

Helicobacter pylori is the strongest risk factor for gastric cancer. Initial interactions between H. pylori and its host occur at the epithelial cell surface, and this activates signaling pathways that drive oncogenesis. This manuscript defines strain-specific gastric epithelial proteomic changes induced by H. pylori in vivo that are critical for initiation of the gastric carcinogenesis. Protein targets were validated in human gastric epithelial cells in vitro, primary human gastric epithelial monolayers, and H. pylori-infected gerbil and human tissue in vivo.
Keywords: Tandem Mass Spectrometry, iTRAQ, Pathway Analysis, Immunohistochemistry, Microbiology, Molecular Biology, Infectious Disease, Inflammatory Response, Gastrointestinal Disease, Cancer Biomarker(s), G3BP2, Helicobacter pylori, RABEP2
Graphical Abstract

Highlights
H. pylori dysregulates the in vivo gastric proteome of gerbils in a strain-specific manner.
H. pylori increases RABEP2 and G3BP2 levels in cell culture.
H. pylori upregulates RABEP2 and G3BP2 in gerbil and human gastric epithelium.
Levels of RABEP2 and G3BP2 increase with severity of malignant lesions in vivo.
Abstract
Helicobacter pylori is the strongest risk factor for gastric cancer. Initial interactions between H. pylori and its host originate at the microbial-gastric epithelial cell interface, and contact between H. pylori and gastric epithelium activates signaling pathways that drive oncogenesis. One microbial constituent that increases gastric cancer risk is the cag pathogenicity island, which encodes a type IV secretion system that translocates the effector protein, CagA, into host cells. We previously demonstrated that infection of Mongolian gerbils with a carcinogenic cag+ H. pylori strain, 7.13, recapitulates many features of H. pylori-induced gastric cancer in humans. Therefore, we sought to define gastric proteomic changes induced by H. pylori that are critical for initiation of the gastric carcinogenic cascade. Gastric cell scrapings were harvested from H. pylori-infected and uninfected gerbils for quantitative proteomic analyses using isobaric tags for relative and absolute quantitation (iTRAQ). Quantitative proteomic analysis of samples from two biological replicate experiments quantified a total of 2764 proteins, 166 of which were significantly altered in abundance by H. pylori infection. Pathway mapping identified significantly altered inflammatory and cancer-signaling pathways that included Rab/Ras signaling proteins. Consistent with the iTRAQ results, RABEP2 and G3BP2 were significantly up-regulated in vitro, ex vivo in primary human gastric monolayers, and in vivo in gerbil gastric epithelium following infection with H. pylori strain 7.13 in a cag-dependent manner. Within human stomachs, RABEP2 and G3BP2 expression in gastric epithelium increased in parallel with the severity of premalignant and malignant lesions and was significantly elevated in intestinal metaplasia and dysplasia, as well as gastric adenocarcinoma, compared with gastritis alone. These results indicate that carcinogenic strains of H. pylori induce dramatic and specific changes within the gastric proteome in vivo and that a subset of altered proteins within pathways with oncogenic potential may facilitate the progression of gastric carcinogenesis in humans.
Gastric adenocarcinoma is the third leading cause of cancer-related death worldwide and accounts for greater than 720,000 deaths annually (1). The strongest known risk factor for this malignancy is chronic gastritis induced by the microbial pathogen Helicobacter pylori (2). H. pylori colonizes greater than 50% of the world's population (3); however, only a fraction of infected individuals ever develop cancer. The specific mechanisms by which H. pylori initiates gastric carcinogenesis are not completely understood, but disease outcomes are mediated through complex interactions between H. pylori strain-specific virulence determinants and host cell signaling responses. Initial interactions between H. pylori and the host originate at the gastric epithelial cell interface, and contact between H. pylori and gastric epithelial cells activates signaling pathways that drive oncogenesis. One microbial constituent that increases the risk for gastric cancer is the cag pathogenicity island, which encodes a bacterial type IV secretion system (T4SS)1 that translocates the effector protein, CagA, into host gastric epithelial cells. Intracellular CagA can become phosphorylated (4–6) or remain unphosphorylated. In either form, CagA affects multiple host pathways that lead to alterations in cell morphology, signaling pathways, and inflammatory responses (7–11). Transgenic mice that overexpress CagA develop gastric epithelial cell hyperproliferation and gastric adenocarcinoma (12), further implicating CagA as a bacterial oncoprotein.
We previously demonstrated that infection of Mongolian gerbils with a carcinogenic cag+ H. pylori strain, 7.13, rapidly induces gastric inflammation and carcinogenesis (13, 14). Intestinal-type gastric cancer in humans progresses through a series of well-defined pathological stages from normal gastric mucosa to superficial nonatrophic gastritis, to premalignant lesions including atrophic gastritis, spasmolytic-expressing metaplasia (SPEM), intestinal metaplasia, dysplasia, and finally gastric adenocarcinoma (15). Gerbils exhibit a similar stepwise progression of discrete histopathologic stages along the gastric carcinogenic cascade, including the development of SPEM (16), dysplasia, and adenocarcinoma within the context of mucosal inflammation (13, 14). Defining the mechanisms by which H. pylori initiates this cascade has important clinical implications for disease prevention and novel therapeutic interventions. Thus, we hypothesized that H. pylori induces gastric cell-specific proteomic changes critical for initiation of gastric carcinogenesis in the Mongolian gerbil model which are also important in gastric cancer progression in humans. To test this hypothesis, gastric tissue cell scrapings were harvested from uninfected gerbils and gerbils infected with the carcinogenic cag+ H. pylori strain, 7.13. Samples were isolated from gerbils prior to the development of any premalignant lesions to directly assess early proteomic changes that may initiate and drive progression along the entire gastric carcinogenic cascade. Gastric samples from two biological replicate experiments were subjected to quantitative proteomic analysis using isobaric tags for relative and absolute quantitation (iTRAQ). Significant H. pylori-induced proteomic alterations were analyzed relative to uninfected controls and subjected to canonical signaling and disease pathway mapping using Ingenuity Pathway Analysis (IPA) to identify critical signaling pathways altered during the early stages of H. pylori infection. Proteomic changes were then validated in in vitro and ex vivo human gastric epithelial cell-H. pylori cocultures, in vivo gerbil gastric epithelium, and in vivo human gastric tissue sections.
EXPERIMENTAL PROCEDURES
Mongolian Gerbil Model
Outbred male Mongolian gerbils were purchased from Charles River Laboratories (Wilmington, MA) and housed in the Vanderbilt University Animal Care Facilities. Wild-type carcinogenic cag+ H. pylori strain 7.13 was minimally passaged on trypticase soy agar plates with 5% sheep blood (BD Biosciences, San Jose, CA) and in Brucella broth (BD Biosciences) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals, Flowery Branch, GA) for sixteen hours at 37 °C with 5% CO2. Gerbils were orogastrically challenged with sterile Brucella broth (negative control), wild-type cag+ H. pylori strain 7.13, or a cagE− isogenic mutant, and gerbils were euthanized 6 weeks post-challenge, as previously described (14). The Vanderbilt University Institutional Animal Care and Use Committee (IACUC) approved all experiments and procedures.
Quantitative H. pylori Culture
Linear strips of gastric tissue, extending from the squamocolumnar junction through the proximal duodenum were harvested and homogenized in sterile phosphate-buffered saline (PBS, Corning, Corning, NY). Following serial dilution, samples were plated on selective trypticase soy agar (TSA, Remel) plates with 5% sheep blood (Hemostat Laboratories, Dixon, CA) containing vancomycin (Sigma-Aldrich, St. Louis, MO, 20 μg/ml), nalidixic acid (Sigma-Aldrich, 10 μg/ml), bacitracin (Calbiochem, San Diego, CA, 30 μg/ml), and amphotericin B (Sigma-Aldrich, 2 μg/ml) for selection, isolation and quantification of H. pylori, as previously described (14). Plates were incubated for 3 to 5 days at 37 °C with 5% CO2. Colonies were identified as H. pylori based on characteristic spiral morphology, Gram stain (Becton, Dickinson and Company, Franklin Lakes, NJ), and urease and oxidase enzyme activities (Becton, Dickinson and Company). Colony counts were expressed as log colony-forming units (CFU) per gram of gastric tissue.
Histopathology and Steiner Stain
Linear strips of gastric tissue, extending from the squamocolumnar junction through the proximal duodenum, were fixed in 10% neutral-buffered formalin (Azer Scientific, Morgantown, PA), paraffin-embedded, and stained with hematoxylin and eosin (H&E) as well as with a modified Steiner stain for detection of H. pylori. A single pathologist (MBP), blinded to treatment groups, assessed and scored indices of inflammation and injury and topography of colonization by Steiner stain. Severity of acute and chronic inflammation was graded on a scale from 0–3 (absent (0), mild (1), moderate (2), or marked inflammation (3)) in both the gastric antrum and corpus, leading to a maximum cumulative score of twelve, as previously described (14).
Isobaric Tags for Relative and Absolute Quantitation (iTRAQ)
Gastric cell scrapings were harvested from linear strips of gerbil gastric tissue, extending from the squamocolumnar junction through the proximal duodenum, and were solubilized in RIPA buffer (50 mm Tris, pH 7.2; 150 mm NaCl; 1% Triton X-100; and 0.1% SDS). Gastric cell scrapings were used to examine proteins from the heterogeneous cell populations of the gastric mucosa. Gastric proteins samples from H. pylori-infected tissues (n = 3) were pooled together, as were gastric proteins samples from uninfected tissue (n = 3) for each independent replicate experiment to achieve statistical power to detect significant differences, as previously performed (17). An equivalent amount of protein was taken from each biological sample, such that the pooled sample contained 100 μg of protein in total. Gastric protein samples were precipitated with ice-cold acetone overnight at −20 °C. Following precipitation, samples were centrifuged at 18,000 × g at 4 °C, and precipitates were washed with cold acetone, dried, and reconstituted in 8 m urea in 250 mm triethylammonium bicarbonate buffer (TEAB, pH 8.0). Samples were reduced with 5 μl of 50 mm tris (2-carboxyethyl) phosphine (TCEP), alkylated with 2.5 μl of 200 mm methyl methanethiosulfonate (MMTS), diluted with TEAB to obtain a final solution containing 2 m urea, and digested with sequencing-grade trypsin (Promega, Madison, WI) overnight. To facilitate quantitative analysis, peptides were labeled with iTRAQ reagents (AB Sciex, Concord, Ontario, Canada), according to the manufacturer's instructions. For each 50 μg of protein, one unit of labeling reagent was used. Labeling reagent was reconstituted in ethanol, such that each protein sample was labeled at a final concentration of 90% ethanol, and labeling was performed for two hours. Pooled lysates from H. pylori-infected or uninfected gastric tissue were labeled with 4-plex iTRAQ reagent 117 or 115, respectively. Two-plex iTRAQ comparisons were then conducted. The resulting labeled peptides were desalted by a modified Stage-tip method, as previously described (18). iTRAQ-labeled samples were mixed and acidified with trifluoroacetic acid (TFA). A disc of C18 extraction membrane (C18 SPE Empore disk, Chrom Tech Inc., Apple Valley, MN) was cored with a 16-gauge needle, and the cored piece of membrane was fitted tightly into a 200 μl pipette tip. Three mg of C18 resin (Jupiter C18, 5 μm particle size, Phenomenex, Torrance, CA) were suspended in 200 μl of methanol and loaded into the pipette tip containing the cored C18 membrane. The C18 material was packed into the tip using centrifugation to form a resin-packed C18 clean-up tip (resin tip). Resin tips were equilibrated with 0.1% TFA in HPLC-grade water (Fisher Scientific, Waltham, MA), labeled peptides were loaded into the tip by centrifugation, washed with 0.1% TFA, and eluted with 100 μl of 80% acetonitrile (ACN) containing 0.1% TFA. Eluted peptides were dried by speed vacuum centrifugation, and then peptides were reconstituted in 0.1% formic acid and analyzed by 2-dimensional liquid chromatography-coupled tandem mass spectrometry (2D LC-MS/MS). Peptides were loaded onto a self-packed biphasic C18/SCX MudPIT column using a Helium-pressurized cell (pressure bomb).
The MudPIT column consisted of 360 × 150 μm i.d. fused silica, which was fitted with a filter-end fitting (IDEX Health & Science, Oak Harbor, WA) and packed with 6 cm of Luna SCX material (5 μm, 100Å) followed by 4 cm of Jupiter C18 material (5 μm, 300Å, Phenomenex). Once samples were loaded, the MudPIT column was connected using an M-520 microfilter union (IDEX Health & Science) to an analytical column (360 μm × 100 μm i.d.), equipped with a laser-pulled emitter tip and packed with 20 cm of C18 reverse phase material (Jupiter, 3 μm beads, 300Å, Phenomenex). Using a Dionex Ultimate 3000 nanoLC and autosampler, MudPIT analysis was performed with 13 salt steps (0, 25, 50, 75, 100, 150, 200, 250, 300, 500, 1 m, 2 m, and 5 m ammonium acetate). Following each salt pulse delivered by the autosampler, peptides were gradient-eluted from the reverse analytical column at a flow rate of 350 nL/min. Mobile phase solvents (HPLC-grade) consisted of 0.1% formic acid, 99.9% water (solvent A) and 0.1% formic acid, 99.9% acetonitrile (solvent B). For the peptides from the first 11 SCX fractions, the reverse phase gradient consisted of 2–50% B in 80 min, followed by a 10-min equilibration at 2% solvent B. For the last 2 SCX-eluted peptide fractions, the peptides were eluted from the reverse phase analytical column using a gradient of 2–98% solvent B in 80 min, followed by a 10-min equilibration at 2% solvent B. A Q Exactive Plus mass spectrometer (Thermo Scientific, Waltham, WA), equipped with a nanoelectrospray ionization source, was used to mass analyze the eluting peptides. The Q Exactive instrument was operated in data-dependent mode acquiring higher energy collisional disassociation (HCD) MS/MS scans (r = 17,500) after each MS1 scan on the 20 most abundant ions using an MS1 ion target of 3 × 106 ions and an MS2 target of 1 × 105 ions. The HCD-normalized collision energy was set to 30, dynamic exclusion was set to 30 s, and peptide match and isotope exclusion were enabled. Mass spectra were processed using the Spectrum Mill software package (version B.04.00, Agilent Technologies) and were searched against a database containing the Mus musculus subset of the UniprotKB protein database (www.uniprot.org, UniProtKB Release 2012_06, 16,651 protein entries). MS/MS spectra acquired on the same precursor m/z (±0.01 m/z) within ±1 s in retention were merged. MS/MS spectra of poor quality, which did not have a sequence tag length >1, were excluded. A minimum matched peak intensity requirement was set to 50%. Additional search parameters included: trypsin enzyme specificity with a maximum of three missed cleavages, ±20 ppm precursor mass tolerance, ±20 ppm (HCD) product mass tolerance, and fixed modifications including MMTS alkylation of cysteines and iTRAQ labeling of lysines and peptide N termini. Oxidation of methionine was allowed as a variable modification. Autovalidation was performed such that peptide assignments to mass spectra were designated as valid following an automated procedure during which score thresholds were optimized separately for each precursor charge state, and the maximum target-decoy-based false-discovery rate (FDR) was set to 1.0% (19). If peptide sequences were contained in multiple protein entries present in the database, proteins were groups together and the protein accession number of the highest scoring protein was provided in supplemental Tables S2 and S3, as previously reported (19). To obtain iTRAQ protein ratios, the median was calculated for all peptides assigned to each protein. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (https://www.ebi.ac.uk/pride/archive/) with the data set identifier PXD009583. For statistical analysis, log2 protein ratios were fit to a normal distribution using nonlinear (least squares) regression. The calculated mean derived from the Gaussian fit was used to normalize individual log2 ratios for each quantified protein. The normalized log2 ratios were then fit to a normal distribution, and the mean and standard deviation values derived from the Gaussian fit of the normalized ratios were used to calculate p values using Z score statistics. Subsequently, p values were corrected for multiple comparisons by the Benjamini-Hochberg (BH) method (BH FDR p < 0.05) (20). After applying the multiple comparisons by the BH method, we allowed all quantified proteins with significant p values less than the BH correction. This resulted in the quantification of 166 proteins significantly altered in abundance following H. pylori infection (supplemental Table S1).
RABEP2 and G3BP2 peptides identified from the Mus musculus UniprotKB protein database were queried using the Protein BLAST® (Basic Local Alignment Search Tool) algorithm accessed through the NCBI, National Library of Medicine (https://blast.ncbi.nlm.nih.gov/Blast.cgi). For BLAST queries, peptide sequences were individually queried and matched to predicted protein sequences from the Mongolian gerbil (Meriones unguiculatus). For both RABEP2 and G3BP2 proteins, all peptides identified following Mus musculus database searches conducted using Spectrum Mill aligned with 100% identity to the predicted peptide sequences from Meriones unguiculatus.
Ingenuity Pathway Analysis (IPA®, Qiagen, Redwood City, CA) was used to determine canonical signaling pathways, disease categories, and networks that were significantly changed with H. pylori infection. Proteins that were significantly changed following iTRAQ quantitative analysis, totaling 166 proteins (supplemental Table S1), were used as input for IPA analysis, the Ingenuity Knowledge Base was used as the reference set, and relationships considered for affected networks included both direct and indirect relationships. For Ingenuity Pathway Analysis, a p value was calculated in IPA using Fisher's exact test, and the p value was then converted to a pathway score by converting the p value to the negative log of the p value for each pathway. In order to map proteins to disease categories, IPA was used to determine specific cellular functions that are significantly associated with the input protein dataset. The p value for each category was calculated using Fisher's Exact test in IPA and was used as a measure of the relationship between a set of proteins and the corresponding function and disease.
Immunohistochemistry Analysis of Gerbil Gastric Tissue
To assess RABEP2 and G3BP2 protein expression in gerbil gastric tissue, immunohistochemistry (IHC) analysis was performed on deparaffinized gastric tissue sections using rabbit polyclonal anti-RABEP2 (1:1000, Abcam, Cambridge, MA) and G3BP2 antibodies (1:1000, Abcam). A single pathologist (MBP), blinded to treatment groups, scored epithelial RABEP2 and G3BP2 immunohistochemistry staining in the gastric sections. The percentage of positive epithelial cells was assessed and the intensity of epithelial staining was graded on a scale of 0–3 (absent (0), weak (1), moderate (2), or strong (3)). Immunohistochemistry data are represented as epithelial RABEP2 and G3BP2 scores, which were determined by multiplying the RABEP2 or G3BP2 staining intensity by the percentage of positively stained cells, as previously described (22).
Flow Cytometry Analysis of Gerbil Gastric Tissue
Gastric epithelial cells were isolated from frozen uninfected or H. pylori-infected gerbil gastric tissue using a dissociation and dispersion technique, as previous described (21, 22). Briefly, gastric tissue was treated with 10 mm DTT at room temperature for 30 min. Samples were then transferred to ice and incubated with 1.0 mm EDTA for 1 h, shaking every 10 min to dissociate and disperse cells. Dispersed cells were then filtered through a 70 μm filter (BD FalconTM, Franklin Lakes, NJ) to isolate single cells. Cells were fixed with 0.1% paraformaldehyde (Fisher Scientific) and then permeabilized with ice-cold methanol (Fisher Scientific). Cells were incubated with a rabbit polyclonal anti-RABEP2 antibody (0.4 μg/ml, Thermo, Waltham, MA) or a rabbit polyclonal anti-G3BP2 antibody (0.4 μg/ml, Thermo) at room temperature for 20 min. Cells were washed and stained with goat anti-rabbit secondary antibody conjugated with Alexa Fluor 488 (1:400, BD Biosciences) and a mouse monoclonal anti-pan-cytokeratin antibody conjugated with phycoerythrin (PE, 0.5 μg/ml, Abcam). Cells were acquired using a Guava EasyCyte Flow Cytometer (EMD Millipore, Burlington, MA) and PE pan-cytokeratin-positive cells were analyzed for RABEP2 and G3BP2 expression by using FlowJo (Tree Star Inc., Ashland, OR).
H. pylori Strains and Growth Conditions
Wild-type carcinogenic cag+ H. pylori strain 7.13, a 7.13 isogenic cagE− (cag secretion system ATPase) mutant, or a 7.13 isogenic cagA− (cag secretion system effector protein) mutant were cultured on trypticase soy agar with 5% sheep blood agar plates (BD Biosciences) for in vitro passage. Wild-type cag+ strain PMSS1 or its PMSS1 cagE− isogenic mutant were cultured on trypticase soy agar with 5% sheep blood agar plates (BD Biosciences) for in vitro passage. Isogenic mutants were also cultured on Brucella agar (BD Biosciences) plates containing 20 μg/ml kanamycin (Sigma) to confirm presence of the kanamycin antibiotic resistance cassette. H. pylori strains were then cultured in Brucella broth (BD Biosciences) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals) for 16 to 18 h at 37 °C with 5% CO2.
Human Gastric Epithelial Cell Culture and H. pylori Coculture
AGS, MKN28, and SNU1 human gastric epithelial cells were grown in RPMI 1640 (AGS and MKN28 cells, Life Technologies, Carlsbad, CA) or DMEM (SNU1 cells, Life Technologies) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals), l-glutamine (2 mm, BD Biosciences), and HEPES buffer (1 mm, Cellgro, Corning, NY) at 37 °C with 5% CO2. Wild-type H. pylori strain 7.13 or 7.13 isogenic mutants were cocultured with gastric epithelial cells at a multiplicity of infection (MOI) of 100:1 for 24 h. Gastric epithelial cocultures were harvested for Western blot analysis.
Transfection of Human Gastric Epithelial Cells with Small Interfering RNA (siRNA)
AGS human gastric epithelial cells were transiently transfected using DharmaFECT siRNA transfection reagents (DharmaconTM, Lafayette, CO), in accordance with the manufacturer's instructions. Briefly, transfection reagent was mixed with 5 μm of nontargeting (NT) siRNA (DharmaconTM), RABEP2-targeting siRNA (DharmaconTM), or G3BP2-targeting siRNA (DharmaconTM) in reduced serum OPTI-MEM cell culture medium (Life Technologies). Cells were incubated with a final concentration of 25 nm siRNA transfection mixtures for 24 h, during which time cells were cocultured with or without H. pylori strain 7.13 at a MOI of 100:1 for 24 h. Transfected cells were harvested for Western blotting and subjected to various functional assays, including cellular viability, proliferation, apoptosis, migration, and invasion.
Primary Human Gastric Organoid 2D Monolayers
Human fundus was collected during sleeve gastrectomies according to an approved University of Cincinnati IRB protocol (IRB protocol number: 2015–4869). Gastric tissue was washed, digested, and isolated glands were incubated in Matrigel, as previously described (23). Primary human gastric organoids were then transferred to 2D epithelial cell monolayers, as previously described (23). Briefly, Matrigel was removed and 3D gastric organoids were plated on collagen-coated plates (23). Primary 2D gastric monolayers were then cocultured with wild-type cag+ H. pylori strains 7.13 or PMSS1, or their isogenic cagE− mutants at a multiplicity of infection (MOI) of 100:1 for 24 h. Gastric organoid cocultures were then harvested for Western blot analysis.
Western Blot Analysis
Whole cell protein lysates from human gastric epithelial cell and primary human gastric monolayer cocultures were harvested using RIPA buffer (50 mm Tris, pH 7.2; 150 mm NaCl; 1% Triton X-100; and 0.1% SDS) containing protease (Roche, Basel, Switzerland) and phosphatase (Sigma) inhibitors, and protein concentrations were determined by a bicinchoninic acid (BCA) assay (Pierce, Waltham, MA). Proteins (20 μg) were separated by SDS-PAGE and transferred (Bio-Rad, Hercules, CA) to polyvinylidene difluoride membranes (PVDF, Millipore). Human RABEP2 and G3BP2 protein expression was quantified using a rabbit polyclonal anti-RABEP2 antibody (1:500, Abcam) or a rabbit polyclonal anti-G3BP2 antibody (1:500, Abcam or 1:500, Thermo). RABEP2 and G3BP2 expression were standardized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using a mouse polyclonal anti-GAPDH antibody (1:5000, Millipore). Primary antibodies were detected using goat anti-rabbit or goat anti-mouse horseradish peroxidase (HRP)-conjugated secondary antibodies (1:10,000, Promega, Madison, WI). Protein levels were visualized by Western Lightning Chemiluminescence Reagent Plus (PerkinElmer, Waltham, MA), according to the manufacturer's instructions and then quantified by densitometry using the ImageJ Software (NIH, Bethesda, MD). Protein expression was normalized to the protein expression levels of GAPDH to ensure equal protein loading. Protein expression is represented as fold over uninfected control.
Cell Viability, Proliferation, and Apoptosis Assays
AGS human gastric epithelial cells were plated in 96-well plates and then left untreated or transfected with nontargeting siRNA, RABEP2-targeting (NT) siRNA, or G3BP2-targeting siRNA. After 24 h, cells were then cocultured with H. pylori strain 7.13 and then analyzed using an MTT Cell Proliferation Assay (Sigma) to assess cell viability and proliferation or a Cytochrome c Oxidase Assay (Sigma) to assess apoptosis, according to the manufacturer's instructions.
Cell Migration and Invasion Assays
AGS human gastric epithelial cells were plated in a migration/invasion chamber within 24-well plates and then were left untreated or transfected with nontargeting (NT) siRNA, RABEP2-targeting siRNA, or G3BP2-targeting siRNA. During the transfection, cells were then cocultured with H. pylori strain 7.13 and harvested for analysis using a Chemotaxis Cell Migration Assay (Sigma) to asses cell migration or a Cell Invasion Assay (Sigma) to assess cell invasion, both according to the manufacturer's instructions.
Immunohistochemistry Analysis of Human Gastric Tissue
The Committees on Ethics of Universidad del Valle and Hospital Departamental de Nariño in Colombia, the Committees on Ethics of Pontificia Universidad Catolica de Chile, and the Institutional Review Board (IRB) of Vanderbilt University Medical Center approved this protocol. Gastric tissue samples from patients residing in regions of high gastric cancer risk in Chile or in the Colombian Andean mountains were used for immunohistochemistry analysis (24). Immunohistochemistry was performed on paraffin-embedded tissue samples from H. pylori-infected patients with nonatrophic gastritis, multifocal atrophic gastritis, intestinal metaplasia, dysplasia, or gastric cancer. Tissue sections were deparaffinized and stained with a rabbit polyclonal anti-RABEP2 antibody (1:1000, Abcam) or a rabbit polyclonal anti-G3BP2 antibody (1:1000, Abcam). A single pathologist (MBP), blinded to treatment groups, scored epithelial RABEP2 and G3BP2 immunohistochemistry staining in the gastric sections. The percentage of positive epithelial cells was assessed, and the intensity of epithelial staining was graded on a scale of 0–3 (absent (0), weak (1), moderate (2), or strong (3)). The immunohistochemistry score was determined by multiplying the staining intensity by the percentage of positively cells stained, as previously described (22).
Experimental Design and Statistical Rationale
Proteomic iTRAQ experiments were performed on two independent occasions with two independent biological replicate experiments containing three individual animals per group. For statistical analysis of iTRAQ protein ratios, log2 protein ratios for all quantified proteins were fit to a normal distribution using nonlinear (least squares) regression. The calculated mean derived from the Gaussian fit was used to normalize individual log2 ratios for each quantified protein. The normalized log2 ratios were then fit to a normal distribution, and the mean and standard deviation values derived from the Gaussian fit of the normalized ratios were used to calculate p values using Z score statistics. Subsequently, p values were corrected for multiple comparisons by the Benjamini-Hochberg method (BH FDR p < 0.05) (20). After applying the multiple comparisons by the BH method, we allowed all quantified proteins with significant p values less than the BH correction. This resulted in the quantification of 166 proteins significantly altered in abundance following H. pylori infection (supplemental Table S1). For Ingenuity Pathway Analysis, a p value was calculated in IPA using Fisher's exact test, and the p value was then converted to a pathway score by converting the p value to the negative log of the p value for each pathway. For in vitro, ex vivo, and in vivo data, mean values with standard error of the mean were determined from experiments performed on at least three independent occasions. ANOVA and Mann-Whitney U tests were used for statistical comparisons. All experiments were performed on at least three independent occasions, and p < 0.05 was considered statistically significant.
Study Approval
All animal studies have been approved by the Vanderbilt University Institutional Animal Care and Use Committee (IACUC). All human studies were conducted in accordance with the Declaration of Helsinki principles and have been approved by the Committees on Ethics of Universidad del Valle and Hospital Departamental de Nariño in Colombia, the Committees on Ethics of Pontificia Universidad Catolica de Chile, the University of Cincinnati, and the Vanderbilt University Institutional Review Board (IRB).
RESULTS
H. pylori Colonizes the Gastric Epithelium of Mongolian Gerbils and Induces Significant Levels of Gastric Inflammation
Our previous studies demonstrated that infection of Mongolian gerbils with a carcinogenic cag+ H. pylori strain, 7.13, recapitulates key features of H. pylori-induced gastric inflammation and carcinogenesis in humans (13, 14). To define host gastric proteomic changes that occur early following H. pylori infection in a controlled in vivo environment, Mongolian gerbils were challenged with Brucella broth (negative control) or with H. pylori strain 7.13. Gerbil gastric tissue was harvested 6 weeks post-challenge, prior to the development of premalignant lesions, to assess H. pylori colonization, inflammation, and global gastric proteomic changes in vivo. Colonization efficiency was 100% for all H. pylori-challenged gerbils (data not shown). Colonization density was not significantly different among H. pylori-infected gerbils from the two independent biological replicates, and, as expected, H. pylori were not isolated from uninfected (UI) gerbils (Fig. 1A). To assess the topography of H. pylori throughout the entire gastric mucosa, Steiner staining was performed on gastric tissue sections from uninfected and H. pylori-infected gerbils. Steiner staining confirmed that H. pylori were distributed throughout the gastric glands of H. pylori-infected gerbils, whereas no H. pylori were detected among uninfected gerbils, as expected (Fig. 1B). To assess the severity of gastric inflammation, gastric tissue sections were stained with H&E and then scored for acute and chronic inflammation within the antrum and corpus of the stomach. Mongolian gerbils infected with H. pylori exhibited significantly higher levels of gastric inflammation compared with uninfected animals (Fig. 1C), and, at this stage of infection, most of the inflammation was acute and localized to the antrum.
Fig. 1.

H. pylori strain 7.13 colonizes gerbils and induces inflammation. A, Gastric tissue from uninfected (UI) and H. pylori-infected gerbils was homogenized and plated on selective trypticase soy agar plates with 5% sheep blood for isolation of H. pylori. Plates were incubated for 3–5 days, and colonization density was determined and expressed as log colony-forming units (CFU) per gram of gastric tissue. Each data point represents colonization density from an individual animal. B, Linear strips of gastric tissue, extending from the squamocolumnar junction through the proximal duodenum, were fixed in 10% neutral-buffered formalin, embedded in paraffin, and stained with Steiner stain to identify H. pylori topography within gastric tissue sections. White arrows designate regions with H. pylori colonization. C, Linear strips of gastric tissue, extending from the squamocolumnar junction through the proximal duodenum, were fixed in 10% neutral-buffered formalin, embedded in paraffin, and stained with hematoxylin and eosin. A pathologist (MBP), blinded to the treatment groups, assessed indices of inflammation. Severity of acute and chronic inflammation was graded 0–3 (absent (0), mild (1), moderate (2), or marked (3) inflammation) in both the gastric antrum and corpus. Each data point represents inflammation scores from an individual animal. Mann-Whitney U test was used to determine statistical significance between uninfected and infected groups. ***, p < 0.005.
H. pylori Selectively Dysregulates the Gastric Proteome In Vivo, Which Significantly Alters Inflammatory- and Cancer-Signaling Pathways
To define the effects of H. pylori on the host gastric proteome, Mongolian gerbil cell scrapings were harvested and pooled from gastric tissue isolated from H. pylori-infected gerbils or uninfected gerbils for quantitative proteomic analyses using isobaric tags for relative and absolute quantitation (iTRAQ) and then quantified targets were validated in in vitro human gastric epithelial cells, in vivo gerbil gastric tissue, ex vivo primary human gastric monolayers, and in vivo human tissue from patients at high risk for gastric cancer. This workflow is diagramed in Fig. 2. The use of gastric cell scrapings allowed for analysis of heterogeneous population of cells that not only included the gastric epithelium, but also included inflammatory infiltrates within the epithelium and submucosa. Based on the experimental design in Fig. 2, pooled lysates were digested and labeled with iTRAQ reagents, and quantitative analyses were performed following 2D LC-MS/MS on a Q Exactive Plus mass spectrometer. Mass spectra were searched against a database containing the Mus musculus subset of the UniprotKB protein database because a complete annotated Meriones unguiculatus protein database is unavailable, and there is high homology between Mus musculus proteins and predicted Meriones ungulculatus genome sequences (25). Quantitative proteomic analysis from two biological replicate experiments quantified a total of 2764 proteins, 166 of which were significantly altered in abundance following H. pylori infection, compared with uninfected samples (supplemental Table S1). Among these significantly altered proteins, 43 proteins were significantly up-regulated in gastric tissue from H. pylori-infected gerbils, whereas 123 proteins were significantly downregulated (supplemental Table S1). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (https://www.ebi.ac.uk/pride/archive/) with the dataset identifier PXD009583. For the full list of quantified proteins and all identified peptides from the two biological replicate experiments, please refer to supplemental Tables S2 and S3 and supplemental Table S4 and S5, respectively.
Fig. 2.

Experimental workflow to identify and validate host proteins that are significantly altered by H. pylori infection and which are associated with gastric carcinogenesis in humans. Mongolian gerbils were challenged with Brucella broth (negative control, n = 3) or with carcinogenic H. pylori strain 7.13 (n = 3). Two biological replicate experiments were performed. Gerbil gastric tissue was harvested 6 weeks post-challenge to assess H. pylori colonization, inflammation, and global gastric proteomic changes in vivo. Proteins from pooled lysates from gastric tissue were digested, peptides were labeled with iTRAQ reagents, and quantitative analyses were performed following 2D LC-MS/MS on a Q Exactive Plus mass spectrometer. Data were searched and proteins were quantified using Spectrum Mill. Quantitative proteomic analysis from two biological replicate experiments quantified a total of 2764 proteins, 166 of which were significantly altered in abundance following H. pylori infection, compared with uninfected samples. Ingenuity Pathway Analysis (IPA) was used to identify biologically relevant canonical signaling pathways and disease pathways significantly altered by H. pylori. Protein targets were selected and validated in in vitro human gastric epithelial cells and ex vivo primary human gastric monolayers by Western blot analysis. Further, these targets were validated in in vivo gerbil gastric tissue and human gastric tissue specimens from patients at high risk for gastric cancer by immunohistochemistry.
We next used Ingenuity Pathway Analysis (IPA) to identify biologically relevant canonical signaling pathways significantly altered by H. pylori. supplemental Table S6 contains a comprehensive list of the 337 canonical pathways that were significantly altered following H. pylori infection. Categorization of significantly altered pathways into biologically relevant subgroups revealed numerous immune-mediated pathways altered by H. pylori, the majority of which are involved in inflammatory signaling and immune regulation (n = 83, Fig. 3). There were also 22 pathways related to proliferation, differentiation, and apoptosis (Fig. 4A) and 12 pathways related to cell cycle regulation (Fig. 4B). Other relevant pathways identified included those related to tight junction, cytoskeletal, and extracellular matrix signaling (n = 16, Fig. 5A) as well as MAPK (n = 15, Fig. 5B) and PI3K (n = 9, Fig. 5C) signaling pathways. Table I includes all of the quantified proteins significantly altered in gastric tissue following H. pylori infection that specifically mapped to the most pertinent biological pathways and networks, including inflammatory signaling pathways (Fig. 3), proliferation, differentiation, apoptosis, and cell cycle signaling pathways (Fig. 4), cell-cell junction, MAPK, PI3K signaling pathways (Fig. 5), and networks (Fig. 6) (Table I). Sixty-four (Table I) of the 166 quantified proteins significantly altered in gastric tissue following H. pylori infection (supplemental Table S1) specifically mapped to these pathways and networks (Table I). In addition to the biological pathways, 20 different disease pathways related to cancer were also identified (data not shown). Importantly, when we assessed the disease pathways related to cancer, 145 (87%) of the 166 significantly altered proteins also mapped to cancer pathways.
Fig. 3.

Inflammatory signaling pathways were significantly altered following H. pylori infection. Ingenuity Pathway Analysis (IPA) was used to determine canonical signaling pathways that were significantly altered in H. pylori-infected animals, as compared with uninfected controls. Categorization of significantly altered pathways into biologically relevant subgroups revealed 83 inflammatory signaling pathways, the majority of which are involved in proinflammatory immune responses and immune regulation. Canonical pathways are derived from the IPA nomenclature. Statistical significance is shown in the x-axis, and each canonical pathway is listed on the y-axis.
Fig. 4.

Proliferation, differentiation, apoptosis, and cell cycle pathways were significantly altered following H. pylori infection. Ingenuity Pathway Analysis (IPA) was used to determine canonical signaling pathways that were significantly altered in H. pylori-infected animals, as compared with uninfected controls. A, There were 22 pathways significantly altered with H. pylori infection that were related to proliferation, differentiation, and apoptosis. B, There were 12 pathways significantly altered with H. pylori infection that were related to cell cycle regulation. Canonical pathways are derived from the IPA nomenclature. Statistical significance is shown in the x-axis, and each canonical pathway is listed on the y-axis.
Fig. 5.

Cell-cell junction, MAPK, and PI3K signaling pathways were significantly altered following H. pylori infection. Ingenuity Pathway Analysis (IPA) was used to determine canonical signaling pathways that were significantly altered in H. pylori-infected animals, as compared with uninfected controls. Biologically significant pathways identified included 16 related to tight junction, cytoskeletal, and extracellular matrix signaling (A), 15 MAPK signaling pathways (B) and 9 PI3K signaling pathways (C). Canonical pathways are derived from the IPA nomenclature. Statistical significance is shown in the x-axis, and each canonical pathway is listed on the y-axis.
Table I. Quantified proteins significantly altered in gerbil gastric tissue following H. pylori infection that map to significantly altered biological pathways and networksa.
| Entry name | Gene | Accession number | Normalized fold change | log2 iTRAQ median | P value | BHb |
|---|---|---|---|---|---|---|
| Fatty acid-binding protein 1, liver | FABP1 | P12710 | 4.567 | 1.983 | 0.00E+00 | 0.000015 |
| Amine oxidase A | MAOA | Q64133 | 2.422 | 1.068 | 8.78E-07 | 0.000489 |
| Rho guanine nucleotide exchange factor 12 | ARHGEF12 | Q8R4H2 | 2.273 | 1.249 | 0.00E+00 | 0.000010 |
| Protein unc-13 homolog D | UNC13D | B2RUP2 | 2.135 | 0.886 | 2.08E-05 | 0.000756 |
| Spermine synthase | SMS | P97355 | 1.795 | 0.636 | 7.85E-04 | 0.001467 |
| Rab GTPase-binding effector protein 2 | RABEP2 | Q91WG2 | 1.485 | 0.635 | 8.44E-06 | 0.000301 |
| 60S ribosomal protein L31 | RPL31 | P62900 | 1.436 | 0.587 | 4.06E-05 | 0.000341 |
| Interleukin-1 receptor antagonist protein | IL1RN | P25085 | 1.411 | 0.561 | 9.01E-05 | 0.000391 |
| DNA-(apurinic or apyrimidinic site) lyase | APEX1 | P28352 | 1.402 | 0.552 | 1.18E-04 | 0.000451 |
| Zyxin | ZYX | Q62523 | 1.398 | 0.548 | 1.32E-04 | 0.000461 |
| Casein kinase II subunit beta | CSNK2B | P67871 | 1.386 | 0.536 | 1.88E-04 | 0.000472 |
| BRCA1-A complex subunit BRE | BABAM2 | Q8K3W0 | 1.385 | 0.534 | 1.99E-04 | 0.000482 |
| Paired amphipathic helix protein Sin3a | SIN3A | Q60520 | 1.372 | 0.521 | 2.86E-04 | 0.000522 |
| Myosin-14 | MYH14 | Q6URW6 | 1.362 | 0.510 | 3.88E-04 | 0.000562 |
| Peptidyl-prolyl cis-trans isomerase | FKBP3 | Q62446 | 1.354 | 0.502 | 4.82E-04 | 0.000582 |
| Serine/threonine-protein phosphatase 2A | PPP2R5A | Q6PD03 | 1.350 | 0.498 | 5.36E-04 | 0.000602 |
| Regulatory-associated protein of mTOR | RPTOR | Q8K4Q0 | 1.347 | 0.494 | 5.96E-04 | 0.000632 |
| Phospholipase A2 | PLA2G1B | Q9Z0Y2 | 0.680 | −0.491 | 1.48E-05 | 0.000321 |
| Acidic mammalian chitinase | CHIA | Q91XA9 | 0.636 | −0.589 | 4.45E-07 | 0.000211 |
| Signal recognition particle 14 kDa protein | SRP14 | P16254 | 0.624 | −0.615 | 1.61E-07 | 0.000201 |
| Fatty acid-binding protein 5, epidermal | FABP5 | Q05816 | 0.595 | −0.685 | 8.64E-09 | 0.000130 |
| Membrane-associated guanylate kinase | MAGI3 | Q9EQJ9 | 0.594 | −0.686 | 8.27E-09 | 0.000120 |
| Proteasome activator complex subunit 4 | PSME4 | Q5SSW2 | 0.582 | −0.988 | 1.75E-03 | 0.001749 |
| Pyruvate carboxylase | PKLR | Q05920 | 0.582 | −0.989 | 1.72E-03 | 0.001734 |
| cAMP-dependent protein kinase catalytic subunit beta | PRKACB | P68181 | 0.581 | −0.991 | 1.68E-03 | 0.001719 |
| Mitogen-activated protein kinase 12 | MAPK12 | O08911 | 0.580 | −0.993 | 1.64E-03 | 0.001689 |
| Mitogen-activated protein kinase 8 | MAPK8 | Q91Y86 | 0.580 | −0.993 | 1.64E-03 | 0.001704 |
| Myosin light chain kinase 2 | MYLK2 | Q8VCR8 | 0.580 | −0.993 | 1.64E-03 | 0.001675 |
| Protein kinase C delta type | PRKCD | P28867 | 0.579 | −0.996 | 1.58E-03 | 0.001645 |
| Inhibitor of nuclear factor kappa-B kinase subunit alpha | CHUK | Q60680 | 0.578 | −1.000 | 1.51E-03 | 0.001630 |
| PDZ and LIM domain protein 3 | PDLIM3 | O70209 | 0.574 | −1.009 | 1.35E-03 | 0.001571 |
| ADP-ribosylation factor 3 | ARF3 | P61205 | 0.573 | −1.011 | 1.32E-03 | 0.001556 |
| Mitogen-activated protein kinase 9 | MAPK9 | Q9WTU6 | 0.563 | −1.038 | 9.42E-04 | 0.001512 |
| Glycosylphosphatidylinositol anchor attachment 1 protein | GPAA1 | Q9WTK3 | 0.561 | −0.769 | 1.81E-10 | 0.000050 |
| Ephrin type-B receptor 3 | EPHB3 | P54754 | 0.554 | −1.061 | 7.01E-04 | 0.001452 |
| Cyclin-dependent kinase 17 | CDK17 | Q8K0D0 | 0.542 | −1.091 | 4.72E-04 | 0.001304 |
| Twinfilin-1 | TWF1 | Q91YR1 | 0.540 | −1.096 | 4.42E-04 | 0.001260 |
| Polyadenylate-binding protein 1 | PABPC1 | P29341 | 0.540 | −1.097 | 4.36E-04 | 0.001245 |
| Eukaryotic translation initiation factor 2 subunit 1 | EIF2S1 | Q6ZWX6 | 0.535 | −1.110 | 3.65E-04 | 0.001186 |
| Signal recognition particle receptor subunit beta | SRPRB | P47758 | 0.530 | −1.125 | 2.97E-04 | 0.001141 |
| 60S ribosomal protein L4 | RPL4 | Q9D8E6 | 0.526 | −1.136 | 2.55E-04 | 0.001111 |
| 40S ribosomal protein S6 | RPS6 | P62754 | 0.521 | −1.149 | 2.13E-04 | 0.001067 |
| Myosin regulatory light chain 12B | MYL12B | Q3THE2 | 0.520 | −1.152 | 2.04E-04 | 0.001052 |
| Methylmalonate-semialdehyde dehydrogenase | ALDH6A1 | Q9EQ20 | 0.515 | −1.165 | 1.69E-04 | 0.001008 |
| Fibrinogen gamma chain | FGG | Q8VCM7 | 0.499 | −1.210 | 8.76E-05 | 0.000948 |
| Polypeptide N-acetylgalactosaminyltransferase 3 | GALNT3 | P70419 | 0.497 | −1.217 | 7.89E-05 | 0.000934 |
| GA-binding protein alpha chain | GABPA | Q00422 | 0.489 | −1.239 | 5.65E-05 | 0.000889 |
| cAMP-dependent protein kinase type II-alpha regulatory subunit | PRKAR2A | P12367 | 0.487 | −1.247 | 5.00E-05 | 0.000874 |
| Myosin regulatory light polypeptide 9 | MYL9 | Q9CQ19 | 0.480 | −1.268 | 3.60E-05 | 0.000845 |
| Proteasome activator complex subunit 1 | PSME1 | P97371 | 0.477 | −1.277 | 3.12E-05 | 0.000771 |
| Cytochrome c | CYCS | P62897 | 0.465 | −1.314 | 1.72E-05 | 0.000726 |
| Alpha-soluble NSF attachment protein | NAPA | Q9DB05 | 0.460 | −1.328 | 1.37E-05 | 0.000711 |
| Wiskott-Aldrich syndrome protein family member 2 | WASF2 | Q8BH43 | 0.454 | −1.346 | 1.01E-05 | 0.000667 |
| Phosphoenolpyruvate carboxykinase 2 | PCK2 | Q8BH04 | 0.454 | −1.347 | 9.98E-06 | 0.000652 |
| 5′-AMP-activated protein kinase catalytic subunit alpha-2 | PRKAA2 | Q8BRK8 | 0.440 | −1.394 | 4.47E-06 | 0.000593 |
| Plasminogen | PLG | P20918 | 0.412 | −1.488 | 8.20E-07 | 0.000474 |
| Fermitin family homolog 2 | FERMT2 | Q8CIB5 | 0.411 | −1.491 | 7.75E-07 | 0.000445 |
| Peroxiredoxin-2 | PRDX2 | Q61171 | 0.390 | −1.568 | 1.76E-07 | 0.000370 |
| 60S ribosomal protein L24 | RPL24 | Q8BP67 | 0.387 | −1.577 | 1.47E-07 | 0.000356 |
| Long-chain-fatty-acid–CoA ligase 5 | ACSL5 | Q8JZR0 | 0.386 | −1.582 | 1.33E-07 | 0.000341 |
| ADP-ribosylation factor 4 | ARF4 | P61750 | 0.343 | −1.752 | 3.69E-09 | 0.000252 |
| Cyclin-dependent kinase 2 | CDK2 | P97377 | 0.337 | −1.779 | 2.01E-09 | 0.000222 |
| Cathepsin D | CTSD | P18242 | 0.294 | −1.976 | 1.78E-11 | 0.000163 |
| Fatty acid-binding protein 4, adipocite | FABP4 | P04117 | 0.272 | −2.089 | 9.31E-13 | 0.000119 |
a 64 of the 166 (Supplemental Table I) quantified proteins significantly altered in gastric tissue following H. pylori infection map to significantly altered biological pathways and networks. These 64 pathways include inflammatory, proliferation, differentiation, apoptosis, cell cycle, cell-cell junctions, MAPK, and PI3K signaling pathways in addition to network analysis.
b p values were corrected for multiple comparisons by the Benjamini-Hochberg (BH) method (BH FDR p < 0.05) (20). After applying the multiple comparisons by the BH method, we allowed all quantified proteins with significant p values less than the BH correction. This resulted in the quantification of 166 proteins significantly altered in abundance following H. pylori infection, 64 of which are shown here.
Fig. 6.

Network analysis identified RABEP2 as an important protein in inflammatory and signaling pathways implication in the development of inflammation and cancer. Ingenuity Pathway Analysis (IPA) was used to determine significant networks that were significantly altered in H. pylori-infected animals, as compared with uninfected controls. RAPEP2 was significantly up-regulated by iTRAQ and mapped to disease pathways and networks directly involved in inflammation, cancer, and gastrointestinal disease. Green designates proteins that were significantly downregulated, whereas red indicates proteins that were significantly up-regulated. Shapes indicate different functional categories of proteins, as defined in the inset. Lines indicate interactions among proteins within the network, as defined in the inset. Arrowheads indicate stimulatory interactions, whereas endcaps indicate inhibitory interactions. Solid lines indicate direct interactions, whereas dashed lines indicate indirect interactions.
Based on the high abundance of inflammatory signaling and cancer-related pathways, we next used IPA to determine the disease-related pathways that were significantly altered by H. pylori. supplemental Table S7 contains a comprehensive list of the 78 disease categories that were significantly altered following H. pylori infection. Among these disease pathways, several important categories were directly related to inflammatory disease pathways, including inflammatory response, cell-mediated immune response, immune cell trafficking, and inflammatory and immunological diseases (supplemental Table S7), pathways consistent with the host immune response to H. pylori infection. Specifically, we identified pathways related to the development of gastric cancer, including organ injury, gastrointestinal disease and cancer. Interestingly, among the cancer-related disease pathways, RABEP2, Rab GTPase-binding effector protein 2, was identified. Based the role of RABEP2 in gastrointestinal cancer (26), we next performed IPA-driven network analysis and were able to identify 12 major networks altered following H. pylori infection; importantly, one network connected RABEP2 to inflammatory signaling pathways related not only to H. pylori infection, but also to the development of cancer (Fig. 6).
H. pylori Significantly Up-regulates RABEP2 and G3BP2 Expression in In Vitro Gastric Epithelial Cells in a cag-dependent Manner
In addition to RABEP2, other Rab/Ras proteins were identified among the iTRAQ results, including G3BP2, Ras GTPase-activating protein-binding protein 2, another protein important in carcinogenesis. We therefore validated expression levels in a subset of the proteomic targets by focusing on these two proteins, RABEP2 and G3BP2, which have been implicated in the development of inflammation and cancer. First, to validate the peptide sequences identified from the Mus musculus UniprotKB protein database search, we performed a BLAST search against the predicted protein sequences derived from Mongolian gerbil, Meriones unguiculatus, sequence data. All RABEP2 (n = 5) and G3BP2 (n = 2) peptides (supplemental Tables S4 and S5) queried had 100% identity between Mus musculus and Meriones unguiculatus.
Next, AGS human gastric epithelial cells were cocultured with the same H. pylori strain 7.13 used for in vivo infection to assess protein expression of RABEP2 and G3BP2 (Fig. 7). In addition, we cocultured gastric epithelial cells with the H. pylori 7.13 cagE− and cagA− isogenic mutants to determine whether up-regulation of these targets was dependent on formation of the cancer-associated cag type IV secretion system or translocation of the effector protein, CagA, respectively. Consistent with our in vivo proteomic results, wild-type carcinogenic H. pylori strain 7.13 significantly up-regulated both RABEP2 and G3BP2, compared with uninfected gastric epithelial cells (Fig. 7). Further, increased expression of RABEP2 and G3BP2 occurred in a cagE−dependent, indicating that H. pylori-induced up-regulation of these proteins requires formation of a functional cag type IV secretion system, but occurred independent of CagA, suggesting that other cag type IV secretion system effectors could contribute to altered expression (Fig. 7). To confirm these results in additional human gastric epithelial cells lines, we cocultured SNU1 and MKN28 gastric epithelial cells with wild-type H. pylori 7.13 and its cagE− isogenic mutant and assessed RABEP and G3BP2 protein expression by Western blot analysis (supplemental Fig. S1). Consistent with the proteomics data and results from AGS cells (Fig. 7), H. pylori significantly up-regulated RABEP2 and G3BP2 expression in a cagE-dependent manner in SNU1 and MKN28 gastric epithelial cells (supplemental Fig. S1).
Fig. 7.

H. pylori up-regulates RABEP2 and G3BP2 in human gastric epithelial cells in vitro. AGS human gastric epithelial cells were cocultured with wild-type cag+ H. pylori strain 7.13, a 7.13 cagE-isogenic mutant, or a 7.13 cagA-isogenic mutant at a multiplicity of infection (MOI) of 100:1 for 24 h. Western blot analysis was used to assess RABEP2 (A) or G3BP2 (B) protein expression relative to GAPDH protein expression. RABEP2 and G3BP2 protein expression levels were standardized to GAPDH expression, and densitometry was used to quantify all independent replicates. Data are represented as fold over uninfected (UI) control. Error bars indicate standard error of the mean from experiments performed on at least three independent occasions, and ANOVAs were used to determine statistical significance among groups. *, p < 0.05; **, p < 0.005; ***, p < 0.0005; ****, p < 0.0001.
H. pylori Significantly Up-regulates RABEP2 and G3BP2 Expression Ex Vivo In Primary Human Gastric Monolayers In a cag-dependent Manner
To next assess RABEP2 and G3BP2 protein expression in a nontransformed model, we harvested primary gastric organoids from human fundus obtained during sleeve gastrectomies (23). Human gastric organoids were then transferred to 2-dimensional (2D) gastric epithelial cell monolayers, as previously described (23). Primary gastric monolayers were then cocultured with wild-type cag+ H. pylori strains 7.13 or PMSS1, or their isogenic cagE− mutants and harvested for Western blot analysis (supplemental Fig. S2). Consistent with the iTRAQ proteomic data and the in vitro data from gastric epithelial cells, wild-type cag+ H. pylori significantly up-regulated RABEP2 and G3BP2 protein expression in primary human gastric monolayers in a cagE-dependent manner (supplemental Fig. S2).
H. pylori Significantly Up-regulates RABEP2 and G3BP2 Expression In Vivo In Gerbil Gastric Epithelium In a cag-dependent Manner
To validate the proteomic data in gerbil tissue and determine cell specificity, we next infected Mongolian gerbils with wild-type carcinogenic H. pylori strain 7.13 and a 7.13 cagE− isogenic mutant. We then assessed epithelial RABEP2 and G3BP2 expression by immunohistochemistry in uninfected, 7.13-infected, and cagE−-infected gerbil gastric mucosa. Consistent with the iTRAQ proteomic data, RABEP2 (Fig. 8) and G3BP2 (Fig. 9) were significantly up-regulated in gastric epithelium following infection with H. pylori in a cag-dependent manner as well as in premalignant lesions.
Fig. 8.
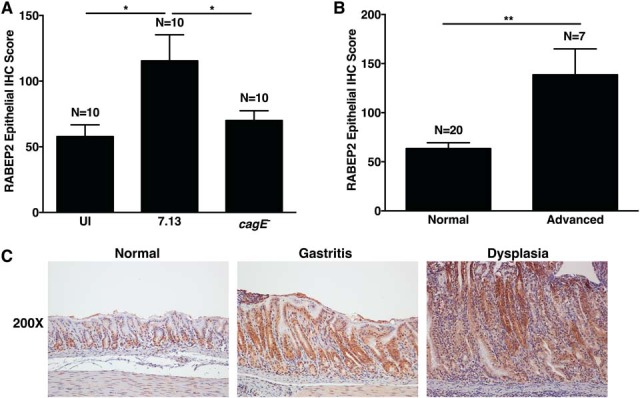
H. pylori up-regulates RABEP2 in vivo in gerbil gastric epithelium in a cag-dependent manner. A, RABEP2 protein expression was evaluated by immunohistochemistry in gerbil gastric tissue sections from uninfected gerbils, and gerbils infected with wild-type cag+ H. pylori strain 7.13 or its cagE-isogenic mutant. A single pathologist assessed the percentage of epithelial RABEP2+ cells and the intensity of RABEP2 staining, as previously described (22). The immunohistochemistry (IHC) score reflects the percentage of cells positive for RABEP2, multiplied by the intensity of staining. Each bar indicates the average epithelial IHC score with standard error of the mean. B, RABEP2 protein expression was evaluated in normal gerbil gastric mucosa (n = 20) or gastric mucosa with advanced lesions (n = 7, advanced), which included gastric dysplasia and adenocarcinoma. Three cases of nonatrophic gastritis were not included in these analyses. C, Representative images of RABEP2 protein expression in normal, gastritis, or dysplastic gastric tissue sections. ANOVA tests were used to determine statistical significance among groups. *, p < 0.05; **, p < 0.005.
Fig. 9.

H. pylori up-regulates G3BP2 in gerbil gastric epithelium in vivo in a cag-dependent manner. A, G3BP2 protein expression was evaluated by immunohistochemistry in gerbil gastric tissue sections from uninfected gerbils, and gerbils infected with wild-type cag+ H. pylori strain 7.13 or its cagE-isogenic mutant. A single pathologist assessed the percentage of epithelial G3BP2+ cells and the intensity of G3BP2 staining. The immunohistochemistry (IHC) score reflects the percentage of cells positive for G3BP2, multiplied by the intensity of staining, as previously described (22). Each bar indicates the average epithelial IHC score with standard error of the mean. B, G3BP2 protein expression was evaluated in normal gerbil gastric mucosa (n = 20) or gastric mucosa with advanced lesions (n = 7, advanced), which included gastric dysplasia and adenocarcinoma. Three cases of nonatrophic gastritis were not included in these analyses. C, Representative images of G3BP2 protein expression in normal, gastritis, or dysplastic gastric tissue sections. ANOVA tests were used to determine statistical significance among groups. *, p < 0.05; **, p < 0.005.
To further characterize cell specificity, we also isolated single cells from gastric tissue sections using a dissociation and dispersion technique that enriches for gastric epithelial cells for flow cytometry. Cells were then stained for an epithelial specific marker, pan-cytokeratin, and either RABEP2 or G3BP2 (supplemental Fig. S3). Epithelial cells were gated to assess expression of RABEP or G3PB2. Expression of both RABEP2 and G3BP2 significantly increased in epithelial cells isolated from H. pylori-infected gerbils, compared with uninfected gerbils. Thus, consistent with the in vitro and ex vivo primary human gastric epithelial cell data, H. pylori up-regulates RABEP2 (Fig. 8) and G3BP2 (Fig. 9) in gerbil gastric epithelium in a cag-dependent manner.
RABEP2 and G3BP2 Contribute to Carcinogenic Phenotypes In Human Gastric Epithelial Cells
To assess the potential functional roles of RABEP2 and G3BP2, we next transfected AGS human gastric epithelial cells with nontargeting (NT), RABEP2 siRNA, or G3BP2 siRNA and cocultured with H. pylori strain 7.13 to assess cellular proliferation, apoptosis, migration, and invasion (supplemental Fig. S4), characteristics associated with carcinogenic phenotypes. Western blot analysis and quantification demonstrated significant siRNA interference that resulted in ∼80 and 73% reductions in RABEP2 and G3BP2 protein expression in H. pylori-infected gastric epithelial cells, respectively (supplemental Fig. S4A). Decreased expression of RABEP2 resulted in a significant decrease in levels of proliferation, suggesting that RABEP2 may play a role in proliferation in response to H. pylori in vitro (supplemental Fig. S4B). However, no differences were observed in the levels of apoptosis among transfected cells infected with H. pylori (data not shown). Reduction in the expression levels of both RABEP2 and G3BP2 also led to nonsignificant decreases in gastric epithelial migration and invasion (supplemental Fig. S4C–S4D), suggesting that these proteins may affect to the ability of cells to migrate and invade in the presence of H. pylori.
H. pylori Mediated Up-regulation of RABEP2 and G3BP2 In Human Gastric Epithelium Parallels the Severity of Gastric Pathology
To extend these findings into the natural niche of infection, we next investigated RABEP2 and G3BP2 expression by immunohistochemistry in human gastric tissue samples isolated from H. pylori-infected patients residing in high-risk gastric cancer regions of Colombia and Chile (Table II, Fig. 10 and 11). From population studies in Colombia and Chile, it has been previously demonstrated that most patients are colonized with cag+ strains of H. pylori. Within the Colombian cohort, over 90% of subjects are colonized with H. pylori (27). de Sablet et al. demonstrated that within the high-risk gastric cancer region of Colombia, 85.7% of strains are cag+ and this is directly associated with the severity of inflammation and disease (27). Within the Chilean cohort, Araya et al. demonstrated that greater than 80% of subjects are colonized with H. pylori (28). Among Chilean subjects with gastritis alone, 49% of the strains were cag+ and this directly correlated with the severity of inflammation and epithelial damage, indicating that presence of the cag pathogenicity island is associated with the severity of gastric damage (28). The subjects from these two cohorts exhibited varying degrees of histologic pathology, including nonatrophic gastritis (NAG), multifocal atrophic gastritis (MAG), intestinal metaplasia (IM), and gastric adenocarcinoma (GC) (Table II). Consistent with the proteomics, in vitro, ex vivo, and in vivo validation data, epithelial protein expression of both RABEP2 (Fig. 10) and G3BP2 (Fig. 11) was significantly increased in parallel with the severity of gastric pathology, such that there was a progressive increase in RABEP2 and G3BP2 from nonatrophic gastritis to premalignant lesions, including multifocal atrophic gastritis, intestinal metaplasia, and dysplasia, as well as malignant lesions, including cases of gastric adenocarcinoma. These data indicate that RABEP2 and G3BP2 are significantly up-regulated in human gastric epithelial cells following acute infection with H. pylori, and that these proteins may also play a role in the progression along the gastric carcinogenesis cascade.
Table II. Human subject information, histological diagnoses, and Helicobacter pylori status.
| Subject | Case | Sample origina | Age | Sex | Histological diagnosisb | GC type | GC Histological Grade | TNM stage | Hp statusc |
|---|---|---|---|---|---|---|---|---|---|
| 1 | NQ3008 | Colombia | 75 | M | NAG | UN | UN | UN | 1 |
| 2 | NQ3017 | Colombia | 61 | M | MAG | UN | UN | UN | 1 |
| 3 | NQ3024 | Colombia | 66 | F | NAG | UN | UN | UN | 1 |
| 4 | NQ3026 | Colombia | 78 | F | MAG | UN | UN | UN | 1 |
| 5 | NQ3028 | Colombia | 55 | M | NAG | UN | UN | UN | 0 |
| 6 | NQ3030 | Colombia | 56 | M | IM | UN | UN | UN | 0 |
| 7 | NQ3037 | Colombia | 58 | F | IM | UN | UN | UN | 1 |
| 8 | NQ3039 | Colombia | 74 | F | IM | UN | UN | UN | 0 |
| 9 | NQ3043 | Colombia | 64 | F | MAG | UN | UN | UN | 1 |
| 10 | NQ3067 | Colombia | 52 | M | IM | UN | UN | UN | 0 |
| 11 | NQ3070 | Colombia | 54 | F | IM | UN | UN | UN | 0 |
| 12 | NQ3082 | Colombia | 54 | M | IM | UN | UN | UN | 0 |
| 13 | NQ3119 | Colombia | 79 | F | NAG | UN | UN | UN | 1 |
| 14 | NQ3122 | Colombia | 66 | F | NAG | UN | UN | UN | 1 |
| 15 | NQ3123 | Colombia | 57 | F | MAG | UN | UN | UN | 1 |
| 16 | NQ3124 | Colombia | 54 | M | NAG | UN | UN | UN | 1 |
| 17 | NQ3138 | Colombia | 64 | F | NAG | UN | UN | UN | 0 |
| 18 | NQ3145 | Colombia | 53 | M | MAG | UN | UN | UN | 1 |
| 19 | NQ3160 | Colombia | 62 | F | IM | UN | UN | UN | 0 |
| 20 | NQ3166 | Colombia | 54 | M | NAG | UN | UN | UN | 1 |
| 21 | NQ3168 | Colombia | 53 | F | IM | UN | UN | UN | 1 |
| 22 | NQ3172 | Colombia | 66 | M | MAG | UN | UN | UN | 1 |
| 23 | NQ3177 | Colombia | 73 | M | IM | UN | UN | UN | 1 |
| 24 | NQ3184 | Colombia | 51 | F | IM | UN | UN | UN | 1 |
| 25 | NQ3190 | Colombia | 77 | M | IM | UN | UN | UN | 0 |
| 26 | NQ3201 | Colombia | 54 | M | NAG | UN | UN | UN | 1 |
| 27 | NQ3270 | Colombia | 57 | F | MAG | UN | UN | UN | 1 |
| 28 | NQ3327 | Colombia | 49 | F | MAG | UN | UN | UN | 1 |
| 29 | NQ3380 | Colombia | 58 | M | MAG | UN | UN | UN | 1 |
| 30 | NQ3396 | Colombia | 51 | M | MAG | UN | UN | UN | 1 |
| 31 | 95-0507 | Chile | 54 | F | GC | Intestinal type | Moderately differentiated | II | UN |
| 32 | 95-0753 | Chile | 68 | M | GC | Intestinal type | Poorly differentiated | II | UN |
| 33 | 95-1721 | Chile | UN | UN | GC | Intestinal type | Moderately differentiated | UN | UN |
| 34 | 95-2982 | Chile | 55 | M | GC | Intestinal type | Poorly differentiated | IIIB | UN |
| 35 | 95-4846 | Chile | 64 | M | GC | Intestinal type | Poorly differentiated | IIIA | UN |
| 36 | 95-5242 | Chile | 74 | M | GC | Intestinal type | Moderately differentiated | II | UN |
| 37 | 95-5501 | Chile | 74 | M | GC | Intestinal type | Moderately differentiated | II | UN |
| 38 | 95-5619 | Chile | 45 | M | GC | Intestinal type | Moderately differentiated | IIIB | UN |
| 39 | 95-5711 | Chile | 67 | F | GC | Intestinal type | Moderately differentiated | IV | UN |
| 40 | 95-6532 | Chile | 69 | M | GC | Intestinal type | Well differentiated | IA | UN |
| 41 | 95-6666 | Chile | 58 | M | GC | Intestinal type | Poorly differentiated | IB | UN |
| 42 | 95-7255 | Chile | 83 | F | GC | Intestinal type | Poorly differentiated | IIIA | UN |
| 43 | 95-1260 | Chile | 70 | F | GC | Intestinal type | Poorly differentiated | II | UN |
| 44 | 95-8565 | Chile | 70 | M | GC | Intestinal type | Moderately differentiated | IA | UN |
| 45 | 96-2083 | Chile | 60 | M | GC | Intestinal type | Moderately differentiated | IB | UN |
a Colombian biopsies were harvested from high-risk gastric cancer patients during follow up endoscopy (24). Chilean samples were harvested during gastrectomy. UN = data unavailable.
b Histological diagnosis represents the most advanced lesion reported and the area scored for IHC. NAG = non-atrophic gastritis; premalignant lesions included MAG = multifocal atrophic gastritis and IM = intestinal metaplasia; and GC = gastric cancer. Chilean subjects with GC did not receive chemotherapy or radiation therapy prior to gastrectomy.
c Hp = H. pylori; 0 = Hp-negative, 1 = Hp-positive as determined by modified Steiner stain (24). All Colombian patients were positive at the time of initial enrollment. H. pylori culture was not performed on Chilean samples (UN = unavailable).
Fig. 10.

RABEP2 expression parallels the severity of gastric premalignant and malignant lesions in H. pylori-infected humans. A, RABEP2 expression was evaluated by immunohistochemistry in human populations at high risk for gastric cancer in Colombia and Chile. Gastric specimens from H. pylori-infected patients with nonatrophic gastritis, multifocal atrophic gastritis, intestinal metaplasia (premalignant) and gastric adenocarcinoma (malignant) were evaluated for RABEP2 immunostaining. Representative images are shown for nonatrophic gastritis, intestinal metaplasia, and gastric adenocarcinoma (200X and 400X magnification). B, A single pathologist assessed the percentage of epithelial RABEP2+ cells and the intensity of RABEP2 staining. The immunohistochemistry (IHC) score reflects the percentage of cells positive for RABEP2, multiplied by the intensity of staining, as previously described (22). Each bar indicates the average IHC score among patients with nonatrophic gastritis, multifocal atrophic gastritis or intestinal metaplasia (premalignant), or gastric adenocarcinoma (malignant) with standard error of the mean from the indicated number of patients evaluated. ANOVA tests were used to determine statistical significance among groups. ****; p < 0.0001.
Fig. 11.

G3BP2 expression parallels the severity of gastric premalignant and malignant lesions in H. pylori-infected humans. A, G3BP2 expression was evaluated by immunohistochemistry in human populations at high risk for gastric cancer in Colombia and Chile. Gastric specimens from H. pylori-infected patients with nonatrophic gastritis, multifocal atrophic gastritis, intestinal metaplasia (premalignant) and gastric adenocarcinoma (malignant) were evaluated for G3BP2 immunostaining. Representative images are shown for nonatrophic gastritis, intestinal metaplasia, and gastric adenocarcinoma (200X and 400X magnification). B, A single pathologist assessed the percentage of epithelial G3BP2+ cells and the intensity of G3BP2 staining. The immunohistochemistry (IHC) score reflects the percentage of cells positive for G3BP2, multiplied by the intensity of staining, as previously described (22). Each bar indicates the average IHC score among patients with nonatrophic gastritis, multifocal atrophic gastritis or intestinal metaplasia (premalignant), or gastric adenocarcinoma (malignant) with standard error of the mean from the indicated number of patients evaluated. ANOVA tests were used to determine statistical significance among groups. ****, p < 0.0001.
DISCUSSION
Although H. pylori is the strongest known risk factor for gastric cancer, the specific mechanisms by which this pathogen initiates gastric carcinogenesis are incompletely defined. It is increasingly apparent that disease outcomes are mediated through complex interactions between H. pylori virulence determinants and host cell responses. Here we have not only defined and validated early gastric proteomic changes that occur in vivo in the Mongolian gerbil model in response to infection with a carcinogenic strain of H. pylori, but we have also further shown that these proteins and associated signaling pathways may be implicated in the later stage transitions to premalignant and malignant gastric lesions in humans.
Using iTRAQ, we identified numerous proteins abundantly altered in the gastric mucosa in response to H. pylori infection in vivo, which likely contribute to important host cell responses driving the initiation and progression of gastric carcinogenesis. Using these proteomic data for pathway mapping, we identified numerous canonical signaling pathways that are commonly activated in response to H. pylori infection. Many inflammatory pathways were altered in response to H. pylori infection in this model, a finding consistent with previous data indicating that chronic inflammation induced by H. pylori is a major contributor to gastric carcinogenesis (2).
Among the inflammatory pathways identified, many of these findings corroborated previous findings in human populations with H. pylori infection. Several cytokine families have been implicated in H. pylori-associated gastric carcinogenesis. For example, the IL-1 family of cytokines are central mediators of mucosal inflammation (29), and IL-1 is induced by H. pylori (30). Furthermore, IL-1 is a major risk factor associated with the development of gastric cancer (31). Specifically, polymorphisms within IL-1β family members are associated with a significantly increased risk for gastric cancer, but only among persons infected with H. pylori (31). Macrophage migratory inhibitory factor (MIF) plays a pivotal role in inflammatory diseases and is also increased with H. pylori infection (32). MIF has also been shown to stimulate cellular proliferative responses (33) and is significantly increased in premalignant lesions and gastric cancer, further implicating its role in gastric carcinogenesis (34, 35). IL-8 is a potent neutrophil chemoattractant and activating factor that mediates robust proinflammatory responses. IL-8 is increased by H. pylori infection in a cag-dependent manner (36), and polymorphisms in IL-8 have been associated with increased risk of chronic atrophic gastritis and gastric cancer (37, 38). IL-6 is an important mediator of inflammation and has been shown to promote a Th17-mediated inflammatory response. IL-6 expression correlates with disease status among patients with H. pylori-associated gastritis (39) as well as gastric cancer (40). IL-17 is a T cell-derived cytokine capable of modulating the Th1 response. IL-17 is up-regulated by H. pylori during infection in vivo (41, 42), and IL-17 genetic polymorphisms have been associated with increased susceptibility to gastric cancer (43). Finally, tumor necrosis factor (TNF) is a cytokine involved in systemic inflammation and the Th1 response, and TNF is increased among patients with H. pylori-associated gastritis (39).
In addition to altering inflammatory signaling pathways, H. pylori has also been shown to disrupt cellular junctional complexes (44) and induce cytoskeletal rearrangements that are reminiscent of unrestrained growth induced by growth factors (45). H. pylori has also been shown to dysregulate the balance between gastric epithelial cell proliferation and apoptosis (46), an effect which may influence gastric transformation. Pathway analysis revealed several alterations in junctional and cytoskeletal complexes (44, 45), MAPK and PI3K signaling (45), as well as proliferation, differentiation, and apoptosis (46). In addition, disease pathways and networks that are directly related to gastrointestinal injury, disease, and the development of cancer were also identified. These identified pathways and networks will allow for future functional analyses of specific proteomic targets that have been previously uncharacterized with respect to either H. pylori infection or gastric carcinogenesis, but now may play an important role in the development of gastric injury and cancer. A more thorough understanding of these networks will ideally enable exploitation of targetable pathways and effectors for clinical benefit and disease prevention.
RABEP2 and G3BP2 were two proteins identified by iTRAQ and pathway mapping that were up-regulated among H. pylori-infected gerbils. These targets were validated in vitro in gastric epithelial cells and ex vivo in primary human gastric monolayers and, consistent with the results in the gerbil gastric tissue samples, these proteins were also significantly up-regulated in human gastric epithelial cells. Further, their up-regulation was mediated by the most intensively studied H. pylori virulence constituent, the cag type IV secretion system. These targets were also validated in gerbil gastric epithelium, where these proteins were significantly increased in a cag-dependent manner and paralleled the severity of gastric lesions. When these targets were validated in human gastric tissues collected from patients at high risk for gastric cancer, expression increased steadily along the gastric carcinogenesis cascade, suggesting that these proteins are up-regulated early during H. pylori infection and that their expression increases with each stage along the gastric cancer cascade. These are novel targets in that they have not been previously identified in conjunction with H. pylori infection or in the development of gastric cancer. However, RABEP2 has been previously demonstrated to play a role in membrane trafficking and in early endosome fusion (47), but more importantly, was identified as a novel genetic susceptibility locus that predisposed to colon tumorigenesis in mice (26). In addition, G3BP2 has been previously predicted to function as a scaffold protein that may be involved in transport (48), but more importantly, is up-regulated in various human cancers (49–56). Furthermore, G3BP2 has been shown to function as a negative regulator of the tumor suppressor, p53 (57) and has also been identified as a downstream target of WNT5A, which is involved in gastric cancer metastasis (58).
In conclusion, by using novel proteomic approaches and pathway analyses, we were able to define changes in the gerbil gastric proteome in response to H. pylori infection. These data mirrored alterations that develop among humans infected with H. pylori, further validating our prior studies that this model recapitulates many aspects of gastric inflammation and disease observed in humans. Importantly, this technique and approach allowed for identification and validation of novel protein targets that appear to play an important role in H. pylori-induced gastric carcinogenesis in individuals at high risk for gastric adenocarcinoma. Indeed, this technique and approach holds promise for accelerating the identification of novel biomarkers, which arise early in the inflammatory and carcinogenic cascade and which may have important applications in therapeutic intervention and disease prevention.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (https://www.ebi.ac.uk/pride/archive/) with the dataset identifier PXD009583.
Supplementary Material
Acknowledgments
We thank Drs. Wael El-Rifai at the University of Miami Health Center, Robert J. Coffey, James R. Goldenring, and Ramona Graves-Deal at Vanderbilt University Medical Center for providing additional gastric epithelial cell lines. We also acknowledge and thank Dr. Kshipra Singh at Vanderbilt University Medical Center for her expertise and assistance with performing flow cytometry.
Footnotes
* We acknowledge the following funding sources that contributed to this work: National Institutes of Health R01CA77955 (RMP), R01DK058587 (RMP), P30DK58404 (RMP), P01CA116087 (RMP, TLC, KTW), R01AI118932 (TLC), P01CA028842 (KTW), U. S. Department of Veterans Affairs BX000627 (TLC), and R01DK083402 (YZ). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- cag T4SS
- cag type IV secretion system
- 2D LC-MS/MS
- 2-dimensional liquid chromatography-coupled tandem mass spectrometry
- ACN
- acetonitrile
- ANOVA
- analysis of variance
- BH
- Benjamini-Hochberg
- BLAST
- Basic Local Alignment Search Tool
- cagA
- cytotoxin associated gene product A
- cag
- T4SS oncogenic effector protein
- cagE
- cytotoxin associated gene product E
- cag T4SS
- ATPase
- CFU
- colony-forming units
- CO2
- carbon dioxide
- FBS
- fetal bovine serum
- FCS
- fetal calf serum
- FDR
- false-discovery rate
- G3BP2
- Ras GTPase-activating protein-binding protein 2
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GC
- gastric cancer
- H&E
- hematoxylin and eosin
- HCD
- higher energy collisional disassociation
- HRP
- horseradish peroxidase
- IACUC
- Institutional Animal Care and Use Committee
- IHC
- immunohistochemistry
- IL-1
- interleukin-1
- Il-6
- interleukin-6
- IL-8
- interleukin-8
- IL-17
- interleukin-17
- IM
- intestinal metaplasia
- IPA
- Ingenuity Pathway Analysis
- IRB
- Institutional Review Board
- iTRAQ
- isobaric tags for relative and absolute quantification
- MAG
- multifocal atrophic gastritis
- MAPK
- mitogen-activated protein kinase
- MIF
- macrophage migratory inhibitory factor
- MMTS
- methyl methanethiosulfonate
- MOI
- multiplicity of infection
- MTT
- (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrasodium bromide)
- NaCl
- sodium chloride
- NAG
- nonatrophic gastritis
- NCBI
- National Center for Biotechnology Information
- PI3K
- phosphoinositide 3-kinase
- PBS
- phosphate-buffered saline
- RABEP2
- Rab GTPase-binding effector protein 2
- SPEM
- spasmolytic-expressing metaplasia
- TCEP
- tris (2-carboxyethyl) phosphine
- TEAB
- triethylammonium bicarbonate
- Th17
- T helper 17
- TNF
- tumor necrosis factor
- TNM
- tumor, node, and metastasis staging
- TSA
- trypticase soy agar
- UI
- uninfected.
REFERENCES
- 1. Bray F., Ren J. S., Masuyer E., and Ferlay J. (2013) Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int. J. Cancer 132, 1133–1145 [DOI] [PubMed] [Google Scholar]
- 2. Polk D. B., and Peek R. M. Jr. (2010) Helicobacter pylori: gastric cancer and beyond. Nat. Rev. Cancer 10, 403–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hooi J. K. Y., Lai W. Y., Ng W. K., Suen M. M. Y., Underwood F. E., Tanyingoh D., Malfertheiner P., Graham D. Y., Wong V. W. S., Wu J. C. Y., Chan F. K. L., Sung J. J. Y., Kaplan G. G., and Ng S. C. (2017) Global Prevalence of Helicobacter pylori infection: systematic review and meta-analysis. Gastroenterology 153, 420–429 [DOI] [PubMed] [Google Scholar]
- 4. Stein M., Bagnoli F., Halenbeck R., Rappuoli R., Fantl W. J., and Covacci A. (2002) c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol. Microbiol. 43, 971–980 [DOI] [PubMed] [Google Scholar]
- 5. Tammer I., Brandt S., Hartig R., Konig W., and Backert S. (2007) Activation of Abl by Helicobacter pylori: a novel kinase for CagA and crucial mediator of host cell scattering. Gastroenterology 132, 1309–1319 [DOI] [PubMed] [Google Scholar]
- 6. Mueller D., Tegtmeyer N., Brandt S., Yamaoka Y., De Poire E., Sgouras D., Wessler S., Torres J., Smolka A., and Backert S. (2012) c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J. Clin. Invest. 122, 1553–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Segal E. D., Cha J., Lo J., Falkow S., and Tompkins L. S. (1999) Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc. Natl. Acad. Sci. U.S.A. 96, 14559–14564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Odenbreit S., Puls J., Sedlmaier B., Gerland E., Fischer W., and Haas R. (2000) Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287, 1497–1500 [DOI] [PubMed] [Google Scholar]
- 9. Mimuro H., Suzuki T., Tanaka J., Asahi M., Haas R., and Sasakawa C. (2002) Grb2 is a key mediator of Helicobacter pylori CagA protein activities. Mol. Cell 10, 745–755 [DOI] [PubMed] [Google Scholar]
- 10. Amieva M. R., Vogelmann R., Covacci A., Tompkins L. S., Nelson W. J., and Falkow S. (2003) Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 300, 1430–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saadat I., Higashi H., Obuse C., Umeda M., Murata-Kamiya N., Saito Y., Lu H., Ohnishi N., Azuma T., Suzuki A., Ohno S., and Hatakeyama M. (2007) Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature 447, 330–333 [DOI] [PubMed] [Google Scholar]
- 12. Ohnishi N., Yuasa H., Tanaka S., Sawa H., Miura M., Matsui A., Higashi H., Musashi M., Iwabuchi K., Suzuki M., Yamada G., Azuma T., and Hatakeyama M. (2008) Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc. Natl. Acad. Sci. U.S.A. 105, 1003–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Franco A. T., Johnston E., Krishna U., Yamaoka Y., Israel D. A., Nagy T. A., Wroblewski L. E., Piazuelo M. B., Correa P., and Peek R. M. Jr. (2008) Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res. 68, 379–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Noto J. M., Gaddy J. A., Lee J. Y., Piazuelo M. B., Friedman D. B., Colvin D. C., Romero-Gallo J., Suarez G., Loh J., Slaughter J. C., Tan S., Morgan D. R., Wilson K. T., Bravo L. E., Correa P., Cover T. L., Amieva M. R., and Peek R. M. Jr. (2013) Iron deficiency accelerates Helicobacter pylori-induced carcinogenesis in rodents and humans. J. Clin. Invest. 123, 479–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Correa P., and Chen V. W. (1994) Gastric cancer. Cancer Surv. 20, 55–76 [PubMed] [Google Scholar]
- 16. Shimizu T., Choi E., Petersen C. P., Noto J. M., Romero-Gallo J., Piazuelo M. B., Washington M. K., Peek R. M. Jr, and Goldenring J. R. (2016) Characterization of progressive metaplasia in the gastric corpus mucosa of Mongolian gerbils infected with Helicobacter pylori. J. Pathol. 239, 399–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Noto J. M., Chopra A., Loh J. T., Romero-Gallo J., Piazuelo M. B., Watson M., Leary S., Beckett A. C., Wilson K. T., Cover T. L., Mallal S., Israel D. A., and Peek R. M. (2017) Pan-genomic analyses identify key Helicobacter pylori pathogenic loci modified by carcinogenic host microenvironments. Gut 67, 1793–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Voss B. J., Loh J. T., Hill S., Rose K. L., McDonald W. H., and Cover T. L. (2015) Alteration of the Helicobacter pylori membrane proteome in response to changes in environmental salt concentration. Proteomics Clin. Appl. 9, 1021–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mertins P., Udeshi N. D., Clauser K. R., Mani D. R., Patel J., Ong S. E., Jaffe J. D., and Carr S. A. (2012) iTRAQ labeling is superior to mTRAQ for quantitative global proteomics and phosphoproteomics. Mol. Cell. Proteomics 11, M111.014423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thissen D., Steinburg L., and Kuang D. (2002) Quick and easy omplementation of the Benjamini-Hochberg procedure for controlling the false positive rate in multiple comparisons. J. Educ. Behav. Stat. 27, 77–83 [Google Scholar]
- 21. Chaturvedi R., Asim M., Romero-Gallo J., Barry D. P., Hoge S., de Sablet T., Delgado A. G., Wroblewski L. E., Piazuelo M. B., Yan F., Israel D. A., Casero R. A. Jr, Correa P., Gobert A. P., Polk D. B., Peek R. M. Jr, and Wilson K. T. (2011) Spermine oxidase mediates the gastric cancer risk associated with Helicobacter pylori CagA. Gastroenterology 141, 1696–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Noto J. M., Khizanishvili T., Chaturvedi R., Piazuelo M. B., Romero-Gallo J., Delgado A. G., Khurana S. S., Sierra J. C., Krishna U. S., Suarez G., Powell A. E., Goldenring J. R., Coffey R. J., Yang V. W., Correa P., Mills J. C., Wilson K. T., and Peek R. M. Jr. (2013) Helicobacter pylori promotes the expression of Kruppel-like factor 5, a mediator of carcinogenesis, in vitro and in vivo. PloS One 8, e54344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Teal E., Bertaux-Skeirik N., Chakrabarti J., Holokai L., and Zavros Y. (2018) Establishment of human- and mouse-derived gastric primary epithelial cell monolayers from organoids. Methods Mol. Biol. 1817, 145–155 [DOI] [PubMed] [Google Scholar]
- 24. Mera R., Fontham E. T., Bravo L. E., Bravo J. C., Piazuelo M. B., Camargo M. C., and Correa P. (2005) Long term follow up of patients treated for Helicobacter pylori infection. Gut 54, 1536–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li C. L., Du X. Y., Gao J., Wang C., Guo H. G., Dai F. W., Sa X. Y., An W., and Chen Z. W. (2016) Phylogenetic analysis of the Mongolian gerbil (Meriones unguiculatus) from China based on mitochondrial genome. Genet. Mol. Res. 15, doi: 10.4238/gmr.15037703 [DOI] [PubMed] [Google Scholar]
- 26. Liu P., Lu Y., Liu H., Wen W., Jia D., Wang Y., and You M. (2012) Genome-wide association and fine mapping of genetic loci predisposing to colon carcinogenesis in mice. Mol. Cancer Res. 10, 66–74 [DOI] [PubMed] [Google Scholar]
- 27. de Sablet T., Piazuelo M. B., Shaffer C. L., Schneider B. G., Asim M., Chaturvedi R., Bravo L. E., Sicinschi L. A., Delgado A. G., Mera R. M., Israel D. A., Romero-Gallo J., Peek R. M. Jr, Cover T. L., Correa P., and Wilson K. T. (2011) Phylogeographic origin of Helicobacter pylori is a determinant of gastric cancer risk. Gut 60, 1189–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Araya J. C., Anabalon L., Roa I., Bravo M., Villaseca M. A., Guzman P., and Roa J. C. (2004) Association between Helicobacter pylori genotype and the severity of gastritis in infected adults. Rev. Med. Chil. 132, 1345–1354 [DOI] [PubMed] [Google Scholar]
- 29. Tran L. S., Chonwerawong M., and Ferrero R. L. (2017) Regulation and functions of inflammasome-mediated cytokines in Helicobacter pylori infection. Microbes Infect. 19, 449–458 [DOI] [PubMed] [Google Scholar]
- 30. Pachathundikandi S. K., Muller A., and Backert S. (2016) Inflammasome activation by Helicobacter pylori and its implications for persistence and immunity. Curr. Top. Microbiol. Immunol. 397, 117–131 [DOI] [PubMed] [Google Scholar]
- 31. El-Omar E. M., Rabkin C. S., Gammon M. D., Vaughan T. L., Risch H. A., Schoenberg J. B., Stanford J. L., Mayne S. T., Goedert J., Blot W. J., Fraumeni J. F. Jr, and Chow W. H. (2003) Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology 124, 1193–1201 [DOI] [PubMed] [Google Scholar]
- 32. Xia H. H., Lam S. K., Huang X. R., Wong W. M., Leung S. Y., Yuen S. T., Lan H. Y., and Wong B. C. (2004) Helicobacter pylori infection is associated with increased expression of macrophage migratory inhibitory factor-by epithelial cells, T cells, and macrophages-in gastric mucosa. J. Infect. Dis. 190, 293–302 [DOI] [PubMed] [Google Scholar]
- 33. Xia H. H., Lam S. K., Chan A. O., Lin M. C., Kung H. F., Ogura K., Berg D. E., and Wong B. C. (2005) Macrophage migration inhibitory factor stimulated by Helicobacter pylori increases proliferation of gastric epithelial cells. World J. Gastroenterol. 11, 1946–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shun C. T., Lin J. T., Huang S. P., Lin M. T., and Wu M. S. (2005) Expression of macrophage migration inhibitory factor is associated with enhanced angiogenesis and advanced stage in gastric carcinomas. World J. Gastroenterol. 11, 3767–3771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. He X. X., Yang J., Ding Y. W., Liu W., Shen Q. Y., and Xia H. H. (2006) Increased epithelial and serum expression of macrophage migration inhibitory factor (MIF) in gastric cancer: potential role of MIF in gastric carcinogenesis. Gut 55, 797–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Crabtree J. E., Farmery S. M., Lindley I. J., Figura N., Peichl P., and Tompkins D. S. (1994) CagA/cytotoxic strains of Helicobacter pylori and interleukin-8 in gastric epithelial cell lines. J. Clin. Pathol. 47, 945–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. de Oliveira J. G., Rossi A. F., Nizato D. M., Cadamuro A. C., Jorge Y. C., Valsechi M. C., Venancio L. P., Rahal P., Pavarino E. C., Goloni-Bertollo E. M., and Silva A. E. (2015) Influence of functional polymorphisms in TNF-alpha, IL-8, and IL-10 cytokine genes on mRNA expression levels and risk of gastric cancer. Tumour Biol. 36, 9159–9170 [DOI] [PubMed] [Google Scholar]
- 38. Wang Y. M., Li Z. X., Tang F. B., Zhang Y., Zhou T., Zhang L., Ma J. L., You W. C., and Pan K. F. (2016) Association of genetic polymorphisms of interleukins with gastric cancer and precancerous gastric lesions in a high-risk Chinese population. Tumour Biol. 37, 2233–2242 [DOI] [PubMed] [Google Scholar]
- 39. Crabtree J. E., Shallcross T. M., Heatley R. V., and Wyatt J. I. (1991) Mucosal tumour necrosis factor alpha and interleukin-6 in patients with Helicobacter pylori associated gastritis. Gut 32, 1473–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu C. W., Wang S. R., Chao M. F., Wu T. C., Lui W. Y., P'Eng F., K, Chi C. W. (1996) Serum interleukin-6 levels reflect disease status of gastric cancer. Am. J. Gastroenterol. 91, 1417–1422 [PubMed] [Google Scholar]
- 41. Luzza F., Parrello T., Monteleone G., Sebkova L., Romano M., Zarrilli R., Imeneo M., and Pallone F. (2000) Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J. Immunol. 165, 5332–5337 [DOI] [PubMed] [Google Scholar]
- 42. Shi Y., Liu X. F., Zhuang Y., Zhang J. Y., Liu T., Yin Z., Wu C., Mao X. H., Jia K. R., Wang F. J., Guo H., Flavell R. A., Zhao Z., Liu K. Y., Xiao B., Guo Y., Zhang W. J., Zhou W. Y., Guo G., and Zou Q. M. (2010) Helicobacter pylori-induced Th17 responses modulate Th1 cell responses, benefit bacterial growth, and contribute to pathology in mice. J. Immunol. 184, 5121–5129 [DOI] [PubMed] [Google Scholar]
- 43. Wang N., Yang J., Lu J., Qiao Q., Bao G., Wu T., and He X. (2014) IL-17 gene polymorphism is associated with susceptibility to gastric cancer. Tumour Biol. 35, 10025–10030 [DOI] [PubMed] [Google Scholar]
- 44. Backert S., Schmidt T. P., Harrer A., and Wessler S. (2017) Exploiting the gastric epithelial barrier: Helicobacter pylori's attack on tight and adherens junctions. Curr. Top. Microbiol. Immunol. 400, 195–226 [DOI] [PubMed] [Google Scholar]
- 45. Tegtmeyer N., Neddermann M., Asche C. I., and Backert S. (2017) Subversion of host kinases: a key network in cellular signaling hijacked by Helicobacter pylori CagA. Mol. Microbiol. 105, 358–372 [DOI] [PubMed] [Google Scholar]
- 46. Meng W., Bai B., Sheng L., Li Y., Yue P., Li X., and Qiao L. (2015) Role of Helicobacter pylori in gastric cancer: advances and controversies. Discov. Med. 20, 285–293 [PubMed] [Google Scholar]
- 47. Gournier H., Stenmark H., Rybin V., Lippe R., and Zerial M. (1998) Two distinct effectors of the small GTPase Rab5 cooperate in endocytic membrane fusion. EMBO J. 17, 1930–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Prigent M., Barlat I., Langen H., and Dargemont C. (2000) IkappaBalpha and IkappaBalpha /NF-kappa B complexes are retained in the cytoplasm through interaction with a novel partner, RasGAP SH3-binding protein 2. J. Biol. Chem. 275, 36441–36449 [DOI] [PubMed] [Google Scholar]
- 49. French J., Stirling R., Walsh M., and Kennedy H. D. (2002) The expression of Ras-GTPase activating protein SH3 domain-binding proteins, G3BPs, in human breast cancers. Histochem. J. 34, 223–231 [DOI] [PubMed] [Google Scholar]
- 50. Rose A. E., Poliseno L., Wang J., Clark M., Pearlman A., Wang G., Vega Y. S. d M. E. C., Medicherla R., Christos P. J., Shapiro R., Pavlick A., Darvishian F., Zavadil J., Polsky D., Hernando E., Ostrer H., and Osman I. (2011) Integrative genomics identifies molecular alterations that challenge the linear model of melanoma progression. Cancer Res. 71, 2561–2571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Winslow S., Leandersson K., and Larsson C. (2013) Regulation of PMP22 mRNA by G3BP1 affects cell proliferation in breast cancer cells. Mol. Cancer 12, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wei S. C., Fattet L., Tsai J. H., Guo Y., Pai V. H., Majeski H. E., Chen A. C., Sah R. L., Taylor S. S., Engler A. J., and Yang J. (2015) Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol. 17, 678–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gupta N., Badeaux M., Liu Y., Naxerova K., Sgroi D., Munn L. L., Jain R. K., and Garkavtsev I. (2017) Stress granule-associated protein G3BP2 regulates breast tumor initiation. Proc. Natl. Acad. Sci. U.S.A. 114, 1033–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ashikari D., Takayama K., Tanaka T., Suzuki Y., Obinata D., Fujimura T., Urano T., Takahashi S., and Inoue S. (2017) Androgen induces G3BP2 and SUMO-mediated p53 nuclear export in prostate cancer. Oncogene 36, 6272–6281 [DOI] [PubMed] [Google Scholar]
- 55. Takayama K. I., Suzuki T., Fujimura T., Takahashi S., and Inoue S. (2018) Association of USP10 with G3BP2 inhibits p53 signaling and contributes to poor outcome in prostate cancer. Mol. Cancer Res. 16, 846–856 [DOI] [PubMed] [Google Scholar]
- 56. Takayama K. I., Suzuki T., Tanaka T., Fujimura T., Takahashi S., Urano T., Ikeda K., and Inoue S. (2018) TRIM25 enhances cell growth and cell survival by modulating p53 signals via interaction with G3BP2 in prostate cancer. Oncogene 37, 2165–2180 [DOI] [PubMed] [Google Scholar]
- 57. Kim M. M., Wiederschain D., Kennedy D., Hansen E., and Yuan Z. M. (2007) Modulation of p53 and MDM2 activity by novel interaction with Ras-GAP binding proteins (G3BP). Oncogene 26, 4209–4215 [DOI] [PubMed] [Google Scholar]
- 58. Katoh M., and Katoh M. (2009) Transcriptional mechanisms of WNT5A based on NF-kappaB, Hedgehog, TGFbeta, and Notch signaling cascades. Int. J. Mol. Med. 23, 763–769 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (https://www.ebi.ac.uk/pride/archive/) with the dataset identifier PXD009583.