question of how tissue-specific PD-1 expression is regulated by an organ-specific cytokine environment constitute an essential piece to better orchestrate therapeutic approaches fighting cancer and infection-related diseases.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Silverman MN, Sternberg EM. Glucocorticoid regulation of inflammation and its functional correlates: from HPA axis to glucocorticoid receptor dysfunction. Ann. N. Y. Acad. Sci. 2012;1261:55–63. doi: 10.1111/j.1749-6632.2012.06633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quatrini L, et al. Endogenous glucocorticoids control host resistance to viral infection through the tissue-specific regulation of PD-1 expression on NK cells. Nat. Immunol. 2018;19:954–962. doi: 10.1038/s41590-018-0185-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Narni-Mancinelli E, et al. Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc. Natl Acad. Sci. USA. 2011;108:18324–18329. doi: 10.1073/pnas.1112064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biron CA, Tarrio ML. Immunoregulatory cytokine networks: 60 years of learning from murine cytomegalovirus. Med. Microbiol. Immunol. 2015;204:345–354. doi: 10.1007/s00430-015-0412-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bai J, et al. Regulation of PD-1/PD-L1 pathway and resistance to PD-1/PD-L1 blockade. Oncotarget. 2017;8:110693–110707. doi: 10.18632/oncotarget.22690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kinter A. L., et al. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J. Immunol.181, 6738–6746 (2008). [DOI] [PubMed]
- 7.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]