

Thermal profiles and melting temperatures have been modeled for over 1700 proteins from the model plant Arabidopsis thaliana using six biological replicates, providing a solid groundwork for future thermal shift studies in the species. Highly significant global correlations were found between melting temperature and several protein characteristics, including molecular weight, secondary structure content, and hydrophobic and charged accessible surface areas.
Keywords: Protein Conformation, Plant Biology, Tandem Mass Spectrometry, Arabidopsis, Quantification, Thermal Proteome Profiling
Graphical Abstract

Highlights
Over 1700 Arabidopsis proteins with thermal models in multiple replicates.
Melting temperature correlates with 1°, 2°, and 3° protein characteristics.
Ligand-induced thermal shifts are evident in complex protein extracts.
Abstract
Modern tandem MS-based sequencing technologies allow for the parallel measurement of concentration and covalent modifications for proteins within a complex sample. Recently, this capability has been extended to probe a proteome's three-dimensional structure and conformational state by determining the thermal denaturation profile of thousands of proteins simultaneously. Although many animals and their resident microbes exist under a relatively narrow, regulated physiological temperature range, plants take on the often widely ranging temperature of their surroundings, possibly influencing the evolution of protein thermal stability. In this report we present the first in-depth look at the thermal proteome of a plant species, the model organism Arabidopsis thaliana. By profiling the melting curves of over 1700 Arabidopsis proteins using six biological replicates, we have observed significant correlation between protein thermostability and several known protein characteristics, including molecular weight and the composition ratio of charged to polar amino acids. We also report on a divergence of the thermostability of the core and regulatory domains of the plant 26S proteasome that may reflect a unique property of the way protein turnover is regulated during temperature stress. Lastly, the highly replicated database of Arabidopsis melting temperatures reported herein provides baseline data on the variability of protein behavior in the assay. Unfolding behavior and experiment-to-experiment variability were observed to be protein-specific traits, and thus this data can serve to inform the design and interpretation of future targeted assays to probe the conformational status of proteins from plants exposed to different chemical, environmental and genetic challenges.
Proteins are fundamental macromolecules involved in all aspects of life, from catalyzing metabolic reactions to providing a scaffold for cellular organization to transmitting external environmental changes into the nuclear transcriptional machinery. Until recently, nearly all large-scale proteomic studies have focused on quantifying changes in protein abundance or degree of post-translational modification to amino acid sidechains. However, changes in protein function result from changes in conformation at the secondary, tertiary, and higher-level structures. Indeed, current evidence suggests that three-dimensional structure is more highly conserved between evolutionarily related proteins than is their primary amino acid sequence (1, 2, 3). Unfortunately, the technology to examine three-dimensional structure at the proteome scale has historically been lacking. Recently, several technologies have emerged that attempt to address this deficiency and have been applied to animal studies, but to our knowledge no such studies have yet been published in the domain of plant research.
It is widely accepted that most cellular interactions involving proteins depend upon and/or induce changes in a protein's three-dimensional conformation. The kinetics and energetics of these changes are closely related to the protein's thermal stability. It is thus reasonable to expect that an organism's proteome will have evolved to minimize the energy needed to maintain any given protein's function at physiological temperature while considering the requirements of any modes of regulation it may undergo. An understanding of global differences in relative thermal stability of proteins may thus provide insight into how different proteins have evolved to function in an energetically efficient way. In addition, given that many plants are exposed to the elements in all seasons and experience large fluctuations in temperature, it is logical to ask whether their proteomes have developed unique characteristics of thermal stability and conformation to preserve their functions under changing environmental conditions.
There are numerous quantifiable attributes of a protein potentially related to its conformation and thermal behavior. One such characteristic is the melting temperature (Tm), typically defined as the temperature at which half of a protein population is unfolded. Until very recently, available techniques for estimation of protein Tm have relied upon measurement of various properties of a purified protein solution in vitro. This generally involves isolating a purified protein of interest and observing changes in physical or chemical properties of the solution across a temperature gradient. High-throughput screens are also possible—for instance, when the measurement technique can be carried out in 96- or 382-well plates—but this still requires purification of individual proteins, a laborious and time-consuming step.
In 2014, an untargeted method called thermal proteome profiling (TPP)1 was introduced which used isotope-encoded multiplexed mass-spectrometry (MS)-based quantification to profile thousands of proteins simultaneously (4). The basis of the technique is like those mentioned above, in that a measure of protein conformation is tracked across a temperature gradient (Fig. 1). In this case, protein solubility is used as a proxy for folding state given that unfolded, denatured proteins precipitate out of solution. After centrifugation to remove proteins precipitated across a temperature gradient, MS/MS of isobarically labeled tryptic peptides derived from the remaining nondenatured proteins is used to measure relative abundance of individual proteins. The resulting temperature-abundance profile for each protein is fit to a standard two-state protein melting model and used to calculate a Tm (Fig. 2). Because the original TPP protocol was published, additional methodologies have been introduced which use different readouts for protein stability and/or different MS-MS based quantification strategies. In the work described herein, we have chosen to use multiplexed isobaric labeling as in the original TPP paper because of the ease of direct relative quantification at all temperature points for every peptide identified.
Fig. 1.

Schematic of TPP workflow. Plants were grown hydroponically and protein was extracted as described in methods. Ten aliquots were incubated over a temperature gradient and the clarified supernatant was subjected to ten-channel isobaric labeling (one tag per temperature). Standard MS/MS-based quantification was used to produce protein-level relative abundance values which were fit to the two-state model as described.
Fig. 2.

Modeling protein melting using TPP. A, MS/MS-based relative abundance data for a protein at ten temperature points is modeled according to the logistic function shown. The melting point (Tm, red dashed line) is defined as the point where half of the protein remains in solution (green dashed line). The parameters are arranged such that m is equal to Tm, k controls the slope of the curve (purple dashed line) for a given value of m, and p defines the lower asymptote. The exponential term in the denominator represents Keq of the unfolding equilibrium. B, An example of a protein profile from real-world Arabidopsis treatment data.
There exists considerable untapped potential for TPP in the plant research community, in which many receptor-ligand pairs and protein-protein interactions remain poorly understood. To that end, we have undertaken a characterization of the thermal proteome of the model plant Arabidopsis thaliana, of which much is already known about the proteome and its modifications. By using a relatively large number of biological replicates and applying extensive offline fractionation, our aim was to produce a large and robust database of untreated Tm measurements that could serve as the groundwork for future targeted work in the species. In particular, it is critical to understand the limitations of a technology in the domain of interest, and the database of melting profiles developed in this work provides a valuable resource of information on which proteins “behave well” in the assay and with what experiment-to-experiment variability, providing researchers with information on their proteins of interest prior to embarking on a TPP experiment.
In the course of this work, the question also arose whether the database of Tm measurements could be used to add to the understanding of more general questions regarding protein structure and thermostability. Such questions are of widespread interest both to basic researchers and to those interested in applying such knowledge to the engineering of novel proteins. To this end, we undertook an analysis of the correlation between the empirical Tms and various possible physicochemical determinants of protein thermostability. We were interested in how such determinants might be preserved or differ between a “poikilothermic” plant proteome and existing data from other kingdoms.
Lastly, we demonstrated the ability, in a complex extract from plant tissue, to detect in vitro conformational changes at the proteome-wide scale caused by a common co-factor, adenosine triphosphate complexed with Mg2+, and correlate these changes with existing knowledge of protein binding sites. Taken together, this initial work establishes a baseline for future studies on wild-type and mutant Arabidopsis and other plant species grown under a variety of environmental, chemical and genetic perturbations.
EXPERIMENTAL PROCEDURES
Experimental Design and Statistical Rationale
For the generation of the core Tm database and analysis of factors affecting thermostability, six untreated biological replicates were used. These replicates were grown and processed at different times over the course of several months to minimize batch effects. The relatively high number of biological replicates was used to overcome the low degree of sample-to-sample overlap in protein IDs which is common in shotgun MS/MS experiments. For the ATP treatment study, single biological replicates were used for treatment and control. The lack of replicates was primarily a cost consideration, and we justified this choice on the basis that (1) this was primarily a proof of principle on the application of a new technology in the plant kingdom, and (2) we were looking for a broad response across a large family of proteins rather than for a reproducible response in any given protein. Additionally, each replicate with eight or more underlying PSMs has 90% confidence intervals applied to both the individual datapoints (vertical error bars) and Tm estimates (horizontal shading) using the bootstrap method described previously (4). The various statistical filters applied and methods used for significance testing are fully described below in the relevant sections. Of note, no multiple testing correction was applied to the GO enrichment p values as the package authors suggest it is redundant with the algorithm used. This agreed with our own observations that Benjamini-Hochberg correction applied to the results seemed to be overly conservative. However, stringent cutoffs (p < 0.002 and 0.005 for lower and upper bins) were applied to the terms reported herein.
Tissue Propagation, Harvesting, and Treatment
Arabidopsis Col-0 seeds were grown in liquid medium (0.5× Murashige and Skoog salts, 1% (w/v) sucrose, 0.05% (w/v) MES, pH 5.7) under constant light for 11 days. Tissue balls were immersed in deionized water and gently spun in a commercial salad spinner to remove adhering solution. For untreated replicates C1, C2, C5, and C6, tissue was flash frozen in liquid nitrogen, homogenized with mortar and pestle, and resuspended in ice-cold homogenization buffer (230 mm sorbitol, 50 mm Tris-HCl pH 7.5, 10 mm KCl, 3 mm EGTA, and the following protease inhibitors added fresh: 1 mm potassium metabisulfite, 1 mm PMSF, 0.5 μg/ml leupeptin, 0.7 μg/ml pepstatin, 1 Roch protease inhibitor tablet). For untreated replicates C3 and C4 and the ATP treated and control samples, homogenization was performed without freezing by placing rinsed tissue balls in 70 μl of homogenization buffer and grinding in an upright tissue blender for 30 s at maximum speed. For all samples, the tissue homogenate was cleared by filtering through two layers of Mira-cloth (Calbiochem, St. Louis, MO), placed in a chilled centrifuge tube and spun at 100,000 × g for 20 min at 4°C. For the in vitro ATP binding experiment, Mg-ATP or mock solution was added to 2 ml of crude extract to a final concentration of 2 mm and incubated for 15 min at room temperature. In this experiment, one biological replicate was performed for treated and control conditions.
Gradient Precipitation
Each sample was aliquoted into ten microfuge tubes in volumes ranging from 0.2 to 1.0 ml for different experiments (to normalize protein concentrations) and placed on equilibrated heating blocks containing mineral oil to facilitate rapid thermal transfer across the tube wall. The following ten-temperature gradients were used for each replicate: 21.0–61.5°C (C1–C2) and 25.0–65.5°C (C3–C6). Tubes were incubated at the given temperature for 10 mins, removed and allowed to cool for 10 mins at room temperature, and placed on ice. Tubes were spun at 17,000 × g for 20 min at 4°C to pellet precipitated protein and the supernatant was carefully transferred to a new microfuge tube. At this stage, 1.0 μg of bovine serum albumin (BSA) was spiked into each tube as an internal standard to facilitate downstream data normalization.
Protein Extraction, Digestion, and Cleanup
Methanol-chloroform protein extraction was performed on the cleared supernatant as described previously (5) and protein pellets were resuspended in 8 m urea. The 660 nm Protein Assay Reagent kit (Pierce, Waltham, MA) was used to quantify proteins in each sample at this stage for later use in orthogonal data normalization. Extracts were diluted to a final concentration of 4 m urea using 50 mm ammonium bicarbonate, reduced with 5 mm DTT for 45 min in a 42°C water bath, and alkylated with 15 mm iodoacetic acid for 45 min in the dark at room temperature. Alkylation was quenched by adding 5 mm DTT for 5 min at room temperature. Protein was digested with LysC (Wako, Richmond, VA) at a 1:60 enzyme:protein ratio at 37°C for 2 h. Samples were diluted to 1.2 m urea and digested with trypsin (Promega, Madison, WI) at a 1:40 enzyme/protein ratio at 37°C overnight. A minimum of 0.1 μg of LysC and 0.2 μg of trypsin was added to all samples. De-salting was performed using OMIX C18 tips (100 μl capacity, Agilent, Santa Clara, CA) as follows. Digests were acidified to pH ¡3 using 20% formic acid (∼4 μl per 140 μl digest). OMIX tips were equilibrated with 3 × 100 μl rinses of 75% acetonitrile (ACN) followed by 4 × 100 μl rinses of 0.1% TFA. Samples were bound to resin by pipetting up and down ten times, washed 2× with 100 μl 0.1% TFA, washed 1× with 100 μl 0.01% TFA, and eluted with 75 μl of 75% ACN and 0.1% formic acid into a low-protein-binding microfuge tube. Vacuum centrifugation was used to reduce sample volume for isobaric labeling.
Isobaric Labeling
Digests were resuspended in 25 μl of 150 mm TEAB, 5% ACN. TMT-10plex reagents (Thermo Fisher Scientific, Waltham, MA) were resuspended in 75 μl 100% ACN to a concentration of 10.7 μg/μl. TMT reagents and protein digests were mixed to achieve a 3:1 label:protein ratio in a 40 μl volume at 60% TEAB and 40% ACN. The actual label and protein concentration in each tube varied as higher temperature fractions contained less protein, but a minimum label concentration of 1.33 μg/μl was used. The specific isobaric tag used for each temperature was varied from replicate to replicate (see supplemental Materials for exact assignments). Samples were labeled for 2 h at room temperature and quenched by adding 5 μl of 5% hydroxylamine solution for 15 min at room temperature. All ten temperature fractions for each sample were then pooled.
Offline Fractionation
Samples were vacuum centrifuged to remove ACN and subjected to offline high-pH RP-HPLC fractionation using a Waters 2795 Separation Module HPLC, Gemini C18 5 μm 110A 4.6 mm×250 mm column (Phenomenex, Torrance, CA), and a Gibson model 201 fraction collector. The HPLC conditions were as follows: Buffer A (10 mm ammonium formate); Buffer B (10 mm ammonium formate, 80% ACN); 35 min total gradient time at a flow rate of 1.0 ml/min; 5–60% B from 3–23 min; 100% B wash from 25–26 min; 0% B for all other time periods. Fractions were collected every minute and fractions 15–27 were used for downstream analysis. Samples were dried in a vacuum centrifuge and resuspended in 0.1% FA for LC-MS injection.
LC-MS/MS
Samples were analyzed on an Orbitrap Elite mass spectrometer (Thermo). Inline nanoflow HPLC was performed on a C18 column at a flow rate of 300 nL/min using the following 2-hr gradient: solvent A (0.1% FA); solvent B (95% ACN, 0.1% FA); 0% B at min 0–30; 3% B at min 31; 30% B at min 108; 50% B at min 113; 95% B at min 118; 0% B at min 123–126. MS/MS spectral data were acquired using the following settings: MS1 acquisition at 120,000 resolving power and a mass range of 380–1800 m/z. The top ten precursor ions for each scan period, subject to dynamic exclusion, were isolated for MS2 using a 2.0 m/z isolation window width and 200 ms maximum injection time. HCD fragmentation was used to produce product ions for analysis in the Orbitrap at 30,000 resolving power and over a dynamic mass range starting at 100 m/z and bounded at the upper end relative to the precursor mass.
Data Analysis
Thermo RAW files were converted to centroided mzML using msconvert (6) version 3.0.7494 with vendor-supplied peak-picking. A search database was generated from the TAIR10 representative protein sequences concatenated with the GPM cRAP database of common contaminants (http://www.thegpm.org/crap/) which includes the BSA spike-in sequence. A set of decoy sequences generated by reversing the original sequences was added, and the protein sequence order of the resulting database was randomized. The final database contained 55064 target and decoy sequences and is available in the supplemental data repository. The MS2 spectra were searched against this database using the comet search engine (7) version 2016.01 rev. 3 with the following settings: trypsin cleavage (max 1 missed cleavage, min 2 tryptic termini), variable Asn/Gln deamidation, variable Met oxidation, static Cys carbamidomethylation, static N-term/Lys TMT labeling, 0.03 fragment bin tolerance, 0.00 fragment bin offset, 10 ppm precursor mass tolerance, b/y ion series with NH3/H2O neutral loss. The exact configuration file used is available in the supplemental data repository. PeptideProphet (TPP v4.8.0) was used to combine alkaline fractions and calculate posterior probabilities for spectral matches (accurate mass binning, nonparametric model) (8). PSMs were filtered to a 1% FDR based on the per-charge probability ROC cutoffs reported by PeptideProphet. Protein identification was performed using ProteinProphet (TPP v4.8.0) with default settings (9). Quantification of the TMT channels from each matching spectrum was performed using tmt_quant version 0.010 (https://github.com/jvolkening/ms_bin), using two-step run-specific recalibration of the channel windows and performing isotope interference calculation as previously described (10). Full quantification data in tab-delimited format is available in the supplemental data repository.
Protein-level quantification, normalization, curve-fitting and Tm estimation were performed on the filtered PSM tables using our publicly available R package mstherm version 0.4.8 (https://cran.r-project.org/package=mstherm). Bootstrap-based 95% confidence intervals were calculated as previously described (4) for all proteins matching the following filtering criteria: minimum total PSMs: 10; minimum distinct peptides: 2; maximum co-isolation interference: 0.3; maximum model slope: −0.03; minimum model R2: 0.7. Protein-level quantification was performed based on summed channel intensities across spectra. Loess smoothing was performed on the data prior to model fitting.
Protein primary characteristics (molecular weight, GRAVY, isoelectric point, CvP, etc) were calculated using ms-perl (http://github.com/jvolkening/p5-MS). Predicted secondary structure features were calculated using the GOR algorithm as implemented in garnier version 6.6.0.0 (11, 12). Protein abundance values were extracted from the PaxDB Arabidopsis integrated whole-plant data set (13). Two statistical tests were used to test for significant patterns among thermostability bins. The Mann-Whitney U test was used to compare the lowest (unstable) and highest (stable) bins for difference in mean whereas Kendall's tau rank correlation was performed using ordinal bin numbers to test for correlation across all bins. All the results reported regarding correlation of protein features with thermostability were calculated after removing the ribosomal proteins, which were highly abundant in the data and were observed to be skewing the results because of the specific amino acid composition of that protein class. Gene set enrichment analysis was performed with the R package topGO version 2.28.0 (14) using the Fisher exact test and the elim algorithm.
For tertiary structure calculations, all available Arabidopsis protein structures and sequences were downloaded from the RCSB Protein Data Bank. Redundant chains (chains from the same structure with identical sequences) were collapsed to a single sequence, and the resulting database was clustered with the TAIR10 representative protein database using CD-HIT version 4.6, requiring a minimum identity of 0.98 and a minimum length overlap of 0.9. PDB structures with matches to TAIR10 proteins were retained for further analysis. The VADAR structural prediction server (version 1.5) (15) was used to calculate surface area values for each structure, and values from structures in the same CD-HIT cluster were averaged by mean. Correlation analysis was performed as described above using only those proteins with both structures and modeled Tms in the HC set. Compactness was calculated as 3 − , where ISA is the surface area of a sphere of the same volume.
RESULTS
Protein Quantification and Modeling
A total of 1.4 million MS2 spectra were collected from 74 offline high-pH RP-HPLC fractions of six biological replicates. Of these spectra, 313,710 were matched to tryptic peptides at a 1% peptide FDR, representing 4246 identified proteins at a minimum ProteinProphet probability of 0.9 (min. two distinct peptides per protein) (supplemental File S1). After modeling and filtering as described above, a total of 2953 unambiguous protein groups were assigned estimated Tm values in at least one replicate. After filtering to remove proteins represented in more than one group, 2073 proteins remained. These proteins were further filtered at two levels of confidence. Group HC (“high confidence”; n = 922) contained proteins modeled in three or more replicates with σx < 1.5°C and from at least 3 distinct peptides, and group MC (“medium confidence”; n = 1707) contained proteins modeled in two or more replicates with σx < 1.8°C and from 2 or more distinct peptides. All further analyses refer to group HC, except where noted. The distributions of Tms in all six replicates as well as the median distribution are shown in Fig. 3. The Tukey five-number summary for the HC median Tm distribution was: 36.9, 41.5, 45.6, 50.9, 63.3. Melting curve plots for all 2953 protein groups modeled are available in supplemental File S2 and plots for HC proteins only are available in supplemental File S3.
Fig. 3.
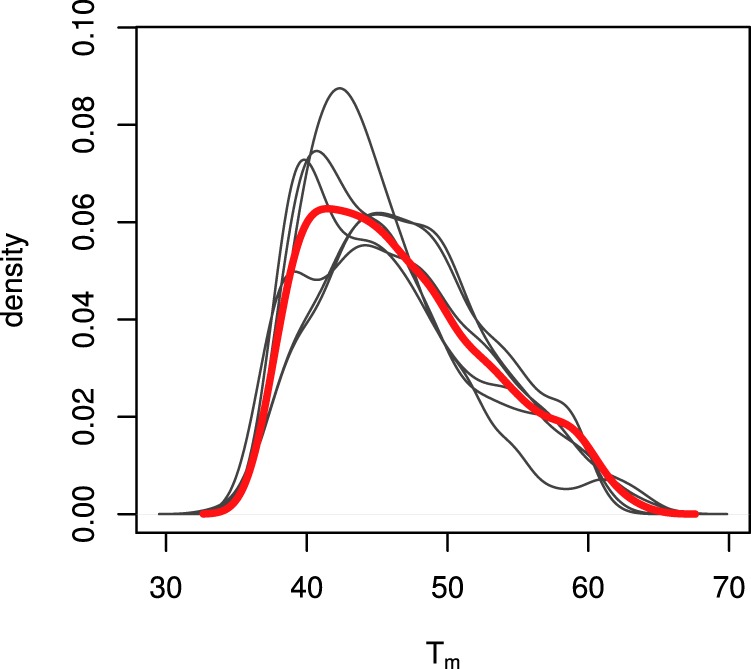
Distribution of melting temperatures in the Arabidopsis proteome. Shown are Tm distributions from six biological replicates (gray lines) along with the distribution of median Tms (solid red line). Distributions represent data from the HC data set (922 proteins modeled in three or more replicates with σx < 1.5°C).
Features of Thermostability
Eight chemical and structural features for each quantified protein were examined for potential correlation with thermostability: molecular weight, protein abundance, aliphatic index (AI), isoelectric point (pI), relative hydrophobicity (GRAVY), charged versus polar residue bias (CvP), predicted secondary structure composition, and relative composition of each of the 20 standard amino acids. All these features were either calculated or predicted directly from primary amino acid sequence or found in published databases. Proteins in the HC group were partitioned into four bins with equal membership and the bins were tested for statistically significant differences in feature distribution. Of the above features, molecular weight, hydrophobicity, CvP bias, and α-helix and β-sheet composition showed highly significant correlation with relative thermostability (all p < 3.226 × 10−4 for both tests) (Fig. 4A). Molecular weight was observed to decrease with increasing Tm, in agreement with previous observations (16, 17), as did CvP bias. We observed a statistically significant increase in relative hydrophobicity with increasing Tm as calculated by the GRAVY index (18), albeit with a small magnitude of change. Correlation with secondary structure showed an increase in the proportion of residues residing in predicted β-sheets and a decrease in α-helix residues with increasing Tm. When examining specific amino acid composition, we found that the charged residues glutamic acid and arginine were highly depleted in thermostable proteins, whereas the polar residue serine is significantly enriched (Fig. 5). These three amino acids alone likely account for the strong correlation with CvP observed above. The full table of replicate Tms, mean Tms and variances for the HC protein set, along with all calculated covariates, is available in supplemental File S4.
Fig. 4.

Correlation between protein melting temperature and potential covariates. A, displays five covariates with highly statistically significant correlation (all p < 3.226 × 10−4) based on both the Mann-Whitney U test on lower and upper bins and the Spearman rank correlation test. B, shows four covariates with no significant correlation. Data is divided into four bins of equal membership, and colors indicate relative thermostablity from unstable (blue, left) to stable (red, right). All values are calculated directly from primary amino acid sequence (see Methods for more detail).
Fig. 5.
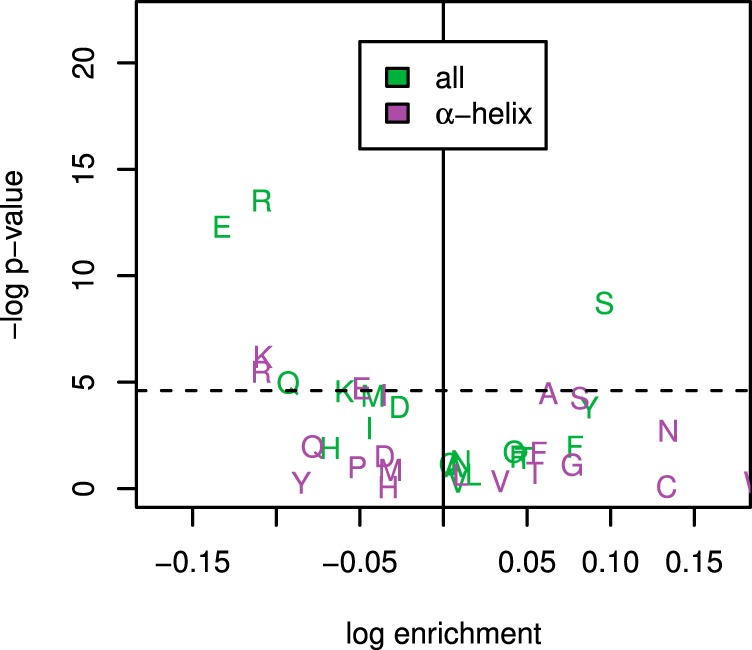
Residue-specific enrichment in stable versus unstable protein bins. The x axis shows log ratio of the median residue proportion in the most stable versus least stable bins, whereas the y axis shows statistical significance by the Mann-Whitney U test. The values in green are for full protein sequences whereas the values in purple are limited to amino acid composition of predicted alpha helices. The dashed horizontal lines marks the 0.05 significance level.
Tertiary Features of Thermostability
Of the 215 nonredundant Arabidopsis protein structures compiled, 61 had modeled Tms in the HC protein set. The following features were extracted from the VADAR output: nonpolar accessible surface area (ASA) relative to total ASA; relative polar ASA; relative charged ASA; total volume; and compactness (as described in the Methods). Quartile binning and correlation analysis were carried out as for 1° and 2° features above. Of the features tested, nonpolar ASA was positively correlated with thermostability at the 0.05 level based on the Mann-Whitney U test (p = 0.015) (Fig. 6). Charged ASA was seen to be negatively correlated with thermostability (p = 0.014). Other features, including protein compactness, did not show any significant trend.
Fig. 6.
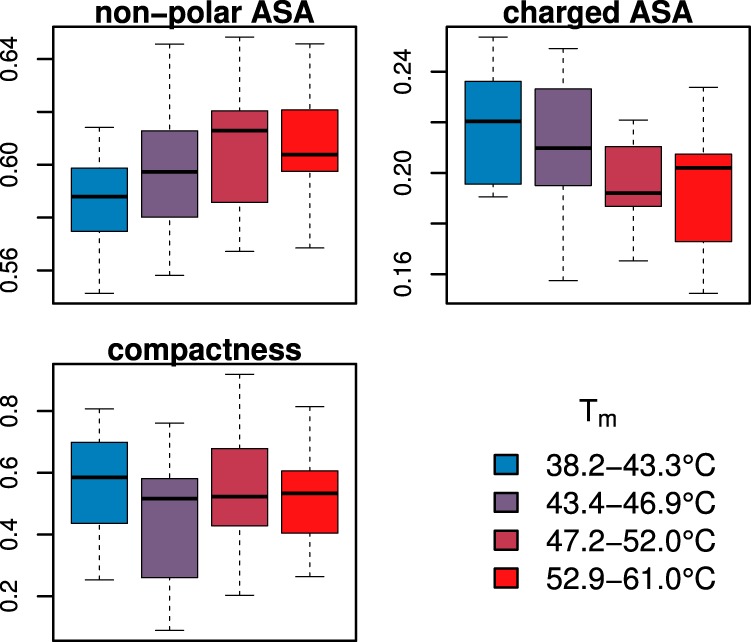
Correlation between protein melting temperature and tertiary structure. Two covariates with statistically significant correlation (based on Mann-Whitney U test) are shown at top (nonpolar ASA: p = 0.015; charged ASA: p = 0.014). The lower plot shows compactness relative to thermostability, where compactness is calculated as 3 − and ISA is the surface area of a sphere of the same volume as the protein. Although hypothesized to affect thermostability, no correlation is seen (p = 0.599). Data is divided into four bins of equal membership, and colors indicate relative thermostablity from unstable (blue, left) to stable (red, right). A total of 61 proteins with known structures and thermal models were analyzed.
Thermostability-associated Functional Enrichment
Gene set enrichment analysis was performed on the lowest (unstable) and highest (stable) quartile bins of the HC Tm data set to test whether specific bins were associated with functional classes. The results are presented in Tables I and II. Within the unstable bin, enrichment in the three ontologies primarily involved ribosomal proteins/nucleic acid binding, proteasomal proteins, and cytoskeletal proteins. Within the stable bin, enrichment was found in terms relating to protein folding, carbon fixation, and the proteasome. Proteins involved in carbon fixation were highly enriched in the thermostable bin and included two PEP carboxylases, three RuBisCo subunits, and several other Calvin cycle proteins. Full result tables for all terms tested are available in supplemental File S5.
Table I. Gene Ontology terms enriched in proteins with the lowest Tms (38.2–43.3°C). MF = molecular function; BP = biological process; CC = cellular compartment. Shown are all terms with Fisher's p < 0.002. No multiple testing correction was applied as per the algorithm recommendations.
| Category | Term | Annotated | Observed | Expected | Fisher's p |
|---|---|---|---|---|---|
| MF | structural constituent of ribosome | 83 | 76 | 19.0 | 0.00E+00 |
| MF | RNA binding | 106 | 54 | 24.3 | 2.50E−06 |
| MF | rRNA binding | 7 | 7 | 1.6 | 3.20E−05 |
| MF | nucleotide binding | 390 | 122 | 89.4 | 1.10E−04 |
| MF | GTP binding | 16 | 11 | 3.7 | 1.10E−04 |
| MF | structural constituent of cytoskeleton | 6 | 6 | 1.4 | 1.40E−04 |
| MF | nucleic acid binding | 143 | 71 | 32.8 | 4.70E−04 |
| MF | translation initiation factor activity | 22 | 12 | 5.0 | 1.19E−03 |
| BP | translation | 172 | 113 | 40.0 | 0.00E+00 |
| BP | RNA methylation | 39 | 34 | 9.1 | 1.50E−17 |
| BP | DNA-templated transcription, elongation | 25 | 17 | 5.8 | 2.10E−06 |
| BP | ribosome biogenesis | 99 | 42 | 23.0 | 1.00E−04 |
| BP | regulation of protein catabolic process | 6 | 6 | 1.4 | 1.50E−04 |
| BP | lignin biosynthetic process | 15 | 10 | 3.5 | 4.00E−04 |
| BP | DNA endoreduplication | 9 | 7 | 2.1 | 8.10E−04 |
| BP | cytoskeleton organization | 66 | 27 | 15.4 | 8.40E−04 |
| BP | embryo development ending in seed dorman … | 91 | 34 | 21.2 | 1.29E−03 |
| BP | translational initiation | 17 | 10 | 4.0 | 1.64E−03 |
| BP | pyrimidine ribonucleotide biosynthetic p … | 37 | 17 | 8.6 | 1.78E−03 |
| CC | cytosolic large ribosomal subunit | 29 | 28 | 6.7 | 1.30E−17 |
| CC | nucleolus | 60 | 42 | 13.8 | 3.30E−15 |
| CC | cytosolic small ribosomal subunit | 20 | 19 | 4.6 | 7.90E−12 |
| CC | proteasome regulatory particle, lid subc … | 10 | 10 | 2.3 | 3.70E−07 |
| CC | plastid small ribosomal subunit | 9 | 9 | 2.1 | 1.60E−06 |
| CC | plasmodesma | 185 | 68 | 42.5 | 4.30E−06 |
| CC | proteasome regulatory particle, base sub … | 7 | 7 | 1.6 | 3.20E−05 |
| CC | plastid large ribosomal subunit | 8 | 7 | 1.8 | 2.10E−04 |
| CC | nucleus | 344 | 124 | 79.0 | 3.80E−04 |
| CC | COPI vesicle coat | 5 | 5 | 1.1 | 6.30E−04 |
| CC | cytosol | 619 | 190 | 142.1 | 7.30E−04 |
| CC | ribonucleoprotein complex | 126 | 87 | 28.9 | 8.20E−04 |
| CC | ribosomal subunit | 68 | 65 | 15.6 | 1.56E−03 |
Table II. Gene Ontology terms enriched in proteins with the highest Tms (52.9–61.0°C). MF = molecular function; BP = biological process; CC = cellular compartment. Shown are all terms with Fisher's p < 0.005. No multiple testing correction was applied as per the algorithm recommendations.
| Category | Term | Annotated | Observed | Expected | Fisher's p |
|---|---|---|---|---|---|
| MF | Threonine-type endopeptidase activity | 17 | 17 | 4.4 | 7.10E−11 |
| MF | Hydrolase activity, hydrolyzing O-glycos … | 54 | 25 | 13.9 | 6.80E−04 |
| MF | Acid phosphatase activity | 7 | 6 | 1.8 | 1.52E−03 |
| MF | Electron transfer activity | 29 | 15 | 7.4 | 2.16E−03 |
| MF | Protein serine/threonine phosphatase act … | 10 | 7 | 2.6 | 4.01E−03 |
| MF | 3-chloroallyl aldehyde dehydrogenase act … | 10 | 7 | 2.6 | 4.01E−03 |
| MF | Epoxide hydrolase activity | 4 | 4 | 1.0 | 4.30E−03 |
| BP | Protein refolding | 7 | 7 | 1.8 | 6.50E−05 |
| BP | Organic hydroxy compound catabolic proce … | 4 | 4 | 1.0 | 4.10E−03 |
| BP | Cell wall macromolecule catabolic proces … | 4 | 4 | 1.0 | 4.10E−03 |
| BP | Carbon fixation | 12 | 10 | 3.0 | 4.70E−03 |
| BP | Reductive pentose-phosphate cycle | 6 | 5 | 1.5 | 4.90E−03 |
| CC | Extracellular region | 275 | 115 | 70.3 | 2.60E−11 |
| CC | Proteasome core complex, alpha-subunit c … | 10 | 10 | 2.6 | 1.10E−06 |
| CC | Proteasome core complex | 17 | 17 | 4.3 | 6.00E−05 |
| CC | Vacuole | 182 | 66 | 46.5 | 4.50E−04 |
| CC | Plant-type cell wall | 44 | 20 | 11.2 | 2.97E−03 |
The Arabidopsis Proteasome
It was observed during gene set enrichment analysis that proteasome subunits were enriched in both the lowest and highest bins. We therefore looked carefully at the Tm distributions of each of the proteasomal proteins and found a marked difference in thermostability between core and regulatory subunits (Fig. 7). Many proteasomal subunits exist as multiple paralogs in the Arabidopsis genome, and at least one homolog of most subunits was modeled in our data. All modeled core subunits had Tms at the upper end of the proteome range, whereas all regulatory subunit Tms were in the lower bin. This is line with observations from other labs regarding co-precipitating protein complexes and is discussed further below.
Fig. 7.
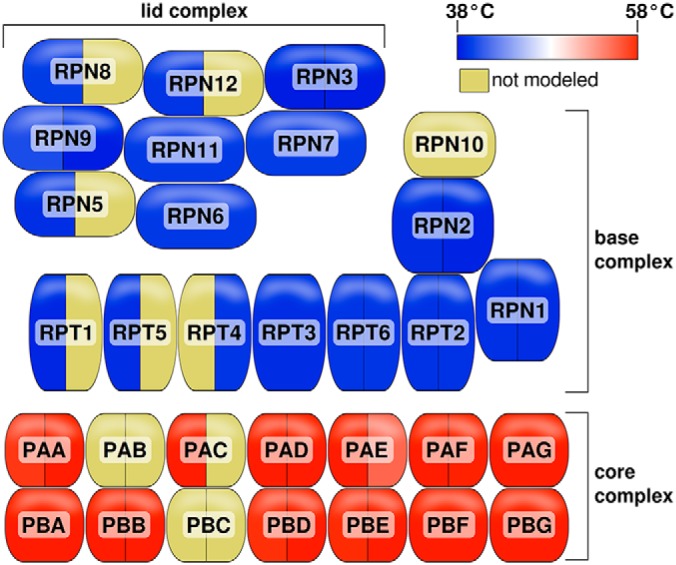
Melting temperatures of the 26S proteasome subunits. Shading indicates median Tm for that subunit (or yellow for unmodeled). Subunits with paralogous family members are indicated by a vertical divider. The MC data set was used to generate the Tm values in order to maximum subunit coverage.
Mg-ATP-induced Thermal Stability Shifts
In order to demonstrate the suitability of TPP-based thermal shift assays to detect treatment-induced conformational changes in complex plant extracts, we performed TPP on Arabidopsis lysates treated in vitro with Mg-ATP or mock solutions and compared calculated ΔTm shifts upon treatment with existing protein functional annotations. The results of this comparison are shown in Fig. 8. There is a significant increase in Mg-ATP-induced ΔTms among annotated kinases when compared with all other proteins (p = 0.015). There is also an apparent enrichment in treatment-induced thermal upshifts in the more general annotated ATP-binding protein set, although the statistical significance was weak (p = 0.103). Possible reasons for this weak effect are discussed in more detail below. Lastly, as Mg-ATP was used in the treatment, we also compared annotated magnesium-binding proteins versus all other modeled proteins and observed a highly significant stabilizing effect in this class (p = 0.004). Gene set enrichment analysis on the ΔTm values greater than 4°C in either direction showed statistically significant enrichment (p < 0.01) in stabilized proteins of terms related to known ATP binding classes, including “adenosine kinase activity” and “glucose-1-phosphate adenylyltransferase activity.” Interestingly, among the most significant enrichments in destablized proteins was the class “heterocylic compound binding,” a parent term of ATP-binding proteins, suggesting that ATP binding can have both stabilizing and destablizing effects depending on the protein class (supplemental File S6).
Fig. 8.

Thermostability shifts upon Mg-ATP treatment. Shown are ΔTm distributions for treated versus control replicates. Proteins are grouped according to: A, annotated as ATP-binding; B, annotated as kinases; C, annotated as magnesium-binding. Included in the analysis were 653 proteins, of which 56 were annotated as ATP-binding, 12 were annotated as kinases, and 15 were annotated as Mg-binding. Mann-Whitney p values for difference of means for the three comparisons were 0.103 (ATP-binding), 0.015 (kinases), and 0.004 (Mg-binding).
DISCUSSION
Thermostability of a Plant Proteome
We have presented here the first in-depth look at the thermal proteome of a model plant. Out of the roughly 4,000 proteins identified in the samples, 922 were modeled in three or more replicates with σx < 1.5°C, representing high-confidence Tm estimates and errors, and 1707 protein Tms were estimated at the lower thresholds of the MC group. This database (in the form of estimated Tm, melting temperature profiles, protein-specific measurement variances, and behavior of low-abundance proteins) represents a valuable resource for Arabidopsis researchers planning a TPP study. It is critical to recognize from the start of such an experiment that all proteins do not behave identically under the assumptions being made, and researchers can thus benefit from prior knowledge as to whether their proteins of interest are likely to “behave well” within the confines of the assay. In our experience, some proteins have highly consistent melting curves from experiment to experiment, whereas others have consistently high variance and many do not appear to behave according to the two-state model at all (Fig. 9 and supplemental File S2). Although biological replicates are critical in any downstream experimental design in order to infer shifts in Tm at the individual protein level, proteins which display reproducible curves across the six replicates presented here, prepared at different times using multiple isolation techniques, will be better candidates for detecting small shifts in Tm in future experiments than those with higher inherent variation.
Fig. 9.

Protein-specific variance in Tm estimates. Most proteins behave relatively reproducibly between replicates and experiments (A). Higher measurement variance is often associated with lower abundance but not always, as some proteins are consistent at low PSM counts (B) whereas others have consistently high variance even at higher abundance (C).
To be considered valid, the database presented here should be consistent with existing knowledge and expected results. Gene set enrichment was used both to validate the approach and to search for novel patterns in plant protein stability. The thermo-labile bin was found to be enriched in cytoskeletal proteins. Actin is known to respond to moderate heat stress in Arabidopsis and to readily adopt multiple conformations, so it is not surprising that its subunits would overwhelmingly fall into the unstable bin (19, 20). Likewise, it would be expected that proteins involved in protein re-folding would require higher thermal stability to retain function under heat stress, and we observed enrichment for terms related to protein-folding in the stable bin. In addition, we observe higher thermostability in proteins involved in carbon fixation, including the three subunits of Rubisco modeled in the experiment. At the same time, the Rubisco activase protein RCA (AT2G39730) is among the least thermally stable proteins modeled (Tm = 38.8°C). This is consistent with the current understanding of the relationship between photosynthesis and heat stress, where activated Rubisco has been found to increase in activity over increasing temperatures while its activase loses activity relatively quickly (21, 22).
Another observation arising from the gene set enrichment analysis was the segregation of core and regulatory 26S proteasomal subunits into different extremes of thermostability. Little has been published on the thermostability of the plant proteasome, but the results observed are perhaps not surprising. The proteasome proteolytic core is a highly structured unit composed of rings of α and β isomers, whereas the regulatory base and lid are composed of proteins with a range of functions which serve in various aspects of ubiquitin recognition and ATP-dependent protein transport into the core. It is therefore not surprising that the regulatory complex would require a higher degree of conformational flexibility and thus unfold at lower temperatures. It is also possible that the role of the proteasome in stress response (including heat stress) may make thermal stability of the core proteasome important, as the core 20S proteasome itself is capable of degrading unfolded and damaged proteins in the absence of the regulatory complex.
The observation that the main proteasomal complexes share Tm profiles among subunits is also not surprising. Because our initial observations, this finding has been confirmed in human cells as part of a larger observation that tightly bound protein complexes tend to denature as a unit (23). In fact, this tendency has been used as the basis of a technique to study protein associations (the 'interactome') via conserved Tm and ΔTm profiles between complexed proteins (24).
Correlation with Protein Properties
Significant past effort has gone into discovering determinants of protein thermostability, defined herein as the relative ability of a protein to maintain a native conformation under increasing temperature. For the most part, this past work has focused on comparing protein structure (1° and higher-level) between orthologs from mesophilic, thermophilic, and hyperthermophilic prokaryotes (25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40). In some cases a small number of experimental Tms were known (for example, taken from the ProTherm database) (32, 37, 40), but often it has been assumed that characteristics that differentiate mesophilic from thermophilic primary protein sequences would also be general determinants of thermostability, justifying purely in silico approaches on sequenced genomes (28, 29, 38). More recent work by Leuenberger et al. used mass spectrometry together with limited proteolysis to estimate Tms for a large number of proteins in representative prokaryotic, fungal, and animal species (17) and found correlations between a number of chemical and structural characteristics of proteins and their relative degree of thermostability.
Our observations on the correlation between fundamental protein characteristics and thermostability bolster many of the previously published observations but also differ in a few key aspects. Molecular weight was seen to be inversely correlated with Tm, which agrees with previous observations (16, 17). In comparative studies of mesophiles versus thermophiles, this observation usually comes with the caveat that group-specific trends in protein size may be caused by evolutionary pressures other than thermostability. However, our observation of a strong similar trend within a single plant proteome lends credence to the idea of a causal relationship between the two factors. We also observed a statistically significant increase in relative hydrophobicity with increasing Tm. Previous mesophile/thermophile studies have come to mixed conclusions regarding this relationship, with Kumar et al. (29) finding no correlation and McDonald (38) finding a positive correlation, in agreement with our results. Correlation with secondary structure was also in agreement with Leuenberger et al., with an increase in the proportion of residues residing in predicted β-sheets and a decrease in α-helix residues with increasing Tm. Most published work examining differences in mesophilic and thermophilic proteomes report an increase in α-helix residency in proteins of thermophilic organisms, suggesting that other evolutionary factors may be in play in those organisms (29, 26, 27). It is important to note that β-sheets require special consideration, given that the readout of our assay involves protein precipitation upon unfolding and that β-sheets are known to affect nonspecific protein aggregation. However, any such effect would tend to bias the results in the opposite direction as that observed, suggesting that any bias because of β-sheet content is negligible.
Existing mesophile/thermophile literature suggests that charged-versus-polar bias (CvP, i.e. the relative proportion of D, E, K and R versus N, Q, S and T) is a robust predictor of mesophilic and thermophilic orthologs and thus is thought to be involved in protein thermostability, with an increase in global CvP corresponding with an increase in optimum growth temperature (30). However, in our own work with a plant extract we find a strong negative correlation between the two values in our data at the single protein level (Fig. 4). Indeed, D, E, K, and R are among the most statistically significant depleted amino acids in thermostable proteins, whereas S is strongly enriched (Fig. 5). This is partly in agreement with Leuenberger et al., who found a depletion in aspartic acid in thermostable proteins of E. coli, although their observation of an enrichment in lysine does not agree with our results. Furthermore, we observed in a smaller subset of proteins with known tertiary structures that the depletion in charged residues extends to the protein surface. At the same time, nonpolar residues are enriched on the surface of thermostable proteins. Clearly these features (for instance, overall charged residue composition and charged surface area) are interdependent, but the tertiary analysis strengthens the case for the importance of these specific amino acids in determining protein thermal stability.
Other protein features examined (abundance, aliphatic index, isoelectric point, unstructured content), which in various reports have been correlated with protein thermostability, do not show statistically significant correlation in our data (Fig. 4B). Leuenberger et al. reported a “clear” positive correlation between protein abundance and thermostability, but we observe no correlation or trend in our data. Abundance is a challenging trait to interpret, as the proteins most readily modeled in the assay are strongly biased toward the most abundant proteins in the proteome (this is true of most tandem MS studies). Additionally, there is a danger of specific classes of very abundant proteins skewing the results. This was seen in our data in the case of ribosomal proteins, which are highly enriched in the most unstable bin and which have specific chemical and structural attributes which may be completely unrelated to thermostability but which skew the results of feature correlation (they were removed prior to the final analysis).
Ribosomal Thermostability
Although most of our observations correlate with those from other kingdoms published previously, the Arabidopsis ribosomal complexes showed marked thermal instability, in contrast with observations in Leuenberger et al. and Becher et al. (17, 23) in animal cells. Although it is tempting to interpret this as a plant-specific behavior, it is also possible that extraction conditions play a role. Given the highly conserved nature of the ribosome, this is perhaps a more likely explanation. As with the 26S proteasome, it was not surprising to observe the ribosomal complex precipitating as a unit. We also see strong enrichment for eukaryotic initiation factors in the same thermo-labile bin as ribosomal subunits, suggesting co-aggregation of these proteins as part of the ribosomal complex. Of interest was the fact that the 60S acidic subunits displayed a markedly different melting profile, suggesting that they are less tightly associated with the ribosomal complex than the other subunits observed (Fig. 10). Clearly, further experimental work is needed to explain the observed behavior of plant ribosomal proteins in the assay as either a unique trait or a technical artifact.
Fig. 10.
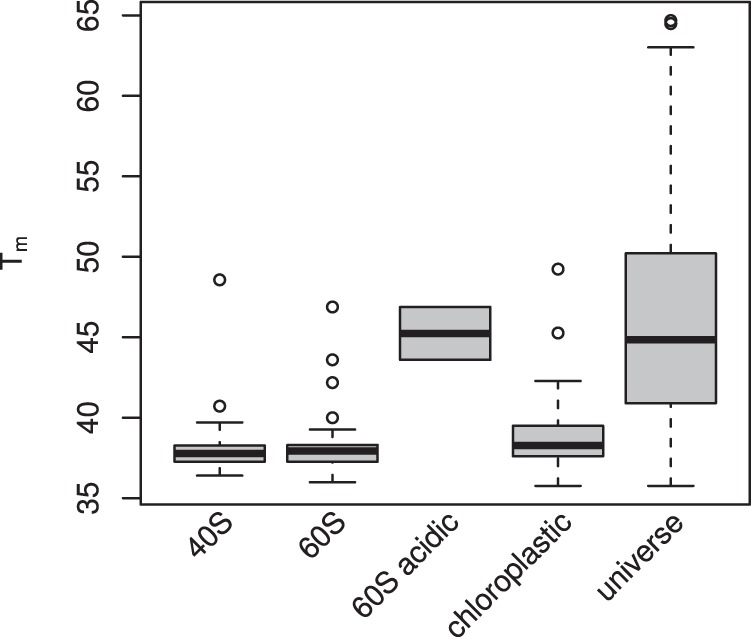
Distribution of Arabidopsis ribosomal protein Tms. 60S acidic proteins are a subset of the 60S set and thus are included twice. Data is taken from the MC protein set. The rightmost plot (“universe”) shows the Tm distribution for all modeled and filtered proteins in the MC data set for the sake of comparison.
Thermal Shifts Upon ATP Treatment
Using a broad-spectrum in vitro treatment, we have demonstrated the suitability of TPP for probing changes in plant protein stability upon perturbation, with Mg-ATP as a positive control as used previously in other organisms (4). Although the lack of replicates prevents reliable interpretation of the data at the individual protein level, it is possible to examine differences in ΔTm distribution among global groups of proteins with statistical significance. The distribution of ΔTms in annotated ATP-binding proteins appears bimodal, with a subset behaving like non-ATP-binding proteins and a subset with significant thermo-stabilization. This suggests a complex mechanism of stabilization upon ATP binding that acts differently on different classes of proteins. There is also a possibility that the level of occupancy of the ATP binding pocket in different protein groups prior to treatment can partly or fully explain the bimodal nature of the observed distributions. Indeed, when annotated kinases alone were analyzed, the distribution took on a more unimodal shape with a significant upshift in ΔTm. The sharpest and most significant shift of all was observed among annotated magnesium-binding proteins, a side-effect of using Mg-ATP as the treatment, despite the generally weak binding affinity of magnesium to proteins compared with other metal ions (41). It is possible that the more marked effect observed in putative Mg-binding proteins is because of lower occupancy of the binding sites pre-treatment compared with ATP-binding proteins. As described above, gene set enrichment analysis of the data was consistent with an effect of ATP binding on thermal stability. Although unreplicated data should be interepreted with great care at the single-protein level, plots of all proteins modeled are provided for inspection in supplemental File S7 and proteins with large shifts (abs(ΔTm) > 4) used in the GO analysis are included in supplemental File S8.
CONCLUSIONS
The primary aim of this study was to lay a groundwork for future studies using TPP in plant systems. To this end, we have developed a database of Tm, melting profiles and experimental variance data which will help guide future researchers using this tool. It is important to acknowledge that the results of work carried out in vitro on extracts from plants grown in defined hydroponic media can only be interpreted with care in the context of real-world environmental conditions or even in vivo experiments in the lab. Factors such as cytoplasmic protein concentration, small molecule interactions and cellular localization can be expected to have significant affects on thermal stability and molecular interactions. Difficulties in detecting and modeling less abundant proteins also narrow the scope of usefulness of the assay. With these limitations in mind, however, emerging technologies such as TPP present a wealth of new opportunities for plant researchers to pursue unknown cellular interactions at a large-scale level.
A secondary aim of the study was to leverage the database of thermal profiling data to look for determinants of protein thermal stability, with a focus on characteristics unique to plants and the environments they face. We observed several features significantly correlated with Tm that add to the body of knowledge in this area. We did not, however, observe any patterns that could confidently be interpreted as unique to the plant proteome, and this remains an area of active interest for future work. Lastly, these results provide a demonstration of the suitability of TPP combined with the thermal shift assay to detect conformational changes in the plant proteome. They complement work from other kingdoms and open a new avenue of investigation to researchers interested in searching for novel protein-ligand interactions, providing the potential to more readily probe the effects of genetic and environmental perturbations on plant protein conformation.
Data Availability
The mass spectrometry raw data have been deposited to the ProteomeXchange Consortium via the PRIDE [42] partner repository with the data set identifier PXD011200. All input data as well as the LATEX/R/knitr source code to reproduce all results and figures in this manuscript are available at https://github.com/Sussman-Lab/at_thermal_proteome.
Supplementary Material
Acknowledgments
This work was funded by NSF MCB grant 1713899, with additional support for JDV from a Morgridge Graduate Fellowship. We thank Heather Burch, Pei Liu and Greg Sabat for their technical and intellectual assistance in carrying out this work.
Footnotes
 This article contains supplemental material. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental material. The authors declare that they have no conflicts of interest with the contents of this article.
1 The abbreviations used are:
- TPP
- thermal proteome profiling
- AI
- aliphatic index
- ATP
- adenosine triphosphate
- CvP
- charged-versus-polar
- FA
- formic acid
- FDR
- false discovery rate
- GO
- Gene Ontology
- HC
- high confidence
- HCD
- higher-energy collisional dissociation
- MC
- medium confidence
- pI
- isoelectric point
- PSM
- peptide-spectrum match
- SASA
- solvent-accessible surface area
- TEAB
- Triethylammonium bicarbonate
- TMT
- tandem mass tag.
REFERENCES
- 1. Cramer P., Bushnell D. A., and Kornberg R. D. (2001) Structural basis of transcription: RNA polymerase II at 2.8 Ångstrom resolution. Science 292, 1863–1876 [DOI] [PubMed] [Google Scholar]
- 2. Shih Y.-L., and Rothfield L. (2006) The bacterial cytoskeleton. Microbiol. Mol. Biol. Rev. 70, 729–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ingles-Prieto A., Ibarra-Molero B., Delgado-Delgado A., Perez-Jimenez R., Fernandez J. M., Gaucher E. A., Sanchez-Ruiz J. M., and Gavira J. A. (2013) Conservation of protein structure over four billion years. Structure 21, 1690–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Savitski M. M., Reinhard F. B. M., Franken H., Werner T., Savitski M. F., Eberhard D., Molina D. M., Jafari R., Dovega R. B., Klaeger S., Kuster B., Nordlund P., Bantscheff M., and Drewes G. (2014) Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346, 1255784. [DOI] [PubMed] [Google Scholar]
- 5. Minkoff B. B., Burch H. L., and Sussman M. R. (2014) A pipeline for 15n metabolic labeling and phosphoproteome analysis in Arabidopsis thaliana. Methods Mol. Biol. 1062, 353–379 [DOI] [PubMed] [Google Scholar]
- 6. Chambers M. C., Maclean B., Burke R., Amodei D., Ruderman D. L., Neumann S., Gatto L., Fischer B., Pratt B., Egertson J., Hoff K., Kessner D., Tasman N., Shulman N., Frewen B., et al. (2012) A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotech. 30, 918–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eng J. K., Jahan T. A., and Hoopmann M. R. (2013) Comet: An open-source MS/MS sequence database search tool. Proteomics 13, 22–24 [DOI] [PubMed] [Google Scholar]
- 8. Ma K., Vitek O., and Nesvizhskii A. I. (2012) A statistical model-building perspective to identification of MS/MS spectra with PeptideProphet. BMC Bioinform. 13, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nesvizhskii A. I., Keller A., Kolker E., and Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 10. Savitski M. M., Mathieson T., Zinn N., Sweetman G., Doce C., Becher I., Pachl F., Kuster B., and Bantscheff M. (2013) Measuring and managing ratio compression for accurate iTRAQ/TMT quantification. J. Proteome Res. 12, 3586–3598 [DOI] [PubMed] [Google Scholar]
- 11. Rice P., Longden I., and Bleasby A. (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 [DOI] [PubMed] [Google Scholar]
- 12. Garnier J., Osguthorpe D. J., and Robson B. (1978) Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J. Mol. Biol. 120, 97–120 [DOI] [PubMed] [Google Scholar]
- 13. Wang M., Weiss M., Simonovic M., Haertinger G., Schrimpf S. P., Hengartner M. O., and Mering C. v (2012). PaxDB, a database of protein abundance averages across all three domains of life. Mol. Cell. Proteomics 11, 492–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alexa A., Rahnenführer J., and Lengauer T. (2006) Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607 [DOI] [PubMed] [Google Scholar]
- 15. Willard L., Ranjan A., Zhang H., Monzavi H., Boyko R. F., Sykes B. D., and Wishart D. S. (2003) VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res. 31, 3316–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ghosh K., and Dill K. A. (2009) Computing protein stabilities from their chain lengths. Proc. Natl. Acad. Sci. U.S.A. 106, 10649–10654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leuenberger P., Ganscha S., Kahraman A., Cappelletti V., Boersema P. J., Mering C. v, Claassen M., and Picotti P. (2017) Cell-wide analysis of protein thermal unfolding reveals determinants of thermostability. Science 355, eaai7825. [DOI] [PubMed] [Google Scholar]
- 18. Kyte J., and Doolittle R. F. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 [DOI] [PubMed] [Google Scholar]
- 19. Levitsky D. I., Pivovarova A. V., Mikhailova V. V., and Nikolaeva O. P. (2008) Thermal unfolding and aggregation of actin. FEBS J. 275, 4280–4295 [DOI] [PubMed] [Google Scholar]
- 20. Müller J., Menzel D., and Šamaj J. (2007) Cell-type-specific disruption and recovery of the cytoskeleton in Arabidopsis thaliana epidermal root cells upon heat shock stress. Protoplasma 230, 231–242 [DOI] [PubMed] [Google Scholar]
- 21. Crafts-Brandner S. J., and Salvucci M. E. (2000) Rubisco activase constrains the photosynthetic potential of leaves at high temperature and CO2. Proc. Natl. Acad. Sci. U.S.A. 97, 13430–13435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Salvucci M. E., and Crafts-Brandner S. J. (2004) Relationship between the heat tolerance of photosynthesis and the thermal stability of rubisco activase in plants from contrasting thermal environments. Plant Physiol. 134, 1460–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Becher I., Andrés-Pons A., Romanov N., Stein F., Schramm M., Baudin F., Helm D., Kurzawa N., Mateus A., Mackmull M.-T., Typas A., Müller C. W., Bork P., Beck M., and Savitski M. M. (2018) Pervasive protein thermal stability variation during the cell cycle. Cell 173, 1495–1507.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tan C. S. H., Go K. D., Bisteau X., Dai L., Yong C. H., Prabhu N., Ozturk M. B., Lim Y. T., Sreekumar L., Lengqvist J., Tergaonkar V., Kaldis P., Sobota R. M., and Nordlund P. (2018) Thermal proximity coaggregation for system-wide profiling of protein complex dynamics in cells. Science 359, 1170–1177 [DOI] [PubMed] [Google Scholar]
- 25. Ikai A. (1980) Thermostability and aliphatic index of globular proteins. J. Biochem. 88, 1895–1898 [PubMed] [Google Scholar]
- 26. Merkler D. J., Farrington G. K., and Wedler F. C. (1981) Protein thermostability. Correlations between calculated macroscopic parameters and growth temperature for closely related thermophilic and mesophilic bacilli. Int. J. Pept. Protein Res. 18, 430–442 [PubMed] [Google Scholar]
- 27. Vogt G., and Argos P. (1997) Protein thermal stability: hydrogen bonds or internal packing?. Fold. Des. 2, S40–S46 [DOI] [PubMed] [Google Scholar]
- 28. Das R., and Gerstein M. (2000) The stability of thermophilic proteins: a study based on comprehensive genome comparison. Funct. Integr. Genomics 1, 76–88 [DOI] [PubMed] [Google Scholar]
- 29. Kumar S., Tsai C.-J., and Nussinov R. (2000) Factors enhancing protein thermostability. Protein Eng. 13, 179–191 [DOI] [PubMed] [Google Scholar]
- 30. Suhre K., and Claverie J.-M. (2003) Genomic correlates of hyperthermostability, an update. J. Biol. Chem. 278, 17198–17202 [DOI] [PubMed] [Google Scholar]
- 31. Berezovsky I. N., and Shakhnovich E. I. (2005) Physics and evolution of thermophilic adaptation. Proc. Natl. Acad. Sci. U.S.A. 102, 12742–12747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Razvi A., and Scholtz J. M. (2006) Lessons in stability from thermophilic proteins. Protein Sci. 15, 1569–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang G., and Fang B. (2006) Discrimination of thermophilic and mesophilic proteins via pattern recognition methods. Process Biochem. 41, 552–556 [Google Scholar]
- 34. Zeldovich K. B., Berezovsky I. N., and Shakhnovich E. I. (2007) Protein and DNA sequence determinants of thermophilic adaptation. PLOS Comput. Biol. 3, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gromiha M. M., and Suresh M. X. (2008) Discrimination of mesophilic and thermophilic proteins using machine learning algorithms. Proteins: Struct., Funct., Bioinf. 70, 1274–1279 [DOI] [PubMed] [Google Scholar]
- 36. Montanucci L., Fariselli P., Martelli P. L., and Casadio R. (2008) Predicting protein thermostability changes from sequence upon multiple mutations. Bioinformatics 24, i190–i195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ku T., Lu P., Chan C., Wang T., Lai S., Lyu P., and Hsiao N. (2009) Predicting melting temperature directly from protein sequences. Comput. Biol. Chem. 33, 445–450 [DOI] [PubMed] [Google Scholar]
- 38. McDonald J. H. (2010) Temperature adaptation at homologous sites in proteins from nine thermophile-mesophile species pairs. Genome Biol. Evol. 2, 267–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Taylor T. J., and Vaisman I. I. (2010) Discrimination of thermophilic and mesophilic proteins. BMC Struct. Biol. 10, S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pucci F., and Rooman M. (2014) Stability curve prediction of homologous proteins using temperature-dependent statistical potentials. PLoS Comput. Biol. 10, e1003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Foster A. W., Osman D., and Robinson N. J. (2014) Metal preferences and metallation. J. Biol. Chem. 289, 28095–28103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vizcaíno J. A., Csordas A., del Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q.-W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry raw data have been deposited to the ProteomeXchange Consortium via the PRIDE [42] partner repository with the data set identifier PXD011200. All input data as well as the LATEX/R/knitr source code to reproduce all results and figures in this manuscript are available at https://github.com/Sussman-Lab/at_thermal_proteome.