

Abstract
The mechanistic (or mammalian) target of rapamycin complex 1 (mTORC1) controls cell growth, proliferation, and metabolism in response to diverse stimuli. Two major parallel pathways are implicated in mTORC1 regulation including a growth factor‐responsive pathway mediated via TSC2/Rheb and an amino acid‐responsive pathway mediated via the Rag GTPases. Here, we identify and characterize three highly conserved growth factor‐responsive phosphorylation sites on RagC, a component of the Rag heterodimer, implicating cross talk between amino acid and growth factor‐mediated regulation of mTORC1. We find that RagC phosphorylation is associated with destabilization of mTORC1 and is essential for both growth factor and amino acid‐induced mTORC1 activation. Functionally, RagC phosphorylation suppresses starvation‐induced autophagy, and genetic studies in Drosophila reveal that RagC phosphorylation plays an essential role in regulation of cell growth. Finally, we identify mTORC1 as the upstream kinase of RagC on S21. Our data highlight the importance of RagC phosphorylation in its function and identify a previously unappreciated auto‐regulatory mechanism of mTORC1 activity.
Keywords: autophagy, cell growth, mTORC1, phosphorylation, RagC
Subject Categories: Development & Differentiation, Signal Transduction
Introduction
The mechanistic (or mammalian) target of rapamycin (mTOR) is an evolutionarily conserved atypical serine/threonine kinase belonging to the phosphoinositide 3 kinase (PI3K)‐related kinase family. mTOR is found in two structurally and functionally distinct complexes—mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2)—defined by their unique components, in particular raptor (mTORC1) and rictor (mTORC2). Through the coordinated phosphorylation of its downstream effectors, mTORC1 integrates extra‐ and intra‐cellular signal inputs such as amino acids, growth factors (GF), stress, and energy status, to regulate major cellular processes including growth, proliferation, and survival. Underlining its crucial role in cellular and organismal homeostasis, mTORC1 dysregulation occurs in numerous human diseases including cancer, metabolic disorders, and neurodegeneration (Bar‐Peled & Sabatini, 2014; Albert & Hall, 2015; Eltschinger & Loewith, 2016). Growth factors and amino acids both acutely enhance mTORC1 activity, and two different types of small GTPases—Ras‐homolog enriched in brain (Rheb) and the Rag GTPases—cooperatively regulate mTORC1 activity via these two parallel activation mechanisms. Rheb is activated under conditions of high cellular ATP and upstream growth factor signals (Tee et al, 2003). Once activated, Rheb interacts with and activates mTORC1 and is required for mTORC1 activation by all signals, including amino acids. Rag GTPases are considered amino acid‐specific regulators of the mTORC1 pathway (Kim et al, 2008; Sancak et al, 2008). Mammals have four Rag proteins—RagA to RagD—which form obligate heterodimers comprising RagA or RagB together with RagC or RagD. Amino acids cause Rag GTPases to switch to an active conformation, in which RagA/B is GTP‐loaded and Rag C/D is GDP‐loaded. The active Rag heterodimer physically interacts with raptor, recruiting mTORC1 to the lysosome where its activator Rheb resides. Extensive work has revealed several mechanisms implicated in the regulation of Rag activity that enables them to function as nutrient sensors. A common feature among these is the control of Rag nucleotide status, particularly through the activation of guanine nucleotide exchange factors (GEFs) and GTPase‐activating proteins (GAPs). These include the Ragulator (GEF for Rag A/B; Bar‐Peled et al, 2012), the GATOR1 complex (GAP for RagA/B; Bar‐Peled et al, 2013), folliculin (FLCN, GAP for RagC/D; Petit et al, 2013; Tsun et al, 2013), and leucyl‐tRNA synthetase (LeuRS, GAP for RagD; Han et al, 2012). Ubiquitination has also recently emerged as a post‐translational modification (PTM) capable of inhibiting Rag GTPase signaling by recruiting GATOR1 to RagA (Deng et al, 2015; Jin et al, 2015). Importantly, these pathways regulating Rag activity are all amino acid‐dependent, and much less is known about the control of growth factor‐mediated Rag GTPase signaling.
In a recent global mass spectrometry‐based phosphoproteomics study in adipocytes, we observed insulin‐dependent phosphorylation of several highly conserved residues on RagC including S2, S21, and T394 (Humphrey et al, 2013). These data highlight a possible role for the Rag GTPases in mTORC1 growth factor sensing. Here, we demonstrate that both growth factors and amino acids trigger RagC phosphorylation and that phosphorylated RagC potentiates mTORC1 activity and affects mTORC1‐dependent cell growth and autophagy. Moreover, we show that the phosphorylation of RagC at S21 (and likely T394) is catalyzed directly by mTORC1, revealing a novel auto‐regulatory feedback loop within the mTORC1 signaling pathway.
Results
mTORC1 phosphorylates RagC S21
Full mTORC1 activation requires amino acids and growth factors, and it is thought that these two parallel pathways operate autonomously. In a recent large‐scale phosphoproteomic analysis of insulin signaling, we observed dynamic phosphorylation of RagC on multiple sites including S2, S21, and T394, in response to insulin. Moreover, this insulin‐induced phosphorylation was blocked by small molecule inhibitors targeting Akt (MK2206) and PI3K/mTOR (LY294002; Fig EV1A and Humphrey et al, 2013). All three phosphorylation sites are evolutionarily conserved from Danio rerio to mammals (Fig EV1B). Interestingly, except for RagC, none of the other Rag GTPases were found to be phosphorylated following insulin treatment, including RagD which shares 81.1% amino acid sequence identity with RagC.
Figure EV1. Identification of three conserved phosphorylation sites on RagC (related to Fig 1).

-
ARelative abundance of RagC pS2, pS21, and pT394 in 3T3‐L1 adipocytes following insulin stimulation with addition of LY294002 or MK2206. Graphs show mean ± SEM of phosphopeptide abundance fold change (log2). n = 1 for the RagC pS21 (insulin + MK2206/insulin), n ≥ 2 for other conditions.
-
BRagC domain structure and sequence homology of the region surrounding identified phosphorylation sites.
-
CTemporal profiles of RagC pS2 and pT394 generated from SILAC‐MS data.
-
D, EGraphs show mean ± SEM of quantitative analyses of Western blots in Fig 1E and F, n = 3.
The surrounding sequences of all three sites on RagC showed features of the mTOR target consensus motif (Fig 1A), suggesting that mTOR might be an upstream kinase for RagC. Among the three phosphorylation sites, we successfully generated a polyclonal antibody that specifically recognizes the phosphorylated state of RagC S21. Using this phospho‐specific antibody, we found that RagC S21 was directly phosphorylated by active mTOR but not by active Akt in vitro, and this phosphorylation was blocked by the ATP‐competitive mTOR inhibitor Torin1 (Fig 1B). By immunoblotting, we also observed that the timing of RagC S21 phosphorylation in cells in response to both insulin (Fig 1C) and amino acids (Fig 1D) occurs on a timescale similar to that of mTORC1 substrates (4E‐BP1 S65, S6K T389), but distinct from that of mTORC2 (Akt S473) or Akt substrates (TSC2 T1462 and PRAS40 T246). As with RagC S21, our mass spectrometry‐based phosphoproteomics data also revealed that the kinetics of RagC S2 and T394 phosphorylation in response to insulin occurs on a timescale closely resembling that of mTORC1 substrates (Humphrey et al, 2013 and Fig EV1C). To further investigate whether RagC S21 depends on mTORC1 activity in cells, we evaluated the phosphorylation of RagC S21 in inducible raptor knockout MEFs (iRapKO). RagC S21 phosphorylation was abolished in raptor KO MEFs, consistent with mTORC1 (S6K T386, ULK1 S758) but not mTORC2 substrates (Akt S473, NDRG1 T346; Figs 1E and EV1D). Moreover, in contrast to mTORC2 substrates, insulin‐dependent RagC phosphorylation was unaffected in SIN1 KO MEFs (Figs 1F and EV1E), confirming that mTORC2 is not upstream of RagC. Collectively, these results demonstrate that RagC S21 is a novel direct substrate of mTORC1 in vitro and in cells.
Figure 1. mTORC1 phosphorylates RagC S21.

-
AAlignment of the sequence surrounding three phosphorylation sites on RagC with those of known mTORC1 target sites.
-
BThe active fragment of mTOR or active Akt2 was incubated with Flag‐RagC WT purified from HEK‐293E cells. The kinase reaction was performed in the presence or absence of mTOR inhibitor Torin or Akt inhibitor GDC‐0068 (GDC).
-
CHEK‐293E cells were deprived of serum for 2 h followed by insulin (100 nM) stimulation for the indicated time. Lysates were analyzed by Western blot for phosphorylation of RagC and proteins known to belong to the Akt and mTOR pathways. Graph shows mean of quantitative analyses of the Western blots after normalization for total protein or the loading controls.
-
DHEK‐293E cells were deprived of total amino acids for 1 h followed by addition of amino acids for the indicated time. Lysates were analyzed by Western blot for phosphorylation of RagC and proteins known to belong to the Akt and mTOR pathways. Graph shows mean of quantitative analyses of the Western blots after normalization for total protein or the loading controls.
-
EControl or iRapKO MEFs were treated with or without 2 μM 4‐Hydroxytamoxifen (4‐OHT) for 4 days to induce raptor knockout. Treated cells were deprived of serum for 2 h, followed by insulin (100 nM) stimulation for 10 min, and samples were analyzed by Western blot.
-
FSIN1 WT was expressed in SIN1 KO MEFs, and cells were selected by FACS. Cell lines were deprived of serum for 2 h, followed by insulin (100 nM) stimulation for 10 min, and samples were analyzed by Western blot.
-
G, HThe active fragment of mTOR was incubated with Flag‐RagC WT purified from HEK‐293E cells. The kinase reaction was performed in the presence or absence of mTOR inhibitor Torin (T) or rapamycin (R). Samples were analyzed by mass spectrum for pRagC T394 (G) or Western blot for pRagC S21 (H).
Source data are available online for this figure.
To investigate the possibility of mTOR‐dependent phosphorylation of the other sites (S2 and T394), we performed another mTOR in vitro kinase assay, using mass spectrometry as a site‐specific readout. While S2 was not identified in this context, we successfully identified the phosphorylation of T394 on Flag‐RagC, which was increased approximately fourfold following incubation with active mTOR (Fig 1G). This increase was almost completely abolished by the ATP‐competitive mTOR inhibitor Torin1 but was unaffected by the allosteric mTORC1 inhibitor rapamycin (Fig 1G). Western blot analysis of the same samples using phospho‐specific antibodies against S21 confirmed that increased mTOR‐mediated phosphorylation of S21 was also unaffected by rapamycin (Fig 1H). Collectively, these results indicate that both S21 and T394 are rapamycin‐insensitive targets of mTOR (Thoreen et al, 2009).
RagC phosphorylation regulates mTORC1 activity
To investigate the possibility of a functional role of these phosphorylation events toward mTORC1 activity, we created single (T394A), double (S2/21A), and triple (3A) RagC phosphorylation mutants and stably expressed these constructs in HeLa cells. Insulin‐dependent phosphorylation of S6K T389, a well‐established substrate of mTORC1, was substantially blunted in cells expressing the RagC 3A mutant compared with wild‐type RagC and partially impaired in cells expressing the T394A mutant (Fig 2A). This suggests that RagC phosphorylation modulates mTORC1 activity and that all three sites are required to confer this function. This inhibitory effect was similarly observed in HEK‐293E cells (Fig EV2A), suggesting it is likely to be a general phenomenon and not restricted to a single cell type. In contrast to the impaired mTORC1 activity observed with the RagC Ala loss‐of‐function mutant, a RagC phosphomimetic mutant (3E) restored insulin‐dependent mTORC1 signaling (Fig 2B). Moreover, time‐course studies showed that phosphorylation of S6K was attenuated in RagC 3A overexpressed cells at all time points, indicating that the RagC Ala mutant results in attenuation of maximal mTORC1 activity, rather than affecting the kinetics of activation but reaching the same eventual maximal level (Fig 2C).
Figure 2. RagC phosphorylation regulates mTORC1 activity.

- The HA‐RagC HeLa Flp‐In T‐Rex stable cell lines were created (see Materials and Methods for details), and the expression of RagC WT or mutants was induced by addition of tetracycline overnight. Cells were deprived of serum for 2 h followed by insulin (100 nM) stimulation for 20 min. Lysates were analyzed by Western blot for levels of the specified proteins and the phosphorylation state of S6K1. Graphs indicate the mean ± SEM of quantitative analyses of the Western blots after normalization for total protein (*P < 0.05, # no significant difference, two‐tailed Student's t‐test, n = 3).
- Expression of RagC WT, 3A, or 3E was induced by addition of tetracycline overnight. Cells were serum starved for 2 h followed by insulin (100 nM) stimulation for 20 min. Lysates were analyzed by Western blot for the levels of the specified proteins and the phosphorylation state of S6K1. Graphs indicate the mean ± SEM of quantitative analyses of the Western blots after normalization for total protein (*P < 0.05, # no significant difference, two‐tailed Student's t‐test, n = 3).
- Expression of RagC WT and 3A was induced by addition of tetracycline overnight. Cells were serum starved for 2 h followed by insulin (100 nM) stimulation for indicated time. Left: Lysates were analyzed by Western blot for the levels of the specified proteins and the phosphorylation state of S6K1. Right: Graph shows mean ± SEM of quantitative analyses of the Western blots after normalization for total protein, n = 3.
- HEK‐293E cells stably expressing RagC WT, 3A, or 3E were deprived of total amino acids for 1 h followed by addition of amino acids for 10 min. Lysates were analyzed by Western blot for the levels of the specified proteins and the phosphorylation state of S6K1. Graphs indicate the mean ± SEM of quantitative analyses of the Western blots after normalization for total protein (*P < 0.05, **P < 0.01, two‐tailed Student's t‐test, n = 3).
Source data are available online for this figure.
Figure EV2. All three phosphorylation sites are required for full function of RagC (related to Fig 2).
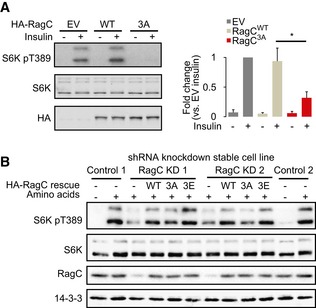
- HEK‐293E cells stably expressing empty vector (EV), RagC WT, or RagC 3A were deprived of serum for 2 h followed by insulin (100 nM) stimulation for 20 min. Lysates were analyzed by Western blot for levels of specified proteins and the phosphorylation state of S6K1. Graphs show mean ± SEM of quantitative analyses of Western blots after normalization for total protein. *P < 0.05, two‐tailed Student's t‐test, n = 3.
- Empty vector (EV), RagC WT, 3A, or 3E was transiently expressed in stable RagC knockdown HEK‐293E cells. Transfected cells were deprived of total amino acids for 1 h followed by addition of amino acids for 10 min. Lysates were analyzed by Western blot.
To test whether RagC phosphorylation affects amino acid‐dependent mTORC1 signaling, we examined the amino acid‐induced phosphorylation of S6K in HEK‐293E cells overexpressing wild‐type RagC (WT) or mutants. Both wild‐type RagC and 3E potentiated amino acid‐dependent mTORC1 activity, but this was not observed in cells expressing the 3A mutant (Fig 2D). To confirm this finding, we performed a rescue experiment in cells in which RagC was stably knocked down and thereby display significantly blunted amino acid‐dependent mTORC1 signaling (Fig EV2B). We isolated two clonal lines for both control and RagC knockdown cell lines. While RagC WT and 3E rescued mTORC1 activity in these stable cells, this was not the case for RagC 3A (Fig EV2B). Collectively, these data demonstrate that ablation of RagC phosphorylation at these sites has a major effect on both growth factor and amino acid‐induced mTORC1 activity.
RagC phosphorylation is required for mTORC1 regulation of cell size and autophagy
We next sought to investigate the role of RagC phosphorylation on cellular functions known to be under the control of mTOR signaling. Cell growth and size are both regulated by mTORC1 via phosphorylation of its substrates S6K and 4E‐BP1. To determine whether loss of RagC phosphorylation affects this aspect of mTORC1 signaling, we examined cell size in HeLa cells expressing either RagC WT or phosphorylation mutants. Cells stably expressing RagC 3A had reduced size compared with cells expressing either wild‐type RagC or RagC 3E (Fig 3A). We further investigated the contribution of RagC phosphorylation to cell size in vivo. Despite changes in surrounding sequence identity, all three residues remain phosphorylatable in Drosophila, with T394 exchanged with serine (S) residues (Fig EV1B). The surrounding sequence of dRagC S2 is exchanged with a tyrosine (Y) residue, which is also a feature of the mTOR target consensus motifs (Fig 1A). We therefore mutated dRagC S2, T10, and S378 to generate dRagC 3A and dRagC 3E. We expressed wild‐type dRagC, dRagC 3A, and dRagC 3E in posterior compartments of the Drosophila wing disk using engrailed‐Gal4 (en‐Gal4; Galagovsky et al, 2014). Expression of dRagC 3A significantly decreased posterior compartment size compared to wild‐type dRagC, while expression of dRagC 3E increased posterior compartment size (Fig 3B). In addition to studying the effects of cell size in transient expression systems, we also investigated the effect of RagC phosphomutants in cells in which RagC was stably knocked down and thereby display reduced cell size (Fig 3C). Re‐expression of either wild type (WT) or the RagC 3E mutant rescued the cell size to that of control cells without RagC knockdown. In contrast, RagC 3A only partially rescued the cell size phenotype, pointing to persistent defective function of RagC in these cells (Fig 3C). Collectively, these results indicate that RagC phosphorylation plays an important role in the mTORC1‐dependent control of cell size.
Figure 3. RagC phosphorylation is required for mTORC1 regulation of cell size and autophagy.

- Expression of RagC WT, 3A, or 3E was induced by overnight addition of tetracycline in HeLa cells followed by cell size analysis using a particle size counter (Horizontal lines, box limits, and whiskers represent the median, the 25th percentile of the 75th percentile and the minimum to the maximum, respectively. *P < 0.05, # no significant difference, one‐way ANOVA with Bonferroni multiple comparisons, n = 4).
- Phospho‐RagC positively regulates wing compartment size in Drosophila. Overexpression (OE) of dRagC WT, 3A, or 3E in wing posterior compartment using en‐Gal4 driver and the posterior compartment sizes were measured in each genotype (Horizontal lines, box limits, and whiskers represent the median, the 25th percentile of the 75th percentile and the minimum to the maximum, respectively. **P < 0.01, ***P < 0.001, ****P < 0.0001, one‐way ANOVA with Bonferroni multiple comparisons, n ≥ 63 flies per condition).
- Empty vector (EV), RagC WT, 3A, or 3E was transiently expressed in stable RagC knockdown HEK‐293E cells followed by cell size analysis using a particle size counter (Horizontal lines, box limits, and whiskers represent the median, the 25th percentile of the 75th percentile and the minimum to the maximum, respectively. *P < 0.05, **P < 0.01, ****P < 0.0001, # no significant difference, one‐way ANOVA with Bonferroni multiple comparisons, n = 2 for C2 and n = 4 for other conditions).
- Empty vector (EV), HA‐RagC WT, 3A, or 3E was expressed in HEK‐293E cells followed 48 h later by incubation with DMEM in the presence or absence of serum for 4 h. Lysates were analyzed by Western blot for phosphorylation of ULK1 and total ULK1.
- Empty vector (EV), HA‐RagC WT, 3A, or 3E was expressed in HEK‐293E cells followed 48 h later by incubation with DMEM in the presence or absence of serum for 4 h. Chloroquine (100 μM) was added to media 30 min prior to harvest. Lysates were analyzed by Western blot for levels of LC3‐II and 14‐3‐3. Graphs show mean ± SEM of quantitative analyses of LC3‐II blotting after normalization for 14‐3‐3. *P < 0.05, two‐tailed Student's t‐test, n = 4.
- GFP‐LC3 was co‐expressed with empty vector, RagC WT, 3A, or 3E in HEK‐293E cells followed 48 h later by serum deprivation for 4 h. Chloroquine (100 μM) was added to media 30 min prior to harvest. LC3 and RagC were monitored by confocal fluorescence microscopy. Scale bar, 10 μm.
- Quantitative analysis of GFP puncta (F) (****P < 0.0001, two‐tailed Student's t‐test; n ≥ 21). Mean value is shown as a horizontal bar.
Source data are available online for this figure.
mTORC1 is also a key controller of the early and late stages of autophagy. This is mediated through phosphorylation of its substrates, ULK1 (Kim et al, 2011) and UVRAG (Kim et al, 2014), respectively. Since RagC phosphorylation regulates mTORC1 activity, we predicted that the RagC phosphomimetic mutant (3E) may maintain mTORC1 activity under conditions of serum starvation. Indeed, RagC 3E expression resulted in elevated ULK1 phosphorylation upon serum starvation, compared to WT‐ and 3A‐expressing cells (Fig 3D). To determine whether RagC phosphorylation affects autophagy, we evaluated serum withdrawal‐induced autophagy in cells expressing RagC WT and the RagC mutants by examining the lipidation status of LC3, an autophagosomal marker. This was similar in cells expressing either RagC WT or RagC 3A, whereas autophagy was significantly blunted in cells expressing the RagC 3E mutant (Fig 3E). Accordingly, we observed less GFP‐LC3 puncta formation in serum‐starved RagC 3E‐expressing cells, indicating that RagC phosphorylation prevents autophagosome formation (Fig 3F and G). Taken together, these results demonstrate that RagC phosphorylation is important functionally for the regulation of cell growth and autophagy.
Phosphorylation of RagC alters mTORC1 stability
We next sought to further probe the mechanistic impact of RagC phosphorylation on mTORC1 behavior. Since Rag GTPases recruit mTORC1 to the lysosome where Rheb resides, we examined a possible role for RagC phosphorylation in (i) localization of RagC to lysosomes, or (ii) the lysosomal recruitment of mTORC1. Wild‐type RagC and mutants (3A and 3E) showed a similar degree of colocalization with the lysosomal marker LAMP1, indicating that RagC localization to lysosomes is not disrupted by phosphorylation at these sites (Fig 4A). To test the second possibility, we examined the colocalization between RagC and mTOR. We again found no difference between the colocalization of RagC 3A and mTOR compared with that of wild‐type RagC (Fig EV3A). We also found that overexpression of either exogenous wild‐type RagC or RagC 3A was able to successfully rescue defective mTOR lysosomal localization in RagC/D knockdown cells (Fig 4B). These results indicate that RagC phosphorylation does not appear to be involved in the recruitment of mTOR to the lysosome.
Figure 4. Phosphorylation of RagC alters mTORC1 stability but not the localization of mTOR .

- Empty vector (EV), HA‐RagC WT, 3A, or 3E was transiently expressed in HEK‐293E cells. RagC and lysosome maker, Lamp1, were monitored by confocal fluorescence microscopy. Scale bar, 10 μm.
- Empty vector (EV), HA‐RagC WT, or 3A was transiently expressed with scramble or RagC and RagD shRNA in HEK‐293E cells. mTOR and HA‐RagC were monitored by confocal fluorescence microscopy. Scale bar, 10 μm.
- Empty vector (EV), Flag‐RagC WT, 3A, or 3E was transiently expressed with HA‐RagA in HEK‐293E cells. Total cell lysates and amounts of mTOR, raptor, and HA‐RagA in the Flag‐RagC immunoprecipitates were analyzed by Western blot. Graphs show mean ± SEM of quantitative analyses of Western blots (*P < 0.05, two‐tailed Student's t‐test, n ≥ 3).
- Expression of HA‐RagC WT, 3A, or 3E was induced by overnight addition of tetracycline in HeLa cells. Total cell lysates (TCL) and amount of mTOR in the raptor immunoprecipitates were analyzed by Western blot. Graphs show mean ± SEM of quantitative analyses of Western blots (***P < 0.001, Student's t‐test, n = 3).
Source data are available online for this figure.
Figure EV3. Phosphorylation of RagC alters mTORC1 stability (related to Fig 4).

- Expression of HA‐RagC WT and 3A was induced by overnight addition of tetracycline in HeLa cells. HA‐RagC and mTOR were monitored by confocal fluorescence microscopy. Scale bar, 10 μm.
- HEK‐293E cells were deprived of total amino acids for 1 h followed by addition of amino acids for 10 min. Lysates and amounts of raptor in the mTOR immunoprecipitates were analyzed by Western blot. TCL: total cell lysate.
- HEK‐293E cells were deprived of serum for 2 h followed by insulin (100 nM) stimulation for 20 min in the presence or absence of Wortmannin (Wort.). Lysates and amounts of raptor in the mTOR immunoprecipitates were analyzed by Western blot. TCL, total cell lysate.
- Flag‐RagC WT, 3A, or 3E was transiently expressed in stable RagC knockdown HEK‐293E cells. Transfected cells were deprived of total amino acids for 1 h followed by addition of amino acids for the indicated time. Total cell lysates (TCL) and amounts of mTOR, raptor, and FLCN in the Flag‐RagC immunoprecipitates were analyzed by Western blot.
- Graphs show mean ± SEM of quantitative analyses of Western blots in (D), n = 3.
It has been reported that the mTOR‐raptor complex exists in two binding states as indicated by changes in the stability of the mTOR‐raptor interaction. In nutrient/growth factor‐poor conditions, mTOR and raptor form an inactive stable complex, whereas in nutrient/growth factor‐rich conditions, mTOR is activated concomitant with destabilization of the complex (Kim et al, 2002; Menon et al, 2014). By immunoprecipitating mTOR, we confirmed that both amino acid and growth factor stimulation result in a weakening of mTOR‐raptor intermolecular contacts (Fig EV3B and C). We hypothesized that RagC phosphorylation may be involved in regulating the stability of the mTOR‐raptor complex, and to evaluate this, we expressed RagA along with wild‐type RagC or phosphomutants in HEK‐293E cells. Immunoprecipitation of RagA co‐purified wild‐type RagC and the RagC phosphomutants equally, suggesting that RagC phosphorylation had no effect on RagA/C heterodimer formation (Fig 4C). Since raptor, not mTOR, is the key mediator of the Rag‐mTORC1 interaction (Sancak et al, 2008), the mTOR that is co‐immunoprecipitated with RagC should be bound to raptor. Therefore, the ratio of mTOR/raptor obtained upon RagC immunoprecipitation reflects the stability of the mTORC1 complex recruited by Rag GTPases. Although not a major effect, the stability of the Rag‐raptor interaction seems to be strengthened with RagC 3E (Fig 4C). Intriguingly, compared to wild‐type RagC, the interaction with mTOR was markedly higher for RagC 3A, indicating that the phosphorylation status of RagC indeed affects stability of the mTOR‐raptor complex (Fig 4C). Since we already demonstrated that RagC mutants influence mTORC1 activity and active mTORC1 has been reported to be less stable (Kim et al, 2002; Menon et al, 2014), it is possible that altered mTORC1 stability reflects differences in the activation state of mTORC1, rather than a direct effect of RagC phosphorylation per se. To address this question, we performed immunoprecipitation of raptor to pulldown mTORC1 from RagC stable cells under conditions of serum starvation where mTORC1 is inactive but still binds to Rag GTPases. Co‐purification of mTOR with raptor was still greatly diminished in RagC 3E stable cells (Fig 4D), while the stability of mTORC1 was not significantly changed in RagC 3A stable cells, indicating a direct effect of RagC phosphorylation on stability of the mTOR‐raptor complex.
Next, we investigated the kinetic impact of RagC phosphorylation on mTORC1 stability during a time‐course of amino acid re‐stimulation, performed after amino acid starvation. Interestingly, the largest effect was observed for RagC 3E, which associates much more strongly with mTOR and raptor during amino acid starvation (0 min), and this effect is strongly destabilized by amino acids (Fig EV3D and E). This suggests that the phosphorylation status of RagC may impact its ability to bind to raptor. Despite the increased total amount of raptor and mTOR co‐immunoprecipitated with RagC, we observed a difference in the ratio of mTOR bound to raptor between the mutants, with RagC 3A able to immunoprecipitate more mTOR relative to the amount of raptor pulled down (Fig EV3E). This effect was markedly diminished by the 3E mutant. Finally, the association between mTOR and raptor under all conditions was progressively less stable upon amino acid re‐feeding. Collectively, these data are consistent with a model that RagC phosphorylation impacts the stability of mTORC1—on the one hand decreasing the overall stability of the mTOR‐raptor complex and on the other hand potentially increasing the tendency for raptor to bind RagC. Recent studies demonstrate that the RagC nucleotide state determines mTORC1 binding to the Rag heterodimer, and folliculin (FLCN) is a GAP for the RagC/D GTPases whose interaction with Rag GTPases is regulated by amino acids (Petit et al, 2013; Tsun et al, 2013). In this assay, although there was no difference in the binding of FLCN between RagC WT and 3E, the RagC 3A mutant displayed increased FLCN binding (Fig EV3D and E). This result may not explain the elevated binding of raptor to RagC 3E observed following amino acid starvation, but it suggests that beyond affecting the stability of mTORC1, the phosphorylation of RagC by mTORC1 may autoregulate mTOR activity by regulating RagC GTP/GDP status.
Discussion
Here, we identified a new auto‐regulatory branch of mTORC1 signaling, involving phosphorylation of the Rag GTPase RagC. This is the first report that Rag GTPase phosphorylation can regulate mTORC1 activity. More importantly, our results confirm that Rag GTPases are not only involved in the amino acid‐sensing mTORC1 pathway, but could also participate in growth factor sensing in the mTORC1 pathway. Although previous studies show that in Rag heterodimers, the GTP/GDP loading of Rag heterodimers plays a dominant role in the interaction between Rag heterodimers and mTORC1 (Kim et al, 2008; Sancak et al, 2008; Tsun et al, 2013), our data indicate that RagC is also a positive regulator of mTORC1 through post‐translational modification. Interestingly, phosphoproteomics data from our laboratory and others suggest that most phosphorylation is concentrated on RagC compared with other Rag GTPases, and S21 is not conserved between RagC and RagD (Fig EV1B), suggesting that RagC is not functionally redundant and potentially has distinct biological functions to RagD (Klammer et al, 2012; Weber et al, 2012; Humphrey et al, 2013; Parker et al, 2015; Mertins et al, 2016).
We established that one of the RagC phosphorylation sites, S21, is a novel rapamycin‐insensitive mTORC1 substrate in vitro and in cells, and the T394 is phosphorylated by mTOR in vitro (Fig 1G). The S2 and T394 sites may also be mTORC1 substrates in vivo, because the kinetics of their phosphorylation resembles that of other bona fide mTORC1 substrates (Fig EV1C and Humphrey et al, 2013) and they also have surrounding sequence features matching the preferred sequence motif of mTORC1 (Fig 1A). These findings indicate the presence of a positive feedback loop between mTORC1 and RagC, which may contribute to the fine‐tuning of mTORC1 activity.
There is evidence that the stability of the raptor‐mTOR complex is related to mTORC activity (Kim et al, 2002; Menon et al, 2014), and our data implicate RagC phosphorylation in the destabilization of mTORC1. This is likely to be a direct effect of RagC phosphorylation, because RagC 3E still destabilized mTOR‐raptor complex under serum starvation (Fig 4D). This is consistent with the observation that RagC 3E causes hyper‐phosphorylation of ULK1 and inhibits autophagy under serum starvation (Fig 3D–G). The next major question is what is the underlying cause of this instability. One possibility is that RagC phosphorylation influences the interaction with other regulators, resulting in “locking” or “opening” of the mTOR‐raptor complex. Interestingly, we observed that RagC 3A bind more FLCN, which is a GAP for RagC/D, and RagC 3E can bind more raptor under both steady and amino acid starvation/re‐fed condition (Figs 4C and EV3D). One possibility is that RagC phosphorylation regulates its nucleotide binding status by modulating the interaction with FLCN. However, we did not observe a substantial difference in FLCN binding between wild‐type RagC and RagC 3E, or raptor binding between wild‐type RagC and 3A. The temporal change in mTOR/raptor stability upon amino acid re‐feeding is similar to that of FLCN/RagC stability, but not with raptor/RagC interaction. A possible explanation is that FLCN has two functions: serving as a GAP for RagC/D and a “lock” for mTORC1. This model could help explain why FLCN releases from Rag GTPases in the presence of amino acids if it is a GAP for RagC/D, which is a positive regulator for mTORC1 activity: After activating RagC/D, FLCN needs to be disassociated from the lysosome to unlock mTORC1, and RagC phosphorylation may affect this process. Further studies will be needed to investigate these possibilities.
We also cannot rule out other explanations for the impact of RagC phosphorylation on impaired mTORC1 activity. For example, it is well established that raptor recruits substrate proteins such as S6K and 4E‐BP1 to mTORC1 so that they can be phosphorylated by mTOR (Hara et al, 2002; Coffman et al, 2014). Therefore, RagC phosphorylation may affect the recruitment of mTORC1 substrates by raptor. Recently, two elegant studies showed that under amino acid or growth factor starvation, the Rag heterodimer binds and recruits TSC2 to lysosomes to inhibit Rheb, resulting in mTORC1 inactivation (Demetriades et al, 2014, 2016). Therefore, a final possibility is that RagC phosphorylation may mediate its effects by acting through TSC2. Future studies into the underlying mechanics of how RagC phosphorylation exerts its effects on mTORC1 signaling are therefore likely to shed light on this newly identified mechanism that sits at the intersection between amino acid sensing and growth factor signaling.
Materials and Methods
Materials and antibodies
Dulbecco's modified Eagle medium (DMEM), fetal bovine serum (FBS), dialyzed fetal bovine serum, and L‐GlutaMAX were from Gibco. Insulin, tetracycline, and chloroquine were from Sigma‐Aldrich (St. Louis, MO). MK‐2206 was from Selleck (Houston, TX). All primary antibodies were diluted 1:1,000 in dilution buffer containing 5% BSA—0.1% Tween 20—0.02% NaN3 in TBS buffer (1.2% Tris–HCl pH 7.4, 8.7% NaCl). The phosphor‐specific RagC S21 antibody was generated by 21st Century Biochemicals. Polyclonal sheep antibody against pThr642 AS160 was described previously (Larance et al, 2005). Pan 14‐3‐3 polyclonal rabbit antibodies were from Santa Cruz Biotechnology. All other primary antibodies were from Cell Signaling Technology. The anti‐mouse horseradish peroxidase (HRP)‐conjugated secondary antibody was from GE Healthcare (Buckinghamshire, United Kingdom), and the anti‐rabbit HRP‐conjugated secondary antibody was from Jackson ImmunoResearch (West Grove, PA). Polyvinylidene difluoride membrane was purchased from Millipore (Billerica, MA). In some cases, IR dye 700‐ or 800‐conjugated secondary antibodies were used, and these were obtained from Rockland Immunochemicals (Gilbertsville, PA).
Plasmids and shRNAs
pOG44 vector for expression of the Flp recombinase was from Invitrogen. Gateway destination vector pGLAP1 was a gift from Peter Jackson (Addgene plasmid # 19702; http://n2t.net/addgene:19702; RRID:Addgene_19702), pLKO.1 RagC shRNA and pLKO.1 RagD shRNA were gifts from David Sabatini (Addgene plasmid # 26630; http://n2t.net/addgene:26630; RRID:Addgene_26630, Addgene plasmid # 30321; http://n2t.net/addgene:30321; RRID:Addgene_30321) (Sancak et al, 2008) and pJFRC‐MUH was a gift from Gerald Rubin (Addgene plasmid # 26213; http://n2t.net/addgene:26213; RRID:Addgene_26213) (Pfeiffer et al, 2010). EGFP of pGLAP1 was replaced by HA to generate pGLAP‐HA. p3xFlag‐CMV‐10 expression vector was from Sigma and converted to a Gateway destination vector p3xFlag‐CMV‐Gateway by ligating a blunt‐ended cassette containing attR sites into the multiple cloning site. pcDNA‐HA‐RagA and RagC WT were generously provided by Kunliang Guan. Primers including the attB1 and attB2 recombination sites were used to amplify RagC. PCR fragments were introduced into plasmid vector pDONR221 (Invitrogen) via the BP recombination reaction to generate the entry clone pDONR‐RagC. pDONR‐RagC S2A/S21A, T394A, S2A/S21A/T394A (3A), and S2E/S21E/T394E (3E) were generated by site‐directed mutagenesis and were verified by DNA sequencing. Entry clones were then used to perform LR recombination reactions with the Gateway system compatible expression plasmid pGLAP‐HA and p3xFlag‐CMV‐Gateway. HA‐RagC WT‐shRNA‐resistant and HA‐RagC 3A‐shRNA‐resistant constructs were generated by site‐directed mutagenesis with specific primer sequences listed below to generate the silent mutations: 5′‐GTTCCCCTGTGGACATGCAGTCTTACGAACTTTGCTGTG‐3′ (sense) and 5′‐CACAGCAAAGTTCGTAAGACTGCATGTCCACAGGGGAAC‐3′ (anti‐sense).
Cell culture, stable cell line, and transfections
Both HeLa and HEK‐293E cells were maintained in Dulbecco modified Eagle medium (DMEM, 4.5 g of glucose/l) with 2 mM L‐GlutaMAX and 10% FBS. iRapKO MEFs and SIN1 KO MEFs were cultured in DMEM with 10% FBS, 2 mM L‐GlutaMAX, non‐essential amino acids, and 1 mM sodium pyrophosphate. 4‐Hydroxytamoxifen (4‐OHT) induction of raptor knockout in iRapKO MEFs cells was performed as previously described (Cybulski et al, 2012). Flp‐In HeLa cells (Invitrogen) were grown in DMEM (4.5 g of glucose/l) with 10% fetal bovine serum (Gibco), 2 μg/ml puromycin, and 100 μg/ml Zeocin. To make stable cell lines, Flp‐In HeLa cells were co‐transfected with pGLAP‐HA empty vector, pGLAP‐HA‐RagC WT, pGLAP‐HA‐RagC 3A, or pGLAP‐HA‐RagC 3E together with pOG44. After a 24‐h recovery period, transfected cells were selected with 2 μg/ml puromycin and 50 μg/ml hygromycin in DMEM supplemented with 10% fetal calf serum. After selecting, the entire polyclonal population is continually grown and used for the experiments. HEK‐293 cells were transfected at approximately 70–75% confluency using Lipofectamine 2000 (Life Technologies) according to manufacturer's instructions, and cells were treated and harvested for experimentation 48 h post‐transfection.
Immunoprecipitation and Western Blotting
For co‐immunoprecipitation experiments, whole‐cell lysates were prepared in 0.3% CHAPS lysis buffer (40 mM HEPES, pH 7.5, 120 mM NaCl, 2 mM EDTA, 10 mM β‐glycerophosphate, 0.3% Chaps) supplemented with EDTA‐free protease inhibitors and phosphatase inhibitors. The total protein concentrations of whole‐cell lysates were measured by the FLUOstar Omega microplate reader (BMG LABTECH) using BCA assay reagent. One mg lysates were incubated with the indicated antibody for 1 h at 4°C followed by 1‐h incubation with protein G or protein A+G Sepharose beads (GE Healthcare). Immunoprecipitates were washed three times with CHAPS buffer followed twice washing with PBS buffer before being resolved by SDS–PAGE and immunoblotted with indicated antibodies.
For Western blotting, proteins were separated by SDS–PAGE and transferred to PVDF membranes. The membranes were incubated in blocking buffer containing 5% skim milk in Tris‐buffered saline (TBS) and immunoblotted with the relevant antibodies overnight at 4°C in blocking buffer containing 5% BSA–0.1% Tween in TBS buffer. After incubation, the membranes were washed and incubated with HRP‐labeled secondary antibodies for 1 h and then detected by SuperSignal West Pico Chemiluminescent Substrate. In some cases, IR dye 700‐ or 800‐conjugated secondary antibodies were used and then scanned at the 700‐ and 800‐nm channels using an Odyssey IR imager.
Cell size determinations
Expression of RagC WT, 3A, or 3E was induced by overnight addition of tetracycline (20 ng/ml) in HeLa cells. To measure cell size, cells were harvested by trypsinization and diluted in PBS, and then subjected to cell size analysis on a particle size counter (Coulter Z2, Beckman Coulter).
Fly stocks and maintenance
Flies were maintained at 25°C and 12‐h light/12‐h dark incubator. For the generation of UAS‐dRagC lines, we amplified the Drosophila RagC cDNA by PCR from the GH16429 cDNA clone (DGRC, Indiana, USA). PCR product was digested with NotI and XbaI and then cloned into pJFRC‐MUH vector (Addgene plasmid # 26213). dRagC 3A and dRagC 3E mutants were generated by site‐directed mutagenesis and were verified by DNA sequencing. The UAS‐dRagC WT, UAS‐dRagC 3A, and UAS‐dRagC 3E constructs were transformed into w1118 attp2 flies (Centre for Cellular and Molecular Platforms, India). The UAS‐dRagC WT, UAS‐dRagC 3A, and UAS‐dRagC 3E flies were further confirmed by sequencing.
Drosophila wing compartment size measurement
The Engrailed (En) Gal4, UAS‐GFP (Neufeld et al, 1998), was obtained from Dr. Kieran Harvey. The expression of UAS‐dRagC WT, UAS‐dRagC 3A, and UAS‐dRagC 3E was driven by En‐Gal4, UAS‐GFP, in the posterior wing. Flies were grown in a density‐controlled manner, and the right wings of anesthetized 4‐ to 7‐day‐old adult males were collected and mounted in 80% glycerol in PBS. Wings were imaged using a Leica stereomicroscope (FM205 FA) with an Axio digital camera. The posterior compartment size was quantified using Fiji (National Institutes of Health). Statistics were performed with Prism software.
Autophagy flux assay
HEK‐293E cells were transfected with pGLAP‐HA empty vector, pGLAP‐HA‐RagC WT, pGLAP‐HA‐RagC 3A, or pGLAP‐HA‐RagC 3E. Transfected cells were split into two dishes 24 h post‐translation. After another 24 h, one dish was fed and the other dish was serum starved for 4 h. Chloroquine (100 μM) was added to media 30 min prior to harvest. Cells were lysed with PBS buffer containing 2% SDS. The amounts of LC3‐I and LC3‐II were analyzed by Western blot.
Cell image analysis
To prepare Matrigel‐coated coverslips, glass coverslips were incubated at room temperature for 120 min with a 1:50 dilution of Matrigel in ice‐cold PBS. Coverslips were washed twice with PBS prior to use. For autophagosome maturation assay, GFP‐LC3 was transiently expressed in HEK‐293E cells with empty vector, pGLAP‐HA‐RagC WT, 3A, or 3E. Twenty‐four hours post‐transfection, the cells were trypsinized and seeded onto Matrigel‐coated glass coverslips. Twenty‐four hours post‐reseeding cells were serum starved for 4 h and chloroquine (100 μM) was added to media 30 min prior to imaging. To analyze the colocalization of RagC with mTOR or Lamp1, HEK‐293E cells were transiently transfected with empty vector, pGLAP‐HA‐RagC WT, 3A, or 3E. For monitoring the mTOR localization on lysosome, Scramble or RagC and RagD shRNAs were transiently expressed in HEK‐293E cells with empty vector, pGLAP‐HA‐RagC WT, or 3A. Twenty‐four hours post‐transfection, the cells were trypsinized and seeded onto Matrigel‐coated glass coverslips. Twenty‐four hours post‐reseeding cells were ready for imaging. Cells were washed twice with PBS and fixed with 3.7% paraformaldehyde in PBS at room temperature for 15 min. Cells were then blocked and permeabilized with blocking buffer containing 0.1% (w/v) saponin and 2% BSA in PBS. Antibody staining was carried out in 2% (w/v) BSA in PBS. Coverslips were mounted on glass slides with ImmunO™ (MP Biomedicals, LLC) and imaged using a Leica laser scanning confocal microscope (TCS SP2; Leica Microsystems).
mTOR and Akt2 in vitro kinase analysis
Purification of unphosphorylated RagC—HEK‐293E cells were transiently transfected at 75% confluence with Flag‐RagC WT. To keep RagC unphosphorylated, cells were deprived of serum for 1.5 h, followed by Torin (100 nM) treatment for half hour. Cells were lysed in NP‐40 buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 1% NP‐40) supplemented with protease inhibitors and phosphatase inhibitors and incubated with monoclonal anti‐FLAG M2 antibody for 1 h at 4°C followed by overnight incubation with protein G Sepharose beads. The beads were washed three times with NP‐40 buffer and once with kinase buffer (50 mM HEPES, pH 7.5, 1 mM EGTA, 10 mM MnCl2, 1 mM DTT, 0.01% Tween 20). To elute FLAG‐tagged RagC, the beads were resuspended and incubated in 30 μl of kinase buffer containing 12 μg of 3× FLAG peptide (Sigma‐Aldrich) with occasional mixing at 4°C for 1 h. In vitro kinase analysis—Five equal aliquots of purified FLAG‐RagC (2 μl) were prepared and incubated with (i) kinase buffer containing 100 μM ATP (control), (ii) active mTOR fragment (Millipore) in kinase buffer containing 100 μM ATP, (iii) active mTOR fragment (Millipore) in kinase buffer containing 100 μM ATP pre‐incubated with 100 nM Torin, (iv) recombinant active Akt2 in kinase buffer containing 100 μM ATP, and (v) recombinant active Akt2 in kinase buffer containing 100 μM ATP pre‐incubated with 10 μM GDC‐0068. Kinase reactions were performed for 20 min at 37°C and quenched with 2× SDS–polyacrylamide gel electrophoresis loading buffer.
Author contributions
Conceptualization: GY and DEJ; Methodology: GY, DSM, DF, SJH, Q‐PW, KCC, and GN; Investigation: GY, DSM, DF, SJH, and Q‐PW; Writing–original draft: GY; Writing–review & editing, GY, SJH, DSM, DF, Q‐PW, GN, and DEJ; Visualization: GY and SJH, Funding Acquisition: DEJ; Supervision: GY and DEJ.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
{kind=link}
Source Data for Figure 2
{kind=link}
Source Data for Figure 3
{kind=link}
Source Data for Figure 4
{kind=link}
Acknowledgements
We thank Dr. Michael Hall for providing iRapKO MEF cells, Dr. Bing Su and Dr. Estela Jacinto for providing SIN1 KO MEF cells, and Dr. Kun‐liang Guan and Dr. Haixin Yuan for providing original RagA and RagC plasmids. This work was supported by National Health and Medical Research Council (NHMRC) project grants (GNT1120201). D.E.J. is an NHMRC Senior Principal Research Fellow, G.Y. and Q.W. were recipients of the NHMRC Early Career Fellowship, and D.S.M. was a recipient of the Whitaker International Fellows and Scholars Program Fellowship.
The EMBO Journal (2019) 38: e99548
References
- Albert V, Hall MN (2015) mTOR signaling in cellular and organismal energetics. Curr Opin Cell Biol 33: 55–66 [DOI] [PubMed] [Google Scholar]
- Bar‐Peled L, Schweitzer LD, Zoncu R, Sabatini DM (2012) Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150: 1196–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar‐Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, Sabatini DM (2013) A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340: 1100–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar‐Peled L, Sabatini DM (2014) Regulation of mTORC1 by amino acids. Trends Cell Biol 24: 400–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman K, Yang B, Lu J, Tetlow AL, Pelliccio E, Lu S, Guo DC, Tang C, Dong MQ, Tamanoi F (2014) Characterization of the Raptor/4E‐BP1 interaction by chemical cross‐linking coupled with mass spectrometry analysis. J Biol Chem 289: 4723–4734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cybulski N, Zinzalla V, Hall MN (2012) Inducible raptor and rictor knockout mouse embryonic fibroblasts. Methods Mol Biol 821: 267–278 [DOI] [PubMed] [Google Scholar]
- Demetriades C, Doumpas N, Teleman AA (2014) Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 156: 786–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetriades C, Plescher M, Teleman AA (2016) Lysosomal recruitment of TSC2 is a universal response to cellular stress. Nat Commun 7: 10662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Jiang C, Chen L, Jin J, Wei J, Zhao L, Chen M, Pan W, Xu Y, Chu H, Wang X, Ge X, Li D, Liao L, Liu M, Li L, Wang P (2015) The ubiquitination of rag A GTPase by RNF152 negatively regulates mTORC1 activation. Mol Cell 58: 804–818 [DOI] [PubMed] [Google Scholar]
- Eltschinger S, Loewith R (2016) TOR complexes and the maintenance of cellular homeostasis. Trends Cell Biol 26: 148–159 [DOI] [PubMed] [Google Scholar]
- Galagovsky D, Katz MJ, Acevedo JM, Sorianello E, Glavic A, Wappner P (2014) The Drosophila insulin‐degrading enzyme restricts growth by modulating the PI3K pathway in a cell‐autonomous manner. Mol Biol Cell 25: 916–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JM, Jeong SJ, Park MC, Kim G, Kwon NH, Kim HK, Ha SH, Ryu SH, Kim S (2012) Leucyl‐tRNA synthetase is an intracellular leucine sensor for the mTORC1‐signaling pathway. Cell 149: 410–424 [DOI] [PubMed] [Google Scholar]
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K (2002) Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110: 177–189 [DOI] [PubMed] [Google Scholar]
- Humphrey SJ, Yang G, Yang P, Fazakerley DJ, Stockli J, Yang JY, James DE (2013) Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab 17: 1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin G, Lee SW, Zhang X, Cai Z, Gao Y, Chou PC, Rezaeian AH, Han F, Wang CY, Yao JC, Gong Z, Chan CH, Huang CY, Tsai FJ, Tsai CH, Tu SH, Wu CH, dos Sarbassov D, Ho YS, Lin HK (2015) Skp2‐mediated RagA ubiquitination elicits a negative feedback to prevent amino‐acid‐dependent mTORC1 hyperactivation by recruiting GATOR1. Mol Cell 58: 989–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument‐Bromage H, Tempst P, Sabatini DM (2002) mTOR interacts with raptor to form a nutrient‐sensitive complex that signals to the cell growth machinery. Cell 110: 163–175 [DOI] [PubMed] [Google Scholar]
- Kim E, Goraksha‐Hicks P, Li L, Neufeld TP, Guan KL (2008) Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 10: 935–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YM, Jung CH, Seo M, Kim EK, Park JM, Bae SS, Kim DH (2014) mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol Cell 57: 207–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klammer M, Kaminski M, Zedler A, Oppermann F, Blencke S, Marx S, Muller S, Tebbe A, Godl K, Schaab C (2012) Phosphosignature predicts dasatinib response in non‐small cell lung cancer. Mol Cell Proteomics 11: 651–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larance M, Ramm G, Stockli J, van Dam EM, Winata S, Wasinger V, Simpson F, Graham M, Junutula JR, Guilhaus M, James DE (2005) Characterization of the role of the Rab GTPase‐activating protein AS160 in insulin‐regulated GLUT4 trafficking. J Biol Chem 280: 37803–37813 [DOI] [PubMed] [Google Scholar]
- Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, Cantley LC, Manning BD (2014) Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 156: 771–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, Kawaler E, Mundt F, Krug K, Tu Z, Lei JT, Gatza ML, Wilkerson M, Perou CM, Yellapantula V, Huang KL et al (2016) Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534: 55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld TP, de la Cruz AF, Johnston LA, Edgar BA (1998) Coordination of growth and cell division in the Drosophila wing. Cell 93: 1183–1193 [DOI] [PubMed] [Google Scholar]
- Parker BL, Yang G, Humphrey SJ, Chaudhuri R, Ma X, Peterman S, James DE (2015) Targeted phosphoproteomics of insulin signaling using data‐independent acquisition mass spectrometry. Sci Signal 8: rs6 [DOI] [PubMed] [Google Scholar]
- Petit CS, Roczniak‐Ferguson A, Ferguson SM (2013) Recruitment of folliculin to lysosomes supports the amino acid‐dependent activation of Rag GTPases. J Cell Biol 202: 1107–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BD, Ngo TT, Hibbard KL, Murphy C, Jenett A, Truman JW, Rubin GM (2010) Refinement of tools for targeted gene expression in Drosophila. Genetics 186: 735–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar‐Peled L, Sabatini DM (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320: 1496–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J (2003) Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase‐activating protein complex toward Rheb. Curr Biol 13: 1259–1268 [DOI] [PubMed] [Google Scholar]
- Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS (2009) An ATP‐competitive mammalian target of rapamycin inhibitor reveals rapamycin‐resistant functions of mTORC1. J Biol Chem 284: 8023–8032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsun ZY, Bar‐Peled L, Chantranupong L, Zoncu R, Wang T, Kim C, Spooner E, Sabatini DM (2013) The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell 52: 495–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber C, Schreiber TB, Daub H (2012) Dual phosphoproteomics and chemical proteomics analysis of erlotinib and gefitinib interference in acute myeloid leukemia cells. J Proteomics 75: 1343–1356 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4