

The affinity antibody purification combined with LC MS/MS was used to investigate the lysine succinylome profile of A. hydrophila ATCC7966. A total of 666 lysine succinylation proteins were identified and analyzed in depth to better understand its regulatory roles. Lysine succinylation modifications on S-ribosylhomocysteine lyase were further studied and shown to regulate its cellular physiology and affect bacterial quorum sensing behavior of A. hydrophila.
Keywords: Post-translational Modifications, Affinity Proteomics, Mass Spectrometry, Microbiology, Metabolomics, Activated Methyl Cycle, Quorum Sensing, S-Ribosylhomocysteine Lyase, Succinylome
Graphical Abstract

Highlights
A global lysine succinylome was investigated in A. hydrophila.
The lysine succinylation modifications play crucial role on various metabolic pathways.
Reversible succinylation on Lys23 and Lys30 regulates the activity of S-ribosylhomocysteine lyase LuxS.
Lysine succinylation modifications of LuxS affect quorum sensing and metabolism.
Abstract
Protein modification by lysine succinylation is a newly identified post-translational modification (PTM) of lysine residues and plays an important role in diverse physiological functions, although their associated biological characteristics are still largely unknown. Here, we investigated the effects of lysine succinylation on the physiological regulation within a well-known fish pathogen, Aeromonas hydrophila. A high affinity purification method was used to enrich peptides with lysine succinylation in A. hydrophila ATCC 7966, and a total of 2,174 lysine succinylation sites were identified on 666 proteins using LC-MS/MS. Gene ontology analysis indicated that these succinylated proteins are involved in diverse metabolic pathways and biological processes, including translation, protein export, and central metabolic pathways. The modifications of several selected candidates were further validated by Western blotting. Using site-directed mutagenesis, we observed that the succinylation of lysines on S-ribosylhomocysteine lyase (LuxS) at the K23 and K30 sites positively regulate the production of the quorum sensing autoinducer AI-2, and that these PTMs ultimately alter its competitiveness with another pathogen, Vibrio alginolyticus. Moreover, subsequent metabolomic analyses indicated that K30 succinylation on LuxS may suppress the activated methyl cycle (AMC) and that both the K23 and K30 sites are involved in amino acid metabolism. Taken together, the results from this study provide significant insights into the functions of lysine succinylation and its critical roles on LuxS in regulating the cellular physiology of A. hydrophila.
Most post-translational modifications (PTMs)1 of proteins are dynamic, reversible, and crucial for regulating the cellular physiology and pathology of various organisms. Several different PTMs have been observed among bacterial species, including phosphorylation, glycosylation, nitrosylation, and acylation (1). Of these, lysine acylation is one of the most important PTMs in bacterial species because of its relative freedom in ionic coordination and the chemical reactivity of the primary amine group (2). The use of affinity enrichment combined with LC-MS/MS technologies have recently advanced the discovery of novel PTMs including modifications of lysine residues such as acetylation in Streptococcus pneumoniae, pupylation in Mycobacterium smegmatis, malonylation in Bacillus amyloliquefaciens, and propionylation in Thermus thermophilus, all of which contribute to diverse biological functions (3–6).
Among the recently described PTMs, modification of lysine by succinylation (Ksucc) has been shown to be widespread in bacteria since its discovery in 2011 (7). As in eukaryotic proteins, bacterial Ksucc are evolutionarily conserved and are involved in core metabolic pathways including the tricarboxylic acid (TCA) cycle, glycolysis, and pyruvate metabolisms (8–11). Thus, Ksucc is likely to have important cellular functions in response to varying physiological conditions. Consequently, a comprehensive identification of Ksucc within organisms is critical to understanding essential biological activities and response mechanisms under diverse physiological conditions. Although the PTM profiles have been identified for some bacterial species, such as Histoplasma capsulatum, Vibrio parahemolyticus, and Escherichia coli, the function of lysine succinylation among prokaryotes, and especially aquatic pathogens, remains largely unknown (10, 12, 13).
Aeromonas hydrophila is a prevalent pathogen of farmed fish that can infect freshwater species, including catfish, salmon, and carp, and causes motile Aeromonas septicemia (MAS) disease (14). Outbreaks of MAS lead to considerable economic losses in aquaculture (15). A. hydrophila also infects domestic animals and can contaminate food products, causing gastrointestinal and extra-intestinal infections, and even death (16). Thus, it is important to understand the mechanisms underlying the pathogenicity of this important bacterial pathogen. The abovementioned observations demonstrate that lysine succinylation dynamically regulates enzymes in key metabolisms that can impact basal bacterial behaviors. Consequently, Ksucc proteins may play important regulatory roles in the biological functions of A. hydrophila. However, to the best of our knowledge, previous investigations of Ksucc in A. hydrophila have not been conducted, which have prevented a complete understanding of protein PTM functions in this pathogen.
Quorum sensing (QS) is a universal bacterial behavior involved in the communication and coordination of social activities via the release and detection of small molecules, including N-acyl-l-homoserine lactones (AHL), autoinducing peptides (AIP), and autoinducer-2 (AI-2). Of these, AI-2 is particularly used for interspecies communication in response to population density changes. The production of 4,5-dihydroxy-2,3-pentanedione (DPD) the precursor for AI-2 (17), is accomplished by the key enzyme S-ribosylhomocysteine lyase (LuxS). Consequently, LuxS activity plays an important role in quorum sensing, biofilm formation, bioluminescence, virulence, and antibiotic resistance (18–20). Moreover, it has an important metabolic function in the activated methyl cycle (AMC), which is an important metabolic pathway for intracellular methylation (21). Motif analysis has shown that LuxS of V. harveyi has a tyrosine kinase phosphorylation site (17), suggesting a role for PTM, and making this enzyme an intriguing target to understand PTMs. However, PTMs have not been previously validated in this enzyme via experimentation.
Proteomics is a recently developed powerful approach for investigating protein PTM sites (22–24). Here, we combined high-affinity purification with high sensitivity mass spectrometry technologies to identify the entire succinylome of A. hydrophila ATCC7966. A total of 2,174 lysine succinylation sites were identified on 666 proteins that were involved in diverse metabolic pathways and biological processes. Motif analysis indicated the presence of eight motifs surrounding the central lysine residues, which were succinylated and conserved across other bacterial species. We validated selected succinylated proteins by Western blotting, and further analyzed the effects of this PTM on the biological functions of LuxS. Our results provide significant insights into the role of lysine succinylation in the cellular physiology and pathology of A. hydrophila.
MATERIALS AND METHODS
Bacterial Strains and Growth Conditions
A. hydrophila ATCC7966 was used for the experiments in this study and was maintained in our laboratory. The strain was routinely grown overnight in fresh Luria Bertani (LB) medium, and cultures were diluted 1:100 in the same medium, followed by incubation at 30 °C with shaking at 200 rpm until OD at 600 nm reached 1.0. Cells were harvested and washed twice with phosphate-buffered saline, PBS (0.1 m Na2HPO4, 0.15 m NaCl, pH 7.5) before further use.
Sample Preparation and In-solution Trypsin Digestion
The cell pellet was re-suspended in lysis buffer (8 m urea, 2 mm EDTA, 5 mm Dithiothreitol (DTT) and 1% (v/v) protease inhibitor mixture (Calbiochem, San Diego, CA), and cells were disrupted by sonication on ice as previously described (25). Proteins (∼10 mg) were precipitated with 20% TCA overnight at 4 °C, and then washed three times with ice-cold acetone. The resultant pellet was dissolved in buffer (8 m urea, 100 mm ammonium bicarbonate, pH 8.0) and proteins were reduced by incubation with 10 mm DTT for 1 h at 37 °C and were alkylated with 20 mm iodoacetamide (IAA) by incubating for 45 min at room temperature. The sample was diluted 8 times with 100 mm NH4CO3 before digestion with trypsin at a 1:50 ratio and incubated for 16 h at 37 °C. The tryptic peptides were dried using a CentriVap Concentrator (Labconco, Kansas City, MO) (26).
Enrichment of Lysine-succinylated Peptides
To enrich for a high abundance of lysine-succinylated peptides, immunoaffinity analysis was performed using agarose-conjugated anti-succinyllysine antibody (PTM Biolabs Inc., Hangzhou, China), as previously described (27). Briefly, the tryptic digested peptides were incubated with anti-succinyllysine agarose beads overnight at 4 °C in NETN buffer (100 mm NaCl, 50 mm Tris-HCl, 1 mm EDTA, and 0.5% Nonidet P-40, pH 8.0), and then eluted with 1% trifluoroacetic acid (TFA). The enriched peptides were desalted with C18 ZipTips (Millipore, Burlington, MA) and then subjected to high-performance liquid chromatography (HPLC)-MS/MS identification.
Protein Analysis by Nano HPLC-MS/MS
Protein identification was performed using the ThermoFisher ScientificTM Q ExactiveTM Plus mass spectrometer (ThermoFisher Scientific, San Jose, CA). Digested peptides were separated using the Acclaim PepMap RSLC C18 capillary reversed-phase analytical column (ThermoFisher Scientific) with a 40 min 6–80% ACN/water gradient containing 0.1% FA at a constant flow rate of 300 nL/min on the EASY-nLC 1000 UPLC system. The eluted peptides were further ionized and sprayed into the nanospray-ionization (NSI) source followed by tandem mass spectrometry (MS/MS) in Q ExactiveTM Plus coupled online to UPLC.
The fragmentation data from two biological replicates were merged and interpreted using the MaxQuant software package (version 1.4.1.2) against the A. hydrophila ATCC 7966 database containing 4150 sequences (downloaded from the Uniprot database on 2/1/2018), with search parameters that included carbamidomethylation of cysteine residues as a fixed modification, methionine oxidation, lysine succinylation (lysine + 100.01604); and acetylation of protein N-terminal as a variable modification, digested by trypsin with at least four missed cleavages, five charges, and five modifications per peptide. The mass error was set to 10 ppm for the first search, 5 ppm for the main search, and 0.02 Da for fragment ions. Identified proteins with lengths of at least seven peptides were used for further analysis. The maximum false discovery rate (FDR) thresholds for protein, peptides, and modification sites were set to 1%. In addition, the site localization probability threshold was specified as > 0.75 and the cut-off score for peptides was set as 40, based on previously described specifications (13). The raw MS files have been submitted to the iProx (Integrated Proteome resources) database under accession numbers IPX0001216000 and PXD009778 (28).
Gene Ontology Categories and Bioinformatics Analyses
Gene Ontology (GO) terms for the identified proteins were assessed by querying the UniProt-GO Annotation database (http://www.ebi.ac.uk/GOA) (29). Amino acid sequence motifs comprising at least 20 amino acids within ± 10 residues of the succinylated lysine sites were analyzed using the web-based Motif-X program (http://motif-x.med.harvard.edu) 30). All of the database protein sequences were used as the background database parameter, whereas the default values were used for the other parameters. Secondary structure analysis was performed using NetSurfP with settings as previously described (31). The PSORTb version 3.0 software package was used for protein subcellular localization prediction (http://www.psort.org/) and protein domain functional annotations were assigned using InterProScan, as implemented in the InterPro domain database (http://www.ebi.ac.uk/interpro/) (32, 33). Protein-protein interactions were predicted with high confidence (0.07) using the STRING software package version 10.1 (http://string-db.org). STRING was also used to identify significantly enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (p value < 0.05), and these correlations were visualized in Cytoscape 3.5.1 (34, 35).
Validation of Proteomic Analysis
To further validate the succinylated lysine modification results from the proteomic analyses, targeted candidate proteins were cloned and overexpressed. Then mice or rabbits were immunized with the expressed proteins to obtain specific antibody sera for co-immunoprecipitation (Co-IP) and Western blotting experiments based on previously described methods with some slight modifications (36–39). Briefly, about 1 ml bacterial cell lysate was incubated with related antibody and pre-immune serum as a negative control, and then gently vortexed at 4 °C overnight, followed by coupling to 50 μl agarose-conjugated protein G beads (Beyotime, Nantong, China) that were prepared in PBS for 4 h at 4 °C. After three washes with PBS, 50 μl of Laemmli sample buffer was added for to the proteins, which were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then electrophoretically transferred to a PVDF membrane for Western blotting. For Western blot analysis of lysine succinylation, Tris-buffered saline (pH 8.0) containing 0.1% (v/v) Tween (TBS-T) with 5% skim milk was used for blocking and TBS-T with 3% bovine serum albumin (BSA) was used to prepare anti-succinyllysine antibody (PTM Biolabs) at a 1:1,000 dilution.
Generation of luxS Knock-out Strains
The strains and plasmids used in this study are shown in supplemental Table S6. The suicide vector pRE112 was used to generate the luxS gene deletion mutant. Approximately 500 bp of sequence upstream and downstream of the luxS gene were amplified from the chromosomal DNA of A. hydrophila ATCC7966 and fused together by overlap extension PCR. The BamHI and SacI restriction sites were used for plasmid construction. Recombinant plasmids were first transformed into E. coli MC1061 (pir) cells, and then subsequently transformed into E. coli S17–1 (pir) cells to select for Cmr. Then the plasmids were transferred via conjugation to wild-type A. hydrophila (Ampr,100 μg/ml) to select for chloramphenicol resistance, Cmr (30 μg/ml) and ampicillin resistance, Ampr (100 μg/ml) single-crossover mutant colonies. Double-crossover mutants resistant to sucrose because of the presence of the sacB gene carried by the plasmid, were isolated by plating onto LB-sucrose agar containing 20% sucrose. Single- and double-crossover mutants were all confirmed by PCR analysis and amplicon sequencing using the primers shown in supplemental Table S7.
Site-directed Mutagenesis of luxS
The luxS complementary strain was constructed using the broad host vector pBBR1-MCS1 and the HindIII and BamHI restriction sites to generate the His-tagged pBBR1-luxS plasmid, as previously described (40). In addition, the corresponding promoter (located about 500 bp upstream of luxS) was also constructed and inserted upstream of luxS to ensure normal expression. Then site-directed mutagenesis of luxS (K23E, K23R, K30E, and K30R) was performed with the corresponding primers (supplemental Table S7) using the Fast Mutagenesis System Kit according to the manufacturer's protocol (Transgen Biotech Co., Beijing, China). All the resulting gene mutations were verified via DNA sequencing.
AI-2 Activity Assay
AI-2 production was measured by site-directed mutagenesis of luxS using the Vibrio harveyi BB170 bioluminescence reporter assay, as previously described (41). All the strains were cultured overnight and cells were pelleted by centrifugation at 12,000 × g for 5 min, followed by filtration of the supernatant through a 0.22-μm sterile filter (Millipore). Then cells were diluted 1:5,000 in fresh AB medium and 180 μl of culture was mixed with 20 μl supernatant in a black 96-well plate (Corning-Costar, Co., Cambridge, MA). Samples were incubated at 30 °C for 12 h and then bioluminescence was measured every hour using the SpectraMax i3 Multi-Mode Detection Platform (Molecular Devices, Toronto, Canada). The overnight cultures of V. harveyi BB170 supernatant and sterile AB medium served as the positive and negative controls, respectively. AI-2 activity (in relative light units) was compared against the bioluminescence of the positive control, and all of the assays were performed in triplicate for each group.
Coculture Experiment
Bacterial competitive ability assays were conducted, as previously described with some modifications (42). Briefly, both the overnight-cultured competitor (Vibrio alginolyticus) and participant (different site-directed mutants of luxS) strains were transferred at 10% into LB media until OD600 values reached 1.0, after which, the cultures were mixed together at equal volumes. The mixture was diluted 1:10 in fresh medium and then co-cultured for 12 h at 30 °C with shaking at 200 rpm. The abundances of V. alginolyticus and A. hydrophila could be easily counted individually on LB plates after co-cultivation because of substantially different colony morphologies. Moreover, additional verification was performed with PCR amplification of the conserved 16S rDNA sequences.
Metabolite Extraction
Metabolite extraction was performed as previously described (43, 44).Briefly, cells grown to an OD600 of 1.0 under the same conditions previously described were harvested and washed twice with PBS buffer. Roughly 50 mg (wet weight) sample from each group was extracted by liquid-liquid extraction using methanol/chloroform at a ratio of 3:1, and then homogenized in a ball mill (Jingxin, Shanghai, China) for 4 min at 45 Hz. Then the samples were treated ultrasonically for 5 min in an ice bath, and the entire procedure was repeated three times. Supernatant was obtained after centrifugation for 15 min at 12,000 rpm at 4 °C. Then, 50 μl substrate from each sample was pooled together as the quality control (QC) sample. In addition, 350 μl supernatant was transferred to 1.5 ml centrifuge tubes and dried in a vacuum concentrator. Then Incubation with 30 μl methoxy amination hydrochloride (20 mg/ml pyridine) was conducted for 30 min at 80 °C, followed by the addition of 40 μl mono(trimethylsilyl)trifluoroacetamide (BSTFA) regent (containing 1% trimethylsilyl chloride, TMCS) to each sample and incubation for 1.5 h at 70 °C. Finally, 5 μl fatty acid methyl ester (FAME) in chloroform was added to the samples when cooled to room temperature prior to analysis.
GC-MS Analysis
Gas chromatography, GC-MS analysis was performed on the Agilent 7890 Gas chromatograph system coupled to the 5975C Mass Spectrometer (J&W Scientific, Folsom, CA). A 1-μl aliquot of the analyte was injected into the GC-MS under the splitless mode. Helium was used as the carrier gas, the front inlet purge flow was set at 3 ml/min, and the gas flow rate through the column was set at 1 ml/min. The initial temperature was maintained at 60 °C for 1 min, then raised to 300 °C at a rate of 5 °C/min, followed by a hold for 15 min at 300 °C. The temperatures of the injection, transfer line, ion source and quad were 280 °C, 280 °C, 230 °C, and 150 °C, respectively. The mass spectrometry data were acquired in scan mode with an m/z range of 33–600, after a solvent delay of 6.5 min.
Data Preprocessing and Analysis
The raw peaks were extracted from the GC-MS analysis using the Chroma TOF 4.3X software (LECO Co., St. Joseph, MI) and the LECO-Fiehn Rtx5 database. The same software was also used for filtering baseline data and calibration of the baseline, peak alignment, deconvolution analysis, peak identification, and integration of peak areas (43). The mass spectra and retention index matches were considered in metabolite identification, and peaks were removed that were detected in < 50% of QC samples or RSD values > 30% in QC samples (45). Then data were normalized to the total sum of the spectrum prior to data analysis. Orthogonal partial least-squares-discriminant analysis (OPLS-DA) was conducted using SIMCA 13.0 software package to visualize the metabolic differences among experimental groups. The statistical significance of metabolite differences between treatment groups and the control group was assessed using the t test and those with a ratio > 1.5 or < 0.667 were used in subsequent analyses. Lastly, pathway analysis was conducted using the MetaboAnalyst 4.0 (http://www.metaboanalyst.ca/) software package, specifying a p < 0.05 threshold (46).
Experimental Design and Statistical Rationale
An immunoaffinity analysis was performed to enrich lysine-succinylated peptides of A. hydrophila and then submitted to identification using mass spectrometry. The fragmentation data from two biological replicates were merged and interpreted by MaxQuant and searched against the A. hydrophila ATCC7966 database. The false discovery rate (FDR) based on a decoy database search for protein, peptides, and modification sites was set to <1%. Succinylated amino acid sequence motifs were analyzed using the motif-X program (http://motif-x.med.harvard.edu) with a significance level of 1e-6 (30). Secondary structure analysis was performed using NetSurfP with p values calculated by Wilcoxon test (31). The protein subcellular localization was predicted by PSORTb version 3.0 and protein domain functional annotations were assigned using InterProScan (32, 33). Protein-protein interactions were predicted with high confidence (0.07) using the STRING version 10.1. STRING was also used to identify significantly enriched KEGG pathways with p value < 0.05, and these correlations were visualized in Cytoscape 3.5.1 (34, 35). Six biological replicates of each group are included in the metabolomics experiments. Statistical analyses were done using SIMCA version 13.0 and GraphPad Prism version 7.0. A student t test was used to calculate the significance of differences between site-directed mutagenesis and the control group. Pathway analysis was conducted by MetaboAnalyst version 4.0 with a p < 0.05 threshold (8). One-way Analysis of Variance (ANOVA) was used to calculate the significance of different AI-2 activities in three biological replicates between different groups.
RESULTS AND DISCUSSION
Identification of Lysine-succinylated Peptides and Proteins in A. hydrophila
To obtain a comprehensive view of the lysine succinylome in A. hydrophila, fresh bacterial lysine-succinylated peptides were immediately enriched after collection using a high affinity purification method and a high specificity succinyl-lysine antibody. Then PTMs within the enriched peptides were identified using a high sensitivity Exactive nano HPLC-Q. The accuracy of mass spectrometry is a critical basis for qualitative analysis of mass spectrometry data. A total of 2174 lysine-succinylated peptides from 666 proteins were identified using a highly conservative threshold (FDR < 1%) resulting in an average peptide score of 99.4 and an average mass error of 0.025 (supplemental Table S1). The near-zero distribution of mass error and that the errors were predominantly < 0.02 Da, indicated a high degree of accuracy for identification of the modified peptides (supplemental Fig. S1A). Most of the enriched lysine-succinylated peptide lengths were in the range of 7–21 segments, which accounted for 96.64% of the total identified peptides. A small number of peptides exhibited lengths of 20–35, which accounted for about 3.36% of the total peptides. These larger peptides are consistent with random cutting by trypsin at lysine and arginine residue sites (supplemental Fig. S1B). These results met the expected requirements and standards for mass spectrometry data that are necessary for constructing a high-quality succinylome of A. hydrophila. Moreover, the proportion of lysine succinylated proteins among the total proteins was about 16%, which is consistent with previous reports in other bacterial species that used the same cut-off scores for identifying PTM peptides. Thus, a considerable number of lysine-succinylated peptides were successfully identified in A. hydrophila.
Most of the succinylated proteins contained between one and four PTM sites. Specifically, 38.44% of the 666 succinylated proteins had one lysine succinylated site, whereas 20.57%, 12.61%, and 6.61% PTM proteins had two, three, and four succinylated sites, respectively (supplemental Fig. S1C). These PTM distributions are like those of V. parahemolyticus and E. coli (7). The number of modified proteins with five or more PTM sites decreased significantly, and proteins were not observed with 16,18, 20, 23, and 25–28 lysine-succinylated sites. The protein A0KQA4, which is the beta subunit of the DNA-directed RNA polymerase, had the highest number (29) of succinylated sites. In addition, numerous other proteins exhibited high abundances (>13) of succinylated sites including the pyruvate metabolism related proteins A0KMQ3 (18 Ksucc sites), A0KPV1 (21), and A0KHY7 (18); the translation process related proteins A0KQ96 (24), A0KQA5 (22), A0KNE3 (19), A0KJ93 (19), and A0KQ95 (17); stress response proteins, including A0KMI6 (17), A0KGL1 (15), and A0KL53 (14); and the major chaperone proteins A0KJU0 (Tig,15), A0KGL1 (GroL,15), and A0KMI6 (DnaK,17).
Based on mass spectrometry identification, many of the PTM proteins were enzymes, which is consistent with the hypothesis that enzyme activities may be precisely regulated by modification via lysine succinylation. For example, isocitrate dehydrogenase (IcD) of E. coli has two succinylated lysine residues (K100 and K242). Indeed, mutagenesis analysis has indicated the importance of these sites in maintaining enzymatic activity, particularly for K100 (7). We identified both PTM sites (K101 and K243) in the isocitrate dehydrogenase of A. hydrophila, in addition to five other succinylated residues in the protein, suggesting that these PTM sites may play roles in the regulation of enzymatic activity in A. hydrophila.
Many translation-related proteins, which undergo PTM inclusive of all 30S ribosome subunits (S1–S21) and most 50S ribosome subunits (except L32 and L36), were highly succinylated in A. hydrophila (47, 48). In addition, elongation factors 4, G, P, Ts, and Tu also exhibited succinylated modifications. These results suggest that PTM may be involved in bacterial protein translation.
Identification of Lysine Succinylation Motifs
To identify sequence motifs that were generally associated with lysine succinylation sites, we examined 21-amino acid sequence windows surrounding central Ksucc residues using the web-based Motif-X program and a significance threshold of p < 0.000001. Eight conserved motifs were identified among the regions surrounding succinylated peptides (Fig. 1A). The motifs were Ksucc****R, Ksucc****K, FKsucc, LKsucc, MKsucc, EKsucc, IKsucc, and QKsucc (where * indicates any amino acid residue). The EKsucc pattern has also been identified in M. tuberculosis XDR and RIMD 2210633, whereas the FKsucc and LKsucc motifs were observed in E. coli BW25113, and the QKsucc and LKsucc motifs were identified in Toxoplasma gondii. In contrast, these motifs differed considerably from those observed in V. parahemolyticus and E. coli DH10B (49–51). The preference for lysine at position +1 in E. coli BW25113, +4 in T. gondii, +5 in A. hydrophila, +6 in M.tuberculosis XDR, and at +7 and +8 in V. parahemolyticus suggests a potential structural domain upstream of succinylated lysine and unmodified lysine, although the patterns were not identical. We generated an intensity map based on the frequency of different amino acid residues that were adjacent to the succinylated lysine residues (Fig. 1B). The results indicated that succinylated sequences were nonrandomly distributed among residues, with higher frequencies in association with nonpolar hydrophobic amino acids, including F (phenylalanine, Phe) and M (methionine, Met) at the −1 site. Two polar, positively charged amino acids, R (arginine, Arg) and K (lysine, Lys), tended to appear at the −6 and +5 sites, respectively. Intriguingly, several polar and positively charged amino acids, including K, R, H (histidine, His), and the negatively charged amino acid, D (aspartate, Asp), exhibited opposite placement at the −1 site (the cut-off value for the log10 transformed Fisher's exact test p value was set at 1). Similar results have been observed in other bacterial species, although the precise positions varied. Lysine and arginine are both alkaline amino acids that share similar molecular structures, but significantly differ from other basic residues like histidine. Thus, the common, conserved motifs between lysine and arginine are reasonable. Overall, the lysine succinylation sequence motif analyses indicated a common preference for substrates among different bacterial species.
Fig. 1.

Properties of lysine-succinylated peptides in A. hydrophila ATCC7966. A, Motif analysis of succinyl-peptides in A. hydrophila. Sequence models comprising amino acids in specific positions of modified-21-mers (10 amino acids upstream and downstream of the site) of all of the protein sequences. All the database protein sequences were used as the background database parameter. Both the foreground and background histograms are shown on the left; B, Heatmap showing the frequency of different amino acid residues around succinylated lysine residues in A. hydrophila; C, Distribution of succinylated-lysines and all lysines in protein secondary structures. The mean secondary structure probabilities of modified lysine residues were compared against the mean secondary structure probabilities of the data set containing all lysine residues from all succinylated proteins identified in this study. p values were calculated using nonpaired Wilcox tests.
Secondary Structural Analysis of Lysine-succinylated Proteins
To further understand the effects of lysine succinylation on protein structure, secondary structural predictions were performed using the NetSrufP algorithm on both the succinylated subset and total lysine residues. Lysine succinylation was more likely to occur on alpha-helix structures than all lysine residues (p value = 4.25 e-10). In contrast, succinylated residues were less likely to be located on the coil (p value = 1.41e-9), and no significant difference between succinylated and all lysine residues was observed on beta-strands (p value = 0.3799) (Fig. 1C, left). These results are consistent with previous studies in other species, including eukaryotes, in which succinylated lysine residues were more frequently found in structured regions (10). Interestingly, the surface accessibility of succinylation sites was significantly lower than that of all the lysine residues (p value = 9.3e-6), indicating that succinylated lysine residues may be preferentially located on the inside of succinylated proteins. This result may also explain the co-localization of hydrophobic amino acids near the Ksucc motifs identified in this study (Fig. 1C, right). Many succinylated lysine residues are involved in the regulation of enzymatic activity, as discussed above. Thus, the Ksucc sites located inside of proteins may be involved in substrate binding and catalysis. For example, two succinyl-lysine residues (K83 and K310) are in the substrate binding pocket of the human ketogenic enzyme 3-hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2), and these residues contribute to the inhibition of HMGCS2 activity (11). Taken together, our results indicate that most lysine succinylations are located on structured regions and may affect protein activities via secondary structure transformations.
Subcellular Predictions and Functional Classifications of Lysine-succinylated Proteins
To better understand the functions of succinylated-proteins within cells, we first used PSORTb software to predict the subcellular localization of Ksucc proteins. Most of the lysine-succinylated proteins (531) were cytoplasmic, accounting for 79.73% of the total PTM proteins (Fig. 2A and supplemental Table S2). The enrichment of PTM proteins in the cytoplasm is consistent with previous studies indicating predominant PTM activities within intracellular metabolic pathways. The prevalence of Ksucc proteins in the cytoplasm indicated that many might be involved in important functions, such as in core energy metabolism, biosynthesis, translation processes, and substrate binding. Other Ksucc proteins of A. hydrophila may be related to transport and resistance to fluctuating conditions in cell envelope proteins, including 12.76%, 4.80%, 2.25%, and 0.45% of the PTM proteins that were in the periplasmic space, inner membrane space, outer membrane, and extracellular space, respectively. Several PTM enzymes, including insertase YidC, pyrroline-5-carboxylate reductase ProC, ATP-dependent zinc metalloprotease FtsH, and Pts system proteins, such as A0KI59 and NagE, are located on the inner membrane, whereas some ribosomal subunits including L11, L31, and L20, were in the periplasm. Interestingly, some outer membrane proteins were also succinylated. For example, OmpAII, an important molecule for initial infection and maintaining bacterial cell envelope structure, contains three lysine-succinylated sites (52). In addition, the agglutination protein A0KLQ6 that is homologous to TolC, is a Type I secretion and efflux outer membrane protein that plays an important role in antibiotic resistance and exhibited two lysine-succinylated sites (53). Thus, these succinylation modifications in nonenzymatic proteins may indicate involvement in multiple cellular physiological processes, including bacterial virulence and pathogenicity.
Fig. 2.

Subcellular prediction of proteins and functional classifications of succinylated-lysine proteins in A. hydrophila ATCC7966. A, Subcellular localization of the identified Ksucc proteins represented as a pie chart. B, KEGG pathway-based enrichment analysis of succinyl-proteins in A. hydrophila. The enrichment of identified proteins against all KEGG database proteins was used to identify enriched pathways via two-tailed Fisher's exact tests. Pathways with corrected p values < 0.05 were considered significantly enriched; C, GO-based enrichment analysis of identified proteins in A. hydrophila. A two-tailed Fisher's exact test was employed to test the enrichment of the identified proteins in each category against all of the database proteins. GOs with corrected p values < 0.05 were considered significantly enriched; D, Enrichment and analysis of domains related to succinylated proteins of A. hydrophila. For each category of proteins, the InterPro database was used to identify domains and a two-tailed Fisher's exact test was employed to assess the enrichment of identified proteins against all database proteins. Correction for multiple hypothesis testing was conducted using standard false discovery rate control methods. Domains with corrected p values < 0.05 were considered significantly enriched.
Previous studies have demonstrated that protein modification is widely distributed in many important central metabolic pathways, including glycolysis, pyruvate metabolism, and the TCA cycle. Thus, we explored the specific metabolic pathways involving the functional proteins observed with PTM. In addition, we assessed the enrichment of KEGG-annotated Ksucc proteins in A. hydrophila. The modified proteins were primarily involved in amphibolic microbial pathways (Fig. 2B and supplemental Table S3) that are important metabolic pathways of catabolism and anabolism. These pathways included the pentose phosphate pathway (∼ 5.18% of the total identified proteins), the TCA cycle (5.83%), 2-oxocarboxylic acid metabolism (4.53%), pyruvate metabolism (7.77%), propanoate metabolism (4.85%), and glycolysis (7.44%). In addition, other important metabolic pathways were represented, including carbon metabolism (19.42%), pyrimidine metabolism (8.74%), and methane metabolism (4.85%), among others. These results suggest that A. hydrophila might directly regulate important metabolic pathways, and other functional proteins, through post-translational modifications. These dynamic and reversible modifications on target proteins may allow the modification of protein/substrate interactions, or activity, in order to adjust bacterial growth and promote adaptation to dynamic, external environments.
Gene ontology analysis was also conducted to investigate the functional enrichment of Ksucc proteins (supplemental Table S4). In the 'biological processes' category, macromolecule biosynthetic processes, cellular macromolecule biosynthetic processes, macromolecule metabolic processes, and translation were primarily enriched (adjusted Fisher's exact test p value < 1e-100) (Fig. 2C, upper panel). Similarly, the three most enriched categories in the molecular function category were structural constituents of ribosomes, structural molecule activities, and rRNA binding (Fig. 2C, lower panel). The modified proteins were mostly prevalent in the cytoplasm, and most were involved in important functions, including core energy metabolism, biosynthesis, translation processes, and substrate binding.
To further identify protein functions associated with PTM, the domains of the identified PTM proteins were annotated with InterProScan (supplemental Table S5). Rossmann-like folds, nucleic acid-binding (OB-fold), and aminoacyl-tRNA synthetases involved in nucleotide binding were the three most enriched folds based on their p values (Fig. 2D). Previous reports have demonstrated that PTM can modify conserved protein domains to regulate biological functions. For example, the phosphorylation of Ser111 within the human TPP1oligonucleotide/oligosaccharide binding (OB) fold is important for cell cycle-dependent telomerase recruitment. In this study, aminoacyl-tRNA synthetases, including A0KQI5, A0KLW3, A0KG39, A0KN87, and A0KKF6, contained Rossmann-like folds were succinylated. These results indicate that the amino acid tRNA ligases may exhibit a common regulatory mechanism via lysine succinylation. In addition, seven ATP-dependent proteases (A0KJU3, A0KJU2, A0KQI8, A0KJC4, A0KQE5, A0KJD7, and A0KNF0) exhibited succinylation modification, suggesting that this type of PTM may also be involved in the regulation of ATPase activities.
Protein-protein Interactions of Lysine-succinylated Proteins
To further understand the roles of lysine-succinylated proteins in metabolic regulatory processes, we evaluated metabolic pathway and protein-protein interaction (PPI) networks involving the observed enrichment of lysine-succinylated substrates. At least night representative KEGG pathways that were significantly enriched as noted by PPIs are shown in Fig. 3. More than 50 ribosomal subunit proteins from the large and small 50S and 30S ribosomal subunit complexes, respectively, were highly enriched (p value = 2.3e-23), indicating the importance of succinylation modification in the regulation of protein translation. In addition, 27 pyrimidine metabolism-related proteins including RpoB, RpoC, PyrG and PyrE were succinylated, and highly enriched (p value = 5.865e-03). Of these, DNA-directed RNA polymerase subunits, including RpoA, RpoB and RpoC play critical roles in transcription, and are also targeted by antibiotics because of their essential functions in maintaining cellular viability (54). Recently, lysine methylation, acetylation, and succinylation modification of these proteins have been observed in other species, suggesting that there may be cross-talk among PTM, and consequently, that the regulatory mechanisms of this enzyme may be very complicated (55). Moreover, proteins involved in export processes, including sec-dependent proteins and tat system proteins that form complexes, exhibited complicated protein-protein interaction networks. These proteins are involved in the injection of virulence proteins into host cells, and exhibited lysine-succinylation modification in A. hydrophila. As expected, the PPI analysis indicated that lysine succinylation in A. hydrophila participates in multiple metabolic pathways including the TCA cycle, the pentose phosphate pathway, glycolysis/gluconeogenesis, and pyruvate metabolism.
Fig. 3.

Analysis of protein-protein interaction networks of lysine-succinylated proteins in A. hydrophila ATCC7966. Different colors display various clusters according to KEGG pathways enrichment.
Lysine Succinylation in A. hydrophila is Involved in Crucial Metabolic Pathways
In addition to the results described above, previous research has suggested that the lysine-succinylation of proteins, especially metabolic enzymes, may play regulatory roles in central metabolic pathways, either in eukaryotes or prokaryotes (26). Lysine succinylated enzymes of A. hydrophila were involved in the TCA cycle, glycolysis/gluconeogenesis, and pyruvate metabolism (Fig. 4), which are all major metabolic pathways of bacteria. Indeed, many of the metabolic enzymes in these pathways were succinylated in A. hydrophila. These lysine modification sites may also exhibit other types of PTM, including acetylation, pupylation, malonylation, and propionylation, and thus, these major metabolisms of bacteria are likely regulated by cross-talk of multiple lysine modifications. Moreover, the regulation of these metabolic pathways may result in numerous vital biological behaviors. For example, recent studies have demonstrated that bacteria may alter metabolic pathways to increase resistance to antibiotics (44, 56, 57). Consequently, the enrichment of lysine-succinylation in crucial metabolic enzymes suggests the presence of potentially novel antibiotic resistance mechanisms, as mediated by PTM.
Fig. 4.
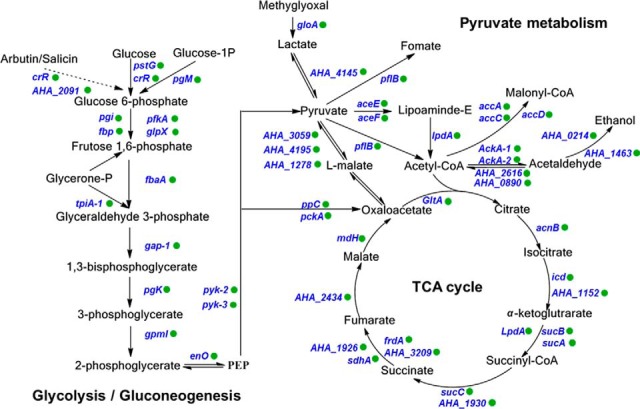
Lysine succinylation is involved in crucial metabolic pathways of A. hydrophila ATCC7966. Identified succinylated proteins are labeled with green circles.
Validation of Selected Lysine-succinylated Proteins with Co-immunoprecipitation and Western Blotting
To confirm the succinylation-enrichment results from proteomics, the lysine-succinylation of six selected proteins were validated with a combination of Co-IP and Western blotting. Specifically, we cloned and purified S-ribosylhomocysteine lyase LuxS (A0KG57), succinyl-CoA ligase subunit beta SucC (A0KJK9), phosphoglycerate kinase PkG (A0KGD3), LysM domain protein (A0KJB5), maltose-binding periplasmic protein A0KIV2, and outer membrane protein (A0KHH2), and then produced specific polyclonal antibodies for each. On the premise of ensuring the specificity of polyclonal antibodies in total protein of A. hydrophila, those candidate proteins were captured by specific antibodies and Western blotting was then performed using anti-related protein antibodies or anti-succinylation antibodies (Fig. 5). The results confirmed that all six candidate proteins exhibited succinylation modifications consistent with the proteomics data.
Fig. 5.

Validation of selected lysine-succinylated proteins in A. hydrophila using Co-Immunoprecipitation and Western blotting. Total proteins of A. hydrophila were used for Western blotting with anti-specific target antibodies as lane inputs (left panel). Candidate proteins were enriched by Co-IP with specific antibodies and followed by Western blotting with specific antibodies (middle panel). Candidate proteins were enriched by Co-IP with specific antibodies and followed by Western blotting with anti-lysine succinylation antibodies (right panel). Pre-immune serum of rabbit antibody was used as the negative controls in both experimental process (left side of each lane in middle and right panel).
Lysine Succinylations of LuxS Positively Regulate the Production of Quorum Sensing AI-2 Molecules
The PTM proteomics indicated that the AI-2 synthetase LuxS protein was modified by succinylation at the K23 and K30 sites (Fig. 6A and 6B). Homology comparisons with other bacterial species indicated that both lysine sites are substantially conserved (Fig. 6C). To better understand the effects of Ksucc on the biological function of this protein, we constructed genetic depletion mutants of luxS and its complement strain. We also used complemented 6× His tag LuxS in ΔluxS and then constructed site-specific mutagenesis mutants for the K23 and K30 sites by replacing lysine with glutamate (E) to mimic lysine succinylation and with arginine (R) to conserve the positive charge on the pBBR1MCS-1 vector, respectively. The subsequent pull down and anti-Ksucc Western blotting indicated decreasing signals of Ksucc on point mutant strains, compared with the rescue strains, suggesting that both lysine residue sites were modified by succinylation (Fig. 6D).
Fig. 6.

Lysine-succinylation of LuxS affects the quorum sensing properties of A. hydrophila. A and B, MS/MS spectral identification of succinyl-peptides on the K23 and K30 sites of A. hydrophila LuxS, respectively; C, Alignment of partial LuxS sequence (1-60 aa) homologs from ten bacterial species, including E. coli, V. alginolytious (WP_017820391.1), V. parahemeolyticus (WP_025792841.1), V. campbellii (WP_012128886.1), V. harveyi (WP_042603130.1), and Shigella flexneri. Conserved sites are shaded in gray/black and residues of interest are highlighted; D, Acetylation levels of purified LuxS and its derivatives were determined by Western blotting using anti-acetyllysine and anti-His tag antibodies, respectively. ΔluxS with the original vector was used as the negative control; E, AI-2 activity of ΔluxS complement strains and derivatives, as determined by V. harveyi BBl70 bioluminescence assays; F, Effect of site-directed mutagenesis of LuxS on bacterial competition between A. hydrophila and Vibrio alginolyticus. Statistical analysis was performed with t tests of different groups. p values < 0.0001 are indicated by *** and p values < 0.05 are indicated by **; Wild type A. hydrophila as the positive control with ΔluxS, and ΔluxS containing the original vector as the negative control; Red color indicates Vibrio alginolyticus, whereas blue indicates A. hydrophila.
To further investigate the effects of Ksucc on the function of LuxS, the capabilities of AI-2 production in the abovementioned site-directed mutagenesis experiments were measured by V. harveyi BB170 bioassays. All the mutants exhibited significantly decreased AI-2 production compared with the positive controls (rescue strain) (Fig. 6E). Moreover, the K to R mutants depressed more AI-2 signal than the K to E mutants. These results suggest that both lysine sites are important for AI-2 production. Moreover, based on comparisons against the K23R and K30R mutant, the related Ksucc modification sites on LuxS (K23E and K30E) are likely to positively regulate its enzymatic function.
The Lysine Succinylations of LuxS Positively Regulate the Competitive Ability of A. hydrophila
Quorum sensing affects communication with other bacterial species. Consequently, we tested the competitive abilities of A. hydrophila luxS mutants via co-culture with V. alginolyticus, another globally distributed aquatic pathogen. The depletion of luxS coincided with sharply decreased bacterial abundances of A. hydrophila, whereas no such decreases were observed with the rescue strain (Fig. 6F). This result indicates that LuxS plays an important role in competition of A. hydrophila with other species. However, when the Ksucc sites of LuxS were substituted by E or R, survival of A. hydrophila fluctuated. Generally, the K23E and K30E mutants exhibited increased survival, especially for K30E, which promoted the growth of V. alginolyticus, whereas K23R resulted in the decreased competitive ability of A. hydrophila. These results indicate that lysine succinylation of LuxS at the K23 and K30 sites may positively regulate A. hydrophila communication with other bacterial species.
Metabolite Profiles of LuxS and Derivatives
It has been well documented that LuxS plays pleiotropic roles in biological functions of bacteria. In addition to quorum sensing, LuxS is also involved in the activated methyl cycle (AMC), which is a major supplier of methyl donors in cells and also affects sulfur and iron metabolism (58, 59). For example, metabolomic analysis of Lactobacillus reuteri luxS mutants revealed that it affects bacterial fermentation, and the metabolism of fatty acids and amino acids (60). Consequently, we further investigated whether LuxS modified by Ksucc is involved in other metabolic pathways. GC-MS based metabolomics analyses were performed to determine the metabolite profiles of LuxS and its derivatives in A. hydrophila. A total of 271 metabolites were identified including 48, 66, 109, and 52 altered metabolites in the K23E, K23R, K30E, and K30R mutants, respectively, when compared with the complement strain, each of the metabolites exhibited fold change (FC) values higher than 1.5 and p values < 0.05 in six biological replicates of each sample (supplemental Table S8). Few altered metabolites were found in common among samples, suggesting little overlap in metabolite profiles (Fig. 7A). Moreover, principal component (PCA) analysis clearly separated LuxS and its derivatives (Fig. 7B), in which the metabolic profile of samples from the K30R mutant was clearly different from the luxS complement strain and the K30E mutant. Significant differentially expressed metabolites were clustered and displayed as a heat map (Fig. 7C). These results indicate the differential regulation of function of Ksucc modified LuxS at the K23 and K30 sites.
Fig. 7.

Metabolic profiles of LuxS and its site-directed mutant derivatives in A. hydrophila. A, Venn diagrams showing the overlap between altered metabolites among K23E, K23R, K30E, and K30R mutants when compared with the LuxS complement strain; B, OPLS-DA plot showing the clustering of samples based on 271 metabolite abundances for LuxS, and its four site-directed mutant derivative samples. The first and second model components discriminate the different groups. The OPLS-DA used one predictive component and four orthogonal components to yield a R2X of 0.519, R2Y of 0.900, and Q2 of 0.706; C, A heat map was used to visualize altered metabolites among luxS mutants when compared with the LuxS complement strain (CK). Blue and red indicate decreased and increased metabolites ratio (log2scale), respectively; D, KEGG pathway enrichment of metabolites among site-directed mutant derivatives of LuxS.
Then the impact of enriched KEGG pathways was further investigated in these four mutants. The succinylation mimic of the lysine 23 site of LuxS (K23E) affected lysine and tyrosine metabolism, whereas the de-succinylated mimic K23 (K23R) was involved in alanine metabolism, aspartate and glutamate metabolism, and unsaturated fatty acid biosynthesis (Fig. 7D). The K30E mutant was enriched in the most KEGG pathways including nitrogen metabolism and the metabolism of various amino acids. Except for sulfur metabolism, the K30R mutant was involved in similar amino acid metabolism as the other mutants. Together with the observation that LuxS affects amino acid, fatty acid, and sulfur metabolisms, our metabolomics data indicate that LuxS may subtly control these central metabolisms via reversible regulation of lysine succinylation status at the K23 and K30 sites.
The Lysine Succinylations of LuxS Affect the Amino Acid and Activated Methyl Cycle Metabolism in A. hydrophila
Based on our metabolomics results, site-directed mutant derivatives of LuxS significantly affected intracellular central metabolism, especially for amino acids and their associated metabolism (Fig. 8). The K23 mutants increased levels of amino acids, including lysine, homoserine, glutamine, tyrosine, phenylalanine, tryptophan, and histidine. In contrast, the K30 mutants decreased their levels, suggesting site-specific effects of K23 and K30 residues of LuxS on bacterial metabolism. Moreover, all of the LuxS derivatives decreased the production of AI-2, which resulted in the accumulation of the toxic metabolite S-adenosylhomocysteine (SAH) through the activated methyl cycle that can be harmful to bacterial growth. Although we failed to detect S-adenosylmethionine (SAM), SAH, and S-ribosylhomocysteine (SRH) metabolites, the concentrations of adenosine and adenine did not change in the mutants. The decrease in methionine in the K30E mutant and its unaltered levels in the K23E, K23R, and K30R mutants suggested that K30E suppresseed the AMC and negatively regulated the methionine metabolite. Concomitantly, the upregulated levels of homocysteine also indicated that metabolic flow is shunted to other amino acid metabolic pathways after suppression of AMC or AI-2 production suppression. This may explain why toxic SAH may not accumulate in cells. Further, we also observed that the metabolism of several amino acids, including serine, arginine, asparagine, isoleucine, alanine, and valine, was not affected by LuxS site-directed mutants. Thus, the metabolite profiles of LuxS and its site-directed mutant derivatives indicate a complicated regulatory network that is mediated by modification via lysine succinylation.
Fig. 8.
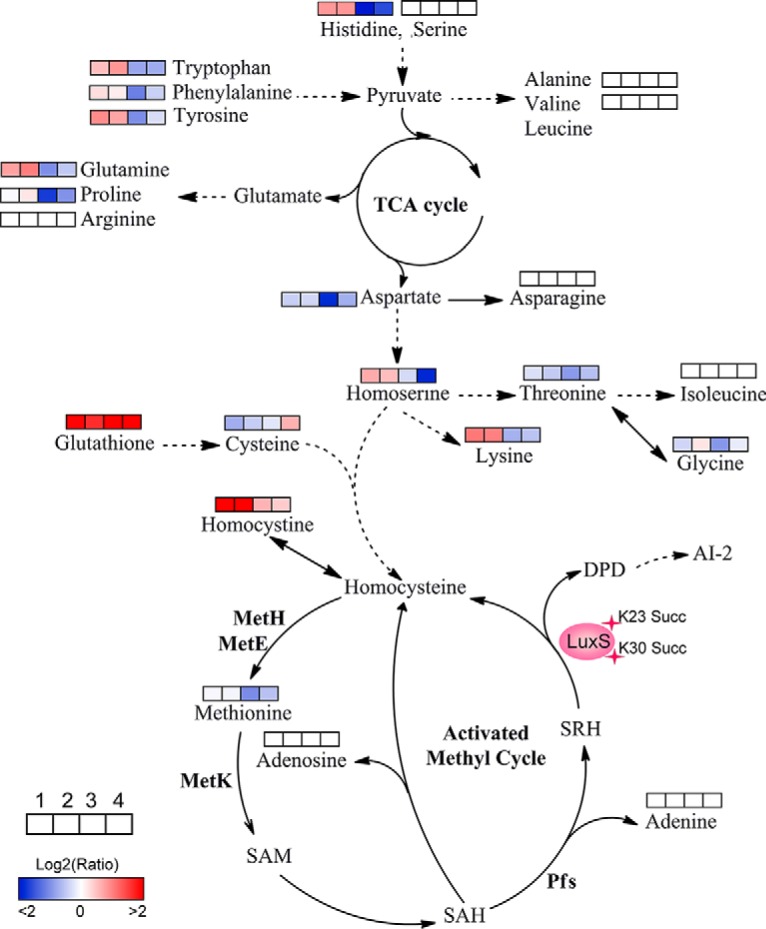
Lysine-succinylation of LuxS affects AMC and amino acid metabolism in A. hydrophila. The AMC- and amino acid-associated metabolic networks involving different metabolites were obtained from comparisons of site-directed mutant derivatives of LuxS to LuxS. Colored boxes labeled 1–4 represent the log2 scale of metabolite ratios of K23E, K23R, K30E, and K30R against the LuxS control, respectively. White colored boxes indicate the lack of a statistically significant change for that metabolite.
CONCLUSIONS
A. hydrophila is a well-known pathogen of fish farms that causes significant economic losses with outbreaks. Moreover, this pathogen can infect animals, including humans, which has led to increasing antibiotic resistance in aquaculture and hospitals. However, the intrinsic biological behaviors of A. hydrophila are largely unknown. A growing body of literature suggests that succinylation of lysine within proteins is involved in the regulation of bacterial physiology and plays a crucial role in multiple biological processes. Here, we investigated the lysine succinylation profile of A. hydrophila ATCC7966 using affinity antibody purification combined with LC MS/MS to better understand the regulatory roles of lysine succinylation in this fish and mammal pathogen. A total of 666 lysine succinylation proteins were identified and the succinylation patterns shared some commonalities with other bacterial species that have been investigated. Further investigation indicated that lysine succinylation modifications on S-ribosylhomocysteine lyase were shown to affect bacterial quorum sensing behavior, as mediated by AI-2, which may influence bacterial community interactions. Moreover, succinylation of lysine was also involved in the regulation of several central metabolic pathways, including AMC and amino acid metabolism. Furthermore, in addition to the Ksucc modification of LuxS, five Ksucc modification sites were identified on S-adenosylmethionine synthase, MetK, which is also involved in AMC through the conversion of methionine to SAM. Lysine acetylation of MetK has also been observed to regulate enzymatic activity in E. coli. The post-translational modifications of LuxS and MetK suggest the presence of a complicated intracellular regulatory network for AMC metabolism, and the cross-talk between both common PTMs requires further investigation. In conclusion, a more complete understanding of the physiological functions of these PTMs in A. hydrophila proteins will aid in disease prevention and the treatment of this important pathogen.
Ethics Statement
This study was approved by Fujian Agriculture and Forestry University Animal Care and Use Committee (Certification Number: CNFJAC0027).
DATA AVAILABILITY
The raw MS files have been submitted to the iProx (Integrated Proteome resources) database under the accessions IPX0001216000 and PXD009778 (http://www.iprox.org/page/PDV014.html?projectId=IPX0001216000).
Supplementary Material
Acknowledgments
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Footnotes
* This work was sponsored by grants from NSFC projects (Nos. 31670129 and 31470238), Program for Innovative Research Team in Fujian Agricultural and Forestry University (No. 712018009), and the Fujian-Taiwan Joint Innovative Center for Germplasm Resources and Cultivation of Crop (FJ 2011 Program, No. 2015-75, China).
 This article contains supplemental Figures and Tables.
This article contains supplemental Figures and Tables.
1 The abbreviations used are:
- PTM
- post-translational modification
- TCA
- trichloroacetic acid
- TFA
- trifluoroacetic acid
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- GC-MS
- gas chromatography mass spectrometry
- KEGG
- Kyoto Encyclopedia of Genes and Genomes
- GO
- Gene Ontology
- PPI
- protein protein interaction
- Co-IP
- Co-Immunoprecipitation
- AI-2
- autoinducer-2
- AMC
- activated methyl cycle
- MAS
- motile Aeromonas septicemia
- LB
- Luria-Bertani
- PBS
- phosphate-buffered saline
- TBS
- Tris-buffered saline
- UPLC
- ultra-performance liquid chromatography
- FDR
- false discovery rate
- BSA
- bovine serum albumin
- Cmr
- chloramphenicol resistance
- Ampr
- ampicillin resistance
- AB
- Agrobacterium
- OPLS-DA
- Orthogonal partial least-squares-discriminant analysis
- QC
- quality control
- OB
- oligosaccharide binding
- SAH
- S-adenosylhomocysteine
- SAM
- S-adenosylmethionine
- SRH
- S-ribosylhomocysteine
- ACN
- acetonitrile
- FA
- formic acid
- OD600
- the optical density at 600 nm.
REFERENCES
- 1. Cain J. A., Solis N., and Cordwell S. J. (2014) Beyond gene expression: the impact of protein post-translational modifications in bacteria. J. Proteomics. 97, 265–286 [DOI] [PubMed] [Google Scholar]
- 2. Azevedo C., and Saiardi A. (2016) Why always lysine? the ongoing tale of one of the most modified amino acids. Adv. Biol. Regul. 60, 144–150 [DOI] [PubMed] [Google Scholar]
- 3. Liu Y. T., Pan Y., Lai F., Yin X. F., Ge R., He Q. Y., and Sun X. (2018) Comprehensive analysis of the lysine acetylome and its potential regulatory roles in the virulence of Streptococcus pneumoniae. J. Proteomics. 176, 46–55 [DOI] [PubMed] [Google Scholar]
- 4. Fascellaro G., Petrera A., Lai Z. W., Nanni P., Grossmann J., Burger S., Biniossek M. L., Gomez-Auli A., Schilling O., and Imkamp F. (2016) Comprehensive proteomic analysis of nitrogen-starved Mycobacterium smegmatis deltapup reveals the impact of pupylation on nitrogen stress response. J. Proteome Res. 15, 2812–2825 [DOI] [PubMed] [Google Scholar]
- 5. Fan B., Li Y. L., Li L., Peng X. J., Bu C., Wu X. Q., and Borriss R. (2017) Malonylome analysis of rhizobacterium Bacillus amyloliquefaciens FZB42 reveals involvement of lysine malonylation in polyketide synthesis and plant-bacteria interactions. J. Proteomics. 154, 1–12 [DOI] [PubMed] [Google Scholar]
- 6. Okanishi H., Kim K., Masui R., and Kuramitsu S. (2014) Lysine propionylation is a prevalent post-translational modification in Thermus thermophilus. Mol. Cell. Proteomics 13, 2382–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang Z. H., Tan M. J., Xie Z. Y., Dai L. Z., Chen Y., and Zhao Y. M. (2011) Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 7, 58–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xie Z. Y., Dai J. B., Dai L. Z., Tan M. J., Cheng Z. Y., Wu Y. M., Boeke J. D., and Zhao Y. (2012) Lysine succinylation and lysine malonylation in histones. Mol. Cell. Proteomics 11, 100–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Park J., Chen Y., Tishkoff D. X., Peng C., Tan M., Dai L., Xie Z., Zhang Y., Zwaans B. M., Skinner M. E., Lombard D. B., and Zhao Y. (2013) SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Molecular Cell 50, 919–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weinert B. T., Scholz C., Wagner S. A., Iesmantavicius V., Su D., Daniel J. A., and Choudhary C. (2013) Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Reports 4, 842–851 [DOI] [PubMed] [Google Scholar]
- 11. Rardin M. J., He W., Nishida Y., Newman J. C., Carrico C., Danielson S. R., Guo A., Gut P., Sahu A. K., Li B., Uppala R., Fitch M., Riiff T., Zhu L., Zhou J., Mulhern D., Stevens R. D., Ilkayeva O. R., Newgard C. B., Jacobson M. P., Hellerstein M., Goetzman E. S., Gibson B. W., and Verdin E. (2013) SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metabolism 18, 920–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xie L. X., Li J., Deng W. Y., Yu Z. X., Fang W. J., Chen M., Liao W. Q., Xie J. P., and Pan W. H. (2017) Proteomic analysis of lysine succinylation of the human pathogen Histoplasma capsulatum. J. Proteomics 154, 109–117 [DOI] [PubMed] [Google Scholar]
- 13. Pan J. Y., Chen R. C., Li C., Li W. Y., and Ye Z. C. (2015) Global analysis of protein lysine succinylation profiles and their overlap with lysine acetylation in the marine bacterium Vibrio parahemolyticus. J. Proteome Res. 14, 4309–4318 [DOI] [PubMed] [Google Scholar]
- 14. Seshadri R., Joseph S. W., Chopra A. K., Sha J., Shaw J., Graf J., Haft D., Wu M., Ren Q., Rosovitz M. J., Madupu R., Tallon L., Kim M., Jin S., Vuong H., Stine O. C., Ali A., Horneman A. J., and Heidelberg J. F. (2006) Genome sequence of Aeromonas hydrophila ATCC 7966T: jack of all trades. J. Bacteriol. 188, 8272–8282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hossain M. J., Sun D., McGarey D. J., Wrenn S., Alexander L. M., Martino M. E., Xing Y., Terhune J. S., and Liles M. R. (2014) An Asian origin of virulent Aeromonas hydrophila responsible for disease epidemics in United States-farmed catfish. MBio 5, e00848–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Janda J. M., and Abbott S. L. (2010) The genus Aeromonas: taxonomy, pathogenicity, and infection. Clin. Microbiol. Rev. 23, 35–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Keersmaecker S. C., Sonck K., and Vanderleyden J. (2006) Let LuxS speak up in AI-2 signaling. Trends Microbiol. 14, 114–119 [DOI] [PubMed] [Google Scholar]
- 18. Meighen E. A. (1993) Bacterial bioluminescence: organization, regulation, and application of the lux genes. FASEB J. 7, 1016–1022 [DOI] [PubMed] [Google Scholar]
- 19. Coulthurst S. J., Kurz C. L., and Salmond G. P. (2004) luxS mutants of Serratia defective in autoinducer-2-dependent 'quorum sensing' show strain-dependent impacts on virulence and production of carbapenem and prodigiosin. Microbiology 150, 1901–1910 [DOI] [PubMed] [Google Scholar]
- 20. Xue T., Zhao L. P., and Sun B. L. (2013) LuxS/AI-2 system is involved in antibiotic susceptibility and autolysis in Staphylococcus aureus NCTC 8325. Int. J. Antimicrob. Agents 41, 85–89 [DOI] [PubMed] [Google Scholar]
- 21. Doherty N., Holden M. T., Qazi S. N., Williams P., and Winzer K. (2006) Functional analysis of luxS in Staphylococcus aureus reveals a role in metabolism but not quorum sensing. J. Bacteriol. 188, 2885–2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou M. L., Xie L. X., Yang Z. Z., Zhou J. H., and Xie J. P. (2017) Lysine succinylation of Mycobacterium tuberculosis isocitrate lyase (ICL) fine-tunes the microbial resistance to antibiotics. J. Biomol. Struct. Dyn. 35, 1030–1041 [DOI] [PubMed] [Google Scholar]
- 23. Atila M., Katselis G., Chumala P., and Luo Y. (2016) Characterization of N-succinylation of L-lysylphosphatidylglycerol in Bacillus subtilis using tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 27, 1606–1613 [DOI] [PubMed] [Google Scholar]
- 24. Okanishi H., Kim K., Fukui K., Yano T., Kuramitsu S., and Masui R. (2017) Proteome-wide identification of lysine succinylation in thermophilic and mesophilic bacteria. Biochim. Biophys. Acta. 1865, 232–242 [DOI] [PubMed] [Google Scholar]
- 25. Liao G. J., Xie L. X., Li X., Cheng Z. Y., and Xie J. P. (2014) Unexpected extensive lysine acetylation in the trump-card antibiotic producer Streptomyces roseosporus revealed by proteome-wide profiling. J. Proteomics. 106, 260–269 [DOI] [PubMed] [Google Scholar]
- 26. Song Y. X., Wang J., Cheng Z. Y., Gao P., Sun J. X., Chen X. W., Chen C., Wang Y. L., and Wang Z. N. (2017) Quantitative global proteome and lysine succinylome analyses provide insights into metabolic regulation and lymph node metastasis in gastric cancer. Sci. Rep. 7, 42053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ren S. L., Yang M. K., Yue Y. W., Ge F., Li Y., Guo X. X., Zhang J., Zhang F., Nie X., Y, Wang S. H. (2018) Lysine succinylation contributes to aflatoxin production and pathogenicity in Aspergillus flavus. Mol. Cell. Proteomics 17, 457–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Deutsch E. W., Csordas A., Sun Z., Jarnuczak A., Perez-Riverol Y., Ternent T., Campbell D. S., Bernal-Llinares M., Okuda S., Kawano S., Moritz R. L., Carver J. J., Wang M., Ishihama Y., Bandeira N., Hermjakob H., and Vizcaino J. A. (2017) The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 45, D1100–D1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shen C. J., Xue J., Sun T., Guo H., Zhang L., Meng Y. J., and Wang H. Z. (2016), Succinyl-proteome profiling of a high taxol containing hybrid Taxus species (Taxus x media) revealed involvement of succinylation in multiple metabolic pathways. Sci. Rep. 6, 21764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. S Schwartz D., and Gygi S. P. (2005) An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol. 23, 1391–1398 [DOI] [PubMed] [Google Scholar]
- 31. Meng X. X., Xing S. H., Perez L. M., Peng X. J., Zhao Q. Y., Redona E. D., Wang C., and Peng Z. H. (2017) Proteome-wide znalysis of lysine 2-hydroxyisobutyrylation in developing rice (Oryza sativa) seeds. Sci. Rep. 7, 17486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu N. Y., Wagner J. R., Laird M. R., Melli G., Rey S., Lo R., Dao P., Sahinalp S. C., Ester M., Foster L. J., and Brinkman F. S. (2010) PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jones P., Binns D., Chang H. Y., Fraser M., Li W., McAnulla C., McWilliam H., Maslen J., Mitchell A., Nuka G., Pesseat S., Quinn A. F., Sangrador-Vegas A., Scheremetjew M., Yong S. Y., Lopez R., and Hunter S. (2014) InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., and Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K. P., Kuhn M., Bork P., Jensen L. J., and von Mering C. (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lin X. M., Yang M. J., Li H., Wang C., and Peng X. X. (2014) Decreased expression of LamB and Odp1 complex is crucial for antibiotic resistance in Escherichia coli. J. Proteomics 98, 244–253 [DOI] [PubMed] [Google Scholar]
- 37. Zhang Y. L., Peng B., Li H., Yan F., Wu H. K., Zhao X. L., Lin X. M., Min S. Y., Gao Y. Y., Wang S. Y., Li Y. Y., and Peng X. X. (2017) C-terminal domain of hemocyanin, a major antimicrobial protein from litopenaeus vannamei: structural homology with immunoglobulins and molecular diversity. Front. Immunol. 8, 611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yamanaka Y., Faghihi M. A., Magistri M., Alvarez-Garcia O., Lotz M., and Wahlestedt C. (2015) Antisense RNA controls LRP1 sense transcript expression through interaction with a chromatin-associated protein, HMGB2. Cell Reports 11, 967–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nguyen D. T. T., Richter D., Michel G., Mitschka S., Kolanus W., Cuevas E., and Wulczyn F. G. (2017) The ubiquitin ligase LIN41/TRIM71 targets p53 to antagonize cell death and differentiation pathways during stem cell differentiation. Cell Death Differentiation 24, 1063–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hu F. Q., Cao Y., Xiao F., Zhang J., and Li H. (2007) Site-directed mutagenesis of Aeromonas hydrophila enoyl coenzyme a hydratase enhancing 3-hydroxyhexanoate fractions of poly(3-hydroxybutyrate-co-3-hydroxyhexanoate). Curr. Microbiol. 55, 20–24 [DOI] [PubMed] [Google Scholar]
- 41. Muras A., Mayer C., Romero M., Camino T., Ferrer M. D., Mira A., and Otero A. (2018) Inhibition of Steptococcus mutans biofilm formation by extracts of Tenacibaculum sp. 20J, a bacterium with wide-spectrum quorum quenching activity. J. Oral Microbil. 10, 1429788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Smalley N. E., An D., Parsek M. R., Chandler J. R., and Dandekar A. A. (2015) Quorum sensing protects Pseudomonas aeruginosa against cheating by other species in a laboratory coculture model. J. Bacteriol. 197, 3154–3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kind T., Wohlgemuth G., Lee D. Y., Lu Y., Palazoglu M., Shahbaz S., and Fiehn O. (2009) FiehnLib: mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal. Chem. 81, 10038–10048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Su Y. B., Peng B., Li H., Cheng Z. X., Zhang T. T., Zhu J. X., Li D., Li M. Y., Ye J. Z., Du C. C., Zhang S., Zhao X. L., Yang M. J., and Peng X. X. (2018) Pyruvate cycle increases aminoglycoside efficacy and provides respiratory energy in bacteria. Proc. Natl. Acad. Sci. U.S.A. 115, E1578–E1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dunn W. B., Broadhurst D., Begley P., Zelena E., Francis-McIntyre S., Anderson N., Brown M., Knowles J. D., Halsall A., Haselden J. N., Nicholls A. W., Wilson I. D., Kell D. B., Goodacre R., and Human Serum Metabolome, C. (2011) Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 6, 1060–1083 [DOI] [PubMed] [Google Scholar]
- 46. Chong J., Soufan O., Li C., Caraus I., Li S., Bourque G., Wishart D. S., and Xia J. (2018) MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Strader M. B., Hervey W. J. t., Costantino N., Fujigaki S., Chen C. Y., Akal-Strader A., Ihunnah C. A., Makusky A. J., Court D. L., Markey S. P., and Kowalak J. A. (2013) A coordinated proteomic approach for identifying proteins that interact with the E. coli ribosomal protein S12. J. Proteome Res. 12, 1289–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Arnold R. J., and Reilly J. P. (1999) Observation of Escherichia coli ribosomal proteins and their posttranslational modifications by mass spectrometry. Anal. Biochem. 269, 105–112 [DOI] [PubMed] [Google Scholar]
- 49. Xie L. X., Liu W., Li Q. M., Chen S. D., Xu M. M., Huang Q. Q., Zeng J., Zhou M. L., and Xie J. P. (2015) First succinyl-proteome profiling of extensively drug-resistant Mycobacterium tuberculosis revealed involvement of succinylation in cellular physiology. J. Proteome Res 14, 107–119 [DOI] [PubMed] [Google Scholar]
- 50. Yang M. K., Wang Y., Chen Y., Cheng Z. Y., Gu J., Deng J. Y., Bi L. J., Chen C. B., Mo R., Wang X. D., and Ge F. (2015) Succinylome analysis reveals the involvement of lysine succinylation in metabolism in pathogenic Mycobacterium tuberculosis. Mol. Cell. Proteomics 14, 796–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li X. L., Hu X., Wan Y. J., Xie G. Z., Li X. Z., Chen D., Cheng Z. Y., Yi X. L., Liang S. H., and Tan F. (2014) Systematic identification of the lysine succinylation in the protozoan parasite Toxoplasma gondii. J. Proteome Res. 13, 6087–6095 [DOI] [PubMed] [Google Scholar]
- 52. Mohan Nair M. K., and Venkitanarayanan K. (2007) Role of bacterial OmpA and host cytoskeleton in the invasion of human intestinal epithelial cells by Enterobacter sakazakii. Pediatr. Res. 62, 664–669 [DOI] [PubMed] [Google Scholar]
- 53. Kessler E., Whalen J., Bradford H., Chavali V., Engle D., Jeng J., Meier T., Merchant G., Narayanan S., Song A., Sundar A., and Lieshout E. V. (2017) TolC: an important component of bacterial efflux pumps and potential target for ameliorating antibiotic resistance. FASEB J. 31, lb266–lb266 [Google Scholar]
- 54. Campbell E. A., Korzheva N., Mustaev A., Murakami K., Nair S., Goldfarb A., and Darst S. A. (2001) Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell 104, 901–912 [DOI] [PubMed] [Google Scholar]
- 55. Azkargorta M., Wojtas M. N., Abrescia N. G., and Elortza F. (2014) Lysine methylation mapping of crenarchaeal DNA-directed RNA polymerases by collision-induced and electron-transfer dissociation mass spectrometry. J. Proteome Res. 13, 2637–2648 [DOI] [PubMed] [Google Scholar]
- 56. Chen X. H., Zhang B. W., Li H., and Peng X. X. (2015) Myo-inositol improves the host's ability to eliminate balofloxacin-resistant Escherichia coli. Sci. Rep. 5, 10720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Peng B., Su Y. B., Li H., Han Y., Guo C., Tian Y. M., and Peng X. X. (2015) Exogenous alanine and/or glucose plus kanamycin kills antibiotic-resistant bacteria. Cell Metab. 21, 249–262 [DOI] [PubMed] [Google Scholar]
- 58. Learman D. R., Yi H., Brown S. D., Martin S. L., Geesey G. G., Stevens A. M., and Hochella M. F. Jr. (2009) Involvement of Shewanella oneidensis MR-1 LuxS in biofilm development and sulfur metabolism. Appl. Environ. Microbiol. 75, 1301–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li L., Xu Z. F., Zhou Y., Li T. T., Sun L. L., Chen H., and Zhou R. C. (2011) Analysis on Actinobacillus pleuropneumoniae LuxS regulated genes reveals pleiotropic roles of LuxS/AI-2 on biofilm formation, adhesion ability and iron metabolism. Microb. Pathog. 50, 293–302 [DOI] [PubMed] [Google Scholar]
- 60. Wilson C. M., Aggio R. B., O'Toole P. W., Villas-Boas S., and Tannock G. W. (2012) Transcriptional and metabolomic consequences of LuxS inactivation reveal a metabolic rather than quorum-sensing role for LuxS in Lactobacillus reuteri 100–23. J. Bacteriol. 194, 1743–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw MS files have been submitted to the iProx (Integrated Proteome resources) database under the accessions IPX0001216000 and PXD009778 (http://www.iprox.org/page/PDV014.html?projectId=IPX0001216000).