

Abstract
Summary
Objective:
The role of neuroinflammation in mesial temporal lobe epilepsy (MTLE), and how it relates to drug resistance, remains unclear. Expression levels of the inflammatory enzymes cyclooxygenase (COX)-1 and COX-2 have been found to be increased in animal models of epilepsy. Knowing the cellular expression of COX-1 and COX-2 is the key to understanding their functional role; however, only 3 studies have investigated COX-2 expression in epilepsy in humans, and there are no reports on COX-1. In addition, previous studies have shown that certain inflammatory proteins up-regulate ATP binding cassette (ABC) transporter expression (thought to be responsible for drug resistance), but this relationship remains unclear in human tissue. This study sought to measure the expression of COX-1, COX-2, and translocator protein 18 kDa (TSPO, an inflammation biomarker acting as a positive control), as well as ABC transporters P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), in brain tissue samples from people with drug-resistant MTLE.
Methods:
Formalin-fixed paraffin-embedded surgical brain tissue was obtained from 33 patients with drug-resistant MTLE. Multiplex immunofluorescence was used to quantify the expression and distribution of COX-1, COX-2, TSPO, P-gp, and BCRP.
Results:
COX-1 was expressed in microglia, and COX-2 and TSPO were expressed in microglia and neurons. BCRP density correlated significantly with TSPO density, suggesting a potential relationship between inflammatory markers and efflux transporters.
Keywords: ABC transporters, cyclooxygenase (COX), epilepsy, inflammation
1. INTRODUCTION
Approximately 30% of all people with epilepsy are drug-resistant, in that their seizures cannot be controlled using antiepileptic drugs.1 Two pathomechanisms associated with drug resistance in epilepsy include neuroinflammation and the overexpression of efflux transporters at the blood-brain barrier. Neuroinflammation is known to play a pathophysiologic role in epilepsies caused by exogenous infectious agents (eg, herpes encephalitis) or autoimmunity (eg, Rasmussen’s encephalitis), and may also play a role in drug- resistant mesial temporal lobe epilepsy (DRMTLE) not immediately linked to infectious or autoimmune mechanisms (for a review see Vezzani et al.2).
Cyclooxygenase-1 and −2 (COX-1 and COX-2) are 2 key enzymes of the prostaglandin-mediated inflammatory pathway; both convert arachidonic acid into several prostanoids.3 Because prostanoids have different and sometimes opposing effects, which prostanoids are synthesized is determined by the specificity of COX-1 and COX-2, which in turn is determined by the effector mechanism in each cell.4 Thus, the type of cell in which COX-1 and COX-2 are expressed, and the availability of intermediate substrates, determines their physiologic role. In humans with DRMTLE, little is known about the cellular expression— defined as the relative expression of the protein among microglia, astrocytes, and neurons—of COX-2, because only a handful of studies have determined the types of cells in which COX-2 is expressed. The few reports of which we are aware used surgically resected tissue from patients with temporal lobe epilepsy (TLE) and found astrocytic5,6 and neuronal expression.5–7 No studies have examined the cellular expression of COX-1 in human TLE tissue.
In this context, another area of interest is the cellular expression of the inflammatory biomarker translocator protein 18 kDa (TSPO). TSPO is known to be primarily located in glia (ie, activated microglia and reactive astrocytes) rather than in neurons.8 Positron emission tomography (PET) studies have shown increased TSPO binding in the epileptogenic focus of patients with TLE,9–11 suggesting that TSPO acts as a potential clinically relevant biomarker of neuroinflammation in TLE.
The overexpression of efflux transporters at the blood-brain barrier may also play a role in DRMTLE. The “transporter hypothesis” of drug-resistant epilepsy (DRE) posits that the overexpression of ATP-binding cassette (ABC) transporters—such as P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP)—at the blood-brain barrier may prevent antiepileptic drugs from reaching their tar- gets.1 Elevated brain levels of P-gp have been reported in animal models of epilepsy, as well as in patients with DRE compared to drug-responsive epilepsy12 and nonepileptic controls.12,13 With regard to inflammation, previous studies found that P-gp up-regulation was in part caused by elevated COX-2 activity,14 and that pharmacologic inhibition of COX-2 allowed for greater uptake of the P-gp substrate phenytoin.15 In more general terms, these studies proposed that drug resistance might be caused by an inflammatory response that would then up-regulate P-gp, resulting in decreased antiepileptic drug uptake near the seizure focus. However, to date, no studies have investigated the cellular expression of COX-1 or COX-2 in brain tissue samples from patients with DRMTLE, or whether any correlative relationship exists with capillary ABC transporter expression.
The aims of this study were 2-fold. First, we identified the expression of neuroinflammatory markers (COX-1, COX-2, and TSPO) in microglia, astrocytes, and neurons in order to determine their cell-specific expression in brain tissue from patients with DRMTLE. Second, we investigated whether a correlation exists between local levels of neuroinflammation and levels of specific ABC transporters (P-gp and BCRP) in capillaries. Immunofluorescent techniques were used to investigate both spatial distribution and quantities of protein immunoreactivity in defined cell types.
2. METHODS
2.1. Human brain tissue
Human brain tissue samples were obtained from 33 patients with DRMTLE undergoing surgical resection of the hippocampus. Patients were referred to the Clinical Epilepsy Section, National Institute of Neurological Disorders and Stroke, NIH, for DRMTLE. All patients were evaluated with ictal video-electroencephalography monitoring, and 3T Philips (Andover, MA, USA) magnetic resonance imaging with fluid-attenuated inversion recovery, T1 (spin-echo and 3D magnetization prepared rapid gradient echo [MPRAGE]), and T2-weighted sequences. The study was approved by the NIH Combined Neurosciences Institutional Review Board, and all patients signed written informed consent forms. Patient demographics and pathologic diagnoses are provided in Table 1. The in vitro experiments were conducted on 2 separate sets of tissues (n = 20 and n = 13, respectively). The tissue was formalin-fixed, blocked in paraffin, and cut into 5 μm sections, which were then mounted onto slides.
TABLE 1.
Patient clinical data
| Set | Number | Status | Sex | Age of onset (y) | Duration (y) | GTCS/SE history | Etiology |
|---|---|---|---|---|---|---|---|
| A | 1 | Non‐MTS | F | 13 | 14 | Yes/No | |
| 2 | Non‐MTS | F | 38 | 5 | None | ||
| 3 | Non‐MTS | F | 1 | 23 | Yes/No | Possible head injury | |
| 4 | Non‐MTS | F | 4 | 12 | Rare/No | Febrile seizure | |
| 5 | Non‐MTS | F | 14 | 7 | None | ||
| 6 | Non‐MTS | F | 14 | 16 | None | ||
| 7 | Non‐MTS | M | 13 | 29 | Yes/No | ||
| 8 | Non‐MTS | F | 39 | 5 | None | ||
| 9 | MTS | M | 3 | 43 | Yes/No | ||
| 10 | MTS | F | 4 | 25 | Yes/No | Cysticercosis | |
| 11 | MTS | M | 17 | 14 | Yes/No | Febrile seizure | |
| 12 | MTS | F | 1 | 21 | Yes/No | Febrile seizure | |
| 13 | MTS | M | 1.5 | 36.5 | Yes/No | Febrile seizure | |
| 14 | MTS | M | 21 | 5 | None | ||
| 15 | MTS | F | 6 | 15 | None | ||
| 16 | MTS | M | 1 | 48 | Yes/No | Febrile seizure | |
| 17 | MTS | F | 6 | 25 | Yes/Several SE episodes | Coma for 2 weeks at age 6 | |
| 18 | MTS | F | 35 | 9 | Yes/No | Head trauma, no LOC | |
| 19 | MTS | M | 26 | 25 | Yes/No | ||
| 20 | MTS | M | 3 | 24 | Yes/No | Head trauma with LOC/febrile seizure | |
| B | 1 | Non‐MTS | M | Childhood | >30 | Yes/No | Possible head injury |
| 2 | Non‐MTS | M | 13 | 6 | Yes/No | ||
| 3 | Non‐MTS | F | 18 | 14 | Yes/No | ||
| 4 | Non‐MTS | M | 25 | 8 | Yes/No | Head injury with LOC | |
| 5 | Non‐MTS | F | 30 | 13 | None | ||
| 6 | Non‐MTS | M | 3 | 30 | Yes/No | TBI with seizures and LOC | |
| 7 | Non‐MTS | M | 7 | 13 | Yes/No | ||
| 8 | Non‐MTS | F | 35 | 10 | None | Possible meningitis | |
| 9 | Non‐MTS | M | 40 | 10 | None | Head injury | |
| 10 | MTS | F | 19 | 14 | Yes/No | Viral encephalitis | |
| 11 | MTS | M | 2 | 38 | Yes/No | Head injury | |
| 12 | MTS | F | 3 | 24 | None | Febrile seizure | |
| 13 | MTS | F | 9 | 19 | Yes/Yes | Varicella‐zoster virus infection at age 9 |
GTCS, generalized tonic–clonic seizures; LOC, loss of consciousness; MTS, mesial temporal sclerosis; SE, status epilepticus; TBI, traumatic brain injury
2.2. Classification of mesial temporal sclerosis
Histopathologic methods were used to stratify each tissue sample into groups based on the degree of mesial temporal sclerosis (MTS, Table 1). MTS, that is, hippocampal sclerosis, refers to pyramidal neuron cell loss and astrogliosis, and can be classified based on severity according to the International League Against Epilepsy.16 Slides were stained with hematoxylin and eosin as well as with antibodies to glial fibrillary acidic protein (GFAP, a marker for astrocytes) and neuronal nuclei (NeuN, a marker for neurons). Patients classified as “non-MTS” included 1 with a cavernous hemangioma, 1 with microdysgenesis, and 13 with nonspecific gliosis.
2.3. Immunohistochemistry
Protein distribution was measured using immunofluorescent techniques, in which the location and quantity of primary antibodies bound to specific target was visualized using fluorophore-conjugated secondary antibodies or the tyramide signal amplification method (Perkin Elmer, Waltham, MA, USA). A BOND-RX automated stainer (Leica Biosystems, Wetzlar, Germany) was used to prepare the slides for staining. The sections were “baked” (30 minutes at 60°C), dewaxed using Bond Dewax Solution (Leica Biosystems, 72°C), and run through a heat-induced epitope retrieval step (EDTA-based solution, pH 9.0, 20 minutes at 100°C).
Following this “pretreatment,” slides were manually washed in phosphate-buffered saline (PBS), incubated in 0.03% H2O2 for 30 minutes to block endogenous peroxidase, and washed again. Primary antibodies (Table S1) were diluted in primary antibody buffer (0.3% TX-100, 0.1% NaN3, PBS) and added to the slides for overnight incubation in a humidified chamber at 4°C. The following day, the slides were washed in Tris-buffered saline (TBS, pH 7.4)-Tween 20 and blocked in Tris-NaCl blocking buffer (TNB) (0.1 mol/L Tris-HCl, pH 7.5, 0.15 mol/L NaCl, 0.5% blocking reagent, Perkin Elmer) for 30 minutes. The secondary antibodies (Table S1) diluted in TNB were then applied to the slides for 90 minutes, followed by a wash in TBS-Tween 20. For the tyramide signal amplification method, horseradish peroxidase-conjugated secondary antibodies were used in conjunction with fluorophore-conjugated tyramide diluted (1:100) in amplification reagent (Perkin Elmer).
For multiplex immunofluorescence staining, antibodies raised in different host species were used if available. For experiments using multiple antibodies raised in the same species, a sequential staining protocol using an intermediate antibody denaturation and elution step was used: the slide underwent a heat-induced epitope retrieval step as described previously (EDTA, pH 9.0, 20 minutes at 100°C), which removed the primary and secondary antibodies without affecting the covalently bound fluorophorelabeled tyramide. This enabled the addition of a second primary antibody without the risk of cross-reactivity with other antibodies from the same species.
To quench lipofuscin autofluorescence in the tissue, slides were counterstained with lipophilic Sudan Black B solution (1% w/v in 70% ethanol, Sigma-Aldrich, St. Louis, MO, USA) for 5 minutes. The slides were then dipped in 70% ethanol followed by a PBS wash and coverslipped using an aqueous mounting medium containing a 4’,6-dia- midino-2-phenylindole (DAPI) counterstain (ProLong Gold Antifade Mountant with DAPI, ThermoFisher Scientific, Waltham, MA, USA). Unless otherwise noted, all steps were executed at room temperature.
Images of the slides were acquired on an automated VSlide slide scanning system (Metasystems, Altlussheim, Germany). Entire pieces of tissue on the slides were imaged with a 10x objective (NA = 0.45, resolution1.8 pixels/μm). Each field of view was captured at 3 z- levels with a 1 μm interval to create an extended focus image. Acquired field of view images were stitched to create a complete overview with microscopic resolution. The emission spectra for the fluorophore-conjugated secondary antibodies were as follows: Hoeschst (420–485 nm), Cy2 (490–530 nm), Cy3 (550–570 nm), Cy3.5 (580–595 nm), and Cy5 (650–670 nm).
2.4. In situ hybridization
In situ hybridization was performed to detect messenger RNA (mRNA) expression of COX-1, COX-2, and TSPO in astrocytes and microglia. Detailed methods can be found in the Supporting Information.
2.5. Antibody validation
To confirm that the COX-1, COX-2, P-gp, and BCRP antibodies were targeting the correct proteins, antibodies were validated using 3 independent methods: pre-adsorption assay, paired antibodies, and in situ hybridization. Detailed methods can be found in the Supporting Information.
2.6. Image analysis
To quantify the expression of proteins detected by immunofluorescence and in situ hybridization, positive pixel intensity was measured using macros created in ImageJ (NIH, Bethesda, MD, USA) and scripts written in Matlab (Mathworks, Natick, MA, USA). Analyses of any differences in the effects of MTS were done in a blinded manner.
2.6.1. ImageJ
From the whole sections of tissue, 500 X 500 pixel (278 X 278 μm) regions of interest (ROIs) were randomly selected (FIGURE 1A). A maximum of 50 ROIs were generated per tissue sample, except in cases of small tissue sample size, in which a maximum of 25 ROIs were generated. ROIs were then visually inspected and excluded from analysis if there were any tears, large arteries, or a high presence of blood. Immunofluorescence signals were captured in 5 separate channels (Hoescht, Cy2, Cy3, Cy3.5, Cy5) and analyzed in each ROI (FIGURE 1B). For the cellular expression experiments, the channels were as follows: DAPI counterstain in Hoescht, GFAP in Cy2, IBA-1 in Cy3, NeuN in Cy3.5, and COX-1, COX-2, or TSPO in Cy5. To minimize possible cross talk between fluorescence signals, nonoverlapping markers for microglia (IBA1) and neurons (NeuN) were stained using Cy3 and Cy3.5, respectively. For these channels, bleed-through signal was calculated based on pixel intensities, and final images were corrected. In addition, we performed a median background correction for the images in the Cy5 channel based on the assumption that most captured pixels would not contain positive immunofluorescence signal.
FIGURE 1.
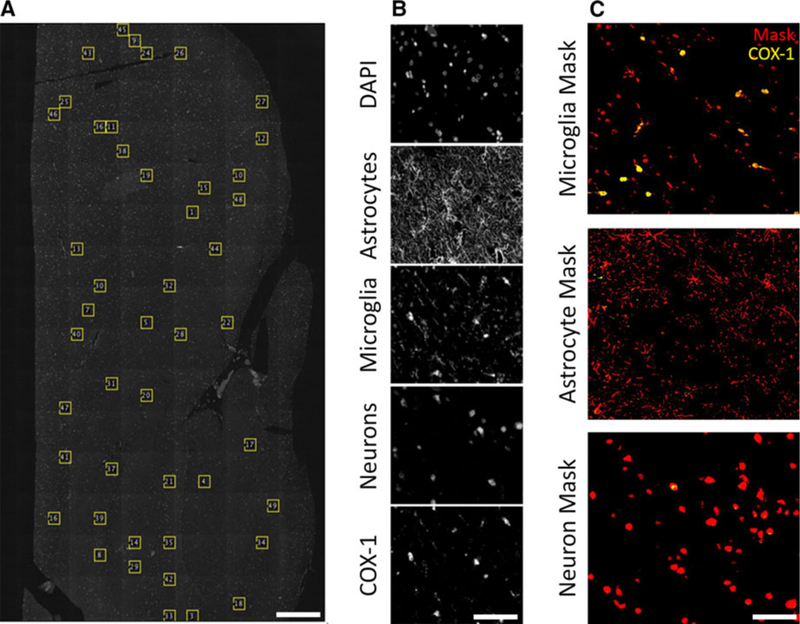
An overview of the methods used to analyze protein expression. Whole pieces of tissue were scanned using a fluorescence slide scanning microscope. A, From each piece of tissue, up to 50 regions of interest (ROIs) were randomly selected using a script written in ImageJ. B, Every ROI contained 5 channels for the inflammation expression experiments, and 4 channels for the ATP‐binding cassette (ABC) transporter correlation experiments. An example set of channels from the former is shown (top to bottom): DAPI, glial fibrillary acidic protein (GFAP), ionized calcium‐binding adapter molecule 1 (IBA‐1), neuronal nuclei (NeuN), and cyclooxygenase‐1 (COX‐1, shown), COX‐2, or translocator protein (TSPO). C, Masks (shown in red) were then generated from the GFAP, IBA1, and NeuN images, and the expression of COX‐1 (shown in yellow), COX‐2, or TSPO was measured within the mask. Scale bar is equal to 1 mm (A), and 60 μm (B, C)
For the efflux transporter experiments, the channels were as follows: DAPI counterstain in Hoescht, glut-1 in Cy2, COX-1, COX-2, or TSPO in Cy3, and P-gp or BCRP in Cy5. For the in situ hybridization experiments, we avoided use of the Cy3 and Cy3.5 channel in the same experiment to minimize possible cross talk between fluorescence signals.
2.6.2. Matlab
Total optic intensity was calculated as a measure of protein density as described previously.17 To measure inflammatory protein concentrations (COX-1, COX-2, and TSPO) within cell types (microglia, astrocytes, and neurons), images were segmented based on expression of cell-type specific markers. To generate the masks, the edges of the cells were detected based on variance using the Sobel method17 and then smoothed to generate a binary mask (FIGURE 1C). The integrated optical densities of COX-1, COX-2, and TSPO were measured in each ROI both within the cell mask and in the whole ROI. Cellular distribution was calculated as mean integrated density within each segmented cell type. For each ROI per tissue sample, the relative distribution between cells for each target (ie, COX-1) within each cell mask (ie, microglia) was calculated by dividing the integrated density of COX-1 in the whole ROI by the total integrated density of COX-1 within the mask. The fraction of integrated density values from all ROIs (≤50) within a piece of tissue were then averaged together. This value was then averaged with all data points from that experiment (ie, COX-1 expression in microglia).
The same method was used to measure ABC transporter expression within capillaries. A glut-1 mask was generated from the Cy2 channel and overlaid onto the Cy5 channel image; the integrated optical densities of P-gp and BCRP were then measured within the mask and reported in arbitrary units. To measure the inflammatory proteins, the Cy3 channel was thresholded based on median pixel intensity, and the total integrated optical density was measured for the whole ROI.
2.7. Statistical analysis
Data were analyzed using SAS software (SAS Institute, Cary, NC, USA). Statistical significance for the expression of the inflammatory proteins among cell types was determined using the Kruskal-Wallis test, followed by the Dunn’s multiple comparison test (α = .05). The data are expressed as percent mean integrated density ± standard deviation (SD). Wilcoxon rank-sum tests were performed to compare protein levels between the MTS and non-MTS groups. Logistic regression analysis was performed to assess the association between MTS and the protein/cell expression with batch effect as a covariate. Pairwise linear correlations between ABC transporter and inflammatory protein expression were evaluated using Pearson correlation coefficients.
3. RESULTS
The immunohistochemical experiments were performed in 2 independent sets of tissues, collected as convenience samples based on tissue availability (Tissue set A, n = 20; Tissue set B, n = 13; Table 1). The ROI count was 44.3 ±5.0 for the samples large enough to generate a maximum of 50 ROIs, and 20.6 ± 3.2 for the 11 samples in which a maximum of 25 ROIs were generated.
3.1. Cellular COX-1, COX-2, and TSPO expression
To increase statistical power, the sets were pooled and analyzed as a whole (see Methods). COX-1 (n = 28) levels (percent mean integrated density ± SD) were significantly higher in microglia (61% ±11) than in astrocytes (4% ± 2, P < .0001) or in neurons (8% ±5, P < .0001) (Figure 2). Expression in neurons was also significantly higher than in astrocytes (P < .05). Upon visual inspection, COX-1 mRNA was found to be greater in microglia than in astrocytes (Figure 3A). COX-2 (n = 29) expression was significantly higher in microglia (37% ±15) and neurons (29% ±12) than in astrocytes (3% ± 2, P < .0001) (Figure 2). COX-2 mRNA expression was observed in microglia, but most of the signal surrounded unlabeled nuclei (Figure 3B). TSPO (n = 33) expression was significantly higher in microglia (50% ±12) and neurons (32% ±16) than in astrocytes (5% ± 3, P < .0001) (Figure 2). In addition, microglial expression was significantly higher than neuronal expression (P < .01). TSPO mRNA expression was greater in microglia than astrocytes (data not shown).
FIGURE 2.
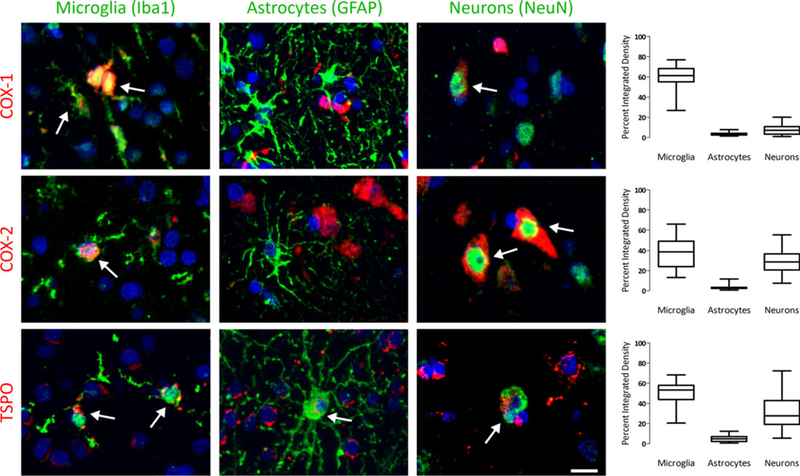
Expression patterns of cyclooxygenase‐1 (COX‐1), COX‐2, and translocator protein (TSPO) among microglia, astrocytes, and neurons. COX‐1 (top row) was primarily expressed in microglia, with minor expression in astrocytes and neurons. COX‐2 (middle row) was expressed in both microglia and neurons. TSPO (bottom row) was also expressed in microglia and neurons. In this image, the protein is shown in red, the cell is shown in green, and the DAPI counterstain is shown in blue. White arrows indicate positive overlap between the protein and the cell. Box plots on the right show the quantification of staining patterns across all tissue samples. Scale bar is equal to 20 μm
FIGURE 3.
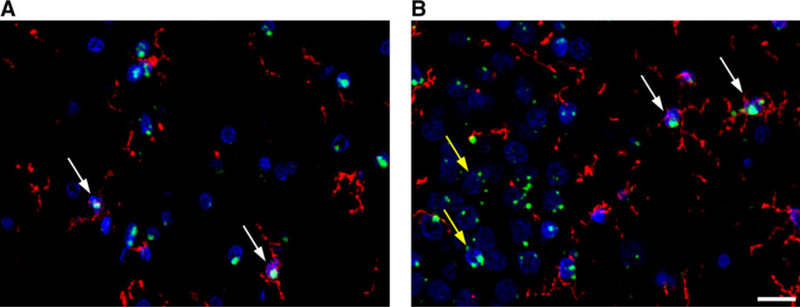
Representative images of cyclooxygenase‐1 (COX‐1) A and COX‐2 B mRNA expression. mRNA expression is shown in green, microglial expression is shown in red, and DAPI (a nuclear counterstain) is shown in blue. White arrows indicate microglial expression and yellow arrows indicate neuronal expression. Although a neuronal antibody could not be included with the mRNA probe, neurons can still be identified based on their larger nucleus size. Scale bar is equal to 20 μm
Upon visual inspection of the images, it was noted that COX-1 was expressed in most microglia, and that decreased evels, measured in an ROI, were due mainly to decreased cellular COX-1 signal rather than an increase in COX-1 negative microglia. In contrast, COX-2 expression in neurons was predominately zonal, in which some neurons contained no COX-2 signal. To determine if a high sampling of white matter could affect neuronal expression results, the tissue samples with the lowest neuronal population were removed as outliers; this had no impact on the outcome (data not shown). In both sets of tissues, cell size variation was minimal and therefore only minimally influenced protein expression outcome (Figure S1).
3.2. Correlation of inflammatory markers with efflux transporters
To measure P-gp and BCRP density, a capillary mask was generated from glut-1 positive structures, and positive P-gp or BCRP pixels were analyzed within the mask. P-gp and BCRP density were thus automatically normalized for capillary density, thereby taking into account any changes in capillary density between groups. Upon visual inspection of the images, no noncapillary P-gp or BCRP expression was observed (Figure S2).
In tissue set A, a positive linear correlation was observed between BCRP expression in capillaries and TSPO immunoreactivity (P = .0003, Pearson r = .72131; Figure 4A). Visual inspection of a subject with a high correlation and one with a low correlation confirmed higher TSPO expression around a BCRP-positive capillary for the former (Figure S3). In addition, pixel values of both capillaries were higher for the capillary from the high- correlation subject (Figure S3). Because TSPO is also expressed in capillaries, this positive linear correlation could be due simply to increased capillary density. However, we found no correlation between TSPO or BCRP density and capillary area (Figure 4A).
FIGURE 4.
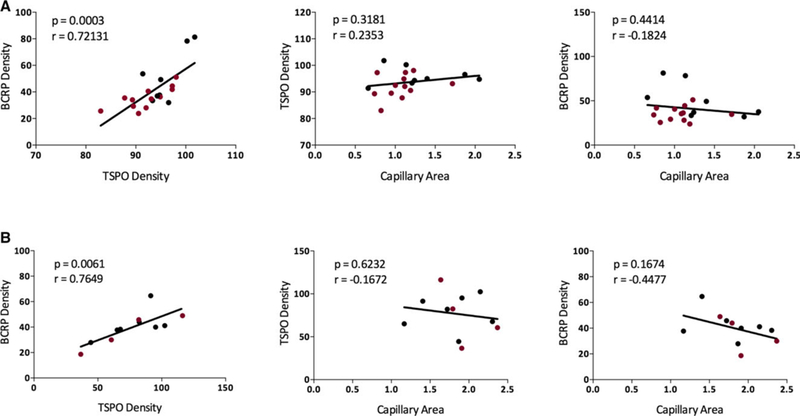
Breast cancer resistance protein (BCRP) density increased with translocator protein (TSPO) density in brain tissue samples from people with drug‐resistant epilepsy (medial temporal sclerosis [MTS] tissue is shown in red). Results were consistent across: A, the first (n = 20, P = .0003, r = .72131), and B, the second (n = 13, P = .0061, r = .7649) set of tissue samples. No positive correlation was observed between BCRP or TSPO density and capillary area
We similarly observed the same positive correlation between BCRP and TSPO density in tissue set B (P = .0061, Pearson r = .76488), neither of which correlated with capillary area (Figure 4B). Results were also statistically significant when data were combined and the batch effect was accounted for (P = .0034, Pearson partial correlation r = .51806). No correlation was observed between P-gp density and COX-1, COX-2, or TSPO, or between BCRP density and COX-1 or COX-2 (Table S3). Because each tissue sample was stained with an ABC transporter antibody and one of the inflammatory protein antibodies, we had no dataset in which the inflammatory protein antibodies were on the same slide; thus we could not determine whether a direct correlation existed between COX-1, COX-2, and TSPO.
3.3. Effect of sclerosis on protein expression
When assessing whether MTS affected protein expression, BCRP was the only protein to show a difference between groups. BCRP density, examined across 4 separate experiments, displayed a trend-level difference in which expression was higher in non-MTS than MTS tissue (Table 2). No differences were found in the expression of COX-1, COX-2, TSPO, or P-gp between the MTS and non-MTS groups (Table 2). In addition, no statistically significant differences were found between the MTS and non-MTS groups in the expression of microglia or neurons (as determined by the area of the cell mask; data not shown). GFAP-astrocyte density was significantly higher in non- MTS tissue in 2 experiments (P < .05), but no difference was seen in the third experiment (each protein was measured independently at least twice). In addition, no change was observed in either capillary density (measured by glut-1 density) or capillary area (measured by the area of the capillary mask) between the groups for either tissue set (data not shown).
TABLE 2.
Influence of MTS on ABC transporter and inflammatory protein
| Tissue set A |
Tissue set B |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Experiment 1 |
Experiment 2 |
Experiment 3 |
Experiment 1 |
|||||||||
| MTS | Non‐MTS | P value | MTS | Non‐MTS | P value | MTS | Non‐MTS | P value | MTS | Non‐MTS | P value | |
| P‐gp | 11.3 | 9.3 | .2363 | 10.4 | 10.6 | .485 | 6.8 | 12.1 | .0165* | ‐ | ‐ | ‐ |
| BCRP | 6.8 | 12.1 | .0165* | 8.8 | 13.1 | .0576 | 8.6 | 13.4 | .0412* | 5.5 | 6.3 | .3939 |
| COX‐1 | 13 | 6.8 | .0101* | 10.3 | 7.1 | .1148 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| COX‐2 | 10.2 | 11 | .396 | 11.4 | 9.1 | .2134 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| TSPO | 8.2 | 10.1 | .2374 | 8.6 | 13.4 | .0412 | ‐ | ‐ | ‐ | 5.5 | 6.3 | .3939 |
ABC, ATP‐binding cassette; BCRP, breast cancer resistance protein; COX, cyclooxygenase; MTS, mesial temporal sclerosis; P‐gp, P‐glycoprotein; TSPO, transloca-tor protein.
P < .05. Data are expressed as average scores from the Wilcoxon rank‐sum test
3.4. Antibody validation
Double-labeling with an antibody from another commercial provider (Table S1) showed identical staining patterns (Figure S4, columns A-C). There was also reasonable agreement between the expression levels seen using the mRNA probe and the antibody (Figure S4, column D), further confirming that the antibody was binding to the correct target. While control slides showed positive staining (Figure S4, column E), incubating the antibodies with their corresponding PrEST antigens abolished the signal (Figure S4, column F), indicating that the antibodies had affinity for their antigen.
4. DISCUSSION
This study had 2 major findings. First, in brain tissue surgically resected from patients with DRMTLE, COX-1 was predominately expressed in microglia while COX-2 and TSPO were found in both microglia and neurons. Second, no correlation was observed between P-gp and any of the investigated inflammatory proteins; however, BCRP immunoreactivity was positively correlated with TSPO protein levels.
Understanding which cells express COX-1 and COX-2 is the key to understanding their function. To the best of our knowledge, this study is the first to use immunofluorescence techniques to assess COX-2 expression in microglia in surgical tissue from people with DRMTLE. The similar expression levels of COX-2 observed here between microglia and neurons contradicts the existing literature, which has traditionally identified COX-2 expression as predominantly neuronal; this discrepancy is likely attributable to the lack of a microglial marker in other studies. However, our results do align with an RNA-Seq database describing gene RNA levels in brain cells in surgical tissue from patients with epilepsy and those with brain tumors.18 Those data show that COX-2 RNA levels are highly expressed in microglia, with moderate expression in neurons and endothelial cells.
Although no studies have investigated the cell-specific expression pattern of TSPO in human TLE brain tissue, a few have used rat epilepsy models and found positive correlations between TSPO autoradiography binding and microglia staining.19–21 The present study similarly found high TSPO expression in microglia, but also noted high neuronal expression (Figure 2). Although neuronal TSPO expression has been reported in various animal and cell culture models,22 to the best of our knowledge this study is the first to report neuronal TSPO expression in the hippocampus of DRMTLE patients. The present findings of low astrocytic TSPO expression may be due to 1 of 3 of the following factors: (1) that the antibody underestimated expression; (2) that astrocytosis was limited in this tissue set; or (3) that astrocyte TSPO expression is low in DRMTLE tissue. We investigated TSPO expression because of its use as a neuroinflammation biomarker in epi- lepsy10; however, our results indicate that COX-1 may serve as a better marker of microglia-induced neuroinflammation in DRMTLE.
With regard to the second aim of this study, we found that capillary BCRP immunoreactivity correlated positively with TSPO protein expression (Figure 3). Because this relationship has remained largely unexplored, this positive correlation provides limited but intriguing insight into the relationship between inflammation and drug resistance. It should be noted that because TSPO is also expressed in capillaries, we investigated whether BCRP or TSPO immunoreactivity was positively correlated with capillary area but found no such correlation (Figure 3). As regards the expression of P-gp, we found no correlation with any of the inflammatory proteins. This somewhat contradicts prior reports showing a connection between COX-2 activity and P-gp expression in rat epilepsy models.14,15 However, these differences are likely attributable to the fact that, in mice, COX-2 RNA is found mainly in endothelial cells.18 It is also possible that the activity of COX-2—not its expression—is what correlates with P-gp expression. This could also explain why we observed no relationship between COX-2 and BCRP expression, in contrast to previous findings from Salvamoser and colleagues.23
We also investigated putative differences in protein expression between the MTS and non-MTS groups. We found no change in the expression of P-gp, COX-1, COX-2, or TSPO between groups. Many studies have reported increased P-gp expression in brain tissue samples from individuals with MTLE compared to control tissue (for a review, see Loscher & Potschka13 and Wang et al24). However, few studies have investigated P-gp expression based on sclerosis. One study found increased P-gp staining in samples with sclerosis compared to those without,25 and another found no correlation between P-gp expression and gliosis.26 For BCRP, we observed a trend-level increase in expression between non-MTS and MTS tissue (Table 2). This contradicts a study that found no change in BCRP expression in TLE tissue with and without hippocampal sclerosis.27 Studies exploring whether BCRP expression is increased in TLE tissue compared to controls have also obtained mixed results. No change in expression was found when comparing tissue samples with hippocampal sclerosis to those with focal cortical dysplasia28 or postmortem12,27 and surgical healthy tissue samples.27 In contrast, BCRP expression was found to be increased in tissue from individuals with DRMTLE as well as in tissue from individuals with focal cortical dysplasia compared to autopsy control tissue.29
Two key benefits of this study were the use of multiplex immunofluorescence and the ability to measure protein immunoreactivity within a cell mask, both of which allowed for more precise quantification of the target protein. Using multiplex immunofluorescence allowed us to measure inflammatory protein expression in cells that would otherwise not be identified based on staining alone. In addition, by using a previously published method of measuring protein expression within a cell mask, we normalized expression of the protein to the cell type, thus accounting for any changes in cell density between groups. This is particularly important when measuring ABC transporter expression, as capillary density can change in certain pathologies.17
Despite these promising preliminary results, the study is associated with 3 limitations of note. The first, which plagues the field, is the lack of suitable control tissue. The ideal control tissue would be a healthy hippocampus that was surgically resected and rapidly processed, which is unavailable. However, because this study sought to determine the cellular protein expression within a group of subjects with DRMTLE, we felt that control tissue was not necessary to achieve these aims. The second limitation is that despite our efforts to use cell-type markers that capture a large part of the cell, the mask antibodies used provided only partial labeling for the 3 target cell types (ie, microglia, astrocytes, and neurons). Thus, it is likely that our results underestimated COX-1, COX-2, and TSPO expression within these cell types. The third limitation concerns the classification of the tissue as either “MTS” or “non- MTS.” Tissue categorization is complicated by the fact that the extent of neuronal loss and gliosis represents a continuum, and diagnosing MTS depends on analyzing the degree and anatomic distribution of cellular changes within the hippocampus.16 In this study, however, the tissue sample sizes varied, as did the degree of structural integrity upon which pathologic diagnoses were made. Thus, although the diagnosis of MTS vs non-MTS was made based on the tissue available, it is likely that variations exist within each group.
In conclusion, we used surgical tissue from patients with DRMTLE to measure the cellular expression of the inflammatory proteins COX-1, COX-2, and TSPO, as well as to assess whether their expression correlated with that of the efflux transporters P-gp and BCRP. Understanding the cellular expression of inflammatory proteins such as COX-1 and COX-2, and how they relate to efflux transporter expression, is particularly important for elucidating the underlying effects of inflammation on DRE.
Supplementary Material
Significance:
To the best of our knowledge, this study is the first to measure the cellular expression of COX-1, COX-2, and TSPO in microglia, astrocytes, and neurons in surgical brain tissue samples from patients with drug-resistant MTLE. Further research is needed to determine the effects of the COX inflammatory pathway in epilepsy, and how it relates to the expression of the ABC transporters P-gp and BCRP.
Key Points
COX-1 is expressed predominately in microglia, whereas COX-2 is expressed in both microglia and neurons
Expression of BCRP, an ABC transporter, correlated positively with the expression of TSPO, an inflammatory biomarker
The correlation between BCRP and TSPO was not due to an increase in capillary density
BCRP was the only protein to show differential expression between mesial temporal sclerosis (MTS) and non-MTS groups
ACKNOWLEDGMENTS
We thank Ioline Henter for invaluable editorial assistance, Dr. Yin Yao for statistical assistance, Drs. Sara Inati and Susumu Sato for evaluation of the patients, and Drs. John Heiss and Kareem Zaghloul for performing the resective surgeries. We also thank the Human Protein Atlas for materials and support.
Funding information
Familjen Erling-Perssons Stiftelse; National Institute of Neurological Disorders and Stroke, Grant/Award Number: 02-N-0014; National Institute of Mental Health, Grant/Award Number: ZIAMH002795
This article has been contributed to by US Government employees and their work is in the public domain in the USA.
Footnotes
DISCLOSURE
This work was supported by the Intramural Research Programs of the National Institute of Mental Health (ZIAMH002795) and the National Institute of Neurological Disorders and Stroke (02-N-0014), as well as by the Karolinska Institutet (KTH Center for Applied Precision Medicine, funded by the Erling-Persson Family Foundation). The authors have no other conflicts of interest to disclose, financial or otherwise. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- 1.Kwan P, Schachter SC, Brodie MJ. Drug-resistant epilepsy. N Engl J Med. 2011;365:919–26. [DOI] [PubMed] [Google Scholar]
- 2.Vezzani A, Aronica E, Mazarati A, et al. Epilepsy and brain inflammation. Exp Neurol. 2013;244:11–21. [DOI] [PubMed] [Google Scholar]
- 3.Aid S, Bosetti F. Targeting cyclooxygenases-1 and −2 in neuroinflammation: therapeutic implications. Biochimie. 2011;93:46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steffel J, Luscher TF, Ruschitzka F, et al. Cyclooxygenase-2 inhibition and coagulation. J Cardiovasc Pharmacol. 2006;47:15–20. [DOI] [PubMed] [Google Scholar]
- 5.Desjardins P, Sauvageau A, Bouthillier A, et al. Induction of astrocytic cyclooxygenase-2 in epileptic patients with hippocampal sclerosis. Neurochem Int. 2003;42:299–303. [DOI] [PubMed] [Google Scholar]
- 6.Holtman L, van Vliet EA, van Schaik R, et al. Effects of SC58236, a selective COX-2 inhibitor, on epileptogenesis and spontaneous seizures in a rat model for temporal lobe epilepsy. Epilepsy Res. 2009;84:56–66. [DOI] [PubMed] [Google Scholar]
- 7.Fiala M, Avagyan H, Merino JJ, et al. Chemotactic and mitogenic stimuli of neuronal apoptosis in patients with medically intractable temporal lobe epilepsy. Pathophysiology. 2013;20:59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Papadopoulos V, Baraldi M, Guilarte TR, et al. Translocator protein (18 kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci. 2006;27:402–9. [DOI] [PubMed] [Google Scholar]
- 9.Banati RB, Goerres GW, Myers R, et al. [11C](R)-PK11195 positron emission tomography imaging of activated microlgia in vivo in Rasmussen’s encephalitis. Neurology. 1999;53:2199–203. [DOI] [PubMed] [Google Scholar]
- 10.Hirvonen J, Kreisl WC, Fujita M, et al. Increased in vivo expression of an inflammatory marker in temporal lobe epilepsy. J Nucl Med. 2012;53:234–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gershen LD, Zanotti-Fregonara P, Dustin IH, et al. Neuroinflammation in temporal lobe epilepsy measured using positron emission tomographic imaging of translocator protein. JAMA Neurol. 2015;72:882–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu JY, Thom M, Catarino CB, et al. Neuropathology of the blood-brain barrier and pharmaco-resistance in human epilepsy. Brain. 2012;135:3115–33. [DOI] [PubMed] [Google Scholar]
- 13.Loscher W, Potschka H. Role of drug efflux transporters in the brain for drug disposition and treatment of brain diseases. Prog Neurobiol. 2005;76:22–76. [DOI] [PubMed] [Google Scholar]
- 14.Bauer B, Hartz AM, Pekcec A, et al. Seizure-induced up-regulation of P-glycoprotein at the blood-brain barrier through glutamate and cyclooxygenase-2 signaling. Mol Pharmacol. 2008;73:1444–53. [DOI] [PubMed] [Google Scholar]
- 15.van Vliet EA, Zibell G, Pekcec A, et al. COX-2 inhibition controls P-glycoprotein expression and promotes brain delivery of phenytoin in chronic epileptic rats. Neuropharmacology. 2010;58:404–12. [DOI] [PubMed] [Google Scholar]
- 16.Blumcke I, Thom M, Aronica E, et al. International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a Task Force report from the ILAE Commission on Diagnostic Methods. Epilepsia. 2013;54:1315–29. [DOI] [PubMed] [Google Scholar]
- 17.Kannan P, Schain M, Kretzschmar WW, et al. An automated method measures variability in P-glycoprotein and ABCG2 densities across brain regions and brain matter. J Cereb Blood Flow Metab. 2016;37(6):2062–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bennett ML, Bennett FC, Liddelow SA, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci USA. 2016;113:1738–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amhaoul H, Hamaide J, Bertoglio D, et al. Brain inflammation in a chronic epilepsy model: evolving pattern of the translocator protein during epileptogenesis. Neurobiol Dis. 2015;82:526–39. [DOI] [PubMed] [Google Scholar]
- 20.Dedeurwaerdere S, Callaghan PD, Pham T, et al. PET imaging of brain inflammation during early epileptogenesis in a rat model of temporal lobe epilepsy. EJNMMI Res. 2012;2:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harhausen D, Sudmann V, Khojasteh U, et al. Specific imaging of inflammation with the 18 kDa translocator protein ligand DPA-714 in animal models of epilepsy and stroke. PLoS One. 2013;8:e69529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rupprecht R, Papadopoulos V, Rammes G, et al. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov. 2010;9:971–88. [DOI] [PubMed] [Google Scholar]
- 23.Salvamoser JD, Avemary J, Luna-Munguia H, et al. Glutamatemediated down-regulation of the multidrug-resistance protein BCRP/ABCG2 in porcine and human brain capillaries. Mol Pharm. 2015;12:2049–60. [DOI] [PubMed] [Google Scholar]
- 24.Wang GX, Wang DW, Liu Y, et al. Intractable epilepsy and the P-glycoprotein hypothesis. Int J Neurosci. 2016;126:385–92. [DOI] [PubMed] [Google Scholar]
- 25.Aronica E, Gorter JA, Ramkema M, et al. Expression and cellular distribution of multidrug resistance-related proteins in the hippocampus of patients with mesial temporal lobe epilepsy. Epilepsia. 2004;45:441–51. [DOI] [PubMed] [Google Scholar]
- 26.Kubota H, Ishihara H, Langmann T, et al. Distribution and functional activity of P-glycoprotein and multidrug resistance-associated proteins in human brain microvascular endothelial cells in hippocampal sclerosis. Epilepsy Res. 2006;68:213–28. [DOI] [PubMed] [Google Scholar]
- 27.Aronica E, Gorter JA, Redeker S, et al. Localization of breast cancer resistance protein (BCRP) in microvessel endothelium of human control and epileptic brain. Epilepsia. 2005;46:849–57. [DOI] [PubMed] [Google Scholar]
- 28.Sisodiya SM, Martinian L, Scheffer GL, et al. Major vault protein, a marker of drug resistance, is upregulated in refractory epilepsy. Epilepsia. 2003;44:1388–96. [DOI] [PubMed] [Google Scholar]
- 29.Banerjee Dixit A, Sharma D, Srivastava A, et al. Upregulation of breast cancer resistance protein and major vault protein in drug resistant epilepsy. Seizure. 2017;47:9–12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.