

Abstract
Accumulation of the protein tau characterises Alzheimer's disease and other tauopathies, including familial forms of frontotemporal dementia (FTD) that carry pathogenic tau mutations. Another hallmark feature of these diseases is the accumulation of dysfunctional mitochondria. Although disease‐associated tau is known to impair several aspects of mitochondrial function, it is still unclear whether it also directly impinges on mitochondrial quality control, specifically Parkin‐dependent mitophagy. Using the mito‐QC mitophagy reporter, we found that both human wild‐type (hTau) and FTD mutant tau (hP301L) inhibited mitophagy in neuroblastoma cells, by reducing mitochondrial translocation of Parkin. In the Caenorhabditis elegans nervous system, hTau expression reduced mitophagy, whereas hP301L expression completely inhibited it. These effects were not due to changes in the mitochondrial membrane potential or the cytoskeleton, as tau specifically impaired Parkin recruitment to defective mitochondria by sequestering it in the cytosol. This sequestration was mediated by aberrant interactions of Parkin with the projection domain of tau. As mitochondria are dysfunctional in neurodegenerative conditions, these data suggest a vicious cycle, with tau also inhibiting the degradation of damaged mitochondria.
Keywords: Alzheimer's disease, autophagy, C. elegans, mitochondria
Subject Categories: Autophagy & Cell Death, Membrane & Intracellular Transport, Neuroscience
Introduction
Mitochondrial dysfunction is characteristic of many neurodegenerative diseases, including Alzheimer's disease (AD) and frontotemporal dementia (FTD). Both AD and the frontotemporal lobar degeneration‐tau subtype of FTD feature an abnormal aggregation of the neuronally enriched protein tau, which normally associates with microtubules. Tau becomes hyperphosphorylated in disease, which is thought to cause its detachment from microtubules. This, together with its de novo synthesis, causes its accumulation in the cytosol (Iqbal et al, 2005; Li & Götz, 2017a). Several cellular and animal models of tauopathy have revealed a myriad of mitochondrial abnormalities caused by the pathological accumulation of tau (Manczak & Reddy, 2012; DuBoff et al, 2013; Eckert et al, 2014). These include increased production of reactive oxygen species, changes in fission, fusion and morphology (mitochondrial dynamics), and impaired oxidative phosphorylation (OxPhos), as evidenced by reduced complex I activity and ATP production (Rhein et al, 2009; DuBoff et al, 2012; Li et al, 2016; Pérez et al, 2017).
Defective mitochondria can be removed by mitochondrial autophagy, a process termed mitophagy, which can be initiated when the mitochondrial membrane potential dissipates as a consequence of functional impairment. This depolarisation activates the PINK1/Parkin pathway, whereby the kinase PINK1 accumulates on the outer mitochondrial membrane, flagging the mitochondrion for removal (Jin et al, 2010; Narendra et al, 2010). PINK1 has been shown to phosphorylate ubiquitin on the outer mitochondrial membrane, which then binds to and activates the ubiquitin ligase Parkin, causing cytosolic Parkin to translocate to the surface of the mitochondrion (Kane et al, 2014; Koyano et al, 2014). Parkin then increases the level of ubiquitination of outer mitochondrial proteins, creating a positive feedback loop. The ubiquitin chains thus generated can interact with autophagy adaptors, recruiting LC3 (microtubule‐associated protein 1A/1B‐light chain 3)‐positive autophagosomal membranes around the damaged mitochondrion, which is finally trafficked to the lysosome for degradation (Lazarou et al, 2015).
Mitophagy in AD has remained largely underexplored. Recently however, Ye et al (2015) demonstrated increased Parkin recruitment to mitochondria in tissue from AD patients as well as in primary neurons from amyloid‐β‐forming amyloid precursor protein (APP) transgenic mice. Similarly, an artificially truncated form of tau, consisting of amino acids 26–230 of its 441 amino acid full‐length human form, was also found to induce mitophagy in primary neurons, coupled with mitochondrial fragmentation and synapse loss (Amadoro et al, 2014). These effects were not observed for full‐length tau. A study in mouse hippocampus reported a full‐length tau‐dependent increase in mitochondrial mass, possibly due to decreased mitophagy (Hu et al, 2016). Although these studies indicate a putative role for tau pathology in mitophagy, the underlying mechanisms are not well understood, nor is it known how accumulated wild‐type and FTD mutant tau might differ in their pathogenic effects.
In the current study, we show that both wild‐type and FTD (P301L) mutant tau accumulation directly inhibit mitophagy in two separate paradigms: neuroblastoma cells and the Caenorhabditis elegans nervous system. Both tau species impaired the translocation of Parkin to mitochondria, without altering the mitochondrial membrane potential compared to control cells. We identify an underlying pathomechanism, wherein tau aberrantly interacts with Parkin in the cytosol, preventing it from correctly translocating to defective mitochondria. These findings add a new dimension to our understanding of tau toxicity, illustrating the existence of a vicious cycle whereby tau inhibits the clearance of damaged mitochondria, even as it contributes to their dysfunction in the first place (David et al, 2005; DuBoff et al, 2012).
Results
Wild‐type and P301L mutant human tau inhibit mitophagy in neuroblastoma cells
Mitochondrial dysfunction occurs in several models of tau pathology, and we therefore sought to determine whether tau impairs the removal of damaged mitochondria via mitophagy. To this end, we used a fluorescent mitophagy indicator (mito‐QC), which consists of an mCherry‐GFP fusion protein targeted to the outer mitochondrial membrane. This dual fluorophore reporter allows the detection of mitochondrial localisation to the lysosomal compartment based on a pH‐induced change in its fluorescent signal (Allen et al, 2013). This is brought about by the acidic environment of the lysosome, which results in decreased GFP fluorescence without affecting mCherry fluorescence (Rosado et al, 2008; Allen et al, 2013). We first cloned the mito‐QC gene under the control of the neuron‐specific human synapsin promoter and verified its mitochondrial localisation by immunostaining for ATP synthase (Fig 1A). Mito‐QC was then expressed in murine neuroblastoma (N2a) cells, in conjunction with myc‐tagged Parkin and either V5‐tagged full‐length (2N4R) human tau (“hTau”), 2N4R P301L mutant human tau (“hP301L”) or an empty vector control. After 24 h, the cells were treated with 8 μM of the protonophore CCCP (carbonyl cyanide 3‐chlorophenylhydrazone) overnight, to induce mitophagy by collapsing the mitochondrial membrane potential (Fig 1B and C). Control cells initiated mitophagy in response to CCCP as represented by a decrease in the ratio of GFP to mCherry fluorescence intensity (Fig 1B and C). However, expression of either hTau or hP301L prevented this change in the GFP/mCherry ratio, indicating that mitophagy was perturbed by the accumulation of each protein variant (Fig 1C). We also examined the response to a mixture of antimycin/oligomycin (AO), which can induce mitochondrial depolarisation by a different means to CCCP, by inhibiting the electron transport chain directly. Overnight treatment with 15 μM AO induced mitophagy in control cells, but not in hTau‐ or hP301L‐expressing cells (Fig 1D and E), mirroring our observation following CCCP treatment. Therefore, a transient transfection of wild‐type or P301L mutant tau resulted in the inhibition of mitophagy of defective mitochondria in a cellular system.
Figure 1. Tau inhibits mitophagy in N2a cells.
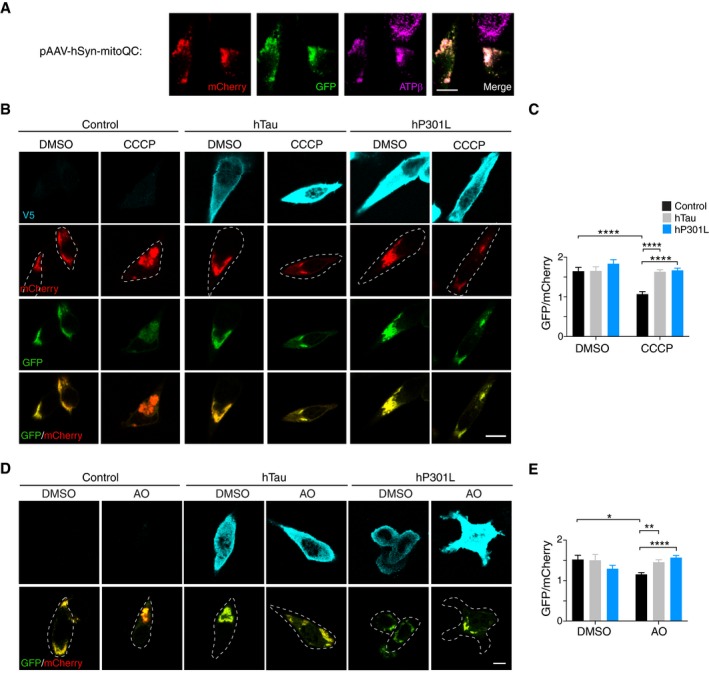
- Immunostaining for the mitochondrial marker ATPβ in N2a cells expressing mito‐QC.
- N2a cells expressing mito‐QC and myc‐Parkin together with hTau‐V5, hP301L‐V5 or an empty vector control. Treatment with CCCP (8 μM, 17 h) induces mitophagy in control cells, as indicated by a decrease in the GFP/mCherry fluorescence ratio, but not in hTau or hP301L cells. Deconvolution (Classic Maximum Likelihood Estimation) was applied to images for display only.
- Quantification of GFP/mCherry fluorescence intensity per cell. Data were analysed by two‐way ANOVA, showing significant main effects of CCCP treatment, F(1, 314) = 15.66, P < 0.0001, and tau expression, F(2, 314) = 12.26, P < 0.0001, and a significant interaction effect, F(2, 314) = 6.137, P = 0.0024, n = 40–73 cells/group.
- Cells expressing mito‐QC and myc‐Parkin together with hTau‐V5, hP301L‐V5 or an empty vector control, treated with a mixture of antimycin and oligomycin (AO, 15 μM, 17 h).
- Quantification of GFP/mCherry fluorescence intensity per cell. Data were analysed by two‐way ANOVA, showing no significant main effects of AO treatment, F(1, 212) = 0.564, P = 0.4534, or tau expression F(2, 212) = 1.579, P = 0.2087, but a significant interaction effect, F(2, 212) = 9.854, P < 0.0001, n = 16–50 cells/group.
To address the possibility that this was due to a general defect of autophagy caused by tau, we assessed autophagic flux in cells expressing GFP‐LC3 and hTau or hP301L (Fig EV1). Cells were starved in HBSS (Hanks’ Balanced Salt Solution) to induce the formation of autophagosomes and treated with bafilomycin A to block the degradation of autophagosomes by lysosomes. DMSO vehicle was used as a control. Both hTau‐ and hP301L‐expressing cells were able to form and degrade autophagosomes to the same extent as control cells (Fig EV1A and B). Together, these results suggest that tau specifically impaired the selective autophagy of damaged mitochondria.
Figure EV1. Transient transfection of Tau does not impair autophagic flux.
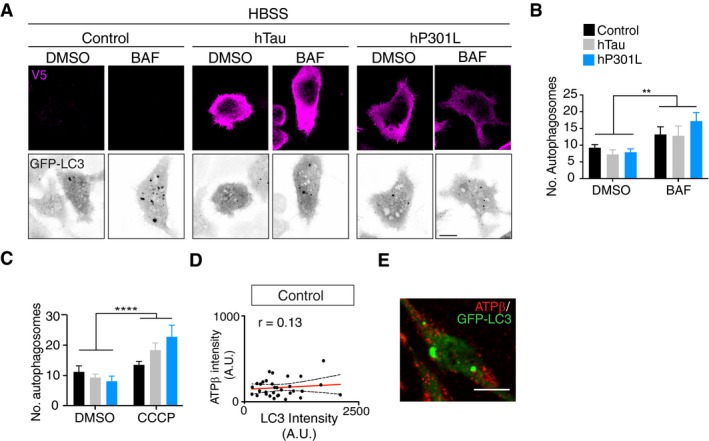
-
ARepresentative images of N2a cells expressing GFP‐LC3 and hTau, hP301L or control vector, that were starved in HBSS and treated with bafilomycin A (BAF), illustrating an increase in LC3‐positive autophagosomes with BAF treatment.
-
BQuantification of LC3‐positive autophagosomes after 24 h of tau expression in starved cells that were treated with DMSO or BAF. Data were analysed by two‐way ANOVA, illustrating a main effect of BAF treatment, F(1, 135) = 10.3, P = 0.0017, but no main effect of tau expression, F(2, 135) = 0.5425, P = 0.5826 and no significant interaction effect F(2, 135) = 0.6667, P = 0.5151, n = 19–35 cells/group.
-
CQuantification of LC3‐positive autophagosomes after 24 h of tau expression and 4 h of CCCP treatment, revealing no inhibition of autophagosome formation by tau. Data were analysed by two‐way ANOVA, illustrating a main effect of CCCP treatment, F(1, 158) = 17.81, P < 0.0001, but no main effect of tau expression, F(2, 158) = 0.7444, P = 0.4767 and no significant interaction effect F(2, 158) = 2.939, P = 0.0558, n = 19–34 cells/group.
-
D, ELC3 fluorescence intensity does not correlate with the mitochondrial signal, indicating a lack of colocalisation (r = 0.125, P = 0.4867), n = 33 cells.
Parkin translocation to mitochondria is impaired by tau following depolarisation of mitochondria
A key step in the canonical mitophagy pathway is the translocation of Parkin from the cytosol to mitochondria, which can be visualised by the accumulation of bright fluorescent clusters of Parkin that usually colocalise with mitochondrial markers (Fig 2A). We therefore asked whether tau impaired this process. To address this, N2a cells were co‐transfected with GFP‐Parkin and either hTau or hP301L, or an empty vector control. After 24 or 48 h, the cells were treated with CCCP for 4 h and assessed for Parkin translocation to mitochondria by manually selecting regions of interest (ROIs) around individual instances of translocation, defining Parkin clusters (Fig 2A). CCCP treatment induced Parkin translocation in control N2a cells; however, the number of Parkin clusters was significantly decreased in hP301L cells at 24 h (Fig 2B and C). We also verified that there was no Parkin translocation in control N2a cells treated with DMSO (Fig 2B). As Parkin ubiquitinates mitochondrial outer membrane proteins once translocated (Geisler et al, 2010; Chen & Dorn, 2013), we also assessed whether there was reduced ubiquitination of mitochondrial targets following tau expression at 24 h (Fig EV2). To this end, cells expressing either hTau, hP301L, or control vector and myc‐Parkin were treated with CCCP and then co‐immunostained for ubiquitin and the mitochondrial matrix protein SOD2 (which should be protected from Parkin‐mediated proteolysis). Ubiquitin fluorescence intensity was then assessed in mitochondrial regions of interest and expressed as fold change of whole‐cell ubiquitin levels, to account for differences in the overall available ubiquitin. Control cells exhibited striking localisation of ubiquitin to mitochondria following CCCP treatment, which remained robust in hTau‐expressing cells at this 24‐h time point (Fig EV2A and B). In contrast, mitochondrial ubiquitin was significantly decreased in hP301L‐expressing cells compared to control cells following CCCP treatment (Fig EV2A and B), in line with our findings of reduced Parkin translocation in this group (Fig 2C).
Figure 2. CCCP‐induced Parkin translocation to mitochondria is impaired in tau‐expressing cells.
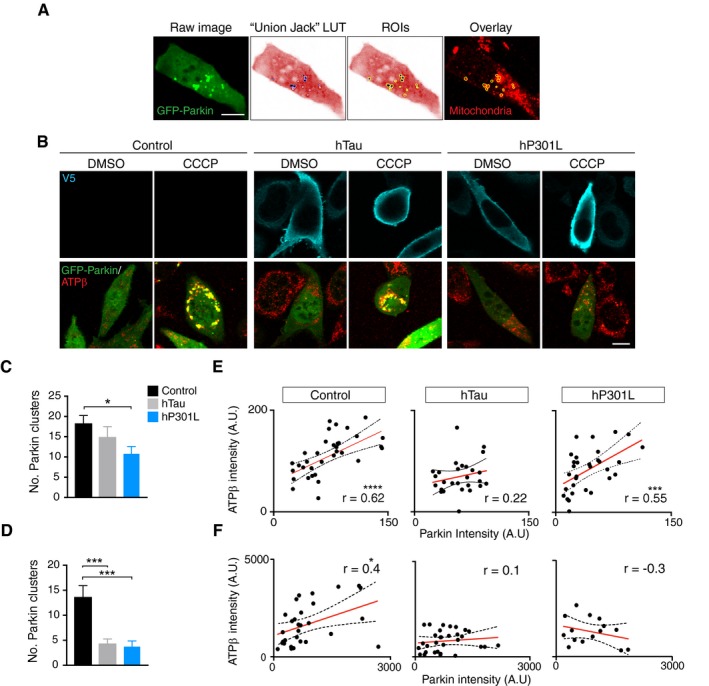
- Illustration of the image analysis process. The inverted “Union Jack” look‐up table (LUT) was used to better visualise GFP‐Parkin clusters, which were then manually selected as regions of interest (ROIs) and the corresponding mitochondrial fluorescence intensity was measured.
- Representative images of N2a cells expressing GFP‐Parkin and hTau, hP301L or empty vector (Control) for 24 h, that were then treated with 10 μM CCCP for 4 h.
- Quantification of Parkin clusters/cell at 24 h post‐transfection. Data were analysed with a Kruskal–Wallis test, χ2(2) = 8.18, P = 0.0168, followed by Dunn's post hoc analysis, n = 33–38 cells/group.
- Quantification of Parkin clusters/cell at 48 h post‐transfection. Data were analysed with a Kruskal–Wallis test, χ2(2) = 19.2, P < 0.0001, followed by Dunn's post hoc analysis, n = 37–55 cells/group.
- To assess colocalisation of Parkin clusters and mitochondria at 24 h post‐transfection, Pearson correlations between the mean normalised fluorescence intensity of the Parkin clusters and the mitochondrial signal following CCCP treatment were calculated. Control r = 0.616, P = 0.0001; hTau r = 0.217, P = 0.2771; hP301L r = 0.548, P = 0.0014 (n = 27–34 cells/group).
- Pearson correlations between the mean normalised fluorescence intensity of the Parkin clusters and the mitochondrial signal following CCCP treatment at 48 h post‐transfection. Control r = 0.434, P = 0.0167; hTau r = 0.128, P = 0.4941; hP301L r = −0.299, P = 0.2794 (n = 15–31 cells/group). Note the different intensity scales between (E) and (F) reflect different bit depths of the images.
Figure EV2. Ubiquitination of mitochondrial targets is reduced by hP301L.
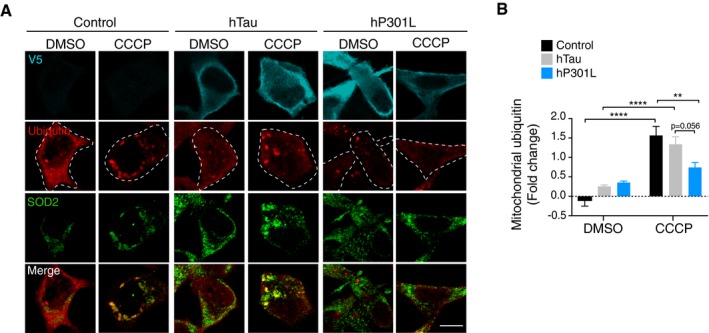
- Representative images of N2a cells transfected with hTau, hP301L, or empty vector control and myc‐Parkin for 24 h and treated with CCCP for 4 h. Immunostaining was performed for mitochondria (SOD2) and ubiquitin. Scale bar = 10 μm.
- Mitochondrial ubiquitin was quantified by assessing its fluorescence intensity in mitochondrial ROIs and calculating the fold change over whole‐cell ubiquitin intensity. Data were analysed by two‐way ANOVA, revealing a main effect of CCCP treatment F(1, 124) = 58.7, P < 0.0001, no main effect of tau species F(2, 124) = 1.21, P = 0.3004, and a significant interaction effect F(2, 124) = 7.16, P = 0.0011. Data are given as mean and SEM, **P < 0.01, ****P < 0.0001 for simple effects, n = 15–29 cells/group.
Source data are available online for this figure.
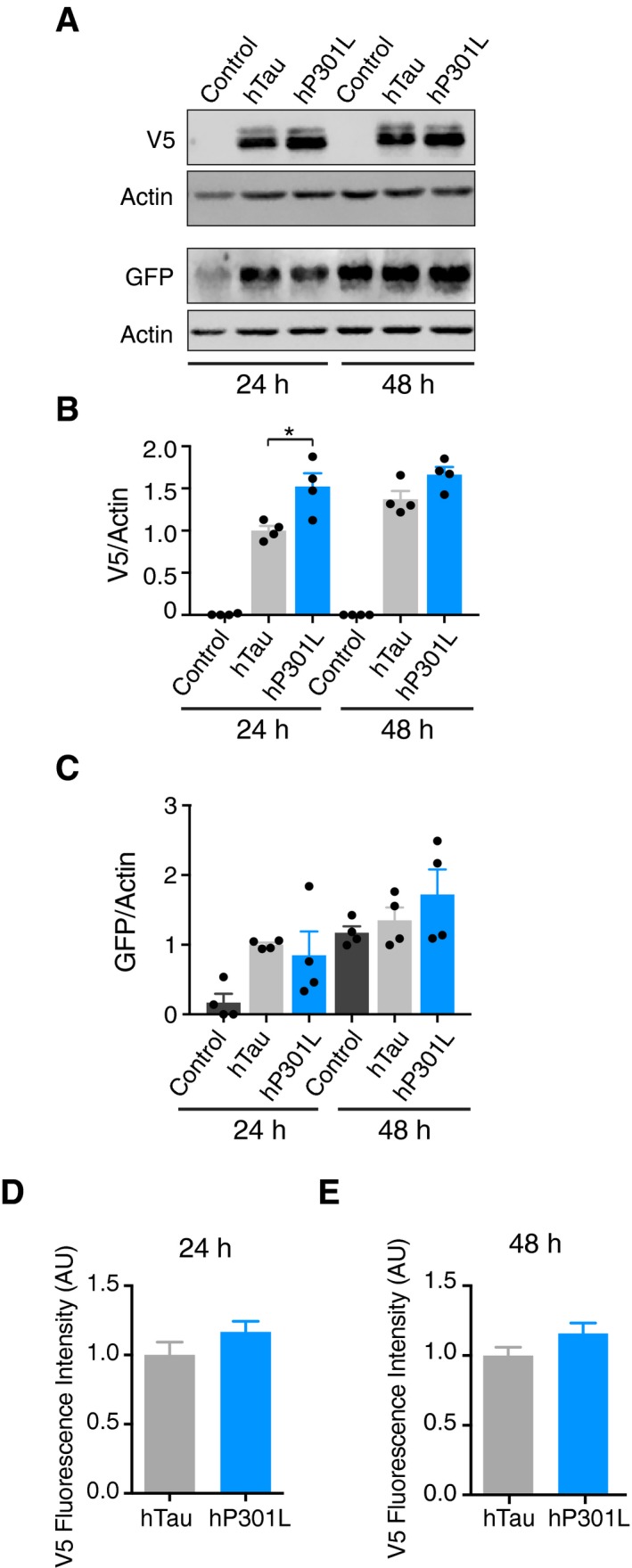
At 48 h post‐transfection, the presence of either hTau or hP301L significantly impaired Parkin translocation (Fig 2D), indicating a time‐dependent effect of tau expression. To assess whether this was due to greater protein levels of tau at 48 h, we performed immunoblots and found that levels were not significantly higher at 48 h (Fig EV3A and B). We also ensured that expression of tau did not decrease the levels of GFP‐Parkin, with co‐transfection actually boosting the expression of GFP‐Parkin (Fig EV3A and C). At 24 h post‐transfection, hP301L was expressed at higher levels than hTau (Fig EV3A and B). Therefore, to ensure that the increased toxicity of hP301L was not simply an effect of greater expression level compared to hTau, we assessed tau expression levels in all captured cells at 24 and 48 h, by measuring the intensity of V5 staining (Fig EV3D and E). Importantly, there were no significant differences between expression levels of hTau and hP301L in the analysed cells, pointing to a specific toxicity of the P301L mutation.
Figure EV3. Tau and Parkin expression levels.
-
AImmunoblots illustrating population expression level of Tau‐V5 and GFP‐Parkin at 24 and 48 h post‐transfection.
-
BQuantification of human tau levels from immunoblots. Tau levels were compared using two‐way ANOVA, with the control group excluded from the statistical analysis, revealing a main effect of expression time, F(1, 12) = 5.865, P = 0.0322, a main effect of tau species, F(1, 12) = 14.66, P = 0.0024 and no interaction effect F(1, 12) = 1.187, P = 0.2972.
-
CQuantification of GFP‐Parkin levels from immunoblots. Parkin levels were compared using two‐way ANOVA, revealing a main effect of expression time, F(1, 18) = 16.29, P = 0.0008, a main effect of tau species, F(2, 18) = 4.249, P = 0.0.308 and no interaction effect F(2, 18) = 1.176, P = 0.3313.
-
D, ETau expression level in cells analysed for Parkin translocation, as represented by V5 fluorescence intensity following immunostaining, at 24 h (unpaired t‐test t = 1.412, P = 0.1692) and at 48 h (t = 1.673, P = 0.0979).
CCCP treatment is known to result in the translocation of diffusely distributed cytosolic Parkin to the surface of mitochondria (Ding et al, 2010; Drew et al, 2014). We therefore assessed the degree of colocalisation of each Parkin cluster (defined by individual ROIs, Fig 2A) with the corresponding mitochondrial signal by correlating the fluorescence intensities (Fig 2E and F). At 24 h post‐transfection, CCCP treatment resulted in a significant correlation between Parkin fluorescence intensity in the clusters (normalised to background fluorescence) and the mitochondrial fluorescence intensity at a whole‐cell level, suggesting Parkin–mitochondria co‐distribution in control cells (Fig 2E). Interestingly, this correlation was absent in hTau‐transfected cells, indicating a disturbance of Parkin translocation to mitochondria, even though the number of Parkin clusters was similar at this time point (Fig 2C and E). In hP301L cells, a significant positive correlation was observed (Fig 2E). At 48 h post‐transfection, CCCP treatment again resulted in a positive correlation between the Parkin and mitochondrial intensities in control cells, but this was completely absent in both hTau and hP301L cells (Fig 2F), indicating disrupted Parkin recruitment to mitochondria in these groups.
Because CCCP has been shown to induce autophagosome formation independent of Parkin‐mediated mitophagy (Kwon et al, 2011), as an additional control, we tested if this process was affected by tau. We treated GFP‐LC3 and tau‐expressing cells with CCCP and quantified the number of GFP‐LC3 autophagosomes per cell. CCCP treatment indeed induced an increase in the number of autophagosomes in the three groups, and while there was a clear trend towards higher numbers of autophagosomes in hP301L‐expressing cells following CCCP treatment, there was no statistically significant interaction effect (P = 0.06, Fig EV1C). At this time point (4 h) after CCCP‐induced depolarisation, autophagosomes did not yet colocalise with mitochondria (Fig EV1D and E) which explains why there was no matching effect of the fewer Parkin clusters to result in fewer mito‐autophagosomes. Therefore, cells expressing either variant of tau were able to form autophagosomes after CCCP treatment, suggesting that tau must affect a separate molecular process. This is compatible with our data showing that tau impairs Parkin clustering and recruitment to defective mitochondria (Fig 2B–F), which is required for autophagosome recognition of these organelles.
Impaired mitophagy is not due to changes in the mitochondrial membrane potential
We next determined whether tau reduced Parkin translocation onto mitochondria by acting upstream in the classical mitophagy pathway. Here, a severe decrease in membrane potential is considered to be the initiating step of the mitophagy cascade (Matsuda et al, 2010). Previous reports have suggested that tau has a hyperpolarising effect on mitochondria, which could counteract the depolarising signal required to initiate mitophagy (Schulz et al, 2012; Hu et al, 2016). We therefore assessed whether tau altered the mitochondrial membrane potential in our model, using the membrane potential‐sensitive dye Rhodamine 123 and performing FACS analysis. We detected no difference in fluorescence intensity between control cells and hTau‐ or hP301L‐transfected cells at baseline (without CCCP treatment, Fig 3A) or after CCCP treatment (Fig 3B). In each experiment, a positive or negative depolarisation control sample was run in parallel (CCCP‐ and DMSO‐treated cells, respectively). There was also no difference in the number of Rhodamine 123‐positive cells observed post‐CCCP treatment between groups (Fig 3C). Therefore, tau did not impair mitophagy by modulating the mitochondrial membrane potential in this model.
Figure 3. Mitochondrial membrane potential is unchanged in tau‐expressing cells.
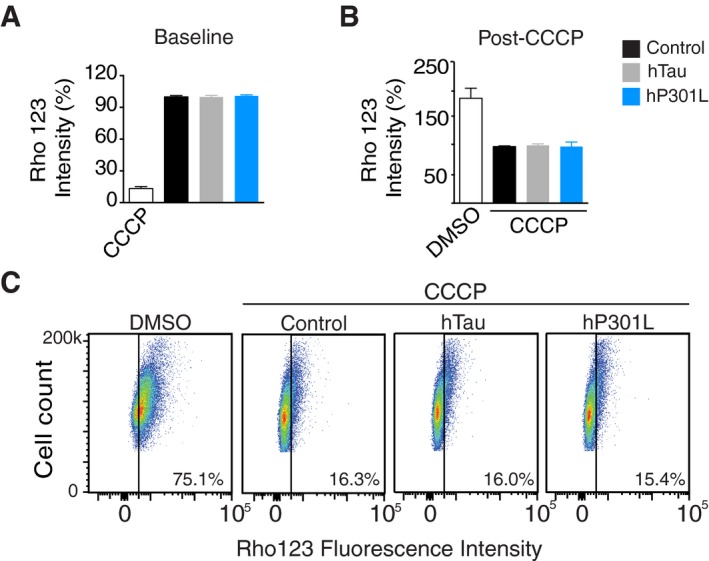
- FACS analysis of cells loaded with Rhodamine 123. The geometric mean fluorescence was calculated and expressed as % of the empty vector control. A CCCP‐treated cohort was included as a positive control, but not included in the statistical analysis. Data were analysed by one‐way ANOVA, F(2, 14) = 0.196, P = 0.8245.
- FACS analysis of cells treated with CCCP after 24 h of tau expression. Data were analysed by one‐way ANOVA, F(2, 15) = 0.434, P = 0.6555. The DMSO negative control is shown but was not analysed statistically.
- Representative dot plots of cells loaded with Rhodamine 123. The gates (black line) were set in unstained cells to distinguish Rhodamine‐positive cells (right half) from Rhodamine‐negative cells (left half).
Pharmacologically mimicking the stabilising effect of tau on the cytoskeleton does not lead to reduced Parkin translocation
An intact microtubule network has been shown to be required for Parkin clustering on mitochondria (Vives‐Bauza et al, 2010), and tau can stabilise microtubules (Fig 4A). It is therefore plausible that an altered microtubule state caused by tau was responsible for the reduced Parkin clustering. To determine whether this was the case, we mimicked the microtubule‐stabilising effect of tau by treating cells with paclitaxel (Taxol, 2 μM, 4 h), prior to CCCP treatment. We found that Taxol treatment stabilised microtubules in a manner similar to tau expression (Fig 4B). However, in contrast to the effect of tau, Taxol treatment significantly increased the number of Parkin clusters (Fig 4C), indicating that tau's effect on microtubule dynamics is unlikely to impair Parkin clustering. We also examined acetylation and tyrosination of microtubules, which are two common post‐translational modifications of tubulin, following tau expression (Song & Brady, 2015). We did not detect any changes in either form of tubulin (relative to total α‐tubulin) in tau‐transfected cells compared to control cells (Fig 4D and E), suggesting that altered modifications of microtubules are not implicated in the current experiments. Tau has also been reported to stabilise actin under certain conditions (Fulga et al, 2007; DuBoff et al, 2012). We therefore used Jasplakinolide, an actin‐stabilising compound, to pre‐treat N2a cells (5 nM, 2 h), before adding CCCP. Similar to Taxol, Jasplakinolide treatment increased the number of Parkin clusters induced by CCCP (Fig 4F), suggesting that tau also did not impair Parkin clustering by stabilising the actin network. Together, these data indicate that an indirect, cytoskeleton‐mediated mechanism is unlikely to be responsible for the inhibitory effect of tau on Parkin translocation.
Figure 4. Stabilising the cytoskeleton pharmacologically does not mimic the effect of tau on Parkin translocation.
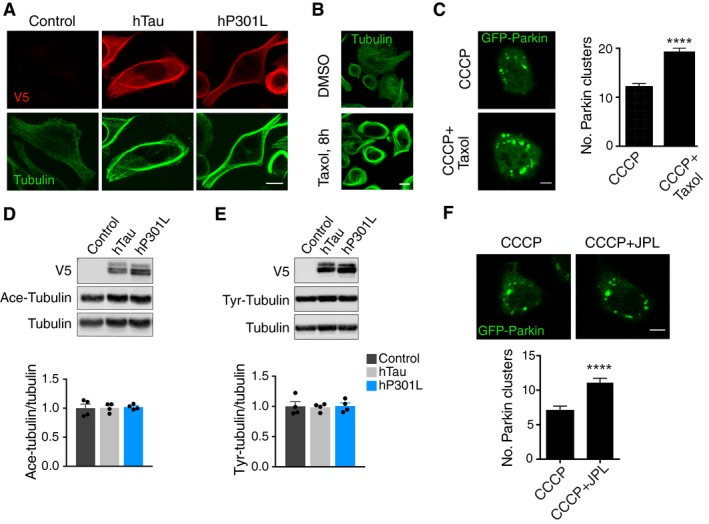
- Immunostaining for microtubules after transient expression of hTau and hP301L.
- Taxol induces microtubule bundling to a similar extent to tau expression.
- Representative images and quantification of Parkin clusters in N2a cells following CCCP and Taxol treatment (Mann–Whitney U‐test, U = 22,448, P < 0.0001, n = 224, 273 cells/group for CCCP only and CCCP + Taxol, respectively).
- Immunoblot analysis of acetylated tubulin (Ace‐Tubulin) following expression of hTau and hP301L for 24 h [one‐way ANOVA, F(2, 9) = 0.0432, P = 0.9579].
- Immunoblot analysis of tyrosinated tubulin (Tyr‐Tubulin) following expression of hTau and hP301L for 24 h [one‐way ANOVA, F(2, 9) = 0.0402, P = 0.9608].
- Representative images and quantification of Parkin clusters in N2a cells following treatment with CCCP and the actin stabiliser Jasplakinolide (JPL; Mann–Whitney U‐test, U = 16,069, P < 0.0001, n = 224, 199 cells/group for CCCP only and CCCP + JPL, respectively).
Pathological tau aberrantly interacts with Parkin, trapping it in the cytosol
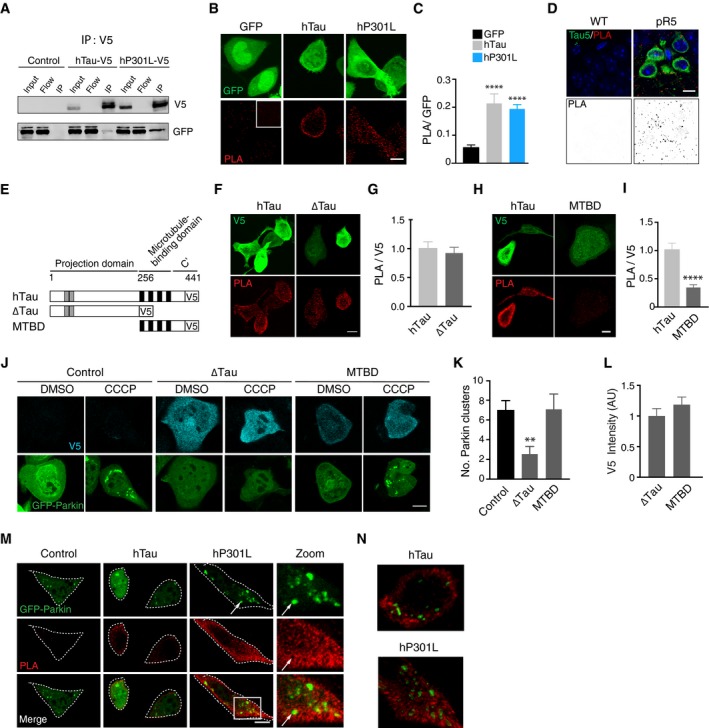
Based on the above results, we hypothesised that tau may exert a direct effect on Parkin, at a site different to the mitochondrial surface, thereby preventing it from correctly translocating to damaged mitochondria. We therefore assessed whether different tau species were able to bind Parkin in N2a cells. To this end, we performed co‐immunoprecipitations using cells transfected with GFP‐Parkin and hTau‐V5 or hP301L‐V5 (Fig 5A). Following pull‐down with a V5 antibody, we were able to detect GFP‐Parkin in the immunoprecipitated fractions of both hTau and hP301L, but not in control cells transfected with an empty vector (Fig 5A). We next confirmed tau‐Parkin interactions by another method, the proximity ligation assay (PLA), which generates a fluorescent signal based on the proximity of primary antibodies to their potential interacting partners, via the in situ hybridisation of oligonucleotides attached to secondary antibodies (Söderberg et al, 2008). We expressed hTau‐GFP and hP301L‐GFP, using GFP as control, and performed PLAs for tau and endogenous Parkin. As the PLA signal is dependent on the amount of protein that is present in the cell, we controlled for differences in expression levels by normalising the PLA fluorescence intensity to the intensity of the tau‐tag (GFP or V5). No interactions were detected between endogenous tau and endogenous Parkin in the control condition, but endogenous Parkin interacted with both species of transfected human tau (Fig 5B and C). To determine whether tau could also interact with Parkin in vivo, we next performed PLAs on brain sections from 14‐month‐old pR5 mice, which express hP301L mutant tau primarily in the hippocampus and amygdala. Unlike in wild‐type mice, we found that tau and Parkin interacted in the pR5 mouse brain (Fig 5D), indicating that tau overexpression resulted in abnormal interactions between these two proteins. As this interaction was absent for the endogenous proteins (Fig 5A–D), it may be specific—and therefore pathophysiologically relevant—to disease contexts, where tau accumulates at abnormal levels in the cytosol (as modelled here by overexpression).
Figure 5. Exogenous tau interacts with Parkin, trapping it in the cytosol.
-
ACo‐immunoprecipitations of V5‐tagged tau and GFP‐Parkin. The V5 tag was used to pull down tau, and the membrane was probed for V5 (using an antibody raised in a different species) and GFP. IP = immunoprecipitation, Flow = flow‐through lysate following removal of beads.
-
BMaximum projection images of proximity ligation assays (PLAs) for tau and endogenous Parkin in cells expressing hTau‐GFP, hP301L‐GFP or GFP. Inset = negative control (antibodies to tau and histone H3, which are not known binding partners).
-
CQuantification of PLA signals in maximum projections normalised to the GFP signal. Results were analysed with a Kruskal–Wallis test, χ2(2) = 27.2, P < 0.0001, followed by Dunn's post hoc analysis to test for differences from the GFP control, n = 17 cells in GFP condition, 14 cells in hTau condition and 27 cells in hP301L condition.
-
DPLA for Parkin and hP301L in an aged pR5 mouse and a littermate control.
-
ESchematic of tau constructs used in PLA experiments.
-
F, GPLA for Parkin and ∆Tau, revealing interactions via the projection domain of tau. Data were analysed with a Mann–Whitney U‐test (U = 185, P = 0.9224, n = 21, 18 cells/group for hTau and ∆Tau, respectively).
-
H, IPLA for Parkin and the microtubule‐binding domain construct (MTBD). Data were analysed with a t‐test (t = 5.226, P < 0.0001, n = 16, 17 cells/group for hTau and MTBD, respectively).
-
J, KCells expressing tau constructs together with GFP‐Parkin (24 h) with and without CCCP treatment. Parkin clusters were counted, illustrating that Parkin translocation was impaired by ∆Tau, but not MTBD. Data were analysed with a Kruskal–Wallis test χ2(2) = 10.4, P = 0.0056, followed by Dunn's post hoc, n = 67, 42, 53 cells/group for hTau, ∆Tau and MTBD, respectively.
-
LThe V5 fluorescence intensity in each cell analysed in (K) (Mann–Whitney U‐test, U = 953, P = 0.2332, n = 42, 53 cells/group for ∆Tau and MTBD, respectively).
-
MCells were transfected with GFP‐Parkin and hTau‐V5, hP301L‐V5 or an empty vector control, and treated with CCCP. PLAs were performed for tau and Parkin. Notably, at Parkin clusters (arrow), no interactions between tau and Parkin were evident.
-
N3D reconstructions in IMARIS, illustrating a lack of PLA signals in Parkin clusters.
To determine whether tau‐Parkin interactions were involved in impairing Parkin translocation to mitochondria, we next conducted PLAs for Parkin and two tau deletion constructs (Fig 5E). More specifically, we examined whether the projection domain or the microtubule‐binding domain (MTBD) of tau (Li & Götz, 2017b) was necessary for its inhibition of mitophagy. To investigate this, we expressed either an amino‐terminal fragment of tau, truncated before the microtubule‐binding domain (“∆Tau”, Fig 5F and G) or a tau fragment containing the microtubule‐binding domain (“MTBD”, Fig 5H and I) in N2a cells, and compared the Parkin PLA signal to that for full‐length hTau. We found that Parkin was able to interact with ∆Tau, but not MTBD‐Tau (Fig 5F–I). As ∆Tau lacks the MTBD and is therefore primarily cytosolic in distribution, we hypothesised that diffuse forms of tau may trap Parkin in the cytosol, preventing it from translocating to mitochondria. To test this, we transfected cells with ∆Tau, MTBD‐Tau or an empty vector control, as well as GFP‐Parkin, and quantified the number of Parkin clusters that occurred in response to CCCP treatment (Fig 5J and K). ∆Tau, but not MTBD‐Tau, significantly reduced the number of Parkin clusters (Fig 5K). Given that modified forms of tau protein may differ in their stability, we ensured that the mean expression levels of the different versions of tau were equivalent (Fig 5L). Our finding indicates that the interaction between tau and Parkin is indeed a factor mediating tau's impairment of this process, providing evidence that it is the cytosolic pool of tau that is responsible for inhibiting Parkin translocation.
If tau traps Parkin in the cytosol, then tau‐Parkin interactions should be absent in cases where Parkin translocation has already occurred in response to mitochondrial damage. To explore this, we expressed GFP‐Parkin and V5‐tagged tau constructs in N2a cells and treated them with CCCP to induce Parkin translocation. PLAs revealed that tau and Parkin did not interact on the surface of mitochondria under these conditions (Fig 5M and N). In some instances, a clear exclusion of the PLA signal in Parkin clusters was observed (Fig 5M, arrows). To better visualise this exclusion, we reconstructed the Parkin clusters in 3D with the surface tool in IMARIS (Fig 5N). This observation lends further support to the hypothesis that tau sequesters Parkin in the cytosol, as tau did not interact with Parkin that was already localised to mitochondria.
Tau inhibits mitophagy in C. elegans neurons
To verify our results in vivo, we made use of a recently published mitophagy reporter in C. elegans (Palikaras et al, 2015). This reporter is a version of mito‐QC, called mito‐Rosella, which we cloned under a pan‐neuronal promoter to restrict its expression to the nervous system (Fig 6A), as opposed to muscle, where it was employed previously (Palikaras et al, 2015). We also confirmed that the reporter maintained its localisation to mitochondria following the change of promoter from muscle to neurons (Fig EV4A). These animals were crossed with transgenic strains expressing hTau or hP301L pan‐neuronally (Kraemer et al, 2003). The worm‐specific mito‐Rosella is similar to the mammalian mito‐QC construct described above, except that the fluorophore dsRed instead of mCherry and an alternative outer membrane targeting sequence (derived from Tom20 instead of from Fis‐1) was used (Palikaras et al, 2015). To induce mitophagy, worms were treated with the mitochondrial complex IV inhibitor sodium azide (NaN3; 8 mM, 1 h) or CCCP (15 μM, 2 h), and the ratio of GFP to dsRed fluorescence intensity in mitochondrial ROIs was analysed in neurons of the ventral nerve cord (Fig 6B, Appendix Fig S1). Both treatments induced mitophagy in the wild‐type strain (no exogenous tau) compared to vehicle (Fig 6C and D), illustrating the ability of neurons to initiate mitophagy in response to mitochondrial stress in this in vivo model.
Figure 6. Tau inhibits mitophagy in Caenorhabditis elegans neurons.
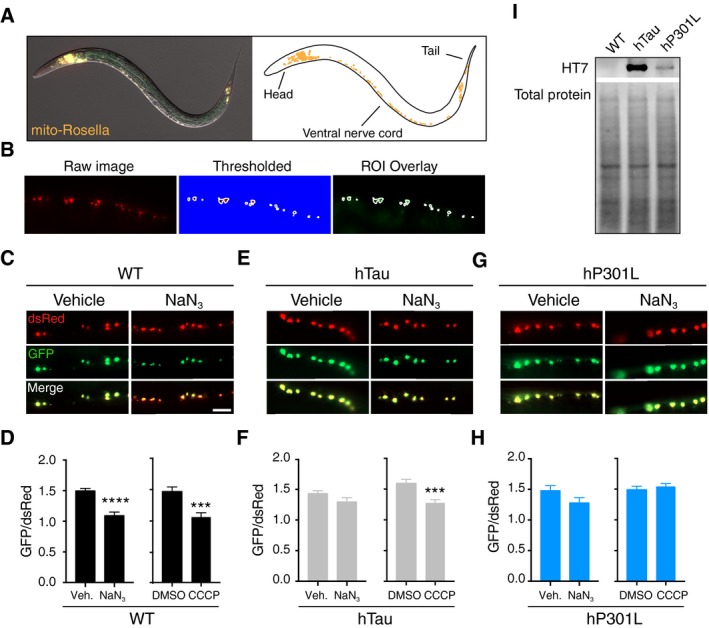
-
AExpression pattern of the mito‐Rosella biosensor in the nervous system.
-
BAnalysis pipeline of mitochondria in the C. elegans ventral nerve cord. Mitochondria were selected by choosing a threshold in the dsRed channel. This threshold was used to create ROIs around single mitochondria, and the corresponding GFP fluorescence intensity was then measured.
-
C, DAnalysis of mitophagy in worms transgenic for mito‐Rosella but not tau (WT), that were treated with NaN3 (8 mM, 1 h) or CCCP (15 μM, 2 h). Data were analysed by unpaired t‐test, NaN3 t = 5.56, P < 0.0001; CCCP t = 3.92, P = 0.0002.
-
E, FAnalysis of mitophagy in worms expressing hTau in response to NaN3 treatment (unpaired t‐test, t = 1.62, P = 0.1126) and CCCP treatment (Mann–Whitney U‐test, U = 113, P = 0.002).
-
G, HAnalysis of mitophagy in worms expressing hP301L in response to either NaN3 (Mann–Whitney U‐test, U = 126, P = 0.2711) or CCCP (Mann–Whitney U‐test, U = 375, P = 0.3697) treatment.
-
ITau expression in worm lysates, as detected using the human tau‐specific antibody HT7.
Figure EV4. Characterisation of mito‐Rosella Caenorhabditis elegans and the effect of hV337M‐Tau on mitophagy in C. elegans neurons.
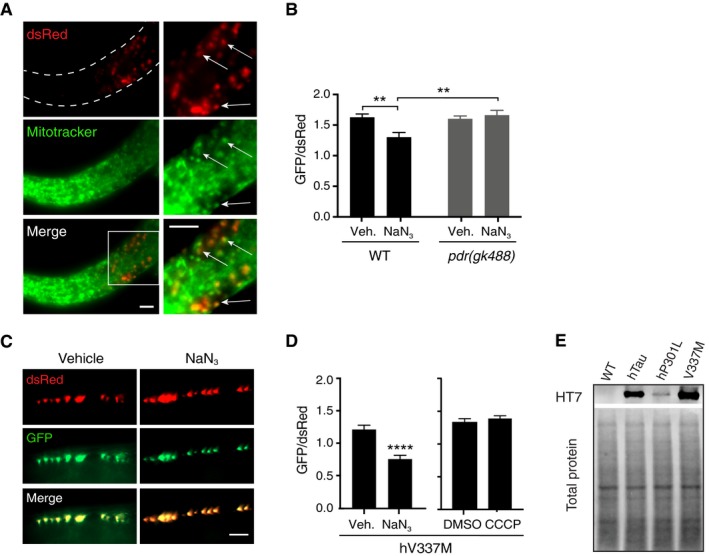
-
AMitotracker Deep Red staining illustrating colocalisation between Mitotracker (pseudo‐coloured green for better visualisation) and mito‐Rosella (dsRed). Scale bars = 5 μm.
-
BAnalysis of mitophagy in wild‐type worms and pdr‐1(gk488) mutant animals in response to NaN3. Data were analysed using two‐way ANOVA, revealing a main effect of genotype F(1, 106) = 6.415, P = 0.0128; a main effect of NaN3 treatment F(1, 106), P = 0.047; and a significant interaction effect F(1, 106), P = 0.0041; n = 26–29 animals/group.
-
C, DAnalysis of mitophagy in worms expressing hV337M‐Tau in response to NaN3 (Mann–Whitney U‐test, U = 25, P < 0.0001, n = 16, 19 for vehicle and NaN3, respectively), and CCCP treatment (unpaired t‐test, t = 0.8113, P = 0.4211, n = 25, 27 for DMSO and CCCP, respectively).
-
ETau expression in worm lysates detected with the human tau‐specific antibody HT7 (extended immunoblot from Fig 6I).
To assess whether this response required the C. elegans Parkin homologue PDR‐1, mito‐Rosella animals were crossed to pdr‐1(gk488) mutant animals, lacking a functional PDR‐1 protein. Worms mutant for PDR‐1 did not undergo mitophagy following NaN3 treatment (Fig EV4B), illustrating that the mitophagy was PDR‐1 dependent. In contrast to neuronal mitochondria in wild‐type animals, those in hTau worms only underwent mitophagy in response to CCCP, but not NaN3 (Fig 6E and F), and in the hP301L strain, the phenotype was even more pronounced, as we could not detect mitophagy in response to either treatment (Fig 6G and H). This suggests that expression of tau in vivo impairs stress‐induced neuronal mitophagy and that P301L mutant tau is more detrimental than wild‐type tau. This difference was found despite there being much higher hTau levels than hP301L levels in each respective strain (Fig 6I), illustrating the hyper‐toxicity conferred by the hP301L mutation. We also tested worms transgenic for V337M tau (another mutation found in familial cases of FTD), to determine whether specific mutations had different effects on mitophagy. These animals displayed an intermediate mitophagy impairment, which was detectable in response to NaN3 treatment, but not CCCP treatment (Fig EV4C and D), despite higher tau expression (Fig EV4E). This suggests that the toxicity of different tau mutations on mitophagy varies, a notion that is supported by the diverse pathological features observed in patients and animal models carrying different FTD mutations (Hong et al, 1998; Gauthier‐Kemper et al, 2011; Bodea et al, 2016).
Discussion
Mitochondrial impairment is a key feature of AD, and abnormal mitochondria have been shown to accumulate in the brains of AD patients and mouse models of AD and FTD (Baloyannis et al, 2004; Calkins et al, 2011; Kopeikina et al, 2011). Under physiological conditions, cells can cope with mitochondrial damage through a number of well characterised quality control systems, with severely damaged mitochondria being eliminated via mitophagy (Rugarli & Langer, 2012; Youle & van der Bliek, 2012). In AD, however, a “system overload” has been described, such that mitochondrial damage cannot be dealt with effectively (Ye et al, 2015). Given that disease‐associated tau is known to be detrimental to mitochondrial function and dynamics, we investigated specifically whether the deleterious effect of wild‐type and P301L mutant tau would extend to mitophagy, thereby exacerbating the damage to mitochondrial homeostasis. Here, we provided evidence in N2a cells and in the nervous system of C. elegans that tau impairs mitophagy in response to acute mitochondrial stress. We also showed that tau does not inhibit Parkin translocation via modulation of the mitochondrial membrane potential or changes to the cytoskeleton. Rather, we demonstrated that tau aberrantly interacts with Parkin in the cytosol, thereby preventing it from translocating to mitochondria following mitochondrial depolarisation.
A recent study reported increases in baseline levels of mitochondrial proteins in the presence of hTau, indirectly suggesting that mitophagy may be decreased, although specific inhibition of this process was not shown (Hu et al, 2016). The authors described a mechanism whereby hTau expression increased the mitochondrial membrane potential. In contrast, we found a pathological interaction of Parkin with human wild‐type and P301L mutant tau in the cytosol, a compartment in which tau is enriched under pathological conditions. This prevented the translocation of Parkin to defective mitochondria when overexpressed, in the absence of changes to the mitochondrial membrane potential. More specifically, we showed that Parkin interacts with tau via the projection domain of tau and that this Parkin‐interacting domain is required for tau‐mediated inhibition of mitophagy. In support of our data, Parkin and tau have also been demonstrated to co‐immunoprecipitate in other cell types (Petrucelli et al, 2004; Moore et al, 2008). We hypothesised that by interacting with Parkin, tau sequesters the protein in the cytosol, a concept that was supported by the lack of interactions between tau and Parkin on mitochondrial Parkin clusters. Interestingly, although hTau also interacted with Parkin, it did not inhibit Parkin translocation at 24 h post‐transfection. As both hTau and hP301L were expressed at similar levels in the analysed cells, this points to an accelerated pathology in the mutant tau model, a feature also reflected by animal models (Götz & Ittner, 2008). This finding also highlights the notion that although the tau‐Parkin interaction is required for the detrimental effect of tau on Parkin translocation, this interaction alone is not sufficient. The finding that tau specifically trapped Parkin in the cytosol could explain the increased toxicity of hP301L, as this mutation impairs microtubule binding and therefore may increase the cytosolic pool of tau that is available to inhibit Parkin translocation (Dayanandan et al, 1999). A similar mechanism of tau toxicity has been observed in the K369I FTD tau mutant mouse, where K369I tau was shown to bind to and trap the kinesin motor adapter JIP1 thus disrupting the transport of mitochondria and other distinct cargos (Ittner et al, 2008, 2009).
Interestingly, a recent report describes lower ubiquitination of mitofusins in hTau‐expressing HEK293 cells (Li et al, 2016). We have shown reduced mitochondrial ubiquitination in response to CCCP treatment in cells expressing hP301L‐Tau. As Parkin is known to ubiquitinate the mitofusins, causing their degradation and contributing to mitochondrial fission (Tanaka et al, 2010; Glauser et al, 2011), it is possible that altered mitofusin levels are also a result of the impaired Parkin recruitment described in our study.
One limitation of our experiments in N2a cells is that Parkin was overexpressed, possibly exaggerating the importance of Parkin‐dependent mitophagy in this system. This does not rule out the possibility that mitophagy can also be achieved by alternative pathways that operate in a Parkin‐independent manner (Chu et al, 2014; Lazarou et al, 2015). However, the mitophagy we observed in C. elegans was dependent on the Parkin homologue PDR‐1, so it is likely that inhibition of Parkin/PDR‐1 translocation was one of the contributing mechanisms, as determined in the cell culture model.
Although basal mitophagy has been recently demonstrated to occur in neurons in vivo both in Drosophila and mouse (Cornelissen et al, 2018; Lee et al, 2018; McWilliams et al, 2018), evidence for neuronal mitophagy specifically in response to acute mitochondrial stress remains sparse (Cummins & Götz, 2017; Whitworth & Pallanck, 2017). Consequently, we used C. elegans to test the effect of tau on mitophagy in a live nervous system. We were able to demonstrate for the first time that in worms, neuronal mitochondria can undergo mitophagy in response to mitochondrial depolarisation. Notably, this occurred without overexpressing Parkin or its C. elegans homologue PDR‐1. In support of our in vitro results, we also found that both hTau and hP301L inhibited mitophagy, but that the P301L mutation was more potent, as it prevented mitophagy at lower expression levels. Interestingly, the P301L mutation was also more toxic than the V337M mutation, illustrating differences in the pathogenicity of different tau mutations.
Our work significantly extends the current understanding of tau‐induced mitochondrial pathology. Tau is known to negatively impact three major aspects of mitochondrial function: (i) oxidative phosphorylation, in part by reducing complex I activity, which leads to decreased ATP production and oxidative stress (Stamer et al, 2002; Rhein et al, 2009; Li et al, 2016); (ii) mitochondrial dynamics, with mitochondrial elongation described in mutant tau flies and mice partly due to impaired localisation of the fission protein Drp‐1 to the outer mitochondrial membrane and increased levels of the fusion proteins OPA1 and the mitofusins (DuBoff et al, 2012; Li et al, 2016); and (iii) impaired mitochondrial transport, which has been documented in tau transgenic mice and cellular models, and can lead to abnormal mitochondrial distribution such that local energy requirements are not met (Ebneth et al, 1998; Stamer et al, 2002; Ittner et al, 2008; Shahpasand et al, 2012). Here, we have shown that a fourth aspect of mitochondrial function, mitophagy, is also inhibited by tau in vivo. Our results suggest that tau can exacerbate its own mitochondrial toxicity by interfering with a neuron's ability to selectively remove the organelles it itself has damaged. Neurons are energetically demanding cells and the exacerbation of damage to the mitochondrial pool could have lasting implications not only for cellular energy requirements, but also for oxidative stress levels and redox balance (Grimm & Eckert, 2017). In the case of tau‐associated diseases such as AD and FTD, impaired mitophagy and the associated accumulation of dysfunctional mitochondria are likely to comprise an important pathomechanism of neurodegeneration. Indeed, the importance of a functional PINK1/Parkin pathway in neuronal health is illustrated by the incidence of mutations in these proteins in familial Parkinson's disease (Matsumine et al, 1997; Valente et al, 2001). Modulating mitophagy is therefore a potential therapeutic avenue to ameliorate tauopathies and other neurodegenerative disorders.
Materials and Methods
Reagents and antibodies
Carbonyl cyanide 3‐chlorophenylhydrazone (CCCP) and Jasplakinolide were obtained from Abcam. Oligomycin, antimycin A, bafilomycin A, NaN3, Taxol, DMSO and Rhodamine 123 were from Sigma‐Aldrich. For immunostaining and proximity ligation assays (PLAs), the following antibodies were used: alpha‐Tubulin mouse (Sigma, T6074, 1:500), ATP synthase beta subunit mouse (Abcam, Ab14730, 1:700), Parkin rabbit (Abcam, Ab15954, 1:500), SOD2 rabbit (Abcam, Ab13533, 1:500), Tau 5 mouse (Millipore, 577801, 1:500), 4R‐Tau (1E1) mouse (Millipore, 05‐804, 1:500), phospho‐H3 rabbit (Millipore, 06‐570), Ubiquitin mouse (Enzo Life Sciences, BML‐PW8810, 1:100), V5 rabbit (Sigma, V8137, 1:500), V5 mouse (Invitrogen, R960‐25, 1:500) and V5 chicken (Abcam, Ab9113, 1:500–700). For immunoblotting, antibodies used were as follows: β‐Actin mouse (Abcam, Ab8226, 1:3,000), GFP rabbit (Millipore, 3080P, 1:2,000), acetylated Tubulin mouse (Sigma, T6074, 1:2,000), tyrosinated Tubulin mouse (Sigma, T9028, 1:2,000), alpha‐Tubulin mouse (Sigma, T6074, 1:2,000) and V5 rabbit (Sigma, V8137, 1:2,500–5,000).
The plasmid containing the mCherry‐GFP‐Fis1 (“mito‐QC”) mitophagy sensor was obtained from the MRC Protein Phosphorylation and Ubiquitylation Unit, The University of Dundee (Allen et al, 2013). To express mito‐QC in N2a cells, the GFP cDNA was excised from a pAAV‐hSyn‐EGFP plasmid (Addgene plasmid # 50465) using the KpnI and EcoRI restriction sites. Primers with the corresponding restriction site sequences were generated to the mito‐QC fragment, and the resulting PCR product was ligated into the AAV plasmid.
Mouse strains and ethics
Male tau transgenic pR5 mice carrying the P301L mutation found in familial cases of FTD and wild‐type littermates were housed in standard conditions, in group cages with ad libitum access to food. Mice were used at 14–15 months of age (Götz et al, 2001). Animal experimentation was approved by the Animal Ethics Committee of the University of Queensland (approval numbers QBI/412/14/NHMRC and QBI/312/14/NHMRC). Mice used for PLA were euthanised by intraperitoneal injections of Lethabarb (Virbac) and intracardially perfused with cold PBS and 4% PFA.
Cell culture and immunostaining
Mouse neuroblastoma (N2a) cells were grown on coverslips (No 1.5) and maintained in Dulbecco's modified Eagle's medium, supplemented with 10% FBS and 1% penicillin/streptomycin. N2a cells were transfected using Lipofectamine LTX and PLUS reagents (Invitrogen) with the following constructs: pAAV‐hSyn‐mito‐QC, myc‐Parkin, pcDNA6.3 empty vector, EGFP‐Parkin (human, gift from E. Fon, Addgene plasmid # 45875), EGFP‐LC3 (human, gift from K. Kirkegaard, Addgene plasmid # 11546) and previously generated tau constructs containing the V5 tag at the C‐terminus (Xia et al, 2015).
To depolarise mitochondria, cells were incubated with 10 μM CCCP or DMSO (as negative control) for 4 h, or with 8 μM CCCP or 15 μM antimycin/oligomycin overnight for mito‐QC experiments, and then fixed with 4% paraformaldehyde (pH = 7 for mito‐QC experiments).
To assess autophagic flux, transfected N2a cells were starved with HBSS containing 100 nM bafilomycin A or DMSO for 7 h before fixation.
For immunostaining, cells were permeabilised for 10 min with PBS containing 0.2% Triton X‐100 and incubated in 5% bovine serum albumin in PBS blocking solution for 30 min. They were then incubated with primary antibodies at 4°C overnight, washed three times with PBS for 5 min each and incubated with secondary fluorescent antibodies (Alexa 488/555/647, Invitrogen) at 1:500 dilution for 1 h at room temperature, washed 3× with PBS again and finally mounted with Vectashield (Vector, H‐1000) for imaging.
Proximity ligation assay
For proximity ligation assays (PLAs, Duolink, Sigma, DUO92102), N2a cells were treated as for immunostaining until day 2, after which the manufacturer's instructions were followed. Each experiment included a negative control (2 proteins known not to interact, e.g. Parkin and histone H3) and a positive control (known binding partners Parkin and alpha‐tubulin). PLAs were performed on fixed, free‐floating pR5 and wild‐type (WT) littermate brain sections. WT sections served as the negative control and were tested for Parkin and α‐tubulin as the positive control.
Confocal microscopy and image analysis
Imaging of N2a cells and mouse tissue was conducted on a Zeiss LSM 710 confocal microscope, running the Zen Black software (2012), with a 63 × 1.4NA oil objective, using high‐sensitivity BiG (GaAsP) detectors where required. Parkin translocation at 24 h was originally imaged at 8 bit, but remaining experiments were imaged at 12 bit. Each dataset was imaged in a single session, with the same imaging settings maintained throughout the session. For display, images were adjusted for brightness and contrast, with channel minimum and maximum values kept consistent between images, in FIJI (version 2.0.0; Schindelin et al, 2012).
To quantify mitophagy using the mito‐QC biosensor, tile scans (single optical sections) were performed to capture multiple cells across the coverslip. A blinded investigator drew regions of interest (ROIs) around single cells in FIJI, selected in the 633 nm channel containing the V5 (tau) signal or mCherry channel in control cells. The fluorescence intensity of mCherry and GFP was measured above a set threshold to exclude background pixel values, and a ratio of GFP to mCherry fluorescence was generated for each cell.
To quantify Parkin/LC3 punctae, cells were selected in the V5 channel and single optical sections were taken. Image analysis was conducted blinded in FIJI as follows: the inverted “union jack” look‐up table was applied to the 488 nm channel containing Parkin/LC3 fluorescence to render areas of increased intensity more visible (Fig 2A). These areas (“Parkin clusters”/autophagosomes) were manually encircled to generate ROIs, in which the fluorescent signal (mean grey value) was measured. These ROIs were then transferred to the 561 nm channel to measure the fluorescence intensity of the mitochondrial signal in the same ROIs. For each cell, a region in the cytosol was measured as background fluorescence. Fluorescence intensity values were normalised to this background, in order to account for differences in expression levels. Similarly, the fluorescence of the 633 nm channel containing the V5 staining for tau‐expressing cells was measured in an area in each cell to give a measure of the tau overexpression level. Once this detailed analysis revealed that the number of Parkin clusters was the most informative measure, Parkin clusters and LC3 autophagosomes were manually counted for the remaining experiments (without generating single ROIs), while blinded to treatment group.
Ubiquitin localisation to mitochondria was assessed blinded by creating mitochondrial ROIs for individual cells based on the SOD2 staining, using the “Otsu” threshold in FIJI. The mitochondrial ROIs were overlayed onto the ubiquitin channel, and the mean fluorescence intensity was measured. To account for differences in basal (whole cell) ubiquitin levels, we also measured the mean fluorescence intensity of total cellular ubiquitin. This enabled calculation of the fold enrichment of ubiquitin on mitochondria (cellular ubiquitin fluorescence subtracted from mitochondrial ubiquitin fluorescence, divided by cellular ubiquitin fluorescence).
For PLA assays in N2a cells, eight z slices, 1 μm apart, were generated, which was enough to capture an entire cell. The PLA signal was quantified from maximum intensity projections, by drawing an ROI around each transfected cell and measuring the mean grey value. The expression level was accounted for by measuring the mean intensity of the GFP/V5 signal in the cell ROI and this was used to normalise the PLA signal in every cell.
Images were adjusted in FIJI for display only. All adjustments were kept constant for each channel in each figure.
Co‐immunoprecipitation
N2a cells were grown in 6‐well plates and transfected with 2 μg of GFP‐Parkin and hTau‐V5, hP301L‐V5 or control vector. At 48 h post‐transfection, cells were collected into lysis buffer consisting of PBS + 0.5% Triton X‐100 and protease/phosphatase inhibitors (Roche), based on a previously described protocol (Moore et al, 2008). Lysates were centrifuged at 12,000 g for 1 min, and the supernatant was incubated with a mouse anti‐V5 antibody (1:200, Sigma V8012) by rotating for 1 h at 4°C. Protein G sepharose beads (40 μl, GE Healthcare, 17‐0618) were added to the lysates before incubation overnight at 4°C. Beads were washed 3× with PBS, before resuspension in Laemmli sample buffer and boiling for 10 min. Samples were subjected to immunoblotting and probed with rabbit antibodies to V5 and GFP.
Immunoblotting
Protein content was measured with a BCA assay (Thermo Scientific), and proteins (10–20 μg) were boiled in Laemmli sample buffer before being separated in a gel containing 10% acrylamide with SDS–PAGE. Following semi‐dry transfer of proteins to an LF PVDF membrane (Millipore), immunoblotting was performed in Odyssey blocking buffer (LI‐COR). The signal was visualised with fluorescent secondary antibodies goat anti‐mouse IR680/800 (LI‐COR) and goat anti‐rabbit IR680/800 (LI‐COR) at 1: 20,000 dilutions, with the Odyssey CLx Infrared Imaging System (LI‐COR). Caenorhabditis elegans total protein was visualised with the REVERT total protein stain (LI‐COR), according to the manufacturer's instructions.
Mitochondrial membrane potential analysis
Twenty‐four hours after transfection and preceding CCCP treatment, cells were loaded with Rhodamine 123 (0.05 mg/ml, 15 min). Following treatment, the cells were collected for fluorescence‐activated cell sorting (FACS) on the BD LSR II Analyser, with excitation and emission settings of 488 and 530 nm, respectively. Thirty thousand events were recorded per sample, and the geometric mean of the fluorescence intensity values was analysed.
C. elegans strains and genetics
Caenorhabditis elegans strains were handled and maintained under standard conditions (Brenner, 1974). The following strains were used: N2 wild‐type Bristol isolate, CK10 bkIs10 [aex‐3::tau4R1N(V337M) + myo‐2p::gfp], CK12 Is[aex‐3::tau4R1N(P301L) + myo‐2p::gfp] and APD178 apdIs11 [aex‐3p::tau4RIN + coel::gfp]. CK10, CK12 and APD178 were kindly provided by Dr H. Nicholas (The University of Sydney, Sydney, Australia). The strain VC1024 [pdr‐1(gk488) III] was obtained from the Caenorhabditis Genetics Centre, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440), backcrossed 3 times and then crossed to SJZ42 foxEx3 [rgef‐1p::TOMM‐20::Rosella].
C. elegans constructs and molecular cloning
The mitochondrially targeted Rosella plasmid under control of a pan‐neuronal promoter, pSZ15 (rgef‐1p::TOMM‐20::Rosella), was generated via PCR cloning. The “pNT953” plasmid containing the first two exons of the C. elegans tomm‐20 gene fused to the Rosella biosensor coding sequence (myo‐3p::TOMM‐20::Rosella) was kindly provided by Dr N. Tavernarakis (Institute of Molecular Biology and Biotechnology, Heraklion, Greece). The pan‐neuronal C. elegans promoter rgef‐1 was isolated from the plasmid pSJ0962 by PCR using the following primers F: CACCTAGGCAGGAACAGCTATGA and R: CTGGCGGCCGTTACTAGTGGATCCTTTT TCTACCGGTACCCTCAAGGGTC. The primers F: ATGTCGGACACAATTCTTGGTTTC and R: CATCACCGAAACGCGCGAGAC were used to amplify the “pNT953” plasmid, excluding the myo‐3 promoter. The PCR to combine the rgef‐1 promoter and the construct containing the mitochondrial targeted Rosella (TOMM‐20::Rosella) was performed with the primers F: CGTTTCCGATACCCCCTTATATCAGCAC and R: GGAAACAGTTATGTTTGGTATATTGGG.
Transgenic worm strains
Transgenic lines were obtained by microinjection into the gonadal arms of adult C. elegans, as per standard methods (Mello et al, 1991). 10 ng/μl of the pSZ15 construct and 50 ng/μl of the co‐injection marker odr‐1p::DsRed were injected. The tau strains previously described (CK10, CK12 and APD178) were all crossed with the transgenic Rosella strain SJZ42 foxEx3.
Mitochondrial stress assays in C. elegans
For sodium azide (NaN3) stress assays, synchronised 1‐day‐old adult (1DOA) hermaphrodites were exposed to 8 mM NaN3 in M9 buffer (with 0.01% Triton X‐100) for 1 h on a shaking incubator at 20°C. Following NaN3 exposure, animals were washed with M9 buffer and imaged. For CCCP stress assays, synchronised 1‐day‐old adult (1DOA) hermaphrodites were exposed to 15 μM CCCP in M9 buffer (with 0.01% Triton X‐100) for 2 h on a shaking incubator at 20°C. Following CCCP exposure, the animals were washed with M9 buffer and imaged.
C. elegans imaging and analysis
Animals were mounted on 2% agarose pads on glass slides and immobilised with 1 mM tetramisole hydrochloride (Sigma) before imaging. Imaging was performed using a Zeiss AxioImager 2 with a Zeiss Axiocam 506 mono camera and Zen2 software (version 2.0.0.0). All images were acquired under the same exposure conditions, and each experiment was imaged in one session. Fluorescence intensity was quantified using FIJI. A threshold was manually set in the dsRed channel to detect individual mitochondrial ROIs. Two investigators independently analysed subsets of images and compared results to ensure the reproducibility of the analysis. The fluorescence intensity for both the dsRed and GFP signals in each mitochondrion along the ventral nerve cord in an image was measured and a GFP‐to‐dsRed ratio calculated per mitochondrion. These ratios were averaged for each animal. As the tau strains had different injection makers, the background fluorescence intensity of GFP differed across these strains. We therefore only statistically compared treated to vehicle worms within each strain.
Mitotracker staining of C. elegans
Mixed stage animals were incubated in 200 μl M9 buffer containing 5 μM Mitotracker Deep Red (Thermo Fisher) for 40 min rotating in the dark. Animals were washed three times with M9 buffer before being mounted on agarose pads and imaged.
C. elegans protein extraction and quantification
Approximately 10,000 mixed stage animals were harvested and washed three times in M9 buffer, and once in ddH2O. Animals were resuspended in RIPA buffer [10 mM Tris–HCl (pH 7.4), 140 mM NaCl, 1% Triton X‐100, 0.1% sodium deoxycholate and 0.1% SDS, 1 mM EDTA] supplemented with protease inhibitors (Sigma, S8830). The animals were lysed by douncing followed by sonication for 30 s, five times (amplitude: 40%; Sonics VibraCell VCX130). Following the addition of 4× Laemmli sample buffer [250 mM Tris pH 6.8, 4% SDS, 45% glycerol, 715 mM β‐mercaptoethanol, 50 mM DTT, 0.02% (w/v) bromophenol blue], the sample was boiled for 5 min, and centrifuged at 20,000 g for 30 min before immunoblotting.
Statistical analysis
Data were tested for normality with the D'Agostino‐Pearson omnibus test and for homogeneity of variance with the Brown–Forsythe test. Parametric data were analysed by Student's t‐tests or analyses of variance (ANOVAs) followed by Tukey's post hoc tests. In cases where two‐way ANOVAs revealed significant interactions, we further tested for simple effects. Non‐parametric data were analysed with the Mann–Whitney U‐test (for two groups) or the Kruskal–Wallis test (multiple groups) followed by Dunn's post hoc. Correlations were tested for with Pearson's r. Statistics were performed Prism 6.0 (GraphPad), with alpha levels set at 0.05. Details of the statistical analyses can be found in figure legends.
Author contributions
NC, AT, SZ, JB‐G and JG designed and interpreted the experiments; NC and AT performed the experiments; NC analysed the data; AT and NC analysed C. elegans data; NC and JG wrote the manuscript; and SZ and JB‐G edited the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank Tishila Palliyaguru for general technical assistance and Geoffrey Osborne and Virginia Nink for assistance with FACS. Imaging was performed at the Queensland Brain Institute's Advanced Microscopy Facility, generously supported by the Australian Government through the ARC LIEF grant LE 30100078. This study was supported by the Estate of Dr Clem Jones AO, the State Government of Queensland and the Federal Government of Australia (ACT900116), and by grants from the Australian Research Council (DP130101932, LE130100078) and the National Health and Medical Research Council of Australia (GNT1037746, GNT1003150, GNT1127999) to JG. NC was supported by an Australian Government Research Training Program Scholarship. SZ was supported by an NHMRC grant (APP1128381) and a Stafford Fox Senior Research Fellowship. We thank Rowan Tweedale for critically reading the manuscript.
The EMBO Journal (2019) 38: e99360
References
- Allen GFG, Toth R, James J, Ganley IG (2013) Loss of iron triggers PINK1/Parkin‐independent mitophagy. EMBO Rep 14: 1127–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadoro G, Corsetti V, Florenzano F, Atlante A, Ciotti MT, Mongiardi MP, Bussani R, Nicolin V, Nori SL, Campanella M, Calissano P (2014) AD‐linked, toxic NH2 human tau affects the quality control of mitochondria in neurons. Neurobiol Dis 62: 489–507 [DOI] [PubMed] [Google Scholar]
- Baloyannis SJ, Costa V, Michmizos D (2004) Mitochondrial alterations Alzheimer's disease. Am J Alzheimers Dis Other Demen 19: 89–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodea L‐G, Eckert A, Ittner LM, Piguet O, Götz J (2016) Tau physiology and pathomechanisms in frontotemporal lobar degeneration. J Neurochem 138: 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans . Genetics 77: 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH (2011) Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum Mol Genet 20: 4515–4529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dorn GW (2013) PINK1‐phosphorylated mitofusin 2 is a parkin receptor for culling damaged mitochondria. Science 340: 471–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CT, Bayır H, Kagan VE (2014) LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons. Autophagy 10: 376–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelissen T, Vilain S, Vints K, Gounko N, Verstreken P, Vandenberghe W (2018) Deficiency of parkin and PINK1 impairs age‐dependent mitophagy in Drosophila . eLife 7: e35878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins N, Götz J (2017) Shedding light on mitophagy in neurons: what is the evidence for PINK1/Parkin mitophagy in vivo? Cell Mol Life Sci 75: 1151–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David DC, Hauptmann S, Scherping I, Schuessel K, Keil U, Rizzu P, Ravid R, Dröse S, Brandt U, Müller WE, Eckert A, Götz J (2005) Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J Biol Chem 280: 23802–23814 [DOI] [PubMed] [Google Scholar]
- Dayanandan R, Van Slegtenhorst M, Mack TGA, Ko L, Yen S‐H, Leroy K, Brion J‐P, Anderton BH, Hutton M, Lovestone S (1999) Mutations in tau reduce its microtubule binding properties in intact cells and affect its phosphorylation. FEBS Lett 446: 228–232 [DOI] [PubMed] [Google Scholar]
- Ding W‐X, Ni H‐M, Li M, Liao Y, Chen X, Stolz DB, Dorn GW, Yin X‐M (2010) Nix is critical to two distinct phases of mitophagy, reactive oxygen species‐mediated autophagy induction and parkin‐ubiquitin‐p62‐mediated mitochondrial priming. J Biol Chem 285: 27879–27890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew BG, Ribas V, Le JA, Henstridge DC, Phun J, Zhou Z, Soleymani T, Daraei P, Sitz D, Vergnes L, Wanagat J, Reue K, Febbraio MA, Hevener AL (2014) Hsp72 is a mitochondrial stress sensor critical for parkin action, oxidative metabolism, and insulin sensitivity in skeletal muscle. Diabetes 63: 1488–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBoff B, Götz J, Feany MB (2012) Tau promotes neurodegeneration via Drp‐1 mislocalization in vivo . Neuron 75: 618–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBoff B, Feany M, Götz J (2013) Why size matters – balancing mitochondrial dynamics in Alzheimer's disease. Trends Neurosci 36: 325–335 [DOI] [PubMed] [Google Scholar]
- Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E‐M, Mandelkow E (1998) Overexpression of tau protein inhibits kinesin‐dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J Cell Biol 143: 777–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert A, Nisbet R, Grimm A, Götz J (2014) March separate, strike together — role of phosphorylated TAU in mitochondrial dysfunction in Alzheimer's disease. Biochim Biophys Acta Mol Basis Dis 1842: 1258–1266 [DOI] [PubMed] [Google Scholar]
- Fulga TA, Elson‐Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT, Feany MB (2007) Abnormal bundling and accumulation of F‐actin mediates tau‐induced neuronal degeneration in vivo . Nat Cell Biol 9: 139–148 [DOI] [PubMed] [Google Scholar]
- Gauthier‐Kemper A, Weissmann C, Golovyashkina N, Sebö‐Lemke Z, Drewes G, Gerke V, Heinisch JJ, Brandt R (2011) The frontotemporal dementia mutation R406W blocks tau's interaction with the membrane in an annexin A2‐dependent manner. J Cell Biol 192: 647–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (2010) PINK1/Parkin‐mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12: 119–131 [DOI] [PubMed] [Google Scholar]
- Glauser L, Sonnay S, Stafa K, Moore DJ (2011) Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J Neurochem 118: 636–645 [DOI] [PubMed] [Google Scholar]
- Götz J, Chen F, Barmettler R, Nitsch RM (2001) Tau filament formation in transgenic mice expressing p301 l tau. J Biol Chem 276: 529–534 [DOI] [PubMed] [Google Scholar]
- Götz J, Ittner LM (2008) Animal models of Alzheimer's disease and frontotemporal dementia. Nat Rev Neurosci 9: 532–544 [DOI] [PubMed] [Google Scholar]
- Grimm A, Eckert A (2017) Brain aging and neurodegeneration: from a mitochondrial point of view. J Neurochem 143: 418–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, Zhukareva V, Vogelsberg‐Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM‐Y (1998) Mutation‐specific functional impairments in distinct tau isoforms of hereditary ftdp‐17. Science 282: 1914–1917 [DOI] [PubMed] [Google Scholar]
- Hu Y, Li X‐C, Wang Z, Luo Y, Zhang X, Liu X‐P, Feng Q, Wang Q, Yue Z, Chen Z, Ye K, Wang J‐Z, Liu G‐P, Hu Y, Li X‐C, Wang Z, Luo Y, Zhang X, Liu X‐P, Feng Q et al (2016) Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 7: 17356–17368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Alonso Adel C, Chen S, Chohan MO, El‐Akkad E, Gong C‐X, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke‐Iqbal I (2005) Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta Mol Basis Dis 1739: 198–210 [DOI] [PubMed] [Google Scholar]
- Ittner LM, Fath T, Ke YD, Bi M, van Eersel J, Li KM, Gunning P, Götz J (2008) Parkinsonism and impaired axonal transport in a mouse model of frontotemporal dementia. Proc Natl Acad Sci USA 105: 15997–16002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Götz J (2009) Phosphorylated Tau interacts with c‐Jun N‐terminal kinase‐interacting protein 1 (JIP1) in Alzheimer disease. J Biol Chem 284: 20909–20916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191: 933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205: 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopeikina KJ, Carlson GA, Pitstick R, Ludvigson AE, Peters A, Luebke JI, Koffie RM, Frosch MP, Hyman BT, Spires‐Jones TL (2011) Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer's disease brain. Am J Pathol 179: 2071–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe J‐F, Saeki Y, Tanaka K, Matsuda N (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: 162–166 [DOI] [PubMed] [Google Scholar]
- Kraemer BC, Zhang B, Leverenz JB, Thomas JH, Trojanowski JQ, Schellenberg GD (2003) Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc Natl Acad Sci USA 100: 9980–9985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon K‐Y, Viollet B, Yoo OJ (2011) CCCP induces autophagy in an AMPK‐independent manner. Biochem Biophys Res Commun 416: 343–348 [DOI] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JJ, Sanchez‐Martinez A, Zarate AM, Benincá C, Mayor U, Clague MJ, Whitworth AJ (2018) Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J Cell Biol 217: 1613–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X‐C, Hu Y, Wang Z, Luo Y, Zhang Y, Liu X‐P, Feng Q, Wang Q, Ye K, Liu G‐P, Wang J‐Z (2016) Human wild‐type full‐length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci Rep 6: 24756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Götz J (2017a) Somatodendritic accumulation of Tau in Alzheimer's disease is promoted by Fyn‐mediated local protein translation. EMBO J 36: 3120–3138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Götz J (2017b) Tau‐based therapies in neurodegeneration: opportunities and challenges. Nat Rev Drug Discov 16: 863 [DOI] [PubMed] [Google Scholar]
- Manczak M, Reddy PH (2012) Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer's disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet 21: 2538–2547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou Y, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189: 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumine H, Saito M, Shimoda‐Matsubayashi S, Tanaka H, Ishikawa A, Nakagawa‐Hattori Y, Yokochi M, Kobayashi T, Igarashi S, Takano H, Sanpei K, Koike R, Mori H, Kondo T, Mizutani Y, Schäffer AA, Yamamura Y, Nakamura S, Kuzuhara S, Tsuji S et al (1997) Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2‐27. Am J Hum Genet 60: 588–596 [PMC free article] [PubMed] [Google Scholar]
- McWilliams TG, Prescott AR, Montava‐Garriga L, Ball G, Singh F, Barini E, Muqit MMK, Brooks SP, Ganley IG (2018) Basal mitophagy occurs independently of pink1 in mouse tissues of high metabolic demand. Cell Metab 27: 439–449 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V (1991) Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J 10: 3959–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DJ, West AB, Dikeman DA, Dawson VL, Dawson TM (2008) Parkin mediates the degradation‐independent ubiquitination of Hsp70. J Neurochem 105: 1806–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen D‐F, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) Pink1 is selectively stabilized on impaired mitochondria to activate parkin. PLoS Biol 8: e1000298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palikaras K, Lionaki E, Tavernarakis N (2015) Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans . Nature 521: 525–528 [DOI] [PubMed] [Google Scholar]
- Pérez MJ, Vergara‐Pulgar K, Jara C, Cabezas‐Opazo F, Quintanilla RA (2017) Caspase‐cleaved tau impairs mitochondrial dynamics in Alzheimer's disease. Mol Neurobiol 55: 1–15 [DOI] [PubMed] [Google Scholar]
- Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, Lucia MD, McGowan E, Lewis J, Prihar G, Kim J, Dillmann WH, Browne SE, Hall A, Voellmy R, Tsuboi Y, Dawson TM, Wolozin B, Hardy J, Hutton M (2004) CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet 13: 703–714 [DOI] [PubMed] [Google Scholar]
- Rhein V, Song X, Wiesner A, Ittner LM, Baysang G, Meier F, Ozmen L, Bluethmann H, Dröse S, Brandt U, Savaskan E, Czech C, Götz J, Eckert A (2009) Amyloid‐β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer's disease mice. Proc Natl Acad Sci USA 106: 20057–20062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado C, Mijaljica D, Hatzinisiriou I, Prescott M, Devenish RJ (2008) Rosella: a fluorescent pH‐biosensor for reporting vacuolar turnover of cytosol and organelles in yeast. Autophagy 4: 205–213 [DOI] [PubMed] [Google Scholar]
- Rugarli EI, Langer T (2012) Mitochondrial quality control: a matter of life and death for neurons. EMBO J 31: 1336–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J‐Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz KL, Eckert A, Rhein V, Mai S, Haase W, Reichert AS, Jendrach M, Müller WE, Leuner K (2012) A new link to mitochondrial impairment in tauopathies. Mol Neurobiol 46: 205–216 [DOI] [PubMed] [Google Scholar]
- Shahpasand K, Uemura I, Saito T, Asano T, Hata K, Shibata K, Toyoshima Y, Hasegawa M, Hisanaga S‐I (2012) Regulation of mitochondrial transport and inter‐microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer's disease. J Neurosci 32: 2430–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderberg O, Leuchowius K‐J, Gullberg M, Jarvius M, Weibrecht I, Larsson L‐G, Landegren U (2008) Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 45: 227–232 [DOI] [PubMed] [Google Scholar]
- Song Y, Brady ST (2015) Post‐translational modifications of tubulin: pathways to functional diversity of microtubules. Trends Cell Biol 25: 125–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow E‐M (2002) Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol 156: 1051–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen D‐F, Karbowski M, Youle RJ (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191: 1367–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, Albanese A, Wood NW (2001) Localization of a novel locus for autosomal recessive early‐onset parkinsonism, PARK6, on human chromosome 1p35‐p36. Am J Hum Genet 68: 895–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives‐Bauza C, Zhou C, Huang Y, Cui M, de Vries RLA, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrane J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S (2010) PINK1‐dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA 107: 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitworth AJ, Pallanck LJ (2017) PINK1/Parkin mitophagy and neurodegeneration—what do we really know in vivo? Curr Opin Genet Dev 44: 47–53 [DOI] [PubMed] [Google Scholar]
- Xia D, Li C, Götz J (2015) Pseudophosphorylation of Tau at distinct epitopes or the presence of the P301L mutation targets the microtubule‐associated protein Tau to dendritic spines. Biochim Biophys Acta 1852: 913–924 [DOI] [PubMed] [Google Scholar]
- Ye X, Sun X, Starovoytov V, Cai Q (2015) Parkin‐mediated mitophagy in mutant hAPP neurons and Alzheimer's disease patient brains. Hum Mol Genet 15: 2938–2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM (2012) Mitochondrial fission, fusion, and stress. Science 337: 1062–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6