

PTMiner post-processes the coarse and error-prone results of an open search of MS/MS spectra. It confidently filters and localizes the modifications (mass shifts) using the transfer FDR and an empirical Bayesian method. Evaluated on simulated and synthetic peptide data, PTMiner showed much higher accuracy than two open search engines and the Ascore algorithm. PTMiner was used to comprehensively characterize the PTMs in a draft map of human proteome, resulting in over 1.7 million modifications confidently identified and localized.
Keywords: Post-translational Modifications, Tandem Mass Spectrometry, Quality Control and Metrics, Statistics, Algorithms, Bioinformatics Software, False Discovery Rate, False Localization Rate, Human Proteome Map, Modification Site Localization, Open Search
Graphical Abstract
Highlights
PTMiner software for intelligent post-processing of open-search results.
Unrestrictive modification site localization based on a Bayesian model.
Extended transfer FDR estimation for accurate grouped FDR estimation.
Comprehensive PTM characterization in a draft map of human proteome.
Abstract
The open (mass tolerant) search of tandem mass spectra of peptides shows great potential in the comprehensive detection of post-translational modifications (PTMs) in shotgun proteomics. However, this search strategy has not been widely used by the community, and one bottleneck of it is the lack of appropriate algorithms for automated and reliable post-processing of the coarse and error-prone search results. Here we present PTMiner, a software tool for confident filtering and localization of modifications (mass shifts) detected in an open search. After mass-shift-grouped false discovery rate (FDR) control of peptide-spectrum matches (PSMs), PTMiner uses an empirical Bayesian method to localize modifications through iterative learning of the prior probabilities of each type of modification occurring on different amino acids. The performance of PTMiner was evaluated on three data sets, including simulated data, chemically synthesized peptide library data and modified-peptide spiked-in proteome data. The results showed that PTMiner can effectively control the PSM FDR and accurately localize the modification sites. At 1% real false localization rate (FLR), PTMiner localized 93%, 84 and 83% of the modification sites in the three data sets, respectively, far higher than two open search engines we used and an extended version of the Ascore localization algorithm. We then used PTMiner to analyze a draft map of human proteome containing 25 million spectra from 30 tissues, and confidently identified over 1.7 million modified PSMs at 1% FDR and 1% FLR, which provided a system-wide view of both known and unknown PTMs in the human proteome.
In the common practice of shotgun proteomics, tandem mass spectrometry (MS/MS)1 data are used to identify peptide sequences and the post-translational modifications (PTMs) via sequence-matching using a certain algorithm. The most commonly used peptide identification approach is restrictive protein sequence database search, in which a tight tolerance for peptide precursor masses is used and a few modification types are considered (1). However, this approach fails to detect unspecified or unknown types of modifications which have been shown to widely exist in the proteome data (2–5). In order to detect both known and unknown types of modifications, various unrestrictive approaches for peptide identification have been proposed in recent years (6), including the open (mass-tolerant) database search (7–20), the de novo sequencing (21–23), the spectral clustering (24–26), the curated modification search (27, 28), and the comprehensive variable modification search with speeding-up technologies such as refinement search or ion indexing (29–32). Among them, the most typical way is the open database search strategy, in which a very large peptide precursor mass tolerance, e.g. 500 Da, is allowed and the precursor mass shifts between the experimental spectra and the theoretical spectra of candidate peptides in database are considered as potential unanticipated modifications.
However, the open search strategy has not been widely adopted in current proteomic community, because of two major bottlenecks. The first one is the dramatically reduced search speed because of the greatly expanded search space. This issue was addressed recently by the MSFragger software which uses a fragment-ion indexing technique to speed up the database search (20). Another bottleneck that leaves to be overcome is the lack of appropriate algorithms for automated post-processing of the error-prone search results, such as misidentified peptide sequences or misplaced modification sites. Firstly, the common target-decoy approach (33) to estimating the FDR of peptide-spectrum-matches (PSMs) can be problematic if directly used for quality control of modification identifications. This is mainly because of the different abundances of modified peptides in the spectra and in the search space of candidate peptides, as has been demonstrated in the restrictive search strategy (34–36). In an open search, the abundances of peptides with different mass shifts in the search space are similar but their abundances in the spectra can be dramatically different, resulting in the heterogeneity of FDRs for different mass shifts at the same score level. Kong et al. (20) tried to solve this problem by extending the mass model in the PeptideProphet algorithm, in which mass shifts are first discretized into bins of 1 Da in size, and then their distribution is modeled to estimate the likelihoods of observing a correct versus incorrect identification among all PSMs belonging to a bin. However, in the open search results there are typically many modifications detected in a very small number of spectra, and accurate FDR estimation for them is very challenging and remains unresolved. An effective solution applicable to various search engines is needed.
More importantly, even though the FDRs of PSMs can be properly controlled, determining which residue site on a peptide bears the mass shift is a more challenging task (37, 38). Some open search engines simply localize the mass shift to the site yielding the highest PSM score, e.g. PTMap (13) and MODa(16), whereas others do not localize the mass shifts at all, e.g. SEQUEST (18) and MSFragger (20). For example, when scoring candidate peptides, MSFragger does not look for modified fragments, which is a major reason for its fast search speed but also leaves modifications un-localized. Therefore, dedicated mass-shift localization algorithms are in great need to improve the reliability and interpretability of the open search results. Existing modification localization algorithms were mostly designed for specific types of modifications, e.g. phosphorylation, which are identified by the traditional restrictive search (39–53), but there were few algorithms available for open search results.
Modification localization for open search results is much more challenging than restrictive search results (37, 38). In an open search, every residue site on the peptide can possibly be responsible for the mass shift, and there is only one discriminative b- or y-ion between two adjacent sites. Because of the common absence of some fragment ions and the existence of noise peaks in a spectrum, the determination of modification site could be very difficult and error-prone in many instances. In fact, the intensity information of fragment ion peaks is very useful to distinguish between real and random peak matches but is rarely used by existing localization algorithms (50, 51). For example, an extended version of Ascore, which allows all amino acids to be modified by any mass, is based on a simple probabilistic model of the number of matched peaks and does not take full advantage of the peak intensity information (18). On the chemical basis of protein modification, the knowledge on the modification specificity is another important source of information to discriminate ambiguous sites (37). This prior knowledge is often absent in practice but can be potentially learned from the data of large scale. For example the open search algorithms MS-Alignment (7) and MODa (16) use a “strength in numbers” strategy to move some modifications from rarely localized amino acids to richly localized ones. Similarly, another algorithm PTMClust (54) corrects the modification locations by formally considers the empirical probability of each amino acid to be modified by each type of modification. However, PTMClust depends on existed localization results of search engines and does not make use of spectrum information. The PTMFinder algorithm (55) uses a peptide-level method to re-localize modifications by grouping partly overlapped modified peptide sequences to assess the accuracy of modification sites. Hence, it is limited to modified peptides that occur multiple times in the same data set. In summary, a modification localization algorithm, which is dedicatedly designed for open search results and built on a flexible mathematical model to make full use of multiple types of information, is still lacking.
In this article, we developed a software tool named PTMiner for intelligent post-processing of open search results to produce reliable, localized and annotated PTM identifications. PTMiner mainly includes an extended version of the transfer FDR approach (34) for accurate grouped FDR estimation, and an unrestrictive modification localization algorithm that uses a Bayesian model to provide a site-specific probability score to measure the localization certainty. Validation experiments on simulated and real spectra demonstrated that PTMiner can effectively control the FDRs of different types of modifications and reliably localize their sites, much superior in performance than the two used open search engines (pFind (56–58) and MODa (16)) and the popular localization algorithm Ascore (18). We applied PTMiner to the draft map of human proteome containing 25 million spectra from 30 human tissues (59). More than 3 million modified PSMs were identified at 1% FDR, and the mass shifts of over 1.7 million PSMs were localized at 1% FLR, providing a system-wide view of comprehensive modifications in the human proteome.
EXPERIMENTAL PROCEDURES
PTMiner Algorithm Overview
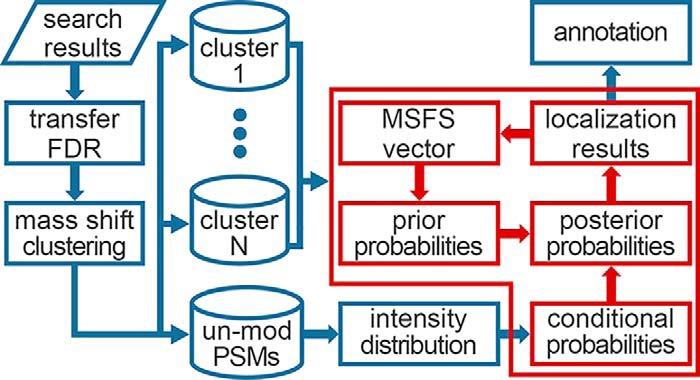
As shown in Fig. 1, given the PSMs from an open search, PTMiner first performs PSM filtering and FDR estimation. A modified version of transfer FDR (34) is used for grouped FDR estimation. Then, for the filtered PSMs, PTMiner conducts clustering of precursor mass shifts, and regards each cluster as one type of modification. Next, the mass shifts in each cluster are localized by estimating the posterior probability of each site on the peptide being the modification location. The posterior probability is computed from two parts, i.e. a prior probability and a conditional probability. The former is derived from the MSFS vector (defined in the Mass Shift Localization section), which measures the probabilities that the modification occurs on different amino acids and is learned iteratively from the data. The latter is computed from an intensity distribution model that is fitted from the matched peaks of unmodified PSMs. Finally, based on the localization results, PTMiner annotates the modification types of mass shifts according to the Unimod modification database (60). Note that PTMiner does not use any information of the Unimod database when localizing mass shifts. The details of each step are given below.
Fig. 1.

Algorithm workflow of PTMiner. The processes in the red block is the localization algorithm, and the red arrows in it represent iterative updating of prior probabilities.
PSM FDR Estimation
The FDRs of unmodified and differently modified peptides are likely to be different at the same score threshold (supplementary Note S1, supplemental Fig. S1). We call the FDR estimated for all PSMs the global FDR, and the FDR estimated separately for a group of PSMs the group FDR. The basic idea of transfer FDR used by PTMiner is to calculate the group FDR from the global FDR that is easy to estimate in general. Here, we group mass shifts into bins that are 1-Da in size and are centered at integer values, e.g. [15.5, 16.5] Da, and follow the same framework as in our previous work (34) to build up the quantitative relationship between group and global FDRs as follows,
| (Eq. 1) |
where the symbol i denotes the i-th bin of mass shifts, N(x) is the number of all target PSMs with scores greater than x, Ni(x) is the number of target PSMs in the i-th bin with scores greater than x, FDR(x) is the global FDR at the score threshold of x, and γi(x) is the probability that a PSM belongs to the i-th bin given that the PSM is random and is scored better than x.
Following our previous work (34), γi(x) is approximated by a linear function of x
| (Eq. 2) |
Because the matches to the decoy sequences are definitely false identifications, we make use of decoy PSMs to estimate the two coefficients a and b. Specifically, we calculate the proportion of decoy PSMs falling into the i-th bin at varying score threshold and use the proportions as training samples to fit the above linear function (example shown in supplemental Fig. S2). Briefly, the principle behind the transfer FDR approach is that because the decoy PSMs observed in a bin might often be too few to perform accurate FDR estimation directly, the transfer FDR uses the fitted function to extrapolate the number (proportion) of decoy PSMs for the effective score threshold for FDR control.
Mass Shift Clustering
There may be more than one types of modifications falling into a 1-Da mass-shift bin, e.g. acetylation (42.010565 Da) and tri-methylation (42.046950 Da) in the bin of [41.5, 42.5] Da. In order to distinguish mass shifts coming from different types of modifications, PTMiner carries out clustering analysis for mass shifts within each bin using the Gaussian mixture model (GMM) and treats each cluster as one modification type in the localization algorithm. First, a discrete convolution algorithm is invented to learn the initial parameters of GMM, i.e. component number K, initial means (Ms) and variances (Vs) (supplementary Note S2). Next, GMM is used to fit the mass shifts with the initial parameters. A component, whose variance is less than 1.5 times of the system measure variance VAR (the variance of mass shifts falling into the interval of [-T, T]) and has mass shifts more than a user-specified number (default is 5), will be regarded as one modification type. These clustered mass shifts will be localized using the iteratively updated prior probabilities whereas un-clustered will not.
Distribution Fitting of Matched-peak Intensities
Before mass-shift localization, each spectrum is preprocessed as follows. First all the peak intensities are divided by the maximal intensity in this spectrum to generate relative intensities, then the peaks with relative intensities less than 0.01 are removed, and finally the relative intensities of remaining peaks are rescaled by taking their square roots. The intensity distribution of matched peaks is fitted based on the matched peaks of unmodified PSMs assuming the distribution form is lognormal (example shown in supplemental Fig. S3).
Mass Shift Localization
We assume that the mass shifts in one cluster come from the same type of modification and do modification localization for these mass shifts in combination. As described below, for clustered mass shifts PTMiner iteratively updates their prior probabilities on different amino acids, but for un-clustered mass shifts, PTMiner does not use the prior probability and takes the conditional probability as the final posterior probability.
Taking one mass shift cluster as example, we suppose there are total N PSMs in this cluster, marked as PSMi, with i ϵ {1, …, N}. PSMi consists of spectrum Speci and matched peptide sequence Pepi with length Li. Let Pepi(j) denote the amino acid in position j of Pepi, with j ϵ {0, 1, …, Li + 1} where Pepi(0) and Pepi(Li + 1) represent the N-terminus and C-terminus of Pepi, respectively, and Pepij denote the modified peptide sequence with modification occurring on Pepi(j). PTMiner uses a Bayesian model to calculate the posterior probability that a modification occurs at the j-th site on Pepi as follows,
| (Eq. 3) |
where Pr(Pepij) denotes the prior probability that this modification occurs at Pepi(j) and Pr(Speci|Pepij) denotes the conditional probability that the Speci is generated from Pepij.
In order to calculate the posterior probability, we need two types of probabilities, i.e. the conditional probability Pr(Speci|Pepij) and the prior probability Pr(Pepij). To evaluate the conditional probability Pr(Speci|Pepij), we first match the experimental peaks of Speci with all the theoretical peaks of the unmodified and all modified forms of fragment ions of Pepi. After matching, we get the corresponding matched peaks in Speci, forming the peak set Peaksi*. We assume that matched peaks are independent, which yields,
| (Eq. 4) |
Let F(x) denote the cumulative distribution function of matched-peak intensity that is learned from unmodified peptides. We define
| (Eq. 5) |
where I is the intensity of Peak, and Peak and Pepij are consistent if Peak matches one of the theoretical peaks of Pepij.
Next, we illustrate how to calculate the prior probability Pr(Pepij). As we know, every modification has its amino acid specificities. For example, phosphorylation mainly occurs on Ser, Thr and Tyr. We define the ratios of amino acid specificities of one modification as modification specificity frequencies (MSF), which are represented by a K-dimensional vector Φ→ = (π1,π2…,πK)T composed of K (= 104) possible combinations of sites (i.e. 20 types of amino acids, and N-/C-terminus) and positions (i.e. N-/C-terminus of peptide/protein, or anywhere on peptide). For example, the MSF vector Φ→ of phosphorylation can be set equally as 1/3 for 'S, Anywhere', 'T, Anywhere' and 'Y, Anywhere', respectively, and zeros for the others. For different protein samples, the frequencies may be very different, e.g. prokaryotic versus eukaryotic samples or enriched versus non-enriched samples (61). To consider the sample dependence, we introduce the term Modification Specificity Frequencies of Sample (MSFS), which we will learn from data using an EM-like algorithm (details given later). We derive prior probability Pr(Pepij) from MSFS as follows,
| (Eq. 6) |
where h is a mapping from the set of all modification specificities to their corresponding indexes in MSFS vector Φ→, i.e. h:x → y, x ϵ {all modification specificities} and y ϵ {1, 2, …, K}.
We regard the site owning the maximal posterior probability, denoted by p*, as the location of the modification. Finally, we estimate the FLR of a set of localized mass shifts by averaging their posterior error probabilities
| (Eq. 7) |
where n is the size of the set.
EM-like algorithm for prior probability updating
We use an EM-like algorithm to estimate the MSFS vector Φ→ as follows,
(0) Set initial values Φ→(0) and set t = 0. Each element is set to be 1/K by default;
(1) Generate the prior probability of each amino acid of Pepi from Φ→(t), that is , for i ϵ {1,2,…,N}.
(2) Calculate the posterior probabilities for all sites using Eq. 3;
(3) Update Φ→(t + 1) using πk(t + 1) = Pr(Pepij|Speci), where k ϵ {1, 2, …, K};
(4) Calculate the change of MSFS, Δπ = . If Δπ is less than a given threshold (0.01 in our study), stop and output; otherwise, set t = t+1 and return to step (1).
Modification Annotation
To determine the identity of a mass shift, PTMiner compares the localized mass shift with each modification in the Unimod database. If the mass shift is matched to the mass of some modification within the precursor mass tolerance of the mass spectrometer, PTMiner further examines whether the localization of the mass shift is consistent with the modification specificity definition. If both the mass and the specificity conform to the modification, the mass shift is regarded as fully annotated. If only the mass is matched but the specificity is not, the mass shift is regarded as partially annotated. Otherwise, the mass shift is regarded as un-annotated.
Because some mass shifts might have been induced by in-source fragmentation, nonspecific digestion or missed cleavages, PTMiner conducts a check procedure for all the PSMs. PTMiner adds amino acids one by one (up to 5) to the peptide N- or C-termini and checks whether the altered mass shift can be fully annotated. Note that the added amino acids come from the corresponding protein sequence in the database. At the same time PTMiner also deletes amino acids one by one (up to 5) from peptide N- or C-termini and checks the altered mass shift.
RESULTS
We used three data sets to evaluate PTMiner, including the simulated data generated by computer program, the chemically synthesized peptide data (62) and the complex proteome data with spiked-in modified peptides. Besides, a draft map of human proteome (59) was used to demonstrate PTMiner utility.
Simulated Data
To evaluate the accuracy of PTMiner in modification localization, some test data with standard answers are needed. For this purpose, we generated a large data set containing 956,550 simulated spectra from random peptide sequences, each of which either had no modification or one of the nine designed types of modifications, including oxidation (M), deamidation (N), methylation (K), phosphorylation (S) and so on (supplementary Note S3). The simulated spectra were divided into eleven subsets, including nine modification subsets, one un-modification subset and one contamination subset (supplemental Table S1). To evaluate how realistic these simulated spectra were, we made a comparison between the simulated and real spectra (supplemental Fig. S4).
Chemically Synthesized Peptide Library Data
The experimental MS/MS spectra in this data set came from part of the ProteomeTools project (62, 63). Briefly speaking, about 5,000 synthesized peptides carrying 21 different modifications including several types of lysine acylation, lysine and arginine methylation, tyrosine phosphorylation and nitration as well as proline hydroxylation were analyzed by mass spectrometry. A total of 25 raw files (including 4 types of unmodified peptides) were downloaded from the PRIDE data repository (https://www.ebi.ac.uk/pride/, dataset identifier PXD009449), and converted to mgf format using the msconvert.exe tool from ProteoWizard (3.0.7069 64-bit version) (64). Conversion was performed using vendor-provided centroiding and default parameters. This data set contained 1,023,540 mass spectra.
Modified-peptide Spiked-in Complex Proteome Data
Six modified peptides were chemically synthesized, each containing one modification site (supplemental Table S2). The synthetic peptide mixture (50 pmol of each one) was mixed with 1 μg trypsin digested HeLa or E. coli whole cell proteins, respectively. Mass spectrometry data were acquired by Q Exactive mass spectrometer in HCD mode. See supplementary Note S4 for details of LC-MS/MS analysis.
Draft Map of Human Proteome
The MS/MS spectra in this data set were from the draft map of human proteome described in Kim et al. (59) and was downloaded from the PRIDE data repository (https://www.ebi.ac.uk/pride/, dataset identifier PXD000561). Briefly, samples from 30 human tissues, including 17 adult tissues, 7 fetal tissues, and 6 hematopoietic cell types, were fractionated at the protein level by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) or at the peptide level by basic reversed-phase liquid chromatography (bRP), and then analyzed on high-resolution Fourier-transform mass spectrometers (LTQ-Orbitrap Elite and LTQ-Orbitrap Velos) using HCD fragmentation, resulting in a total of 24,954,916 MS/MS spectra (59). All these spectra were analyzed in this paper. Thermo RAW files were converted to mgf format using the pParse tool (65) with default parameters except that the co-elute option was set to 0.
pFind Search Engine
MS/MS spectra were searched using pFind (version 2.8.5) (56–58) in the open search mode (Open_KSDP score selected). Open_KSDP treats the precursor mass shift as a potential modification and localizes it to the site that yields the highest peptide score. The simulated, synthesized peptide library and human proteome map data were searched against the random protein sequences, the sequences of chemically synthesized peptides and the Swiss-Prot human protein sequences (downloaded on October 28, 2015), respectively. For the modified-peptide spiked-in proteome data, the combined sequences of the Swiss-Prot human or E. coli proteins (downloaded on June 27, 2018) and the proteins that the spiked-in peptides belongs to were searched. All the sequences were concatenated with their reversed protein sequences for target-decoy based FDR estimation. For simulated data, chemically synthesized peptide data, modified-peptide spiked-in proteome data and the human proteome map data, the precursor mass tolerances were all set to 500 Da and the fragment mass tolerances were set to 0.05 Da, 20 PPM, 20 PPM and 0.05 Da, respectively. For all the data sets, candidate peptides were fully tryptic with up to 2 allowed miss-cleavages, and carbamidomethylation on Cys was considered as a fixed modification except for the simulated data. No variable modification was considered for the simulated data and the human proteome map data, and oxidation on Met was set as variable modification for the chemically synthesized peptide data and the modified-peptide spiked-in E. coli proteome data. For the modified-peptide spiked-in HeLa proteome data, oxidation on Met and acetylation on protein N-terminal were set as variable modifications.
MODa Search Engine
MODa (v1.23) (16) was run in the single-blind mode with a maximum modification size of 500 Da for the simulated and the chemically synthesized peptide data. Parent mass tolerances of the mass spectrometer were set to be 10 PPM and 20 PPM for the simulated and the chemically synthesized peptide data, respectively. Fragment mass tolerances were both set to 0.05 Da. No fixed modification was specified for simulated data set and carbamidomethylation on Cys was specified as fixed modification for the chemically synthesized peptide data. High-resolution MS/MS search was enabled and fully tryptic digestion was specified with at most two missed cleavages for both data set. Other parameters were set as default. The “anal_moda.jar” program bundled with the MODa software was used to estimate the global FDR of PSMs. In order to do separate and transfer FDR estimation, we first used “anal_moda.jar” to export all PSMs of MODa by setting a wrong prefix of decoy proteins, and then estimated the separate and transfer FDRs for the exported PSMs at varying score threshold.
Extended Ascore
The Ascore algorithm was initially designed for localization of phosphorylations to amino acids serine, threonine, or tyrosine (40). To compare it with PTMiner, we extended it to allow for the localization of any modification mass to any type of amino acids as done by Chick et al. recently (18). In order to validate our implementation, we tested the extended Ascore on phosphorylated peptides and compared with the original Ascore software. The phosphorylated peptides and original Ascore localization results came from the original Ascore web site (http://ascore.med.harvard.edu/ascore.php). The comparison results showed that our extended Ascore was almost equivalent to the original Ascore software (supplemental Fig. S5, supplemental Table S3).
Correctness Judgment of Peptide Identification and Modification Localization
(A) The simulated data. A peptide identification is judged as true if it conforms to one of the following three situations, (1) the identified sequence is the same as the real one; (2) the identified sequence is part of the real one; and (3) the identified sequence covers the real one. For modification localization, there are four criteria as follows, (1) if the identified peptide sequence is the same as the real one, we consider the localization is correct when the location is the same as designed; (2) if the identified peptide is part of or covers the real peptide sequence, we also consider the localization is correct when the localized amino acid is the designed modification site. For example, if the real peptide sequence is RM(oxidation)DSSSLRAYSK and the identified peptide is MDSSSLRAYSK, we consider the localization is correct when the mass shift (oxidation + Arg) is localized to the first Met; (3) if a peptide identification is false, then the localization is certainly false; (4) if there are multiple locations (with equal probabilities), we regard all of them as false even when they include the true one. (B) The chemically synthesized peptide data and the modified-peptide spiked-in proteome data. We considered only those PSMs matching to the sequences of synthetic peptides with expected corresponding mass shifts to evaluate PTMiner localization accuracy. If one mass shift was localized at the designed site, then we thought the localization was correct.
Validation Results On Simulated Data
Because the corresponding peptide sequences and modification locations of these simulated spectra were known, the accuracies of PTMiner in FDR estimation and modification localization could be evaluated. We searched these simulated spectra using two search engines pFind (56–57) and MODa (16) in the open search mode, and then analyzed their search results using PTMiner and Ascore (18). In FDR analysis, we compared the transfer FDR with the global FDR (the traditional target-decoy based FDR estimation executed on the whole set of PSMs) and the separate FDR (the traditional target-decoy based FDR estimation executed on the subsets/groups of individual modification types). In modification localization, we compared PTMiner with the search engines themselves and the extended Ascore algorithm.
Performance Comparison of Transfer, Global and Separate FDRs
We compared the transfer, global and separate FDRs on the open search results of pFind and MODa (Table 1, supplemental Table S4, and supplemental Fig. S1). First, we evaluated the performance of global FDR. Our analysis results show that although the global FDR was accurately estimated for the whole set of PSMs (unmodified and various modified peptides), it was too conservative for each of the subsets of designed modifications. For example, on the search result of pFind (Table I), the real global FDR was 0.97%, very close to the control level 1%, but the real group FDRs of both unmodified and modified peptide identifications with designed modification types were almost zero. This overly conservative FDR estimation resulted in a quite low spectral identification rate, i.e. only 14.6% in total. Note that the 0.97% global FDR was from PSMs with mass shifts out of the design set.
Table I. The real FDRs and the numbers of spectral identifications obtained with three FDR estimation approaches (global, separate and transfer FDRs) at 1% FDR control level on the pFind search results of the simulated spectra. “Sub-Correct” means the identified peptide is part of or covers the real peptide sequence.
| Total Spectra | Real FDR |
Identification number |
|||||
|---|---|---|---|---|---|---|---|
| Global | Separate | Transfer | Global | Separate | Transfer | ||
| Unmodified | 800,000 | 0.00% | 0.29% | 0.29% | 127,148 | 601,375 | 601,375 |
| Oxidation | 30,000 | 0.02% | 0.84% | 0.86% | 6,343 | 23,265 | 23,369 |
| Deamidation | 15,000 | 0.00% | 0.86% | 0.56% | 2,240 | 9,506 | 8,940 |
| Tri-Methyl & Acetyl | 10,000 | 0.00% | 1.06% | 1.08% | 1,513 | 5,072 | 5,166 |
| Methyl | 1,000 | 0.00% | 2.30% | 0.45% | 133 | 305 | 222 |
| Di-Methyl | 500 | 0.00% | 1.44% | 1.63% | 70 | 139 | 123 |
| Phospho | 50 | 0.00% | 4.55% | 0.00% | 13 | 22 | 17 |
| Sub-Correct | – | 0.00% | 0.00% | 0.00% | 956 | 1,152 | 681 |
| Other Modifications | – | 100% | 100% | 100% | 1,358 | 951 | 372 |
| Sum | 856,550 | – | – | – | 139,744 | 641,787 | 640,265 |
Next, we compared the two group FDR approaches, i.e. the separate FDR and our transfer FDR. Results show that the separate FDR could control the FDRs for large group but failed for small groups (Table I, supplemental Table S4). For example, on the search result of pFind, the real FDR of phosphorylation group was up to 4.55% at 1% estimated global FDR. On the other hand, our transfer FDR can effectively control the FDRs for all designed modification groups and is therefore a better choice of FDR control method for open search results, in which small groups are very common.
Mass-shift Localization Results
On the PSMs filtered at 1% transfer FDR, we compared the mass shift localization results of different methods, including the search engines themselves (pFind and MODa), the extended Ascore algorithm, and our PTMiner. We sorted the localization results of each method in descending order by their localization score, e.g. the posterior probability in PTMiner, and evaluated the real FLR and the proportion of localized mass shifts at varying score threshold. For search engines, we used the PSM score as the localization score because they do not have a specific localization score to measure the localization reliability. For pFind and MODa, we used the E-value and Probability score, respectively.
Fig. 2A–2B plot the localization proportion against the real FLR for different methods. It is shown that the two search engines performed poorly in localizing the mass shifts, both PTMiner and Ascore greatly improved the localization accuracy, and PTMiner was much superior to Ascore. For example, at the 1% real FLR, the localization proportions were only 0.07% and 0.13% for pFind and MODa, respectively. Ascore increased the localization proportions to 0.52% and 17.43%, whereas PTMiner achieved 93.06% and 79.18%, respectively. These results indicate that the PSM scores of search engines are not suitable for the measurement of localization confidence, and the simple extension of restrictive localization algorithms to open search results is helpful but cannot provide satisfactory sensitivity. In contrast, by full consideration of the features of the open search strategy, PTMiner showed remarkable superiority.
Fig. 2.

PTMiner confidently localized the mass shifts on the simulated data. A–B, PTMiner increased the localization proportions greatly compared with the search engines (A for pFind and B for MODa) and the extended Ascore at the same real FLR thresholds. C–D, The real and estimated FLRs were fitted using linear regressions (C for pFind and D for MODa).
Besides, PTMiner can also estimate the FLR based on the localization probabilities. For the open search results of both pFind and MODa, the estimated FLRs showed good linear relationship with the real FLRs (Fig. 2C–2D). The probability thresholds of for 1% real FLR pFind and MODa search results were 0.837 and 0.775, respectively. The Ascore thresholds for 1% real FLR of pFind and MODa results were 60 and 27, respectively (supplemental Fig. S6).
Validation Results on Chemically Synthesized Peptide Library Data From the ProteomeTools Project
We searched these synthetic peptide spectra using pFind (56, 57) and MODa (16) in the open search mode, and analyzed their search results using PTMiner and Ascore (18).
FDR Estimation
Different from the simulated data, we did not exactly know the peptide sequences of spectra, so we cannot calculate the precise real FDR. The golden criterion for the correctness of a PSM is that the peptide is one of the chemically synthesized peptides and bears the given modification in the ProteomeTools project. However, by using this criterion, we found that the FDR could not be accurately calculated, because some quality control peptides and impurities (byproducts) existed in this dataset. Some of them could be confidently identified by the search engines, e.g. a spiked-in quality control peptide (supplemental Fig. S7A) and a byproduct LQKbiotinQSVVYGGK from the expected product TLQKbiotinQSVVYGGK (supplemental Fig. S7B).
In the pFind search results, global, separate and transfer FDR approaches achieved 949619, 1003346 and 998114 PSMs at 1% FDR, respectively. In the MODa search results, 800534, 805484 and 796297 PSMs were accepted by the three FDR approaches at 1% FDR, respectively. We found that the numbers of filtered PSMs by the three FDR approaches were almost the same, which was mainly because of the similar abundances of unmodified and different modified peptides in the spectra.
Mass-shift Localization Results
For the PSMs filtered at 1% FDR, we first used PTMiner to localize these mass shifts. We selected those results matching to synthetic peptide sequences and mass shifts of the expected modifications to compare PTMiner with the search engines (pFind and MODa) and the extended Ascore algorithm. As similarly did for the simulated data, we used E-value for pFind, Probability score for MODa, Ascore for Ascore algorithm and posterior probability for PTMiner to sort the localization results in descending order to calculate the real FLR and the proportion of localized mass shifts at varying score threshold. At 1% real FLR, the localization proportions for pFind and MODa were almost zero. Ascore increased the proportion to 16% for the results of pFind but remained around zero for the results of MODa, whereas PTMiner increased the proportions to 84% and 71%, respectively, for the results of pFind and MODa (Fig. 3). Therefore, the localization results on chemically synthesized peptide data also indicated that PTMiner behaved better in localizing the mass shifts in open search results than search engines (pFind and MODa) and Ascore.
Fig. 3.

PTMiner confidently localized the mass shifts on the chemically synthesized peptide data. A–B, PTMiner increased the localization proportions greatly compared with the search engines (A for pFind and B for MODa) and the extended Ascore at the same real FLR thresholds.
Validation Results on Modified-peptide Spiked-in Complex Proteome Data
We searched the two modified-peptide spiked-in data sets using pFind (56, 57) in the open search mode and analyzed the search results using the three FDR estimation methods. For the HeLa data set, a total of 267 designed PSMs (both sequences and mass shifts were designed) were obtained, from which 104, 117, and 175 PSMs were kept by the global, separate and transfer FDR approaches at 1% FDR level, respectively. For the E. coli data set, a total of 260 designed PSMs were obtained, from which 126, 142, and 163 PSMs were kept by the global, separate and transfer FDR approaches at 1% FDR level, respectively (Table. 2). These results indicated that transfer FDR performed best among the three FDR estimation approaches. We then used PTMiner and Ascore to localize these mass shifts. At 1% real FLR, PTMiner, Ascore and pFind correctly localized 146, 46, and 3 mass shifts from the 175 designed PSMs of the HeLa data set, respectively. From the 163 designed PSMs of the E. coli data set, PTMiner, Ascore, and pFind correctly localized 91, 35, and 18 mass shifts at the same 1% FLR level, respectively (Fig. 4). This result indicated, again, that PTMiner is significantly more accurate than pFind and Ascore in localizing mass shifts.
Table II. The identification numbers of chemically synthesized peptides from the modified-peptide spiked-in proteome data with three FDR approaches (global, separate and transfer FDRs) at 1% FDR. “PSMs” was the number when no filtering was used. Here “ph,” “me,” “ac,” and “pr” represent phosphorylation, methylation, acetylation, and propionyl, respectively.
| Separate FDR (1%) | P62805 | P0A870 | P0A7K2 | P62807 | A4FNV9 | A4FBI4 |
|---|---|---|---|---|---|---|
| Species | Homo | E. coli | E. coli | Homo | S. erythraea | S. erythraea |
| Sequence | DNIQGITKPAIR | EYAPAEDPGVVSVSEIYQYYK | GATGLGLKEAK | KESKYSVYVYK | TYKLYVGGK | HGGGAFSGKDPSK |
| Mod | 7,T (ph) | 7,D (me) | 9,E (me) | 4,K (ac) | 3,K (pr) | 9,K (pr) |
| HeLa | ||||||
| PSMs | 21 | 86 | 19 | 68 | 41 | 32 |
| Global FDR (1%) | 5 | 48 | 0 | 33 | 4 | 14 |
| Separate FDR (1%) | 0 | 67 | 8 | 33 | 0 | 9 |
| Transfer FDR (1%) | 21 | 61 | 1 | 19 | 41 | 32 |
| E. coli | ||||||
| PSMs | 24 | 77 | 22 | 69 | 37 | 31 |
| Global FDR (1%) | 9 | 52 | 0 | 33 | 17 | 15 |
| Separate FDR (1%) | 9 | 63 | 10 | 17 | 26 | 17 |
| Transfer FDR (1%) | 0 | 56 | 1 | 69 | 22 | 15 |
Fig. 4.
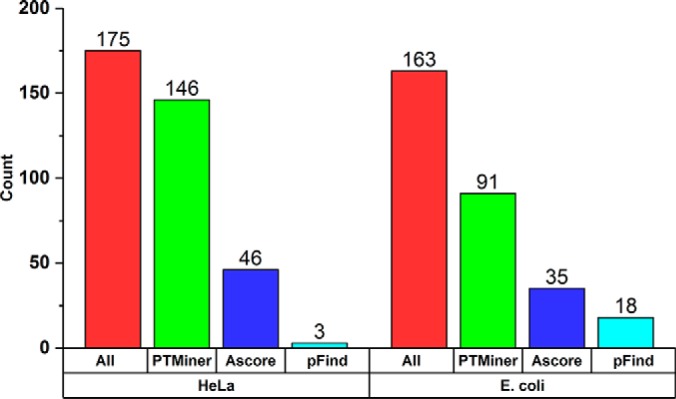
Comparison of PTMiner with pFind and Ascore on the modified-peptide spiked-in proteome data. “All” indicates correct PSMs (both peptide sequence and mass shift were correct) obtained at 1% transfer FDR. “PTMiner,” “Ascore,” and “pFind” indicate correct localization results given by PTMiner, Ascore and pFind at 1% FLR, respectively.
Application to the Draft Map of Human Proteome
We next used a large-scale public-accessible data set, the draft map of human proteome (59) to test the performance of PTMiner. This data set, which contains ∼25 million MS/MS spectra generated from 30 human tissues, was searched using pFind in the open search mode with a 500-Da precursor window. In line with the findings on simulated data, we observed the big FDR heterogeneity among different modification types (examples shown in Supplemental Fig. 8), indicating that global FDR control alone is not reliable for unrestrictive PTM analysis. We used PTMiner to analyze the search results. The FDR of PSMs was controlled at 1% within each 1-Da mass shift bin using the transfer FDR approach. This resulted in a total of 9,272,908 PSMs accepted at 1% FDR, of which 3,347,581 (36.10%) had mass shifts outside [-0.5, 0.5] Da and were potentially modified peptides (Fig. 5A). These mass shifts were then localized by PTMiner to specific amino acids on peptides, and the FLR was estimated. Of the modified PSMs, 1,755,278 (52.43%) were kept at 1% estimated FLR. The histogram of the mass shifts of these PSMs showed heavy clusters (Fig. 5B). For example, 502,315 (28.62%) of them were around 15.996 Da, very close to the mass of oxidation modification.
Fig. 5.

Result summary of unrestrictive modification identification in an open search and modification localization by PTMiner for the draft map of human proteome. A, The numbers of total MS/MS spectra, PSMs with 1% FDR, modified PSMs among them, and modified PSMs with 1% FLR, as well as the proportions of fully annotated (by both mass and location specificity), partially annotated (by mass only) and unannotated PSMs. B, Histogram of 1,755,278 mass shifts with 1% FDR and 1% FLR.
The localized mass shifts were next compared with the modifications in the Unimod database for annotation. In total, 83.51% (1,465,908/1,755,278) of the confidently localized mass shifts were fully annotated by their mass values and amino acid specificities. If one mass shift was annotated by multiple modifications, we manually determined which was the most possible one, and then increased its count by one (supplemental Table S5). Among them 38 types of modifications had >1000 PSMs, including the commonly reported oxidation on Met, formylation on peptide N-terminus, deamidation on Asn, acetylation on protein N-terminus and so on (supplemental Table S6). Their localized sites were consistent with expectation. For example, 97.59% of oxidations were localized to Met, and 89.21% of deamidations were localized to Asp (Fig. 6A). Besides, a total of 148,583 (8.46%) mass shifts were annotated by their mass values only whereas their localized amino acids were not included in the Unimod database, and among them 21 types of modifications had >1,000 PSMs (supplemental Table S7). The remaining 140,787 (8.02%) mass shifts could not be annotated by Unimod, and among them 28 1-Da bins had >1,000 PSMs (supplemental Table S8).
Fig. 6.

Modification analysis of the human proteome data. A, Modification specificity distributions on amino acids and protein/peptide termini for the fully-annotated modifications with 1% FDR and 1% FLR. Normalization was performed so that the sum of modification specificities of each modification was equal to 1. Only fully annotated modifications with more than 1,000 PSMs were shown. B, Modification distributions across the 30 human tissues. For each modification, the number of modified PSMs from each tissue was divided by the total number of the spectra from that tissue, and then the derived ratios were normalized across all tissues such that the sum for this modification was 1. These fully annotated modifications with more than 1,000 PSMs were shown in this figure. C–D, KEGG enrichment analysis results of deamidation and succinylation modifications that were differentially observed in the tissues.
Analysis of Fully Annotated Modifications
Apart from the common artificial modifications probably introduced during sample processing (such as oxidation on Met), we also detected many well-documented but less studied PTMs such as oxidation on Pro, di-oxidation on Met, deamidation on Arg, succinylation on Lys, etc (supplemental Fig. S9, supplemental Fig. S10). We showed the tissue specificities for the fully annotated modifications with more than 1000 PSMs in Fig. 6B. Some of these modifications could possibly be artifacts introduced by sample handling, such as carbamylation and dehydration. Interestingly, we found some PTMs, which showed strong preference to specific tissues, could possibly be biologically meaningful. Notably, deamidation on Arg (supplemental Fig. S10A) was mainly detected in adult spinal cord and adult retina, which has been reported in the central nervous system and is associated with a number of neurological diseases (66), and succinylation on Lys (supplemental Fig. S10B) was mainly observed in adult front cortex and adult liver. In order to understand the possible roles of these PTMs that differentially occurred in the tissues, we performed enrichment analysis using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Fig. 6C–6D). For example, in the KEGG metabolic pathway analysis, the arginine-deamidated proteins were significantly enriched in phototransduction in adult's retina. Phototransduction is important for the visual system, which could convert light into electrical signals in the retina of the eye (67). As for the adult's spinal cord, the arginine-deamidation modified proteins were mostly enriched in neuron cell related bioprocess, such as gap junction, tight junction and axon guidance (68). For the lysine-succinylation, the modification proteins were significantly enriched in chronic neurodegenerative diseases in adult frontal cortex, such as the Parkinson's disease (69). However, in adult's liver, the succinylation modified proteins were mostly enriched in cytochrome P450-involved metabolic pathways, suggesting succinylation could be closely associated with the xenobiotic metabolism role of liver. Therefore, the KEGG pathway enrichment analysis suggested that these two modifications are closely associated with tissue-specific functions (70).
We observed a total of 1,968 mass shifts which were annotated as succinyl:2H(4), succinyl:13C(4) or benzoyl, which were known to be derived from chemical labeling reagents (400 PSMs had E-Value scores <1e-30, one example shown in supplemental Fig. S11). However, no isotopic labeling reagents were used in these samples. A very recent study reported that lysine benzoylation is a new type of protein PTMs (71), which exactly matches to this mass shift we observed here. Therefore, it is likely that some PSMs of this mass shift could possibly be in vivo benzoylation. This case further suggested that some mass shifts annotated as artifacts or chemical labels could be unreported biological protein modifications.
In addition to modifications, some single amino acid variations (SAVs) were also detected and annotated by PTMiner. In order to make sure that they were truly caused by single nucleotide polymorphisms (SNPs), we further compared our results with the UniProt database, and found 3874 of the detected SAVs had SNP annotations. Some SAVs are tissue specific. For example, rs16967510, localized to protein myosin-11 (V1289A), was found mainly in adult colon and adult urinary bladder, rs6085324 localized to protein secretogranin-1 (S93T), was found mainly in adult adrenal gland. The spectra of peptides with these SAVs show excellent consistency with those of corresponding non-variant peptides (without SAVs), justifying the confidence of these SAV identifications by PTMiner (supplemental Fig. S12, supplemental Table S9).
Moreover, we observed that some modifications showed strong difference between two sample preparation methods, i.e. bRP and SDS-PAGE (supplemental Fig. S13). These mass shifts could be method-specific in vitro modifications. For example, methylation on Glu was mainly observed in SDS-PAGE samples whereas dehydrated on Thr was mainly found in bRP samples. Therefore, more careful attention should be paid and more evidences are needed if these mass shifts were considered as endogenous PTMs in future experiment.
Analysis of Partially and Un-annotated Modifications
We noted that in the open search results, the 'mass shifts' might also be resulted from other events than modifications, such as in-source fragmentation, nonspecific digestion and missed cleavages. PTMiner features the detection of some of such events that could not be fully annotated. In brief, PTMiner attempts to consider some amino acid addition to the N- or C-terminus of identified peptide sequence, or deletion of some amino acids from the N- or C-terminus, to check whether such mass shift can be logically explained. From the 289,370 partially and un-annotated PSMs, PTMiner recovered 116,057 (40.11%) new peptide sequences in this way. For example, 4528 (∼76%) mass shifts in the bin of [240.5, 241.5] Da were explained as the loss of two amino acids of Ile/Leu and Lys (samples shown in supplemental Table S10).
We also found that some mass shifts that could not be annotated or explained by Unimod showed amino acid specific preference. For example, the mass shifts falling in [11.99, 12.01] Da, which coincides with the mass of thiazolidine modification, were mainly localized to peptide N-termini (99.39%, 12104/12178) (Fig. 7, supplemental Fig. S14). This modification cannot be added on peptide N-terminus except in formaldehyde treated samples, but there is no mention of such treatment (59). Another example is the 149 mass shifts falling into [33.993, 34.013] Da, which were mainly localized to His (90%, 133/149). However, they could not be annotated by any type of modification in Unimod (Fig. 8, supplemental Fig. S15). They might be probably from some unknown types of modification but deducing the identities of unannotated mass shifts is more than a computational problem and has been outside the scope of this article.
Fig. 7.

The mass shift of 12.0054 Da was localized to peptide N-terminus. MS/MS spectra of modified (top) and unmodified (bottom) forms of the peptide are shown and compared. Red arrows between two spectra indicate the shift of fragment ion peaks. It can be seen that the two spectra are very similar to each other in terms of their peptide fragmentation patterns, justifying the reliability of identification and localization.
Fig. 8.

The mass shift of 34.0061 Da was localized to His. MS/MS spectra of modified (top) and unmodified (bottom) forms of the peptide are shown and compared. Red arrows between two spectra indicate the shift of fragment ion peaks. It can be seen that the two spectra are very similar to each other in terms of their peptide fragmentation patterns, justifying the reliability of identification and localization.
DISCUSSION
We presented in this article a software tool named PTMiner for accurate estimation of modification-specific FDRs and probability-based modification localization to support more reliable PTM detection in an open search. We validated our approach using extensive data sets, including simulated data, chemically synthesized peptide data and modified-peptide spiked-in complex proteome data. We also searched and analyzed a draft map of human proteome to comprehensively characterize the PTMs in human proteome. PTMiner aims at reducing the enormous uncertainty in the data produced in an open search, which is becoming increasingly popular and promising for the field of proteomics.
Particularly, the reliable localization of detected mass shifts can provide great help to verify their correctness and determine their identities, which is critical for protein PTM analysis. For example, some modifications, which have identical or very similar masses but occur on different amino acids, e.g. the amino acid mutation Ala->Ser and oxidation modification (their masses are both 15.994915 Da), cannot be distinguished without localization information. On the other hand, the open search results are very complicated because of the intrinsic complexity of PTMs in the cellular proteome and the simplistic implementation of open search engines, and we cannot address every problem. For example, PTMiner is currently unable to handle multiple modifications existing on a peptide. Also, PTMiner is to some extent dependent on the Unimod modification database which may not be complete and error-free at present. However, as the knowledge on protein modifications increases in the future, the accuracy of our method will be also improved. In summary, we expect that PTMiner, as a first statistical analysis tool for open database search results, will facilitate more reliable discovery-oriented PTM studies in proteomics.
Data and Software Availability
The chemically synthesized peptide data and the draft map of human proteome were downloaded from the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) with the dataset identifiers PXD009449 and PXD000561, respectively. The modified-peptide spiked-in proteome data have been deposited to iProX (a full member of the ProteomeXchange consortium, http://www.iprox.org) with the dataset identifier IPX0001318000. All of the spectra identified as modified peptide have been uploaded to MS-Viewer data sets (72) (the search keys for the simulated data, the chemically synthesized peptide library data, the modified-peptide spiked-in complex proteome data and the draft map of human proteome are 9yxuedofcl, qjgpjpgwsg, nsdxlsv1qu and jr8wrhwar6, respectively).The PTMiner software and the extended Ascore algorithm coded with Matlab are available at http://fugroup.amss.ac.cn/software/ptminer/ptminer.html.
PTMiner now supports spectra generated using collision-induced dissociation (CID) or HCD in 'mgf' format as data input, and the output of pFind, Sequest or MSFragger as the search result input. Besides, PTMiner can also read in tab-delimited files transferred from any search engines.
Supplementary Material
Footnotes
* This work was supported by grants from the National Key Basic Research Program of China (2017YFC0908400, 2017YFC0906600 and 2015CB554406), the Natural Science Foundation of China (81872888, 31670066, 31700724 and 91630314), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB13040600), the International S&T Cooperation Program of China (2014DFB30010), and the NCMIS CAS.
 This article contains supplemental material. We declare no conflict of interest.
This article contains supplemental material. We declare no conflict of interest.
1 The abbreviations used are:
- MS/MS
- tandem mass spectrometry
- PTM
- post-translational modification
- FDR
- false discovery rate
- FLR
- false localization rate
- PSM
- peptide-spectrum match
- MSFS
- modification specificity frequencies of sample
- GMM
- gaussian mixture model
- LC-MS
- liquid chromatography mass spectrometry
- CID
- collision-induced dissociation
- HCD
- higher-energy collision dissociation
- SDS-PAGE
- sds-polyacrylamide gel electrophoresis
- bRP
- basic reversed-phase liquid chromatography
- SNP
- single nucleotide polymorphism
- SAV
- single acid variation
- PPM
- parts per million.
REFERENCES
- 1. Yates J. R. 3rd, Eng J. K., McCormack A. L., and Schieltz D. (1995) Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 67, 1426–1436 [DOI] [PubMed] [Google Scholar]
- 2. Nesvizhskii A. I., Roos F. F., Grossmann J., Vogelzang M., Eddes J. S., Gruissem W., Baginsky S., and Aebersold R. (2006) Dynamic spectrum quality assessment and iterative computational analysis of shotgun proteomic data: toward more efficient identification of post-translational modifications, sequence polymorphisms, and novel peptides. Mol. Cell Proteomics 5, 652–670. [DOI] [PubMed] [Google Scholar]
- 3. Chalkley R. J., Baker P. R., Medzihradszky K. F., Lynn A. J., and Burlingame A. L. (2008) In-depth analysis of tandem mass spectrometry data from disparate instrument types. Mol. Cell. Proteomics 7, 2386–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Griss J., Perez-Riverol Y., Lewis S., Tabb D. L., Dianes J. A., Del-Toro N., Rurik M., Walzer M. W., Kohlbacher O., Hermjakob H., Wang R., and Vizcaíno J. A. (2016) Recognizing millions of consistently unidentified spectra across hundreds of shotgun proteomics datasets. Nat. Methods 13, 651–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nielsen M. L., Savitski M. M., and Zubarev R. A. (2006) Extent of modifications in human proteome samples and their effect on dynamic range of analysis in shotgun proteomics. Mol. Cell. Proteomics 5, 2384–2391. [DOI] [PubMed] [Google Scholar]
- 6. Fu Y. (2016) Data Analysis Strategies for Protein Modification Identification. Methods Mol. Biol. 1362, 265–275. [DOI] [PubMed] [Google Scholar]
- 7. Tsur D., Tanner S., Zandi E., Bafna V., and Pevzner P. A. (2005) Identification of post-translational modifications by blind search of mass spectra. Nat. Biotechnol. 23, 1562–1567. [DOI] [PubMed] [Google Scholar]
- 8. Chalkley R. J., Baker P. R., Hansen K. C., Medzihradszky K. F., Allen N. P., Rexach M., and Burlingame A. L. (2005) Comprehensive analysis of a multidimensional liquid chromatography mass spectrometry dataset acquired on a quadrupole selecting, quadrupole collision cell, time-of-flight mass spectrometer I. How much of the data is theoretically interpretable by search engines? Mol. Cell. Proteomics 4, 1189–1193. [DOI] [PubMed] [Google Scholar]
- 9. Hansen B. T., Davey S. W., Ham A. J., and Liebler D. C. (2005) P-Mod: an algorithm and software to map modifications to peptide sequences using tandem MS data. J. Proteome Res. 4, 358–368. [DOI] [PubMed] [Google Scholar]
- 10. Tang W. H., Halpern B. R., Shilov I. V., Seymour S. L., Keating S. P., Loboda A., Patel A. A., Schaeffer D. A, and Nuwaysir L. M. (2005) Discovering known and unanticipated protein modifications using MS/MS database searching. Anal. Chem. 77, 3931–3946. [DOI] [PubMed] [Google Scholar]
- 11. Havilio M., and Wool A. (2007) Large-scale unrestricted identification of post-translation modifications using tandem mass spectrometry. Anal. Chem 79, 1362–1368. [DOI] [PubMed] [Google Scholar]
- 12. Baumgartner C., Rejtar T., Kullolli M., Akella L. M., and Karger B. L. (2008) SeMoP: a new computational strategy for the unrestricted search for modified peptides using LC-MS/MS data. J. Proteome Res. 7, 4199–4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen Y., Chen W., Cobb M. H., and Zhao Y. (2009) PTMap–a sequence alignment software for unrestricted, accurate, and full-spectrum identification of post-translational modification sites. Proc. Natl. Acad. Sci. U.S.A. 106, 761–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ye D., Fu Y., Sun R. X., Wang H. P., Yuan Z. F., Chi H., and He S. M. (2010) Open MS/MS spectral library search to identify unanticipated post-translational modifications and increase spectral identification rate. Bioinformatics 26, i399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ahrne E., Nikitin F., Lisacek F., and Muller M. (2011) QuickMod: A tool for open modification spectrum library searches. J. Proteome Res. 10, 2913–2921. [DOI] [PubMed] [Google Scholar]
- 16. Na S., Bandeira N., and Paek E. (2012) Fast multi-blind modification search through tandem mass spectrometry. Mol. Cell. Proteomics 11, M111 010199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ma C. W., and Lam H. (2014) Hunting for unexpected post-translational modifications by spectral library searching with tier-wise scoring. J. Proteome Res. 13, 2262–2271. [DOI] [PubMed] [Google Scholar]
- 18. Chick J. M., Kolippakkam D., Nusinow D. P., Zhai B., Rad R., Huttlin E. L., and Gygi S. P. (2015) A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides. Nat. Biotechnol. 33, 743–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu F., Li N., and Yu W. (2016) PIPI: PTM-Invariant Peptide Identification Using Coding Method. J. Proteome Res. 15, 4423–4435. [DOI] [PubMed] [Google Scholar]
- 20. Kong A. T., Leprevost F. V., Avtonomov D. M., Mellacheruvu D., and Nesvizhskii A. I. (2017) MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 14, 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Searle B. C., Dasari S., Turner M., Reddy A. P., Choi D., Wilmarth P. A., McCormack A. L., David L. L., and Nagalla S. R. (2004) High-throughput identification of proteins and unanticipated sequence modifications using a mass-based alignment algorithm for MS/MS de novo sequencing results. Anal. Chem. 76, 2220–2230. [DOI] [PubMed] [Google Scholar]
- 22. Han Y., Ma B., and Zhang K. (2005) SPIDER: software for protein identification from sequence tags with de novo sequencing error. J. Bioinform Comput Biol. 3, 697–716. [DOI] [PubMed] [Google Scholar]
- 23. Shen Y., Toliæ N., Hixson K. K., Purvine S. O., Anderson G. A., and Smith R. D. (2008) De novo sequencing of unique sequence tags for discovery of post-translational modifications of proteins. Anal. Chem. 80, 7742–7754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bandeira N., Tsur D., Frank A., and Pevzner P. A. (2007) Protein identification by spectral networks analysis. Proc. Natl. Acad. Sci. U.S.A. 104, 6140–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Falkner J. A., Falkner J. W., Yocum A. K., and Andrews P. C. (2008) A spectral clustering approach to MS/MS identification of post-translational modifications. J. Proteome Res. 7, 4614–4622. [DOI] [PubMed] [Google Scholar]
- 26. Fu Y., Xiu L.Y., Jia W., Ye D., Sun R.X ., Qian X. H., and He S. M. (2011) DeltAMT: a statistical algorithm for fast detection of protein modifications from LC-MS/MS data. Mol. Cell Proteomics 10, M110 000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shortreed M. R., Shortreed M. R., Wenger C. D., Frey B. L., Schaffer L. V., Scalf M., and Smith L. M. (2015) Global Identification of Protein Post-translational Modifications in a Single-Pass Database Search. J. Proteome Res. 14, 4714–4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li Q., et al. , (2017) Global Post-Translational Modification Discovery. J. Proteome Res. 16, 1383–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Craig R., and Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467. [DOI] [PubMed] [Google Scholar]
- 30. Creasy D. M., and Cottrell J. S. (2002) Error tolerant searching of uninterpreted tandem mass spectrometry data. Proteomics 2, 1426–1434. [DOI] [PubMed] [Google Scholar]
- 31. Han X., He L., Xin L., Shan B., and Ma B. (2011) PeaksPTM: Mass spectrometry-based identification of peptides with unspecified modifications. J. Proteome Res. 10, 2930–2936. [DOI] [PubMed] [Google Scholar]
- 32. Chi H., He K., Yang B., Chen Z., Sun R. X., Fan S. B., Zhang K., Liu C., Yuan Z. F., Wang Q. H., Liu S. Q., Dong M. Q., and He S. M. (2015) pFind-Alioth: A novel unrestricted database search algorithm to improve the interpretation of high-resolution MS/MS data. J. Proteomics 125, 89–97. [DOI] [PubMed] [Google Scholar]
- 33. Elias J. E., and Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- 34. Fu Y., and Qian X. (2014) Transferred subgroup false discovery rate for rare post-translational modifications detected by mass spectrometry. Mol. Cell. Proteomics 13, 1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vaudel M., Burkhart J. M., Zahedi R. P., Oveland E., Berven F. S., Sickmann A.., Martens L., and Barsnes H. (2015) PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol 33, 22–24. [DOI] [PubMed] [Google Scholar]
- 36. Fu Y. (2012) Bayesian false discovery rates for post-translational modification proteomics. Statistics and Its Interface 5, 47–59. [Google Scholar]
- 37. Na S., and Paek E. (2015) Software eyes for protein post-translational modifications. Mass Spectrom Rev. 34, 133–147. [DOI] [PubMed] [Google Scholar]
- 38. Chalkley R. J., and Clauser K. R. (2012) Modification site localization scoring: strategies and performance. Mol. Cell. Proteomics 11, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., and Mann M. (2006) Global in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648. [DOI] [PubMed] [Google Scholar]
- 40. Beausoleil S. A., Villen J., Gerber S. A., Rush J., and Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292. [DOI] [PubMed] [Google Scholar]
- 41. Albuquerque C. P., Smolka M. B., Payne S. H., Bafna V., Eng J., and Zhou H. (2008) A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol. Cell. Proteomics 7, 1389–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wan Y., Cripps D., Thomas S., Campbell P., Ambulos N., Chen T., and Yang A. (2008) PhosphoScan: a probability-based method for phosphorylation site prediction using MS2/MS3 pair information. J. Proteome Res. 7, 2803–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ruttenberg B. E., Pisitkun T., Knepper M. A., and Hoffert J. D. (2008) PhosphoScore: an open-source phosphorylation site assignment tool for MSn data. J. Proteome Res. 7, 3054–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bailey C. M., Sweet S. M., Cunningham D. L., Zeller M., Heath J. K., and Cooper H. J. (2009) SLoMo: automated site localization of modifications from ETD/ECD mass spectra. J. Proteome Res. 8, 1965–1971. [DOI] [PubMed] [Google Scholar]
- 45. Edwards N., Wu X., and Tseng C.-W. (2009) An unsupervised, model-free, machine-learning combiner for peptide identifications from tandem mass spectra. Clinical Proteomics 5, 23. [Google Scholar]
- 46. Phanstiel D. H., Brumbaugh J., Wenger C. D., Tian S., Probasco M. D., Bailey D. J., Swaney D. L., Tervo M. A., Bolin J. M., Ruotti V., Stewart R., Thomson J. A., and Coon J. J. (2011) Proteomic and phosphoproteomic comparison of human ES and iPS cells. Nat. Methods 8, 821–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Taus T., Köcher T., Pichler P., Paschke C., Schmidt A., Henrich C., and Mechtler K. (2011) Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 10, 5354–5362. [DOI] [PubMed] [Google Scholar]
- 48. Baker P. R., Trinidad J. C., and Chalkley R. J. (2011) Modification site localization scoring integrated into a search engine. Mol. Cell. Proteomics 10, M111 008078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lemeer S., Kunold E., Klaeger S., Raabe M., Towers M. W., Claudes E., Arrey T. N., Strupat K., Urlaub H., and Kuster B. (2012) Phosphorylation site localization in peptides by MALDI MS/MS and the Mascot Delta Score. Anal. Bioanal Chem. 402, 249–260. [DOI] [PubMed] [Google Scholar]
- 50. Fermin D., Walmsley S. J., Gingras A. C., Choi H., and Nesvizhskii A. I. (2013) LuciPHOr: algorithm for phosphorylation site localization with false localization rate estimation using modified target-decoy approach. Mol. Cell. Proteomics 12, 3409–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fermin D., Avtonomov D., Choi H., and Nesvizhskii A. I. (2015) LuciPHOr2: site localization of generic post-translational modifications from tandem mass spectrometry data. Bioinformatics 31, 1141–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vaudel M., Breiter D., Beck F., Rahnenführer J., Martens L., and Zahedi R. P. (2013) D-score: a search engine independent MD-score. Proteomics 13, 1036–1041. [DOI] [PubMed] [Google Scholar]
- 53. Saeed F., Pisitkun T., Hoffert J. D., Rashidian S., Wang G., Gucek M., and Knepper M. A. (2013) PhosSA: Fast and accurate phosphorylation site assignment algorithm for mass spectrometry data. Proteome Sci. 11, S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chung C., Liu J., Emili A., and Frey B. J. (2011) Computational refinement of post-translational modifications predicted from tandem mass spectrometry. Bioinformatics 27, 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tanner S., Payne S. H., Dasari S., Shen Z., Wilmarth P. A., David L. L., Loomis W. F., Briggs S. P., and Bafna V. (2008) Accurate annotation of peptide modifications through unrestrictive database search. J. Proteome Res. 7, 170–181. [DOI] [PubMed] [Google Scholar]
- 56. Fu Y., Yang Q., Sun R., Li D., Zeng R., Ling C. X., and Gao W. (2004) Exploiting the kernel trick to correlate fragment ions for peptide identification via tandem mass spectrometry. Bioinformatics 20, 1948–1954. [DOI] [PubMed] [Google Scholar]
- 57. Wang L. H., Li D. Q., Fu Y., Wang H. P., Zhang J. F., Yuan Z. F., Sun R. X., Zeng R., He S. M., and Gao W. (2005) pFind 2.0: a software package for peptide and protein identification via tandem mass spectrometry. Rapid Commun Mass Spectrom 21, 2985–2991. [DOI] [PubMed] [Google Scholar]
- 58. Li D., Fu Y., Sun R., Ling C. X., Wei Y., Zhou H., Zeng R., Yang Q., He S., and Gao W. (2005) pFind: a novel database-searching software system for automated peptide and protein identification via tandem mass spectrometry. Bioinformatics 21, 3049–3050. [DOI] [PubMed] [Google Scholar]
- 59. Kim M. S., Pinto S. M., Getnet D., Nirujogi R. S., Manda S. S., Chaerkady R., Madugundu A. K., Kelkar D. S., Isserlin R., Jain S., Thomas J. K., Muthusamy B., Leal-Rojas P., Kumar P., Sahasrabuddhe N. A., Balakrishnan L., Advani J., George B., Renuse S., Selvan L. D., Patil A. H., Nanjappa V., Radhakrishnan A., Prasad S., Subbannayya T., Raju R., Kumar M., Sreenivasamurthy S. K., Marimuthu A., Sathe G. J., Chavan S., Datta K. K., Subbannayya Y., Sahu A., Yelamanchi S. D., Jayaram S., Rajagopalan P., Sharma J., Murthy K. R., Syed N., Goel R., Khan A. A., Ahmad S., Dey G., Mudgal K., Chatterjee A., Huang T. C., Zhong J., Wu X., Shaw P. G., Freed D., Zahari M. S., Mukherjee K. K., Shankar S., Mahadevan A., Lam H., Mitchell C. J., Shankar S. K., Satishchandra P., Schroeder J. T., Sirdeshmukh R., Maitra A., Leach S. D., Drake C. G., Halushka M. K., Prasad T. S., Hruban R. H., Kerr C. L., Bader G. D., Iacobuzio-Donahue C. A., Gowda H., and Pandey A. (2014) A draft map of the human proteome. Nature 509, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Creasy D. M., and Cottrell J. S. (2004) Unimod: Protein modifications for mass spectrometry. Proteomics 4, 1534–1536. [DOI] [PubMed] [Google Scholar]
- 61. Villen J., Beausoleil S. A., Gerber S. A., and Gygi S. P. (2007) Large-scale phosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. U.S.A. 104, 1488–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zolg D. P., Wilhelm M., Schmidt T., Médard G., Zerweck J., Knaute T., Wenschuh H., Reimer U., Schnatbaum K., and Kuster B. (2018) ProteomeTools: Systematic characterization of 21 post-translational protein modifications by LC-MS/MS using synthetic peptides. Mol. Cell Proteomics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zolg D. P., Wilhelm M., Schnatbaum K., Zerweck J., Knaute T., Delanghe B., Bailey D. J., Gessulat S., Ehrlich H. C., Weininger M., Yu P., Schlegl J., Kramer K., Schmidt T., Kusebauch U., Deutsch E. W., Aebersold R., Moritz R. L., Wenschuh H., Moehring T., Aiche S., Huhmer A., Reimer U., and Kuster B. (2017) Building ProteomeTools based on a complete synthetic human proteome. Nat. Methods 14, 259–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chambers M. C., Maclean B., Burke R., Amodei D., Ruderman D. L., Neumann S., Gatto L., Fischer B., Pratt B., Egertson J., Hoff K., Kessner D., Tasman N., Shulman N., Frewen B., Baker T. A., Brusniak M. Y., Paulse C., Creasy D., Flashner L., Kani K., Moulding C., Seymour S. L., Nuwaysir L. M., Lefebvre B., Kuhlmann F., Roark J., Rainer P., Detlev S., Hemenway T., Huhmer A., Langridge J., Connolly B., Chadick T., Holly K., Eckels J., Deutsch E. W., Moritz R. L., Katz J. E., Agus D. B., MacCoss M., Tabb D. L., and Mallick P. (2012) A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yuan Z. F., Liu C., Wang H. P., Sun R. X., Fu Y., Zhang J. F., Wang L. H., Chi H., Li Y., Xiu L. Y., Wang W. P., and He S. M. (2012) pParse: a method for accurate determination of monoisotopic peaks in high-resolution mass spectra. Proteomics 12, 226–235. [DOI] [PubMed] [Google Scholar]
- 66. Jin Z., Fu Z., Yang J., Troncosco J., Everett A. D., and Van Eyk J. E. (2013) Identification and characterization of citrulline-modified brain proteins by combining HCD and CID fragmentation. Proteomics 13, 2682–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yau K. W., and Hardie R. C. (2009) Phototransduction motifs and variations. Cell 139, 246–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liu N. K., and Xu X. M. (2012) Neuroprotection and its molecular mechanism following spinal cord injury. Neural Regen. Res. 7, 2051–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Moya-Alvarado G., Gershoni-Emek N., Perlson E., and Bronfman F. C. (2016) Neurodegeneration and Alzheimer's disease (AD). What can proteomics tell us about the Alzheimer's brain? Mol. Cell. Proteomics 15, 409–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Uhlen M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A., Odeberg J., Djureinovic D., Takanen J. O., Hober S., Alm T., Edqvist P. H., Berling H., Tegel H., Mulder J., Rockberg J., Nilsson P., Schwenk J.M., Hamsten M., von Feilitzen K., Forsberg M., Persson L., Johansson F., Zwahlen M., von Heijne G., Nielsen J., and Pontén F. (2015) Proteomics Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- 71. Huang H., Zhang D., Wang Y., Perez-Neut M., Han Z., Zheng Y.G., Hao Q., and Zhao Y. (2018) Lysine benzoylation is a histone mark regulated by SIRT2. Nature Communications 9, 3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Baker P. R., and Chalkley R. J. (2014) MS-viewer: a web-based spectral viewer for proteomics results. Mol. Cell. Proteomics 13, 1392–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The chemically synthesized peptide data and the draft map of human proteome were downloaded from the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) with the dataset identifiers PXD009449 and PXD000561, respectively. The modified-peptide spiked-in proteome data have been deposited to iProX (a full member of the ProteomeXchange consortium, http://www.iprox.org) with the dataset identifier IPX0001318000. All of the spectra identified as modified peptide have been uploaded to MS-Viewer data sets (72) (the search keys for the simulated data, the chemically synthesized peptide library data, the modified-peptide spiked-in complex proteome data and the draft map of human proteome are 9yxuedofcl, qjgpjpgwsg, nsdxlsv1qu and jr8wrhwar6, respectively).The PTMiner software and the extended Ascore algorithm coded with Matlab are available at http://fugroup.amss.ac.cn/software/ptminer/ptminer.html.
PTMiner now supports spectra generated using collision-induced dissociation (CID) or HCD in 'mgf' format as data input, and the output of pFind, Sequest or MSFragger as the search result input. Besides, PTMiner can also read in tab-delimited files transferred from any search engines.