

Abstract
Transcription factor TFEB is thought to control cellular functions—including in the vascular bed—primarily via regulation of lysosomal biogenesis and autophagic flux. Here, we report that TFEB also orchestrates a non‐canonical program that controls the cell cycle/VEGFR2 pathway in the developing vasculature. In endothelial cells, TFEB depletion halts proliferation at the G1‐S transition by inhibiting the CDK4/Rb pathway. TFEB‐deficient cells attempt to compensate for this limitation by increasing VEGFR2 levels at the plasma membrane via microRNA‐mediated mechanisms and controlled membrane trafficking. TFEB stimulates expression of the miR‐15a/16‐1 cluster, which limits VEGFR2 transcript stability and negatively modulates expression of MYO1C, a regulator of VEGFR2 trafficking to the cell surface. Altered levels of miR‐15a/16‐1 and MYO1C in TFEB‐depleted cells cause increased expression of plasma membrane VEGFR2, but in a manner associated with low signaling strength. An endothelium‐specific Tfeb‐knockout mouse model displays defects in fetal and newborn mouse vasculature caused by reduced endothelial proliferation and by anomalous function of the VEGFR2 pathway. These previously unrecognized functions of TFEB expand its role beyond regulation of the autophagic pathway in the vascular system.
Keywords: angiogenesis, embryo, membrane trafficking, miRNA transcription, proliferation
Subject Categories: Cell Cycle, Transcription, Vascular Biology & Angiogenesis
Introduction
Transcription factor EB (TFEB) belongs to the microphthalmia family of bHLH‐leucine zipper molecules. It is involved in the biogenesis and function of the endo‐lysosomal compartment, including membrane trafficking and autophagy (Sardiello et al, 2009; Settembre et al, 2011, 2013b; Napolitano & Ballabio, 2016; Raben & Puertollano, 2016). Furthermore, TFEB mutation characterizes a subset of renal cell carcinoma carrying the t(6;11)(p21:q13) translocation, which leads to a TFEB promoter substitution with the 5′ upstream regulatory sequence of the alpha intronless gene (Calcagnì et al, 2016).
TFEB recognizes E‐box‐type DNA sequences (Palmieri et al, 2011) and resides in the cytosol, moving to the nucleus when lysosomes and autophagy are required for cell activities (Martina et al, 2012; Settembre et al, 2012). The functions of TFEB are mainly regulated by mTOR complex 1 (mTORc1), which integrates energy availability with cellular demand. In the presence of nutrients, mTORc1 phosphorylates TFEB and inhibits its transport into the nucleus. Conversely, under starvation conditions, when mTORc1 is inactive, unphosphorylated TFEB rapidly accumulates in the nucleus. In general terms, TFEB is involved in the pathogenesis of lysosomal storage diseases (Xu & Ren, 2015) and, through its connection with mTOR pathway, in the control of energy expenditure both in physiology (Mansueto et al, 2017) and in pathology (Perera et al, 2015; Di Malta et al, 2017).
An increasing number of observations suggest a pivotal role of TFEB in vascular biology. Targeted inactivation of TFEB in mice results in impaired placental vascularization and inhibits the expression of VEGF‐A in labyrinthine cells (Steingrímsson et al, 1998). The embryonic vasculature is unable to invade the placenta, halting the exchange of nutrients and causing lethal hypoxia and embryonic lethality. During atherogenesis, the lysosomal stress induced by the accumulation cholesterol activates a TFEB response, which triggers an anti‐inflammatory (Lu et al, 2017) and anti‐atherogenic response (Emanuel et al, 2014). Finally, the overexpression of Tfeb in endothelial cells (ECs) promotes post‐ischemic angiogenesis through the activation of autophagic flux (Fan et al, 2018).
Using EC‐specific loss‐of‐function mouse mutants (Tfeb EC−/−) and cellular models, we investigated the effect of Tfeb deletion on the vasculature in the embryo and in newborn mice. We found that TFEB positively controls the expression of cyclin‐dependent kinase 4 (CDK4) and its deletion results in the block of cell growth and in a futile attempt to recover this process by targeting vascular endothelial growth factor (VEGF) receptor (R)‐2.
Results
Tfeb is expressed in embryonic and post‐natal vessels
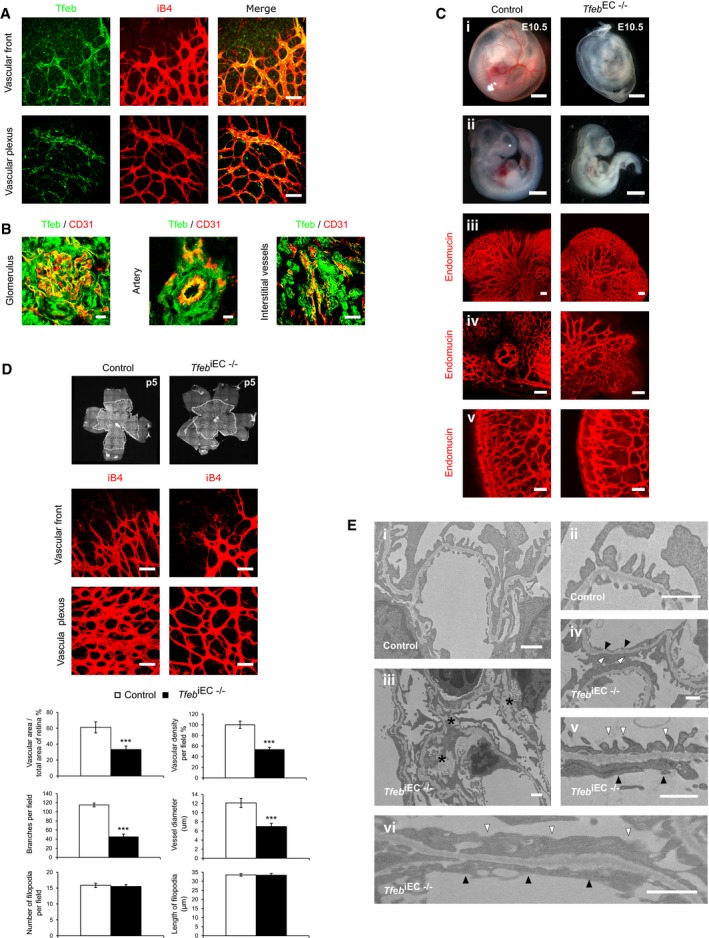
To analyze Tfeb expression in the vasculature, we used constitutive knock‐in Tfeb‐EGFP mice. Tfeb was expressed very early in developing vessels and persisted in newborn pups. The vascular expression was heterogeneous and not generalized to all ECs (Figs 1A and B, and EV1A), suggesting a dynamic role in the vasculature. At E9.5, Tfeb‐EGFP co‐localized with endothelial endomucin in head and in the intersomitic vessels as well as yolk sac capillaries (Fig EV1A). We then examined the expression of Tfeb in retina and kidney, whose vascular beds undergo post‐natal development (Gariano & Gardner, 2005; Little & McMahon, 2012). At p5, Tfeb‐EGFP was present in both large and small retinal vessels at the vascular front and vascular plexus (Fig 1A). The analysis of renal vessels at p17 showed that Tfeb was present in glomerulus, capillaries and some small arteries (Fig 1B). As reported by the whole mRNA expression analysis (Steingrímsson et al, 1998), Tfeb‐EGFP was present in alpha‐smooth muscle actin (SMA)‐positive cells and pericytes of embryo tissues (E9.5; Fig EV1B and C) and retina (p5; Fig EV1D) as well as in renal podocytes (p17; Fig EV1E), as inferred by the use of specific antibodies anti‐SMA, neural/glial antigen‐2 (NG2), and podocin.
Figure 1. Mouse genetic ablation of endothelial Tfeb leads to vascular defects.
-
A, BTfeb expression in the vasculature (mice n = 10). (A) Representative images of retinal vessels (p5) (A) (scale bar: 50 μm) and (B) glomerular, arterial (scale bars: 10 μm) and interstitial vessels (scale bar: 50 μm) of kidney (p17) of Tfeb‐EGFP mice stained with anti‐iB4 (A), anti‐CD31 (B) and anti‐GFP (A,B).
-
CAlterations in the embryonic vasculature in Tfeb EC−/− mice at E10.5 (mice n = 25). Representative images of whole‐mount embryos and yolk sacs (i, ii) (scale bars: 0.5 mm). Vessels of the head (iii), ocular (iv), and intrasomitic regions (v) were stained with anti‐endomucin Ab (scale bars: 100 μm).
-
DAlteration of retinal vascular maturation in Tfeb iEC−/− at p5. Representative images of whole mounts of retina and immunostaining of vascular front and vascular plexus with an anti‐iB4 Ab (scale bars: 50 μm). Bar graphs indicate the percent of vascular area versus total area of the retina, the percent of vascular density, the number of branches per field, vessel diameter, the number of filopodia per field, and the length of filopodia (mice n = 10, mean ± SEM; ***P < 0.0001 versus control mice by Student's t‐test).
-
EAlteration of the glomerular ultrastructure in Tfeb iEC−/− mice at p17 (mice n = 5). Representative transmission electron micrographs of renal tissue. Magnification: (i) ×6,000, (ii) ×20,000), (iii–iv) ×10,000, (v–vi) ×25,000; scale bars: 1 μm). Asterisks indicate the accumulation of extracellular matrix in mesangium; white and black triangles indicate the fusion of podocyte foot processes and the lack of endothelial fenestrae, respectively.
Figure EV1. Different cells types express Tfeb in embryos, yolk sac, retina, and kidney.
-
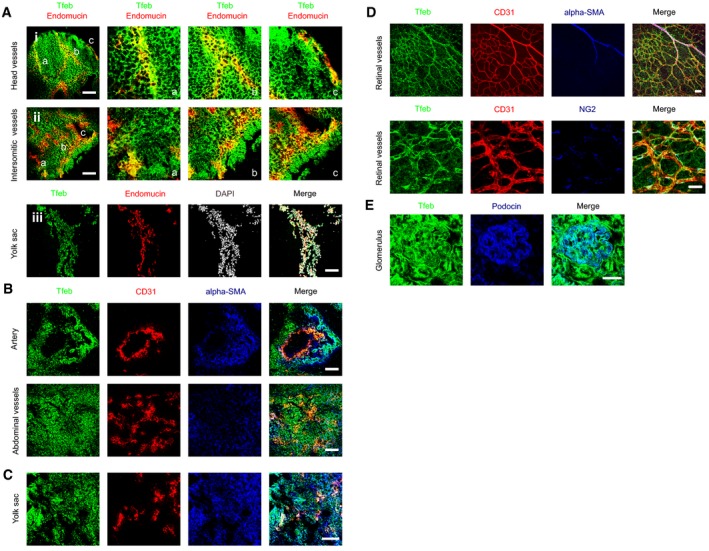
ATfeb expression in embryos at E9.5 (embryos n = 6). Representative images of head (details a, b, c), intrasomitic region (details a, b, c), and yolk sac of Tfeb‐EGFP embryos stained with anti‐GFP and anti‐endomucin Abs (scale bars: 100 μm).
-
B–ETfeb expression in ECs and smooth muscle cells in embryos, retina, and kidney. Representative images of Tfeb‐EGFP embryos (E9.5) (B), yolk sac (E9.5) (C), retina (p5) (D), and renal glomerulus (E) (embryos and mice n = 6) stained with anti‐CD31, anti‐alpha SMA, anti‐NG2, anti‐podocin, and anti‐GFP Abs (scale bars: 100 μm).
Tfeb expression in ECs is essential for vascular development
To investigate the role of Tfeb in embryonic vessel development, we interbred Tfeb floxed mice (Settembre et al, 2013a) with Tie2‐Cre mice, which allows EC‐specific gene targeting from E8.5 (Kisanuki et al, 2001). Tie2‐Cre ˉ /Tfeb floxed (control) and Tie2‐Cre + /Tfeb −/+ (Tfeb EC−/+) embryos survived, while Tie2‐Cre + /Tfeb −/− (Tfeb EC−/−) mice were absent among the different progenies (n = 8), indicating embryonic lethality.
At E9.5, analysis of control, Tfeb EC−/+, and Tfeb EC−/− embryos showed indistinguishable phenotypes from normal vasculature (Fig EV2A). At E10.5, Tfeb EC−/+ mice were similar to control, while Tfeb EC−/− presented an altered vascular phenotype (Fig 1C). Indeed, embryos and yolk sacs from Tfeb EC−/− mice were smaller and paler than controls (Fig 1C), with evident hypoxic areas (Fig EV2B). Whole‐mount (i, ii) and endomucin staining of the head (iii), ocular (iv), and intersomitic regions (v) displayed vascular defects characterized by excessive fusion into irregular dilated vessels, reduced branching, and failure to follow the normal anatomical patterns seen in control mice. Furthermore, the intersomitic region was characterized by reduced vascular invasion into somitic tissues and the presence of avascularized zones (Fig 1C). The percentage of point prevalence of vascular defects in Tfeb EC−/− (n = 12) and control embryos (n = 13) at E10.5 was respectively 83 and 16%.
Figure EV2. Tfeb deletion in embryos and pups.
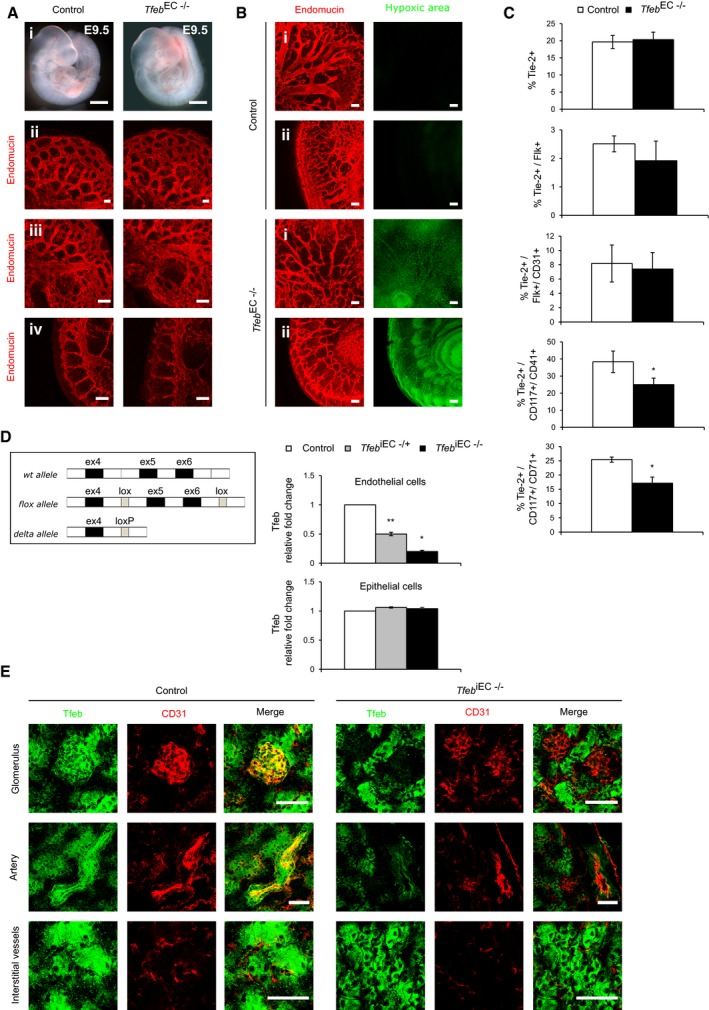
- EC Tfeb deletion does not induce alterations in the embryonic vasculature at E9.5 (embryos n = 6). Representative images of whole‐mount embryos (i) (scale bars: 0.5 mm). Vessels of the head (ii), ocular (iii), and intrasomitic regions (iv) were stained with anti‐endomucin Ab (scale bars: 100 μm).
- EC Tfeb deletion induces embryonic hypoxia at E10.5 (embryos n = 6). Representative images of vessels and tissues of the head (i) and intrasomitic region (ii) of control and Tfeb EC−/− embryos stained with anti‐endomucin (red) and the hypoxic marker pimonidazole (green; scale bars: 100 μm).
- Expression of Tie2 and markers of the endothelial and hematopoietic lineages in TfebEC−/− yolk sacs at E9.5. Bar graphs indicate the percentage of yolk sac Tie2+ cells or Flk+ or CD31+ or CD117+/CD41+ and CD117+/CD71+ within the Tie2+ population (n = 3, mean ± SEM; *P < 0.01 versus control mice by Student's t‐test).
- Genotype of Tfeb iEC mice. Schematic representation of murine Tfeb, the targeted allele and the knockout allele (delta allele), and qPCR analysis of mRNA encoded by Tfeb exon 5–6 in lung ECs and epithelial cells isolated from control, Tfeb iEC−/+, and Tfeb iEC−/− mice. Data are expressed as relative fold‐change compared with the expression in control mice after normalization to the housekeeping gene TBP (mice n = 3, mean ± SEM; *P < 0.01 and **P < 0.001 versus control mice by Student's t‐test).
- Tfeb expression in renal tissue of control and Tfeb iEC−/− mice (p17). Representative images of renal glomerulus, artery, interstitial vessels, and the surrounding tissue of control and Tfeb iEC−/− mice (mice n = 6). Tissues were stained with anti‐CD31 and anti‐GFP Abs (scale bars: 100 μm).
Since Tie2 is also expressed by hematopoietic precursors at E8.5 (Takakura et al, 1998), we evaluated the effects of Tfeb deletion on the maturation of this system. The expression of markers characterizing the hematopoietic and endothelial lineages, together with Tie2, was analyzed in yolk sacs at E9.5. The percentage of Tie2+ cells in the control was similar to that of Tfeb EC−/− yolk sacs as well as the abundance of early endothelial precursors identified as Tie2+/Flk+ and Tie2+/Flk+/CD31+. Interestingly, we observed a slight reduction in the Tie2+/CD117+/CD41+ cells and Tie2+/CD117+/CD71+ cells that represent early erythroid precursors (Fig EV2C).
These data support the concept that TFEB acts in a later phase of vascular development, mostly characterized by the remodeling of the primitive vascular plexus.
Tfeb is involved in retinal and renal vascular maturation after birth
To overcome early embryonic lethality, we generated inducible EC‐specific mutants by crossing Tfeb floxed mice with Cdh5‐CreERT2 mice (Wang et al, 2010), thus allowing generating Cdh5‐CreERT2 + /Tfeb −/+ (Tfeb iEC−/+) and Cdh5‐CreERT2 + /Tfeb −/− (Tfeb iEC−/−) and Cdh5‐CreERT2 ˉ/Tfeb floxed (control) mice.
The successful deletion of Tfeb after in vivo Cre induction by tamoxifen was established by detecting the Tfeb delta allele in genomic DNA after lox site recombination. In Tfeb iEC−/+ and Tfeb iEC−/− mice, Tfeb deletion specifically occurred in endothelium as evidenced by the decrease in the exons 5 and 6 transcript only in ECs but not in epithelial cells isolated from the lungs (Fig EV2D). The recombination efficiency and specificity were further demonstrated by the marked Tfeb reduction in the renal vasculature of Tfeb iEC−/− mice (p17) but not in surrounding tissues (Fig EV2E).
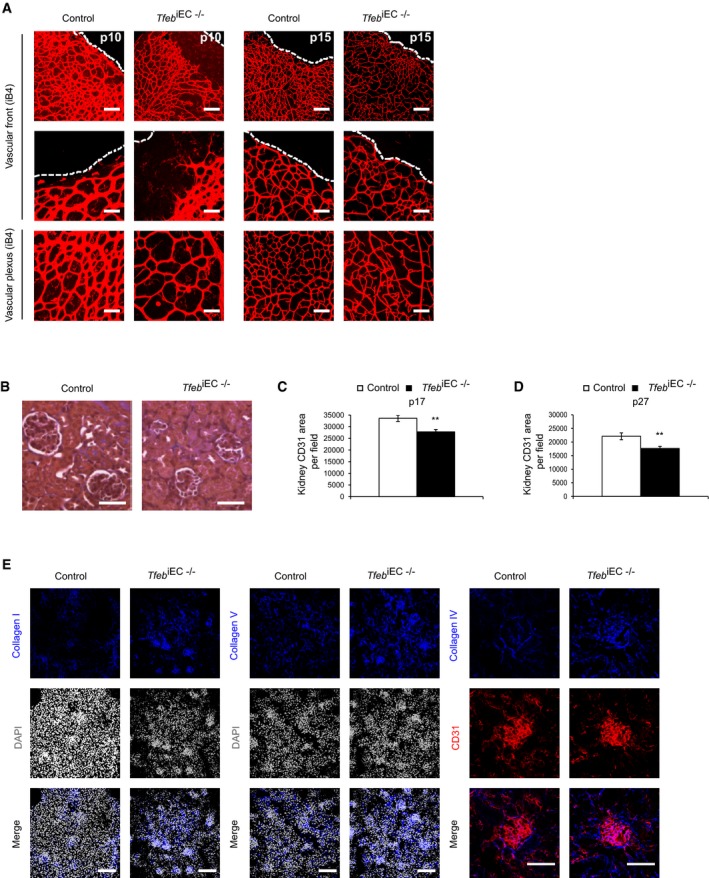
According to the post‐natal maturation of retinal and renal vasculature, we examined the consequences of Tfeb deletion in the ECs of these organs. At p5, retinas from Tfeb iEC−/− mice showed impaired outgrowth of the superficial capillary network (Fig 1D). The retinal vascular mesh was wider with a reduction of the vascular area, vascular density, branching points, and vessel size (Fig 1D). The vascular front did not show any differences in the number and length of filopodia (Fig 1D). The detrimental effect of Tfeb deletion persisted up to p15, when vascular retinal net reaches the maturity. At p10, the net features were similar to those reported at p5. From p10 to p15, the vascular area reached the size observed in control mice but the alterations in vascular density, vessel diameters, and branching points were still present (Fig EV3A).
Figure EV3. Post‐natal maturation of retinal and renal vasculature after endothelium Tfeb silencing.
-
AEC Tfeb deletion compromises retinal vascular maturation at p10 and p15. Representative images of immunostaining of vascular front and vascular plexus of retina of control and Tfeb iEC−/− mice (p10, p15) with an anti‐iB4 Ab (scale bars: 50 μm).
-
B–DEC Tfeb deletion compromises glomerular development and renal vascularization at p17. (B) Representative images of renal trichrome staining of control and Tfeb iEC−/− mice at p17 (original magnification x10, scale bars: 25 μm). (C) Bar graph indicates the CD31+ vascular area in control and Tfeb iEC−/− mice (p17; mice n = 5, mean ± SEM; **P < 0.001 versus control mice by Student's t‐test). (D) EC Tfeb deletion compromises renal vascularization at p27. Bar graph indicates the CD31+ vascular area in control and Tfeb iEC−/− mice (p27; mice n = 5, mean ± SEM; **P < 0.001 versus control mice by Student's t‐test).
-
EEC Tfeb deletion correlates with collagen deposition in kidney at p27. Representative images of immunostaining of kidney of control and Tfeb iEC−/− mice (p27) with anti‐collagen I, anti‐collagen V, and anti‐collagen IV antibodies (scale bars: 50 μm). Quantification area per field (mean ± SEM): collagen I: 26,107.2 ± 3,200.82 μm2 in control mice versus 43,069 ± 9,754.7 μm2 in Tfeb iEC−/− mice, mice n = 4, P = 0.02 versus control mice by Student's t‐test; collagen V: 33,500.4 ± 4,201 μm2 in control mice versus 47,007.9 ± 6,287.7 μm2 in Tfeb iEC−/− mice, mice n = 4, P = 0.04 versus control mice by Student's t‐test; collagen IV: 10,972.12 ± 1,856.8 μm2 in control mice versus 17,525.07 ± 2,106.58 μm2 in Tfeb iEC−/− mice, mice n = 4, P = 0.03 versus control mice by Student's t‐test)
Correct assembly of the glomerular vasculature is required for filtration barrier function (Bartlett et al, 2016). At p17, Tfeb iEC−/− kidneys were smaller (% area of Tfeb iEC−/− mice versus control 72.6 ± 7.9%; mice n = 3, P < 0.01), characterized by poorly developed glomeruli (Fig EV3B) and reduced volumes (20.53 ± 2.50 × 104 μm3 for control mice and 14.43 ± 3.27 × 104 μm3 for Tfeb iEC−/−; mice n = 3, P < 0.002). The altered glomerular development paralleled the reduced CD31+ endothelial area in Tfeb iEC−/− kidneys (p17; Fig EV3C), which persisted up to p27 (Fig EV3D). Transmission electron microscopy showed deep alterations in glomerular structure, with expansion of the mesangium by deposition of extracellular matrix, focal loss of podocyte foot processes, and endothelial fenestration (Fig 1E). According to the increased deposition of extracellular matrix in Tfeb iEC−/− (Fig 1E), we observed a significant accumulation of collagen IV along capillaries, and collagen I in the interstitium, while collagen V was only moderately increased (Fig EV3E), supporting the hypothesis that Tfeb deletion could be instrumental in renal fibrosis.
In Tfeb iEC−/− mice, we did not observe any alterations of the ratio between vascular muscle cells and ECs both in retina (ratio NG2 area/IB4+ vascular area 1.2 ± 0.1 in control mice and 1.2 ± 0.3 in Tfeb iEC−/−; mice n = 4, P = ns) and in renal glomeruli (ratio podocytes/CD31+ vascular area 1 ± 0.1 in control mice and 0.9 ± 0.1 in Tfeb iEC−/−; mice n = 4, P = ns). These data support the role of the endothelial dysfunction in the histological alterations of the retina and kidney observed after Tfeb deletion.
Tfeb deletion reduces the proliferation of ECs
The main processes characterizing vascular development are the proliferation and the migration of ECs and their relationships with extracellular matrix (Carmeliet & Jain, 2011). Therefore, to understand the vascular defects observed in vivo, we studied ECs lacking TFEB by analyzing their growth, motility, and morphogenetic capability when layered on extracellular matrix.
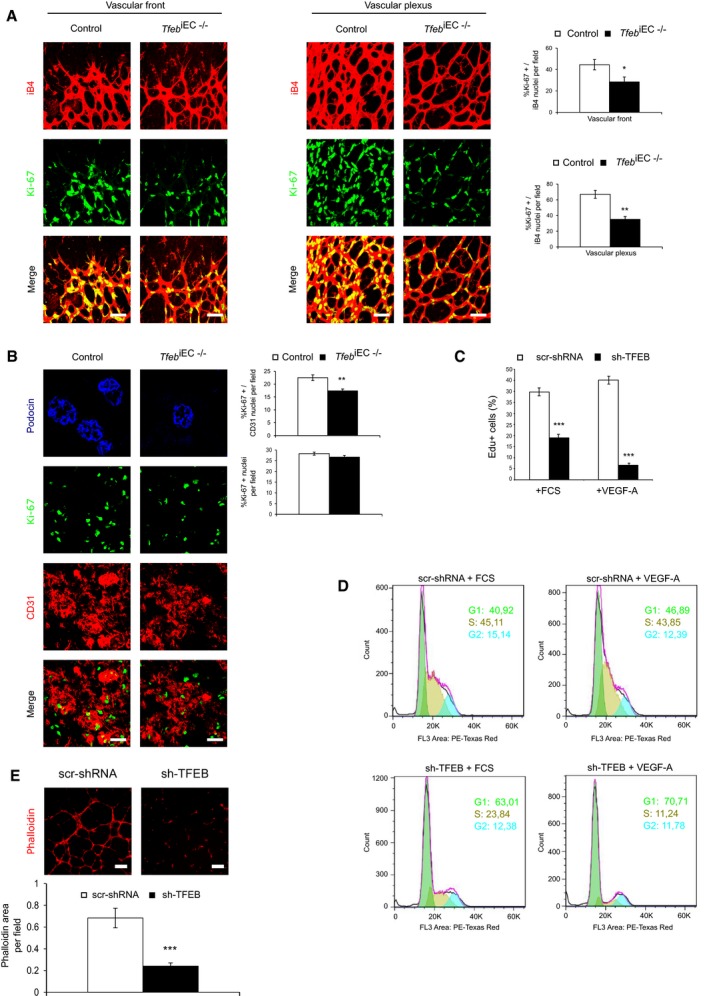
The in vivo analysis of Ki‐67+ EC nuclei at p5 and p17 indicated a marked reduction in proliferating ECs in both the retina (37% in vascular front and 50% in vascular plexus) and kidney vessels (30%) of Tfeb iEC−/− mice (Fig 2A and B). Of note, in kidney the total number of Ki‐67+ cells per field was not modified in Tfeb iEC−/− mice compared to control (Fig 2B), indicating that the proliferation rate of other cell types was not modified and suggesting that the impaired EC growth is instrumental in the alteration of renal maturation.
Figure 2. Tfeb deletion reduces EC proliferation in vivo.
-
A, BReduced EC proliferation in the retina (p5) and kidney (p17) of in Tfeb iEC−/− mice. (A) Representative images of vessels of the vascular front and vascular plexus in the retina of control and Tfeb iEC−/− mice stained with anti‐iB4 and Ki‐67 antibodies (scale bars: 50 μm). Bar graph indicates the percentage of EC Ki‐67+ nuclei versus total nuclei co‐localized with iB4+ vessels (mice n = 6, mean ± SEM; **P < 0.001 and *P < 0.01 versus control mice by Student's t‐test). (B) Representative images of vessels of the kidney in control and Tfeb iEC−/− mice stained with anti‐CD31 and Ki‐67 antibodies (scale bars: 50 μm). Bar graph indicates the percent of EC Ki‐67+ nuclei versus total nuclei co‐localized with CD31+ vessels (mice n = 6, mean ± SEM; **P < 0.001 versus control mice by Student's t‐test). Podocin staining is shown to glomerular localization.
-
C, DTFEB silencing reduced EC proliferation. Representative graph of scr‐shRNA and sh‐TFEB ECs treated for 24 h with FCS (20%) or VEGF‐A (30 ng/ml). (C) DNA incorporation of EdU was detected by flow cytometry. The percentage of proliferating cells is indicated (n = 4, mean ± SEM; ***P < 0.0001 versus scr‐shRNA ECs by Student's t‐test). (D) DNA content was determined by propidium iodide staining and assessed by fluorescence‐activated cell sorter analysis (representative experiment out of 4 with similar results).
-
ETFEB silencing reduced human EC morphogenesis. Representative images of tube‐like structure of scr‐shRNA and sh‐TFEB human ECs stained with phalloidin‐555. Bar graph indicates the percentage of phalloidin+ area in sh‐TFEB and scr‐shRNA ECs (scale bars: 0.25 mm; n = 6, mean ± SEM; ***P < 0.0001 versus scr‐shRNA by Student's t‐test).
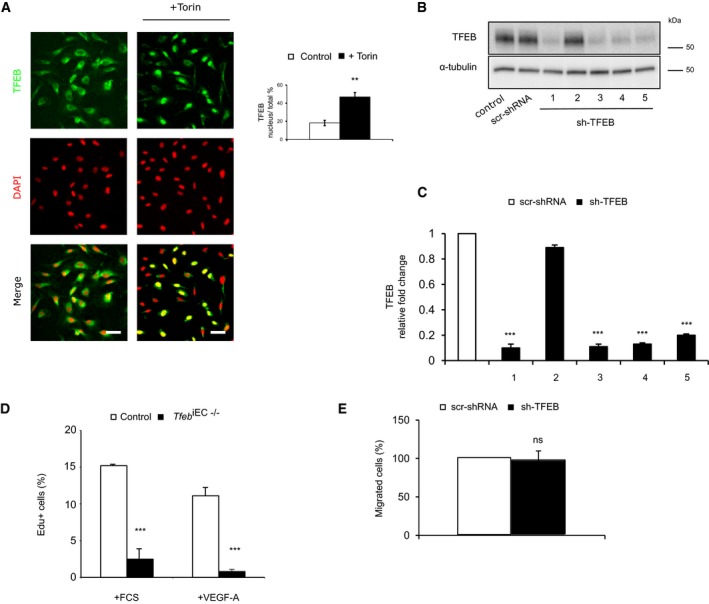
Before evaluating the effect of TFEB deletion in vitro, we experimentally verified whether TFEB mechanism of action in ECs was similar to that described in other cell types. In EC standard culture conditions, we showed a cytosolic and nuclear endogenous expression of TFEB (Fig EV4A). Furthermore, when ECs were treated with Torin, an mTOR inhibitor that mimics starvation conditions and activates TFEB (Settembre et al, 2012), we observed an increase in its nuclear translocation (Fig EV4A). We silenced TFEB via specific short‐hairpin RNA (Fig EV4B and C), and the in vitro ECs’ proliferation rate was evaluated by the count of 5‐ethynyl‐2′‐deoxyuridine (EdU)‐positive cells. The proliferative effect of fetal calf serum (FCS) and vascular endothelial growth factor‐A (VEGF‐A) was significantly impaired in TFEB‐silenced human ECs (sh‐TFEB ECs; Fig 2C) and in ECs from lung of Tfeb iEC−/− mice (Fig EV4D). TFEB silencing in ECs specifically restrained the G1–S cycle transition, as assessed by propidium iodide staining (Fig 2D). In sh‐TFEB ECs stimulated by VEGF‐A or FCS, we evidenced an increased percentage of cells blocked in the G1 phase and a decreased percentage of those progressing in S‐phase (Fig 2D).
Figure EV4. TFEB expression and regulation in human ECs. Additional effects of Tfeb deletion on human ECs.
-
AmTOR inhibition activates TFEB nuclear translocation. Representative images of human ECs under basal conditions or after Torin stimulation (100 nM, 1 h) stained with anti‐TFEB Ab and DAPI (scale bars: 20 μm). Bar graphs indicate the percentage of TFEB positive nuclei versus total nuclei after Torin challenge (n = 3, mean ± SEM; **P < 0.001 versus control cells by Student's t‐test).
-
B, CCharacterization of TFEB shRNA (n = 3). (B) Western blot of TFEB in human ECs carrying five different commercial sh‐TFEB RNAs (1‐5) or the appropriate control (scr‐shRNA) transduced by pLKO lentivirus vector. (C) TFEB mRNA expression analyzed by qPCR in ECs carrying the different TFEB shRNAs (1‐5). Data are expressed as relative fold‐change compared with scr‐shRNA after normalization to the housekeeping gene TBP (n = 3, mean ± SEM; ***P < 0.0001 by Student's t‐test).
-
DTFEB silencing reduced murine EC proliferation. Representative graph of lung ECs isolated from control and Tfeb iEC−/− mice ECs treated for 24 h with FCS (20%) or VEGF‐A (30 ng/ml). DNA incorporation of EdU was detected by flow cytometry. The percentage of proliferating cells is indicated (n = 4, mean ± SEM; ***P < 0.0001 versus ECs derived from control mice by Student's t‐test).
-
ETFEB silencing does not affect human EC migration. Bar graph indicates the percentage of migrated sh‐TFEB and scr‐shRNA ECs stimulated with VEGF‐A (30 ng/ml, 5 h; n = 6, mean ± SEM, P = ns versus scr‐shRNA by Student's t‐test).
Source data are available online for this figure.
Then, we studied the effect of TFEB deletion on EC migration. An indirect in vivo evidence of EC motility is the analysis of filopodia, which characterize migrating cells. As shown in Fig 1D, the number of filopodia at the front of retinal plexus was similar in control and in Tfeb iEC−/− mice, suggesting that Tfeb deletion did not affect EC motility. Accordingly, sh‐TFEB and scr‐shRNA ECs’ in vitro chemotactic response to VEGF‐A was similar, suggesting that TFEB down‐regulation did not interfere with EC motility (Fig EV4E).
Finally, the effect of TFEB deletion was further examined in a morphogenetic assay. As shown in Fig 2E, the absence of TFEB dampened ECs to form tube‐like structures on reduced growth factor Matrigel only after VEGF‐A stimulation.
TFEB modifies the transcriptional landscape in ECs
To describe the genetic program triggered by TFEB in ECs, we investigated the transcriptome modifications induced by TFEB silencing and the DNA promoter regions to which TFEB is recruited. The comparative transcriptome analysis of scr‐shRNA and sh‐TFEB ECs by LIMMA (Smyth, 2002) defined a subset of 502 differentially expressed genes (DEGs; 133 up‐regulated and 369 down‐regulated, |log2FC|> 0.5 and FDR < 0.1; Fig 3A). A volcano plot showed the changes in the log2 fold‐change and P‐values for all the genes in sh‐TFEB ECs compared to scr‐shRNA cells (Fig EV5A). The enriched biological functions of up‐ and down‐regulated genes were determined by DAVID analysis. In sh‐TFEB ECs, down‐regulated genes characterized processes related to “Cell Cycle”, “Cell Division”, “Mitotic cell cycle process”, “G1/S transition of mitotic cell cycle”, “DNA replication”, and “DNA metabolic process”. This result is consistent with the observation of the arrest of cell cycle and proliferation in vivo and in vitro after Tfeb silencing. On the other hand, up‐regulated gene enrichment was limited to “Positive regulation of protein catabolic process” and “Endocytosis” categories (Fig 3B). Gene set Enrichment Analysis (GSEA) revealed the enrichment of the “negative regulation of cellular process”, “negative regulation of proliferation”, and “vesicle‐mediated transport” categories, supporting the DAVID analysis (Fig 3C).
Figure 3. TFEB gene regulation in human ECs.
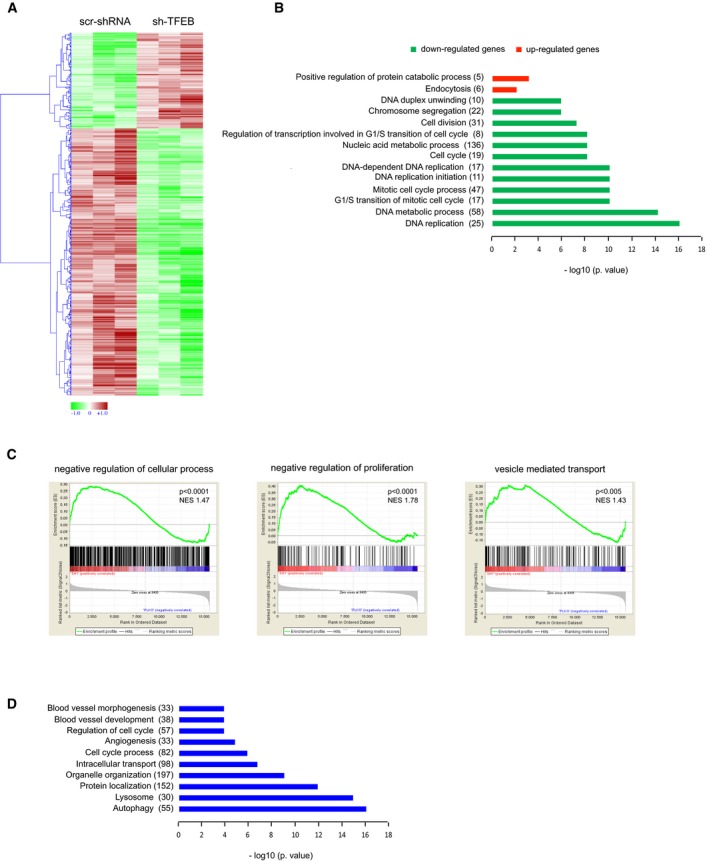
- Heatmap showing unsupervised hierarchical clustering of human differentially expressed genes between human scr‐shRNA and sh‐TFEB ECs. Red: up‐regulated genes; green: down‐regulated genes.
- Selection of enriched functional GO categories by DAVID analysis in differentially expressed genes between human sh‐TFEB and scr‐shRNA ECs. GO analyses were performed individually on down‐ or up‐regulated genes using DAVID tool (biological process). GO terms are ranked by P‐value corrected by Benjamini‐Hochberg method, and the number of genes is indicated.
- Selection enriched of Molecular Pathways by GSEA of microarray data comparing human sh‐TFEB and scr‐shRNA ECs. Normalized enrichment scores (NESs) and P‐values are reported.
- Selection of enriched functional GO categories by DAVID analysis on ChIP‐seq data set performed in human ECs overexpressing TFEBS142A. GO analyses were performed using DAVID tool (biological process). GO terms are ranked by P‐value corrected by BH method, and the number of genes is indicated.
Figure EV5. TFEB gene regulation in human ECs.
-
AVolcano plots of human gene expression showing fold‐change and P‐value data comparing human sh‐TFEB and scr‐shRNA ECs. Red: up‐regulated genes; green: down‐regulated genes.
-
BCharacterization of TFEBS142A overexpression in human ECs. Representative images of human ECs transduced with pTRIPZ‐TFEBS142A inducible lentivirus (scale bars: 20 μm) after transgene induction with doxycycline (0.5 μg/ml, 3 h; TFEBS142A) or not (control) and stained with anti‐TFEB Ab and DAPI. Bar graph indicates the quantity of cytosolic and nuclear TFEB as ratio between TFEBS142A and control ECs (n = 10, mean ± SEM, **P < 0.0001 and ***P < 0.0001 by Student's t‐test).
-
CRepresentative genomic view of the TFEB ChIP‐seq analysis. The TFEB ChIP‐seq plot shows distinct binding peaks with respect to the IgG ChIP‐seq.
-
DComparison of the TFEB‐bound genomic regions with respect to the other indicated ChIP‐seq regions taken from the literature. TFEB correlates with open chromatin (DNAse) and with transcription factors known to be associated with active gene promoters (FOS, JUN, RNA PolII, MYC).
-
EEnrichment of the E‐box DNA‐binding sequence of TFEB (CACGTG) on 70.9% of TFEB target genes in human ECs.
-
FComparison of expression values in human ECs between TFEB‐bound and unbound genes. Box plots are median‐centered. The end of the box shows the upper and lower quartiles. The whiskers line shows the highest and lowest value excluding outliers.
-
GDNA methylation analysis of the TFEB‐bound regions in human ECs using 450K Illumina microarray data from the literature (methylation status of 6,461 CpG has been analyzed).
-
HGenomic localization of TFEB‐bound regions in human ECs.
-
IGSEA analysis of TFEB‐bound genes comparing human sh‐TFEB and scr‐shRNA ECs microarray data. Enrichment plot shows modulated TFEB‐bound genes.
-
JTFEB overexpression increased EC proliferation. Representative graph of control and TFEBS142A ECs treated for 24 h with FCS (20%). DNA incorporation of EdU was detected by flow cytometry. The percentage of proliferating cells is indicated (n = 4, mean ± SEM; *P < 0.01 versus control ECs by Student's t‐test).
-
K, L(K) qPCR and (L) immunoblots showing modulation of CDK4 after TFEB overexpression in ECs. (K) Data are expressed as relative fold‐change compared with the expression in control cells after normalization to the housekeeping gene TBP (n = 3, mean ± SEM; ***P < 0.0001 by Student's t‐test). (L) Immunoblots of total lysates from control and TFEBS142A ECs probed with specific Abs. The bar graph shows the densitometric analysis expressed as % of CDK4 in TFEBS142A versus control ECs after the normalization with α‐tubulin (n = 3, mean ± SEM; ***P < 0.0001 versus scr‐shRNA by Student's t‐test).
Source data are available online for this figure.
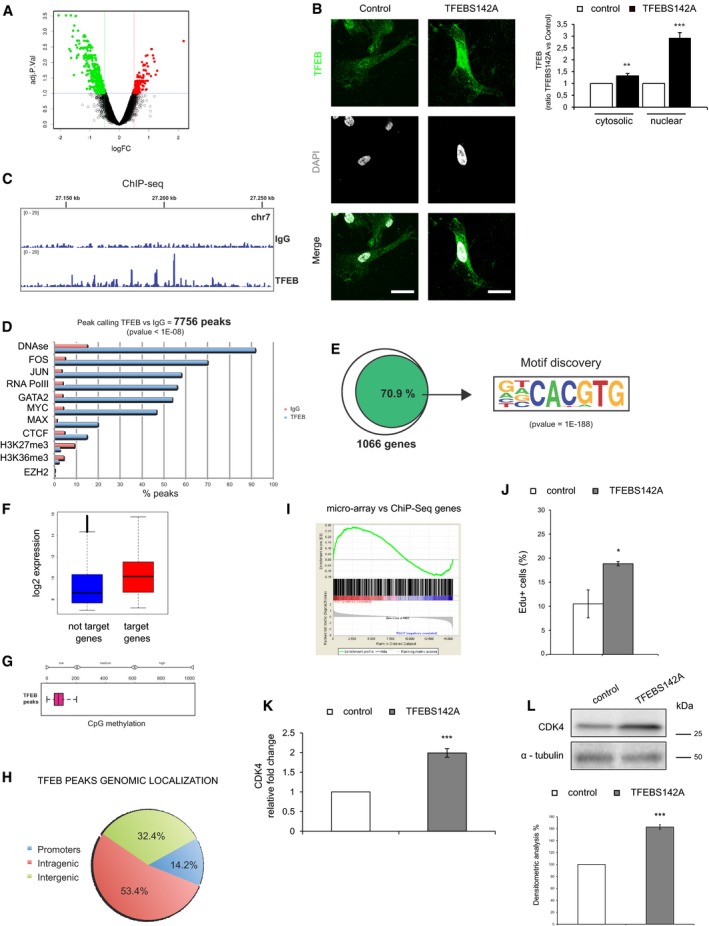
To identify direct TFEB gene targets potentially supporting the transcriptional modulation described above, we performed chromatin immunoprecipitation sequencing (ChIP‐seq) in ECs overexpressing a TFEB mutant (S142A) protein, which is constitutively translocated to the nucleus and biologically active (Settembre et al, 2012; Fig EV5B).
TFEB ChIP‐seq showed several distinct binding events compared with IgG ChIP‐seq (Fig EV5C). TFEB binding regions on DNA correlated with open chromatin regions (DNAse) and with transcription factors known to be associated with active gene promoters (FOS, JUN, RNA PolII, MYC; Fig EV5D).
In particular, we defined a set of 1,066 Ref/Seq protein‐coding genes showing strong TFEB binding enrichment on their core promoter. Of these, 71% contained the canonical TFEB binding sequence CACGTG (Palmieri et al, 2011; Fig EV5E).
Gene‐expression profiling of ECs revealed that TFEB was principally bound to highly expressed genes (Fig EV5F), at hypomethylated regions (Fig EV5G), and that almost 15% of the peaks (P‐value < 10e−8) were located on gene promoters (Fig EV5H).
GSEA was used to investigate the correlation between the regulated genes and the list of TFEB‐bound genes. An enrichment plot showed that both up‐regulated and down‐regulated genes can be TFEB targets (Fig EV5I).
DAVID analysis on ChIP‐seq data set confirmed the known role of TFEB in lysosome/autophagic pathway but also underlined its involvement in cell cycle regulation, angiogenesis, blood vessel development, and morphogenesis (Fig 3D). As shown in Fig EV5L, the overexpression of TFEBS142A increased the proliferation of ECs as previously described for other cell types (Haq & Fisher, 2011; Calcagnì et al, 2016).
Taken together, these data support the in vivo and in vitro results showing that TFEB is involved in EC proliferation by regulating the expression of genes directly involved in the control of cell cycle.
The CDK4 gene is a direct target of TFEB
As evidenced by transcriptome analysis (Fig 4A), qPCR (Fig 4B), and immunoblotting analysis (Fig 4C), TFEB silencing in human ECs negatively regulated the expression of genes involved in cell proliferation, including cyclin‐dependent kinase 4 (CDK4), cyclins (CCNA1, CCNA2), E2F transcription factors (E2F1, E2F2, E2F4), and their targets (MCM5, MCM6, CDC25B, CDCA4, CDCA7, PLK1, PCNA), which are involved in the control of S‐phase and mitosis. These data were further validated in murine lung ECs isolated from control and Tfeb iEC−/− mice (Fig 4D). Therefore, we interrogated the ChIP‐seq data set to identify the direct TFEB targets within these modulated genes and we found that the promoter of CDK4 contains a binding site for TFEB (Fig 4E).
Figure 4. TFEB regulates cell cycle genes.
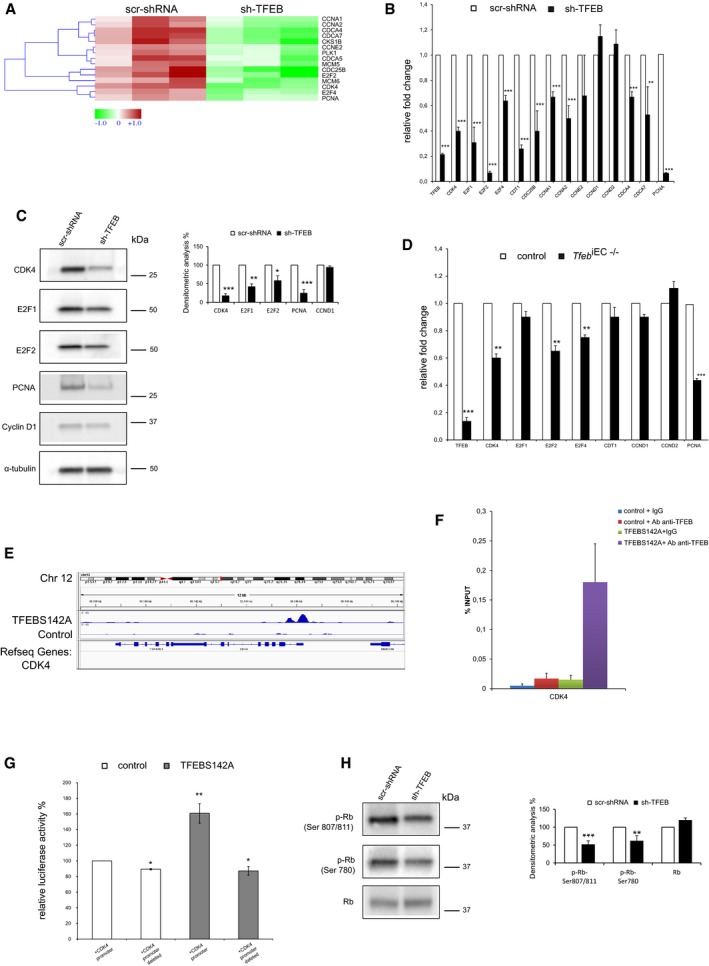
-
A–C(A) Heatmap, (B) qPCR, and (C) immunoblots showing the differentially expressed cell cycle‐related genes between scr‐shRNA and sh‐TFEB ECs. (A) Red: up‐regulated genes; green: down‐regulated genes. (B) Data are expressed as relative fold‐change compared with the expression in scr‐shRNA cells after normalization to the housekeeping gene TBP (n = 3, mean ± SEM; **P < 0.001 and ***P < 0.0001 by Student's t‐test). (C) Immunoblots of total lysates from scr‐shRNA and sh‐TFEB ECs probed with specific Abs. The bar graph shows the densitometric analysis expressed as the ratio between the cell cycle genes and α‐tubulin (n = 3, mean ± SEM; *P < 0.01, **P < 0.001, and ***P < 0.0001 versus scr‐shRNA by Student's t‐test).
-
DModulation of cell cycle genes expression in the lung ECs derived from control and Tfeb iEC−/− mice. Data are expressed as relative fold‐change compared with the expression in ECs of control mice after normalization to the housekeeping gene TBP (n = 3, mean ± SEM; **P < 0.001 and ***P < 0.0001, by Student's t‐test).
-
E–GAnalysis of TFEB binding and modulation of the CDK4 promoter in human ECs. (E) Representative snapshot of TFEB binding on CDK4 in human ECs. (F) ChIP was performed using digested chromatin from control ECs and TFEBS142A ECs incubated with IgG (indicated in the bar graph as “+IgG”) or with Ab anti‐TFEB (indicated in the bar graph as “+Ab anti‐TFEB”), followed by qPCR for CDK4. Bar graph shows the percent enrichment (n = 3, mean ± SD). (G) Analysis of TFEB modulation of CDK4 promoter in human ECs. Bar graph shows the relative luciferase activity % evaluated in control and TFEBS142A human ECs after transfection of CDK4 full promoter and CDK4 promoter deleted of TFEB binding site (n = 3, mean ± SEM; *P < 0.01 and **P < 0.001 by Student's t‐test).
-
HModulation of Rb protein in sh‐TFEB ECs. Immunoblots of total lysates from scr‐shRNA and sh‐TFEB ECs probed with phospho‐Rb and total Rb Abs. The bar graph shows the densitometric analysis of the immunoblotting expressed as % of sh‐TFEB versus scr‐shRNA and the ratio between phospho‐Rb and total Rb in the different conditions (n = 3, mean ± SEM; **P < 0.001, ***P < 0.0001 versus scr‐shRNA by Student's t‐test).
Source data are available online for this figure.
To further confirm the direct activity of TFEB on CDK4 expression, ChIP and promoter‐luciferase assay were performed. As shown in Fig 4F, the chromatin immunoprecipitated with an antibody anti‐TFEB contained the CDK4 promoter identified by PCR using two CDK4‐specific primers. We further investigated the CDK4 promoter activity by luciferase reporter assay in ECs overexpressing TFEBS142A, an active constitutive variant that localizes preferentially in the nucleus (Settembre et al, 2012). TFEBS142A‐carrying luciferase reporter vectors respectively containing CDK4 full‐length promoter and a deleted form lacking 100 bps encompassing the putative TFEB binding site were analyzed. TFEBS142A overexpression resulted in a significant increase in CDK4 promoter activity, which was completely blunted by the deletion of TFEB binding site (Fig 4G). The activation of CDK4 promoter activity was also related to the increase in CDK4 transcription and protein expression (Fig EV5K and L). One of the most important substrates of CDK4 is retinoblastoma (Rb) protein, which associates with E2F transcription factor in quiescent cells. When cells progress in G1 phase, CDK4 phosphorylates Rb, allowing the release of E2F, which migrates into the nucleus and activates the transcription of genes required for S‐phase (Malumbres & Barbacid, 2009). Transcriptomic analysis of sh‐TFEB ECs did not indicate Rb as a DEG and immunoblotting analysis showed that its expression was not altered by TFEB silencing (Fig 4H). On the contrary, we evidenced in sh‐TFEB ECs a down‐regulation of phospho‐Rb (Fig 4H), which paralleled the reduced expression of CDK4 (Fig 4B and C).
These data support the rationale that CDK4‐Rb‐E2F axis is a target of TFEB in ECs and suggest that its impairment could explain the in vivo alteration of vascular development described in Tfeb iEC−/− mice.
VEGFR2 gene is an indirect target gene of TFEB in ECs
The above considerations stimulated us to study other possible mechanisms sustaining the vascular phenotype. We investigated a possible role for VEGF‐A/VEGFR2 pathway, which is definitely a key machinery involved in EC migration, proliferation, and morphogenesis (Simons et al, 2016). We were also intrigued by the observation that full Tfeb −/− mice exhibited a reduced placenta expression of VEGF‐A (Steingrímsson et al, 1998). Furthermore, while the reduced transcription of CDK4 in sh‐TFEB ECs could justify the observed proliferative block, this defect was not necessarily accountable for the morphogenetic alterations observed in vivo (Fig 1D) and in vitro in the Matrigel morphogenetic assay (Fig 2E), which is independent from the proliferation even in the presence of VEGF‐A (Serini et al, 2003).
For these reasons, we investigated whether TFEB deletion could interfere on VEGFR2 expression. The expression of Vegfr2 was increased in the vessels of Tfeb EC−/− embryos (E10.5; Fig 5A) and of retina and kidney of Tfeb iEC−/− mice compared with that in control mice (Fig 5B and C). Since neural cells (Robinson et al, 2001) and podocytes (Bartlett et al, 2016) both express Vegfr2, we examined the amount of receptor co‐localized with the specific endothelial markers iB4 or CD31 in retina and kidney and confirmed the increased level of the receptor in the ECs of Tfeb iEC−/− mice (Fig 5B and C).
Figure 5. Regulation of VEGFR2 by TFEB in ECs.
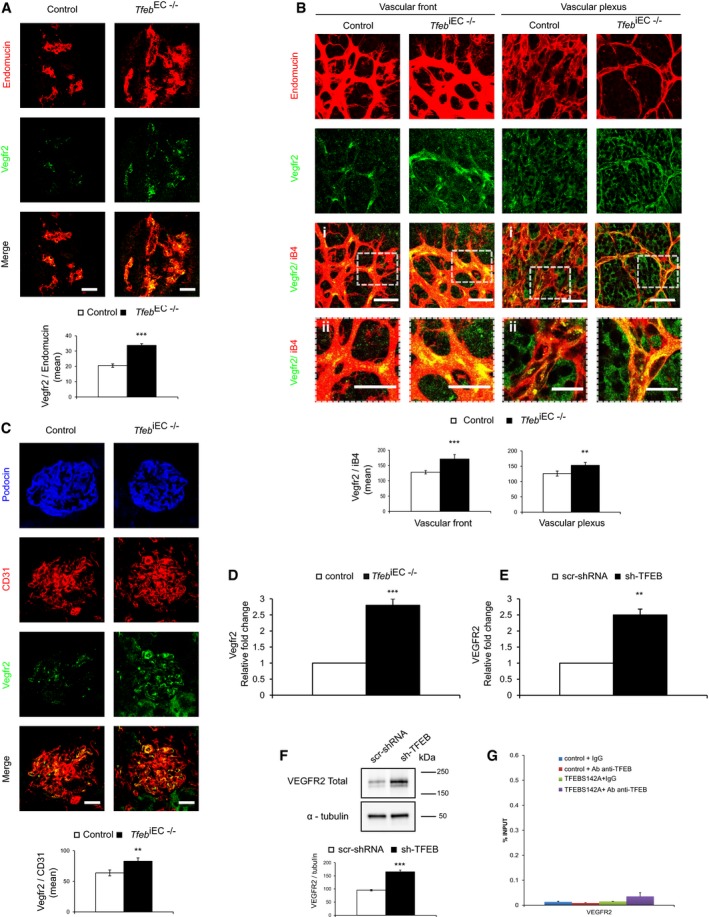
-
A–CIncrease in Vegfr2 expression in the vasculature of Tfeb EC−/− and Tfeb iEC−/− mice. (A) Representative immunostaining images of Tfeb EC−/− embryonic vessels (E10.5) stained with anti‐endomucin and anti‐Vegfr2 Abs (scale bars: 50 μm). Bar graph indicates the Vegfr2 mean intensity only in endomucin+ vessel areas (embryos n = 6, mean ± SEM; ***P < 0.0001 versus control embryos by Student's t‐test). (B) Representative immunostaining images (i) and detail (ii) of the vascular front and vascular plexus of the retina (p5) of control and Tfeb iEC−/− mice with anti‐iB4 and anti‐Vegfr2 Abs (scale bars: 50 μm). Bar graph indicates the Vegfr2 mean intensity only in iB4+ vessel areas (mice n = 6, mean ± SEM; **P < 0.001 and ***P < 0.0001 versus control mice by Student's t‐test). (C) Representative immunostaining images of the glomerulus (p17) of control and Tfeb iEC−/− mice with anti‐podocin, anti‐CD31, and anti‐Vegfr2 Abs (scale bars: 50 μm). Bar graphs indicate the Vegfr2 mean intensity in CD31+ vessel areas (mice n = 6, mean ± SEM; **P < 0.001 versus control mice by Student's t‐test).
-
D–FVEGFR2 expression is regulated by TFEB. (D, E) qPCR of VEGFR2 in lung ECs obtained from control and Tfeb iEC−/− mice (D) and in human sh‐TFEB (E). Data are expressed as relative fold‐change compared with the expression in control cells after normalization to the housekeeping gene TBP (n = 3, mean ± SEM; **P < 0.001 and ***P < 0.0001, by Student's t‐test). (F) Immunoblots of total lysates from scr‐shRNA and sh‐TFEB ECs probed with anti‐VEGFR2 and α‐tubulin Abs. The bar graph shows the densitometric analysis expressed as the ratio between VEGFR2 and α‐tubulin (n = 3, mean ± SEM; ***P < 0.0001 versus scr‐shRNA by Student's t‐test).
-
GAnalysis of TFEB binding to the VEGFR2 promoter in human ECs. ChIP was performed using digested chromatin from human control ECs and TFEBS142A ECs incubated with IgG (indicated in the bar graph as “+IgG”) or with Ab anti‐TFEB (indicated in the bar graph as “+Ab anti‐TFEB”), followed by qPCR for VEGFR2. Bar graph shows the percent enrichment (n = 3, mean ± SD).
Source data are available online for this figure.
Transcriptome data validated by qPCR (Fig 5D and E) and immunoblotting (Fig 5F) showed the up‐regulation of VEGFR2 in murine lung ECs isolated from Tfeb iEC−/− mice and sh‐TFEB ECs. However, this transcriptional effect is independent from a direct binding of TFEB on VEGFR2 promoter, as demonstrated by ChIP‐seq (not shown) and ChIP‐qPCR (Fig 5G) performed in sh‐TFEB ECs.
In sh‐TFEB ECs, the analysis of VEGFR2 localization by FACS (Fig 6A), total internal reflection fluorescence (TIRF) microscopy (Fig 6B), and biotinylation of endothelial plasma membrane (PM; Napione et al, 2012aa; Fig 6C) indicated a strong accumulation of the receptor at the PM. To rescue the effect of TFEB down‐modulation, ECs were co‐infected with sh‐TFEB and pTRIPZ‐TFEBS142A vectors without an evident effect (not shown). This negative result could be explained by the fact that TFEB overexpression (Fig EV5) targeted a wide number of genes. Therefore, the overexpression of exogenous TFEB could activate molecules that mask the expected modification of VEGFR2 and other specific TFEB targets identified by loss‐of‐function strategy.
Figure 6. Role of TFEB in regulating VEGFR2 localization and activity in human ECs.
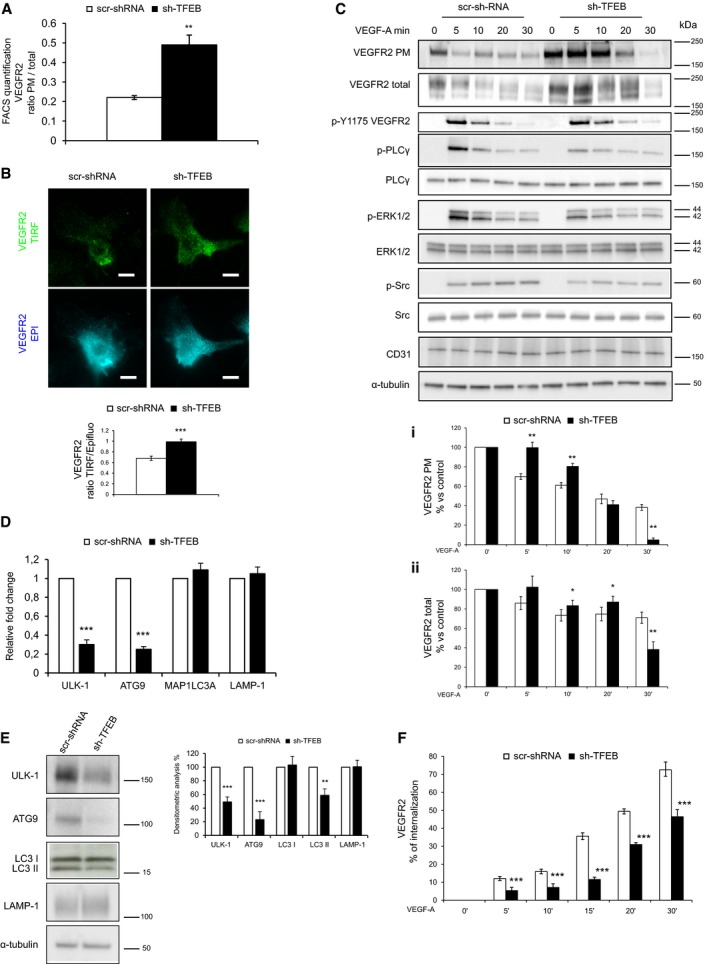
-
A, BSilenced TFEB alters the localization of VEGFR2. (A) FACS analysis of surface VEGFR2 expression on human scr‐shRNA and sh‐TFEB ECs. Bar graph shows the ratio between total and PM VEGFR2 (n = 6, mean ± SEM; **P < 0.001 versus scr‐shRNA by Student's t‐test). (B) Representative TIRF and epifluorescence images of human scr‐shRNA and sh‐TFEB ECs after staining with anti‐VEGFR2 Ab (scale bars: 10 μm). Bar graphs show the ratio of VEGFR2 analyzed in epifluorescence and TIRF mode analyzed by TIRF (n = 40, mean ± SEM; ***P < 0.0001 versus scr‐shRNA by Student's t‐test).
-
CSilenced TFEB alters the localization and the phosphorylation state of VEGFR2 and its signal transduction. Representative immunoblot of PM biotinylated portion and total cell lysates of scr‐shRNA and sh‐TFEB ECs after VEGF‐A stimulation (30 ng/ml). Blots of total or PM cell lysates were probed with anti‐VEGFR2. Blots of total cell lysates were probed with anti‐p‐Y1175‐VEGFR2, anti‐PLCγ, p‐PLCγ, anti‐ERK‐1/2, anti‐pERK1/2, anti‐p‐Src, anti‐Src, anti‐CD31, and α‐tubulin Abs. The bar graphs (i,ii) show densitometric analysis of stimulated versus unstimulated scr‐shRNA and sh‐TFEB ECs expressed as: (i) % of VEGFR2 on PM fraction (n = 3, mean ± SEM; ANOVA P < 0.02; **P < 0.001 versus scr‐shRNA by Bonferroni post‐test), (ii) % of VEGFR2 total (n = 3, mean ± SEM; ANOVA P > 0.05; *P < 0.05 and **P < 0.001 versus scr‐shRNA by Bonferroni post‐test).
-
D, ERegulation of autophagy and lysosome pathway by TFEB silencing. (D) qPCR and (E) immunoblots showing the differentially expressed autophagy‐ and lysosome‐related genes between human scr‐shRNA and sh‐TFEB ECs. (D) Data are expressed as relative fold‐change compared with the expression in control cells after normalization to the housekeeping gene TBP (n = 3, mean ± SEM; ***P < 0.0001 by Student's t‐test). (E) Immunoblots of total lysates from human scr‐shRNA and sh‐TFEB ECs probed with anti‐ULK‐1, anti‐ATG9, anti‐LC3‐I/II, anti‐LAMP‐1, and α‐tubulin Abs. The bar graph shows the densitometric analysis expressed as the ratio between scr‐shRNA and sh‐TFEB ECs (n = 3, mean ± SEM; **P < 0.001, ***P < 0.0001 versus scr‐shRNA by Student's t‐test).
-
FTFEB silencing inhibits VEGFR2 internalization. Bar graphs of VEGFR2 internalization expressed as the percent of internalized VEGFR2 versus PM VEGFR2 after VEGF‐A stimulation (n = 6, mean ± SEM, ANOVA P < 0.0001; ***P < 0.0001 versus scr‐shRNA by Bonferroni post‐test).
Source data are available online for this figure.
In the absence of a direct effect of TFEB on VEGFR2 transcription (Fig 5G), we hypothesized that the altered expression of VEGFR2 and its increased localization on PM might be the result of the combined activity of an altered membrane trafficking and a miR‐dependent post‐transcriptional mechanism.
TFEB deletion alters VEGFR‐2 membrane trafficking
On the basis of the described results, we investigated whether the accumulation of VEGFR2 in PM (Fig 6A–C) could be related to a specific TFEB‐mediated mechanism orchestrating the VEGFR2 membrane trafficking (Simons et al, 2016).
Actually, TFEB deletion negatively modulated the genetic program sustaining lysosome biogenesis, autophagy, vesicles trafficking, and exocytosis (Medina et al, 2011; Napolitano & Ballabio, 2016; Raben & Puertollano, 2016) in many cell types including ECs (Lu et al, 2017). Transcriptomic analysis indicated that the expression of some genes characterized by the presence of the coordinated lysosomal expression and regulation (CLEAR) element (Palmieri et al, 2011) were modified in sh‐TFEB ECs (DEGs down‐regulated: ATP6V0D1, CLCN7, CTSB; DEGs up‐regulated: RRAG). Similarly, TFEB silencing reduced markers of autophagic flux. In particular, the ratio between microtubule‐associated proteins 1A/1B light chain (LC) 3‐II and LC3‐I, and the expression of Unc‐51 like autophagy activating kinase (ULK‐1; Martina et al, 2012; Settembre et al, 2012) and autophagy‐related gene (ATG)9A (Roa et al, 2008) were reduced in sh‐TFEB ECs (Fig 6D and E).
Because lysosomal and autophagy pathways are interconnected with endosomal system (Pavel & Rubinsztein, 2017), we analyzed the dynamics of VEGFR2 trafficking in sh‐TFEB ECs. Whereas PM VEGFR2 started to decrease immediately after VEGF‐A challenge in scr‐shRNA ECs (~30% reduction after 5 min), knocking down TFEB consistently altered VEGFR2 PM clearance that was slower (~20% reduction) and started only after 10 min from stimulus. Concomitantly, the reduction of total amount of VEGFR2, correlated with its degradation, in scr‐shRNA ECs became evident after 5 min of VEGF‐A challenge (~14% versus unstimulated ECs), while this drop was evident later (10 min) in sh‐TFEB ECs (~17% versus unstimulated ECs; Fig 6C).
The perturbing effect of TFEB silencing was further analyzed by studying the co‐localization of VEGFR2 with caveolin‐1 (CAV‐1)‐rich membrane rafts, which represent specific domains that favor the signaling properties of the receptor (Labrecque et al, 2003; Cho et al, 2004). As inferred from co‐localization analysis, TFEB silencing did not significantly alter the expression and localization of CAV‐1 on the PM, but interestingly, it modified the spatial relationship between VEGFR2 and CAV‐1. sh‐TFEB ECs exhibited an approximately 25% reduction in CAV‐1‐associated VEGFR2 compared with scr‐shRNA ECs (Appendix Fig S1B).
The effect of TFEB silencing on VEGFR2 distribution was further evaluated by the specific quantification of receptor endocytosis by a receptor internalization assay (Valdembri et al, 2009). TFEB silencing moderately but significantly altered VEGFR2 internalization time‐course. Whereas VEGFR2 internalization started immediately (5 min) after VEGF‐A challenge in control cells (~12% versus unstimulated ECs), a 10‐min delay was seen in sh‐TFEB ECs suggesting an impairment of internalization process (~5% versus unstimulated ECs, P = ns after 5 min of VEGF‐A incubation and ~7% versus unstimulated ECs, P < 0.01 after 10 min of VEGF‐A incubation; Fig 6F). The inhibition of internalization of VEGFR2 in sh‐TFEB ECs was confirmed by the reduction of co‐localization with Rab5+ endosomes after VEGF‐A stimulation (Appendix Fig S1C).
Because VEGFR2 exocytosis from endosomal compartment to PM participates to properly maintain its signaling properties (Simons et al, 2016), we investigated the co‐localization of the receptor with the Golgi marker Trans‐Golgi Network 46 (TGN46) and Rab4+ exocytic vesicles (Jopling et al, 2014). TFEB silencing did not modify the amount of VEGFR2 localized in Golgi compartment (Appendix Fig S1D), but increased that accumulated in Rab4+ vesicles (170.2 ± 9% compared to scr‐shRNA ECs; n = 4, P < 0.01; Appendix Fig S2E).
These observations prompted us to interrogate the enriched gene set belonging to the Gene Ontology “protein targeting to membrane” in TFEB‐silenced ECs to identify a mechanism responsible for the observed accumulation of VEGFR2 in the PM.
Within the modulated genes (ADORA1, ARL6, CACNB1, ICMT, ATG3, SDCBP, SEC63, ATG4C, MGEA5, MICALL1, MYO1C, TAOK2, NCF1, PRKCI), we focused our attention on myosin 1c (MYO1C), which was involved in VEGFR2 exocytosis (Tiwari et al, 2013). ChIP‐seq (Fig 7A) and ChIP‐qPCR (Fig 7B) indicated that TFEB can bind to the MYO1C promoter. ECs overexpressing TFEBS142A showed reduced activation of MYO1C promoter (Appendix Fig S2A) and consequently a decreased transcription (Appendix Fig S2B). On the contrary, in sh‐TFEB ECs MYO1C was up‐regulated (Fig 7C and D). This phenotype was also observed in the vasculature of retina (p5) and kidney (p17) of Tfeb iEC−/− mice (Fig 7E and F).
Figure 7. Role of MYO1C in the localization of VEGFR2 in TFEB‐silenced ECs.
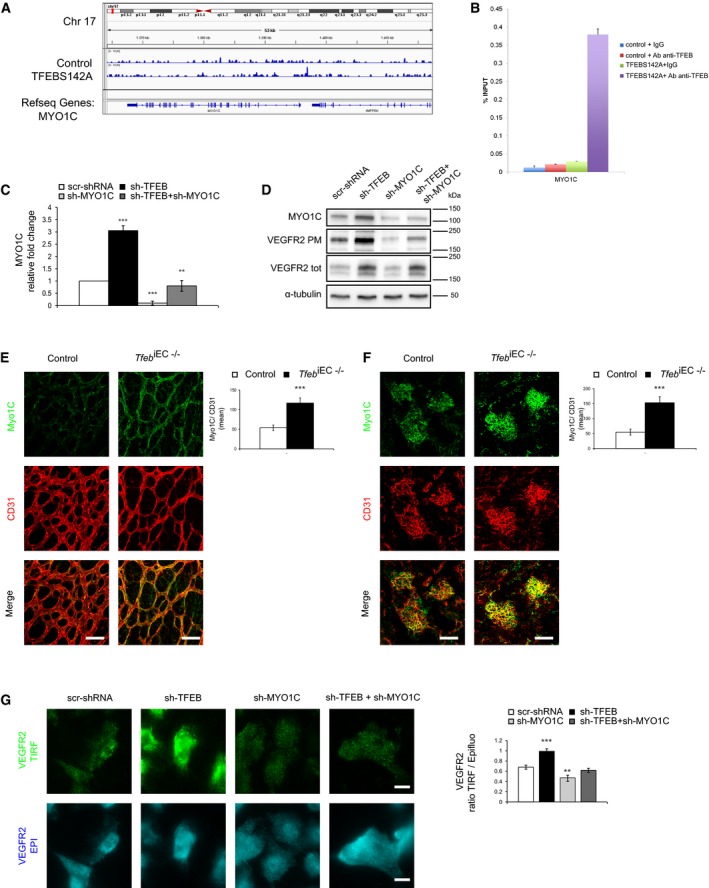
-
A–CAnalysis of TFEB binding and modulation of the MYO1C promoter in human ECs. (A) Representative snapshot of TFEB binding on MYO1C in human ECs. (B) ChIP was performed using digested chromatin from control ECs and TFEBS142A ECs incubated with IgG (indicated in the bar graph as “+IgG”) or with Ab anti‐TFEB (indicated in the bar graph as “+Ab anti‐TFEB”), followed by qPCR for MYO1C. Bar graph shows the percent enrichment (n = 3, mean ± SD). (C) qPCR of MYO1C expression in scr‐shRNA, sh‐TFEB, sh‐MYO1C, and sh‐TFEB+sh‐MYO1C ECs. Data are expressed as relative fold‐change compared with the expression in scr‐shRNA ECs after normalization to the housekeeping gene TBP (n = 3, mean ± SEM; **P < 0.001 and ***P < 0.0001 by Student's t‐test).
-
DMYO1C silencing reverses the effect of TFEB silencing on the up‐regulation of PM VEGFR2. Analyses were performed on human ECs carrying appropriate scr‐shRNA or sh‐TFEB in the presence or absence of sh‐MYO1C. Representative Western blot of MYOC1, total and PM biotinylated VEGFR2 (representative experiment out of 4 with similar results).
-
E, FRepresentative immunostaining images of the vascular plexus of the retina (p5) (E) and glomerulus (p17) (F) of control and Tfeb iEC−/− mice with anti‐CD31 and anti‐MYO1C Abs (scale bars: 50 μm). Bar graphs indicate the Myo1C mean intensity only in vessel areas CD31+ (n = 6, mean ± SEM; ***P < 0.0001 versus control mice by Student's t‐test).
-
GRepresentative TIRF and epifluorescence images of VEGFR2 in scr‐shRNA, sh‐TFEB, sh‐MYO1C, and sh‐TFEB+sh‐MYO1C human ECs (scale bars: 10 μm). Bar graph shows the ratio between PM and total VEGFR2 (n = 40, mean ± SEM; **P < 0.001 and ***P < 0.0001 versus scr‐shRNA by Student's t‐test).
Source data are available online for this figure.
Interestingly, MYO1C‐silencing (Appendix Fig S2C and D) decreased the amount of VEGFR2 co‐localized with Rab4+ vesicles (Appendix Fig S2E). These data suggested that MYO1C deletion reduces the transport of the receptor to Rab4+ endosomes involved in its exocytosis. This result matches the observation that MYO1C silencing in sh‐TFEB ECs reduced specifically the amount of PM VEGFR2, as demonstrated by immunoblotting (Fig 7D), the ratio of the VEGFR2 signal recorded in TIRF and epifluorescence modes (Fig 7G), and the ratio of VEGFR2 on the PM to total expression measured by FACS (Appendix Fig S2F).
On the contrary, MYO1C silencing both in scr‐shRNA and in sh‐TFEB ECs did not affect VEGFR2 transcription (Appendix Fig S2G), total protein expression (Fig 7D; densitometric analysis as % of scr‐shRNA: 220.7 ± 5.2% in sh‐TFEB ECs P < 0.0001; 114.7 ± 7.7% in sh‐MYO1C P = ns; 230.3 ± 3.7% in sh‐TFEB+sh‐MYO1C ECs P < 0.0001, n = 3), and receptor internalization (Appendix Fig S2H).
The inhibitory effect of TFEB deletion on ECs proliferation was not counteracted by MYO1C silencing in human ECs (Appendix Fig S2I). This result is not surprising in view of the deep influence of TFEB on genes related to cell proliferation (Fig 3B and C).
TFEB deletion up‐regulates VEGFR2 by inhibiting a miR‐15a/16‐1‐dependent post‐transcriptional regulatory mechanism
Because TFEB silencing up‐regulated VEGFR2 mRNA independently from a direct action on the promoter (Fig 5G), we further speculated a role for a miR‐dependent post‐transcriptional mechanism (Cora’ et al, 2017), which is widely involved in the control of vascular functions, including VEGFR2 (Chamorro‐Jorganes et al, 2013; Dang et al, 2013; Park et al, 2013).
By crossing data of literature about the genomic location of miRs (Chamorro‐Jorganes et al, 2013; Dang et al, 2013) directly involved in the angiogenic process (Appendix Fig S3A) with our ChIP‐seq data set, we identified the “structural maintenance of chromosome 4” (SMC4) and the “deleted in leukemia‐2” (DLEU2) genes (Appendix Fig S3A) as two putative miR host genes involved in VEGFR2 regulation by TFEB. ChIP‐seq and ChIP‐qPCR indicated that TFEB bound to the DLEU2 but not the SMC4 promoter (Fig 8A). For these reasons, we focused on DLEU2, which is a tumor suppressor gene that is lost early in chronic lymphatic leukemia (Klein et al, 2010). DLEU2 encodes a sterile transcript as well as the miR‐15a/16‐1 cluster, which is located intronic to the gene. This cluster encodes mature miR‐15a‐3p/5p and miR‐16‐1‐3p/5p (Yue & Tigyi, 2010), which regulate the cell cycle and apoptosis and influence vascular function (Sun et al, 2013; Jackstadt & Hermeking, 2015), including VEGFR2 expression (Chamorro‐Jorganes et al, 2011; Chan et al, 2013).
Figure 8. Indirect regulation of VEGFR2 via miR‐15a/16 by TFEB in ECs.
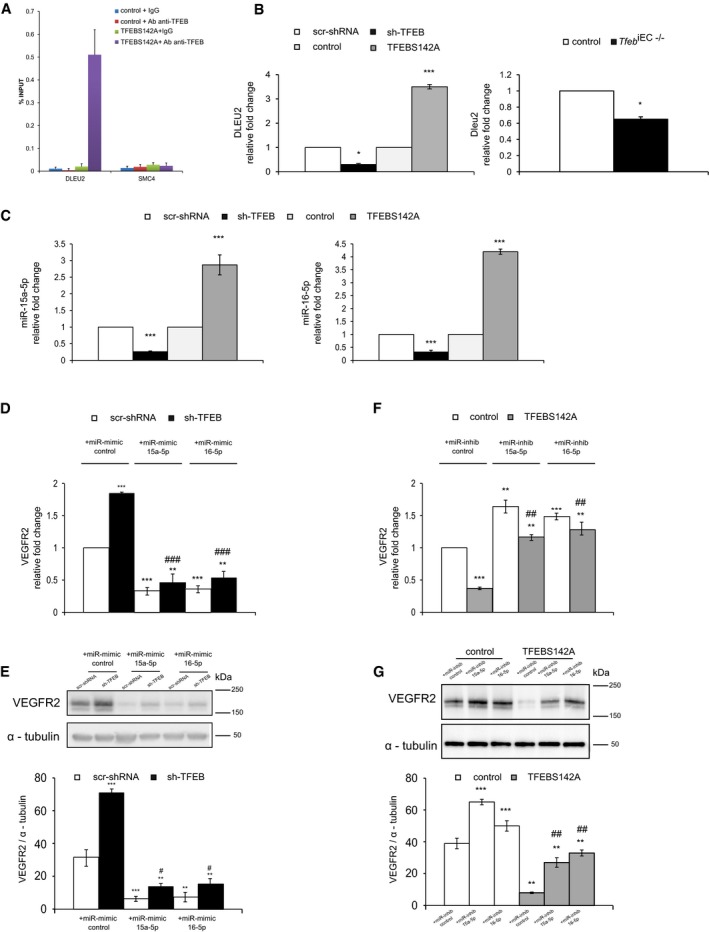
-
AAnalysis of TFEB binding to DLEU2 and SMC4 promoters in human ECs. ChIP was performed using digested chromatin from control ECs and TFEBS142A ECs incubated with IgG (indicated in the bar graph as “+IgG”) or with Ab anti‐TFEB (indicated in the bar graph as “+Ab anti‐TFEB”), followed by qPCR for DLEU2 and SMC4. Bar graph shows the percent enrichment (n = 3, mean ± SD).
-
BDLEU2 expression is regulated by TFEB. qPCR of DLEU2 in human scr‐shRNA, sh‐TFEB, or control and TFEBS142A ECs (left panel) and lung ECs obtained from control and Tfeb iEC−/− mice (right panel). Data are expressed as relative fold‐change compared with the expression in scr‐shRNA and control cells after normalization to the housekeeping gene TBP (n = 3, mean ± SEM; *P < 0.01, and ***P < 0.0001 by Student's t‐test).
-
CHuman miR‐15a‐5p and miR‐16‐5p are regulated by TFEB. qPCR of miR‐15a‐5p (left panel) and miR‐16‐5p (right panel) in sh‐TFEB or TFEBS142A ECs. Data are expressed as relative fold‐change compared with the expression in scr‐shRNA and control cells after normalization to the housekeeping gene RNU44 (n = 3, mean ± SEM; ***P < 0.0001 by Student's t‐test).
-
D–GVEGFR2 expression is regulated by TFEB through a miR‐dependent mechanism. (D, F) qPCR of VEGFR2 in human scr‐shRNA and sh‐TFEB ECs (D, E) and in control and TFEBS142A ECs (F, G) treated with a specific miR‐control, miR‐15a‐5p, and miR‐16‐5p mimics or inhibitors. (D, F) Data are expressed as relative fold‐change compared with the expression in control cells after normalization to the housekeeping gene TBP (n = 3, mean ± SEM; **P < 0.001 and ***P < 0.0001 versus control or scr‐shRNA plus miR‐control and ## P < 0.001 and ## P < 0.0001 versus sh‐TFEB and TFEBS142A plus miR‐control by Student's t‐test). (E, G) Representative Western blot of VEGFR2 expression under the same experimental conditions previously reported. The bar graph shows the densitometric analysis expressed as the ratio between VEGFR2 and α‐tubulin (n = 3, mean ± SEM; **P < 0.001 and ***P < 0.0001 versus control or scr‐shRNA plus miR‐control and # P < 0.01 and ## P < 0.001 versus sh‐TFEB and TFEBS142A plus miR‐control by Student's t‐test).
Source data are available online for this figure.
DLEU2 was down‐ and up‐regulated in sh‐TFEB and TFEBS142A ECs, respectively (Fig 8B). The deletion of Tfeb resulted also in the down‐modulation of Dleu2 in lung ECs isolated from mutant mice (Fig 8B).
Next, we analyzed the expression of isoforms of the miR‐15a/16‐1 cluster in ECs (Fig 8C, Appendix Fig S3B). miR‐15a‐5p and miR‐16‐5p were expressed at high levels, whereas miR‐15a‐3p and miR‐16‐1‐3p levels were negligible and were therefore not considered further.
In parallel with DLEU2, miR‐15a‐5p and miR‐16‐5p were down‐ and up‐regulated in sh‐TFEB and TFEBS142A ECs, respectively (Fig 8C). Similarly, the in vitro Cre‐mediated Tfeb deletion resulted in a marked reduction of miR‐15a‐5p and miR‐16‐5p in lung ECs isolated from mutant mice (Appendix Fig S3B).
The functional connection between DLEU2 and the intragenic mir‐15a‐5p and mir‐16‐5p was further validated in sh‐DLEU2 ECs (Appendix Fig S3C), in which expression of TFEBS142A was unable to increase their expression. In particular, the fold‐change expression of mir‐15a‐5p and mir‐16‐5p was respectively increased of 2.8 ± 0.6 and 4.8 ± 0.7 in ECs carrying TFEBS142A compared to control cells (n = 3, P < 0.0001). The co‐expression of sh‐DLEU2 and the constitutively active TFEB mutant blocked the expression of both miRs (mir‐15a‐5p: 0.2 ± 0.2 relative fold‐change; and mir‐16‐5p: 0.4 ± 0.2 relative fold‐change n = 3, P < 0.0001 versus TFEBS142A ECs).
To determine the possible regulatory role for miR‐15a‐5p and miR‐16‐5p in VEGFR2 expression, we employed gain‐ and loss‐of‐function approaches.
The up‐regulation of VEGFR2 transcription and protein synthesis observed in sh‐TFEB ECs was rescued by transducing the specific mimic of miR‐15a‐5p and miR‐16‐5p (Fig 8D and E). On the contrary, the down‐regulation of VEGFR2 transcription and protein synthesis observed in TFEBS142A ECs was rescued by transduction of the specific inhibitors of miR‐15a‐5p and miR‐16‐5p (Fig 8F and G).
These data suggest a direct role for these miRs in the regulation of VEGFR2 by TFEB.
On the contrary in sh‐TFEB ECs, the rescue of the level of VEGFR2 similar to that presented in scr‐shRNA ECs was not able to overcome the reduced proliferation stimulated by VEGF‐A (Appendix Fig S3D), in agreement with the direct effect of TFEB on CDK4 (Figs 4A–F and EV5K and L).
VEGFR2 signal is reduced in TFEB‐silenced ECs
The above‐described alterations of VEGFR2 behavior in sh‐TFEB ECs prompted us to investigate its signaling properties by analyzing VEGFR2 phosphorylation at Y1175 in total lysates, which is a discrete docking site for PLCγ and one of the most important effector of activated VEGFR2 (Simons et al, 2016). In sh‐TFEB ECs compared with control cells, the degree of receptor phosphorylation after VEGF‐A challenge was smaller relative to the larger amount of total VEGFR2 (Fig 6C, Appendix Fig S1A). We also observed a concomitant decrease in the phosphorylation of PLCγ and ERK‐1/2, which are implicated in EC proliferation in response to VEGF‐A (Takahashi et al, 2001)) (Fig 6C, Appendix Fig S1A). The activation of c‐Src, another tyrosine kinase substrate of VEGFR2 involved in cytoskeletal rearrangements and cell‐matrix adhesion (Ferrando et al, 2012; Fig 6C, Appendix Fig S1A), was reduced in sh‐TFEB challenged with VEGF‐A.
These data indicate that the altered mRNA dynamics connected with a defect of receptor trafficking result in a reduced function of VEGFR2 in ECs lacking TFEB.
Discussion
Here, we demonstrate that the EC‐targeted deletion of Tfeb alters the mid‐ and late phase of vascular development in mouse. By combining gene expression and ChIP‐seq analyses in genetically modified ECs where TFEB is either silenced or overexpressed, we found that this transcription factor positively regulates the expression of CDK4 and miR‐15a/16‐1 cluster, while it exerts a repressor activity on MYO1C. The absence of TFEB results in a reduction of CDK4 and the consequent block of cell cycle in ECs. At the same time, we found that VEGFR2 trafficking and its compartment localization is profoundly modified. Such alterations are mediated by the up‐regulation of MYO1C and the abrogation of miR‐15a/16‐1‐mediated post‐transcriptional control of VEGFR2 expression, respectively. While the abnormal VEGFR2 behavior could be a potential compensation mechanism with respect to the cell cycle hindrance, we demonstrated that the overall signaling activity is impaired.
The effect of TFEB on cell proliferation has been extensively investigated and primarily connected with its effect on autophagic flux. Knockdown of TFEB decreased proliferation of prostate (Blessing et al, 2017) and pancreatic cancer cell lines (Perera et al, 2015). On the contrary, TFEB up‐regulation in renal and in cancer cells resulted in an increased proliferation rate (Calcagnì et al, 2016; Di Malta et al, 2017). The necessary role of TFEB in cell growth is further indirectly supported by the enhanced cell proliferation observed in renal cell carcinoma carrying the t(6;11)(p21:q13) translocation, which leads to a TFEB promoter substitution with the 5′ upstream regulatory sequence of the alpha intronless gene (Calcagnì et al, 2016). The lack of promoter‐mediated physiological control of TFEB expression promotes clonogenic cell growth (Haq & Fisher, 2011). In ECs, besides controlling autophagic flux (Fan et al, 2018; Fig 6D and E) our data indicate that TFEB exerts a more direct effect on cell proliferation. TFEB deletion reduced the expression of genes belonging to the “regulation cell cycle” GO, and most importantly, it directly bound the promoter of CDK4, an activator of G1‐S transition of cell cycle. The reduced availability of CDK4 impaired the phosphorylation of Rb, which relieves the Rb‐mediated inhibition of the transcription factor E2F involved in the expression of cell cycle‐related genes (Malumbres & Barbacid, 2009). Interestingly, E2F is able to activate autophagy genes (Polager et al, 2008), supporting the emerging concept that authophagy and cell cycle are not mutually exclusive processes (Mathiassen et al, 2017). Therefore, the dual positive effects of TFEB on authophagy and cell cycle are not necessarily a paradox, but they may therefore depend on the temporal context and stimuli. It is possible to speculate that when TFEB is active to trigger the clearance of senescent cells by autophagy, it can initiate the machinery involved in the cell renewal.
However, the vascular phenotype observed in Tfeb mutants cannot be simply explained by the down‐modulation of CDK4 and the other cell cycle‐related genes.
VEGFR2 is considered the master gene of vascular development and angiogenesis in adult life (Simons et al, 2016). Therefore, the alterations of VEGFR2 biology here observed can contribute to explain the vascular phenotype of Tfeb mouse mutants. We speculate that in an attempt to compensate the cell cycle defect triggered by TFEB deletion, ECs increased the amount of PM VEGFR2, which, however, showed signaling limitations. In murine and human ECs lacking TFEB, we demonstrated the down‐modulation of the expression of intragenic miR‐15a/16‐1 cluster, which specifically targets the VEGFR2 3′UTR (Chamorro‐Jorganes et al, 2011; Chan et al, 2013) and the subsequent post‐transcriptional stabilization of VEGFR2 mRNA. We also show that TFEB silencing increased the expression of the motor protein MYO1C, which functions as a cargo transporter (Greenberg & Ostap, 2013), and it has been reported to deliver VEGFR2 to PM (Tiwari et al, 2013). Actually, MYO1C silencing restored the effect of TFEB deletion on the co‐localization between VEGFR2 and Rab4+ vesicles, which are involved in the receptor exocytosis. These data are consistent with previous observations that the amount of membrane VEGFR2 is reduced by MYO1C depletion, whereas MYO1C overexpression rescues VEGFR2 at the PM (Tiwari et al, 2013; Jopling et al, 2014; Simons et al, 2016). However, the increased expression of VEGFR2 and its localization at PM were not paralleled by increased receptor signaling.
Actually, in sh‐TFEB ECs we reported the decrease in the phosphorylation of VEGFR2 in Y1175, which represents a docking site for PLCγ (Simons et al, 2016) and of the downstream signal molecules c‐Src and Erk1/2.
This discrepancy can be explained by the effect of TFEB on VEGFR2 membrane localization and trafficking. First in sh‐TFEB ECs, the PM accumulated VEGFR2 defectively co‐localized with CAV‐1‐rich domains, which are hotspots for signaling activity and required for VEGFR2 activation (Cho et al, 2004). Second, when TFEB was knocked down, we observed an alteration of receptor trafficking analyzed by biotinylated VEGFR2 internalization, FACS, and TIRF. Of note, the accumulation of VEGFR2 induced by the inhibition of endocytic (Sawamiphak et al, 2010; Gourlaouen et al, 2013) and recycling (Ballmer‐Hofer et al, 2011; Lanahan et al, 2013) pathways leads to the inhibition of its signaling including the activation of PLCγ and MAP kinases.
The existence of extensive cross‐talks of autophagic and lysosome pathways with the mechanisms fueling endocytosis is well demonstrated (Pavel & Rubinsztein, 2017).
Therefore, it is intriguing to speculate that the canonical effect of TFEB on endothelial autophagic flux (see Fig 6D and E) negatively influences the VEGFR2 endocytosis, which is known to be important in its signaling (Simons et al, 2016).
Altogether, these observations shed light on the TFEB‐mediated cellular mechanisms regulating VEGFR2 expression and allow a better understanding of the observed vascular phenotype in Tfeb mutants.
The in vivo vascular defects observed in Tfeb EC−/− and Tfeb iEC−/− mice validated the concept that one of the major consequences of TFEB deletion is the reduction of EC proliferation and the impairment of VEGFR2 activity. After the establishment of the primitive vascular plexus, the formation of the vascular tree is promoted by sprouting angiogenesis, which is characterized by the presence of either migratory tip cells or proliferating stalk cells. Tip cells guide network expansion, while stalk cells proliferate behind the tip cells to extend the vascular lumen as the sprouts elongate. The crucial role of VEGFR2 in this scenario has been demonstrated by experiments performed with ECs derived from embryonic stem cells, in which cells that are heterozygous for the Vegfr2 allele show defects in stalk–tip dynamics (Jakobsson et al, 2010).
EC Tfeb‐deficient embryos died between E10.5 and E11.5, displaying defects in the patterning of several regions of the vascular tree, with a reduction in larger caliber vessels and the capillary network. The effect of TFEB presumably begins after the formation of primitive vascular plexus because Tfeb deletion did not affect the appearance of hemangioblasts. Furthermore, post‐natal vascular maturation in the retina and kidney was altered in Tfeb mutants, with reduced proliferation. In Tfeb mutants, the retinal vasculature presented a delayed expansion of the vascular plexus and a significant reduction in vessel density. However, the ability of tip cells to extend filopodia was unaffected. This phenotype resembles that caused by the concomitant endothelial ablation of Vegfr2 and Vegfr3, in which the vascular hyperplasia caused by Vegfr3 deletion is counterbalanced by the absence of Vegfr2 (Zarkada et al, 2015), or by enhanced Notch activity in stalk cells (Phng et al, 2009; Guarani et al, 2011).
The deletion of Tfeb in renal ECs impairs the maturation of the glomerulus, a process that is strictly dependent of the VEGF‐A/VEGFR2 axis (Esser et al, 1998). We observed a deep alteration of the cellular structures with the fusion of podocyte foot processes and the disappearance of endothelial fenestrae. Generally, the formation of the filtration barrier is regulated by a paracrine mechanism involving VEGF‐A released by podocytes and VEGFR2 expressed on glomerular ECs. The phenotype observed in Tfeb mutants is similar to that described in whole‐body post‐natal deletion of Vegfr2 and podocyte‐specific Vegfa knockout (Eremina et al, 2003; Sison et al, 2010). Interestingly, pre‐ or post‐natal Vegfa deletion in podocytes promotes nephrotic syndrome or thrombotic microangiopathy, respectively (Eremina et al, 2003, 2008).
Altogether, these data bring new insight into the regulatory effect of TFEB in vascular cells (Lu et al, 2017; Fan et al, 2018) by activating a genetic program regulating cell proliferation and VEGFR2 functions. We speculate that TFEB represents a powerful cellular tool that connects vascular needs with the metabolic state. Recent data indicate that cellular nutrient levels regulate TFEB concentrations through an autoregulatory feedback loop, in which TFEB binds to its own promoter in a starvation‐dependent manner and induces its own expression (Settembre et al, 2013a) and that TFEB negatively regulates the level of hypoxia‐inducible factor‐1 (Hubbi et al, 2013). Furthermore, TFEB controls lipid breakdown, and its overexpression activates fatty acid oxidation (Settembre et al, 2013a), which is necessary for sprouting angiogenesis, by fueling de novo nucleotide synthesis for DNA duplication (Schoors et al, 2015).
Furthermore, the recent observation that shear stress up‐regulates TFEB (Lu et al, 2017) allows hypothesizing its role in regulating the optimal PM amount of VEGFR2, which, through a multimeric complex with VE‐cadherin, PECAM‐1, and VEGFR3 (Baeyens et al, 2016), transduces the frictional force from blood flow into biochemical signals that regulate gene expression and cell behavior.
Materials and Methods
Antibodies
Anti‐GFP and anti‐Ki67 (SP6) from Thermo Fisher Scientific; isolectin‐B4, anti‐MYO1C, and anti‐α‐tubulin (B‐5‐1‐2), anti‐LAMP1, anti‐PCNA (PC10) from Sigma‐Aldrich; anti‐CD31, anti‐FLK1‐APC‐conjugated (Avas12alpha1), anti‐CD71‐FITC‐conjugated (C2), anti‐CD117 (2B8)‐PE‐cy7‐conjugated, anti‐CD31‐FITC‐conjugated (Mec 13.3), anti‐CD102, anti‐IgG2a k isotype‐APC‐conjugated (R3595), anti‐IgG2bk isotype‐PE‐Cy7 conjugated (2B8), anti‐CD326, anti‐Rab4, and anti‐Rab5 from BD Biosciences; anti‐VEGFR2 (55B11), anti‐p‐Tyr‐1175‐VEGFR2 (D5B11), anti‐PLCγ‐1, anti‐p‐Tyr‐783‐PLCγ‐1, anti‐ERK1/2, anti‐p‐ERK‐1/2 (T202/Y204; E10), anti‐Src (36D10), anti‐p‐Src (Tyr416; D49G4), anti‐CDK4 (D9G3E), anti‐Rb (4H1), anti‐p‐Rb (Ser780; C84F6), and anti‐p‐Rb (Ser807/811; D20B12), anti‐PCNA (PC10), anti‐ULK1 (D8H5), anti‐ATG9A (D409D) from Cell Signaling Technology; anti‐endomucin (V.7C7), anti‐Flk‐1 (A‐3), anti‐podocin (G‐20), anti‐caveolin‐1 (N‐20) and anti‐E2F2 (TFE‐25), anti‐E2F1 from Santa Cruz Biotechnology; anti‐human VEGFR2 (89109) from R&D System; anti‐TFEB from MyBiosource; hypoxyprobe‐1‐FITC‐conjugated antibody (Chemicon); anti‐LC3 from Novus Biologicals; anti‐cyclin D1 (SP4), anti‐E2F1, and anti‐TGN46 (2F7.1) from Abcam.
Mice
All animal procedures were approved by the ethics committee of the University of Turin and by the Italian Ministry of Health (Protocol approval no. 864/2015‐PR).
To generate transgenic mice expressing Tfeb‐GFP, the sequence for the open reading frame of EGFP was inserted between the last amino acid and the translation termination codon in exon 9 (NCBI transcript NM_001161722.1). The positive selection marker (puromycin resistance—PuroR) was flanked by FRT sites and was inserted into intron 8. The targeting vector was generated using BAC clones from the C57BL/6J RPCIB‐731 BAC library and was transfected into the TaconicArtemis C57BL/6N Tac ES cell line. Homologous recombinant clones were isolated using positive (PuroR) and negative (thymidine kinase—Tk) selection. The constitutive KI allele was obtained after Flp‐mediated removal of the selection marker. The Tfeb‐EGFP fusion protein was expressed from the endogenous Tfeb promoter. The remaining recombination site was located in a non‐conserved region of the genome.
Endothelium‐specific silencing of Tfeb was achieved by crossing Tfebflox mice (Settembre et al, 2013a) with the following: (i) transgenic mice expressing Cre‐recombinase driven by the Tie2 promoter (Tie2‐Cre; The Jackson Laboratory) to obtain a line with constitutive silencing of Tfeb in the endothelium (Tfeb EC); and (ii) transgenic mice expressing tamoxifen‐inducible Cre‐recombinase (Cre‐ERT2) driven by the vascular endothelial cadherin promoter (Cdh5‐Cre‐ER T2; Tfeb iEC). Tfeb floxed /Cre + mice (indicated respectively as Tfeb EC−/+ or Tfeb EC−/− and Tfeb iEC−/+ or Tfeb iEC−/− in the consideration of Tfeb deletion in heterozygosis or homozygosis) were compared with Tfeb floxed /Cre − (without Cre) mice of the same progenies (indicated as control). Inducible Cre was activated by daily tamoxifen administration from p1 to p3 (1 mg/ml, 50 μl by intragastric injection) or from p5 to p8 (2 mg/ml, 50 μl by i.p. injection; Pitulescu et al, 2010). To exclude tamoxifen pathological effects, both control mice that Tfeb iEC−/+ and Tfeb iEC−/− mice were treated.
The systemic effect of Tfeb deletion was evaluated by analyzing hematological and biochemical parameters in blood after 1 month from the Cre induction (p0‐p8) in Tfeb iEC−/− mice. Mice survival was of 32.4 ± 5.9% (mice n = 24, P = 0.003 versus control mice), without any significant modification of renal and hepatic functions. However, the Tfeb iEC−/− mice only presented an increase of % reticulocytes (950.5 ± 200.8% versus 516.2 ± 107.1% in control mice; mice n = 10; P = 0.04) and % platelets (98.25 ± 27.24% versus 31.75 ± 15.3% in control mice; mice n = 10; P = 0.006).
% point prevalence of vascular alterations was evaluated at E10.5 (our point time). Briefly, after endomucin immunostaining of the vessels, embryos with genotypes blindly analyzed (n = 25) were divided into two groups: “positive embryos” showing vascular alterations and “negative embryos” with absence of vascular defects. Following that, we separated the embryos of the two groups accordingly to their genotype (control, n = 13; Tfeb EC−/−, n = 12) and the % point prevalence in control or Tfeb EC−/− embryos was calculated as:
Cells, genetic manipulation, and biological assays
Lung ECs were isolated from control and Tfeb iEC mice by positive selection with anti‐CD31 and anti‐CD102 Abs conjugated to Dynabeads and maintained in vitro up to passage 2 in M199 with 20% FCS. Lung epithelial cells were isolated with the same procedure by using anti‐CD326 Ab and maintained up to passage 2 in DMEM with 10% FCS. When Tfeb silencing was not induced in vivo (as previously described) but in vitro, lung ECs were incubated with 4‐OH‐tamoxifen (5 μM, 48 or 72 h) to allow Cre activation and Tfeb silencing. The correct recombination of the Tfeb allele was verified by real‐time PCR (Power SYBR Green PCR Master Mix). PCR was performed using specific primers (5′‐GACTCAGAAGCGAGAGCTAACAG‐3′ and 5′‐TGGCCTTGGGGATCAGCATT‐3′) recognizing the exon 5‐6 region of Tfeb.
In vitro experiments were carried out on human endothelial cells (indicated as ECs) isolated from umbilical cord veins maintained as described previously (Napione et al, 2012b). To minimize cell variability, pools of 5 different donors were used. The isolation of primary human ECs was approved by the Office of the General Director and Ethics Committee of the Azienda Sanitaria Ospedaliera Ordine Mauriziano di Torino hospital (protocol approval no. 586, October 22, 2012; and no. 26884, August 28, 2014), and informed consent was obtained from each patient. Cells were tested for mycoplasma contamination by means of Venor GeM Mycoplasma Detection Kit.
The TFEBS142A mutant was generated from TFEB cDNA (Origene, cod. SC122773) by inserting a single point mutation using the Phusion Site‐Directed Mutagenesis Kit. TFEBS142A was cloned into the pTRIPZ inducible vector, and the transgene was induced by doxycycline addition (0.5 μg/ml) for 3 h (for ChIP‐seq experiments) or 24 h (other experiments). These cells were indicated as TFEBS142A. ECs infected with pTRIPZ‐TFEBS142A but not treated with doxycycline were used as control and indicated as “control”. To exclude unspecific doxycycline effects, no infected ECs were also treated.
Loss‐of‐function experiments were carried out with shRNA against TFEB (#1 TRCN0000013111, #2 TRCN0000437429, #3 TRCN0000013110, #4 TRCN0000437246, and #5 TRCN0000440038 NM_007162.2) or MYO1C (Catalog number #1 TRCN0000122925, #2 TRCN0000122927, #3 TRCN0000122928 NM_033375) and DLEU2 (Catalog number #1 TRCN0000072484 and #2 TRCN0000072485 NR_002612) cloned into the pLKO.1‐puro non‐Mammalian vector. In particular, the different experiments were performed with shRNA #1 TRCN0000013111 against TFEB, shRNA #1 TRCN0000122925 against MYO1C, and shRNA #1 TRCN0000072484 against DLEU2. ECs were transduced with specific lentiviral particles (MOI = 1) prepared according to (Follenzi et al, 2000) in the presence of 8 μg/ml polybrene. The medium was replaced after 24 h, and cells stably expressing the lentivirus were selected on puromycin (1 μg/ml) for 24 h. ECs were transfected with the appropriate control (miR‐inhibitor/miR‐mimic control) has‐miR‐15a‐5p and has‐miR‐16‐5p mirVanaTM miRNA inhibitors or mimics (90 nmol/l, 24 h) using Lipofectamine® RNAiMAX Reagent. We verified the degree and the specificity of miRNA inhibition or up‐regulation and the eventually off‐target effects using qPCR (see specific section).
Proliferation rate and DNA content were evaluated by using Click‐iT® EdU Flow Cytometry Cell Proliferation Assay and propidium iodide (PI) staining according to manufacturer's protocol. Briefly, human or lung murine ECs (5 × 106) were starved overnight and then stimulated with 20% FCS or VEGF‐A (30 ng/ml) for 24 h. After 1‐hr incubation with 10 μM thymidine fluorescent analog 5‐ethynyl‐2′‐deoxyuridine (EdU), cell suspensions were processed for EdU detection and cell DNA was labeled by a 3‐h treatment with 50 μg/ml PI and 100 μg/ml ribonuclease A. Data were acquired with a CyAn ADP flow cytometer (Dako) and analyzed with FlowJo software (Tree Star, Ashland, OR, USA).
EC chemotaxis and morphogenesis were assayed and analyzed as described previously (Serini et al, 2003). In particular, in morphogenesis assay ECs (2 × 104) were resuspended with poor MEM and loaded on the top of the growth factor reduced Matrigel. Each conditional group contained 6 wells. Following incubation at 37°C for 6 h with VEGF‐A (30 ng/ml), each well was fixed with 4% paraformaldehyde, permeabilized in PBS 1% Triton X‐100, and stained with phalloidin‐555 and DAPI.
Yolk sac cells analysis
Yolk sacs from control or Tfeb EC embryos were collected at E9.5‐E10.5 and genotyped as described above. Each yolk sac was treated separately to obtain single‐cell suspensions by incubation with collagenase/dispase at 37°C for 1 h with occasional trituration. The samples were then subjected to flow cytometry analysis (see specific section).
Tissue and cell staining and analysis
Whole‐mount samples were prepared (Yokomizo et al, 2012) by fixing tissues in 4% paraformaldehyde for 1 h on ice. Tissues were incubated overnight at 4°C in DPBS, 1% Triton X‐100, containing the indicated Abs. After washes, tissues were incubated for 1 h in the same buffer containing the appropriate Alexa Fluor secondary Abs. After fixation with 4% paraformaldehyde for 30 min at RT, samples were flat‐mounted onto glass coverslips.
Tissue sections were frozen in OCT compound and cut into 6–10‐μm thick sections. Embryo kidney and lung slices were fixed in 4% paraformaldehyde and Zinc fixative, respectively, for 10 min at room temperature, permeabilized, stained as indicated, and then incubated with the appropriate fluorescently labeled secondary Ab.
Hypoxia was detected by the formation of pimonidazole adducts 1 h after intraperitoneal (i.p.) injection of 60 mg/kg pimonidazole hydrochloride in pregnant females. Mice were then sacrificed, and the embryos were harvested and immunostained with hypoxyprobe‐1‐FITC‐conjugated Ab.
Immunofluorescence staining of vessels was manually quantified using ImageJ software after excluding any small‐sized, not‐interconnected objects. Immunoreactivity was calculated as the surface area of each Ab staining co‐localized with the EC markers endomucin, isolectin B4, or CD31 and normalized to the total vascular surface area visualized by the same molecules. The number of branching points per field was quantified as described previously (Samarelli et al, 2014).
To quantify differences in the average length and total number of filopodia between control and Tfeb iEC mice, we used custom‐made image analysis algorithms written in MATLAB. (MATLAB and Image Processing Toolbox, The MathWorks, Inc., Natick, Massachusetts, United States.) Briefly, confocal stacks were maximum projected and then processed to obtain filopodia or the main vascular network. To extract main vascular network, images were median filtered and thresholded, and then, the external perimeter of the vasculature was measured. As for filopodia, images were filtered with a linear rotating kernel filter (Lee et al, 1990) and then processed with a multiscale vessel enhancement filter (Frangi et al, 1998). Quantifications were based on the total length of the segmented filopodia divided by the total perimeter of the main vascular network and average length of major axis of each of the filopodial structure identified.
For transmission electron microscopy, mice were perfused with fixative buffer (2.5% glutaraldehyde and 2% paraformaldehyde in 0.15 M sodium cacodylate buffer). Kidneys were immediately placed into fresh fixative and then cut into small cubes. The analysis was performed at the Department of Cellular and Molecular Medicine, UCSD (La Jolla, CA).
For cell immunofluorescence staining, ECs (7 × 104), grown up on cover slides, were fixed in 4% paraformaldehyde, permeabilized (PBS 0.1% Triton X‐100) and then incubated with the indicated primary Abs and the appropriate Alexa Fluor secondary Abs.
FACS
Cell suspensions from dissociated yolk sacs were incubated with indicated specific Abs and then analyzed on a CyAn ADP Analyzer (Beckman Coulter). DAPI‐positive dead cells were excluded from analysis. Cytosolic and PM VEGFR2 staining was performed using an IntraPrep kit (Beckman Coulter), and the data were analyzed using Summit 4.3 software (Beckman Coulter). The following Abs were used: anti‐VEGFR2‐PE‐conjugated (89106); anti‐IgG1‐PE‐conjugated (11711); anti‐CD202b (Tie2)‐PE‐conjugated (Tek4); anti‐IgG1 k isotype‐PE‐conjugated (RTK2071); anti‐FLK1‐APC‐conjugated (Avas12alpha1); anti‐IgG2a k isotype‐APC‐conjugated (R3595); anti‐CD71‐FITC‐conjugated (C2); anti‐IgG1 k isotype‐FITC‐conjugated (R3‐34); anti‐CD117‐PE‐Cy7‐conjugated (2B8); and anti‐IgG2bk isotype‐PE‐Cy7 conjugated.
TIRF microscopy
TIRF microscopy was performed using a Leica AM TIRF MC system mounted on a Leica AF 6000LX workstation with a 63× oil‐immersion objective and a laser penetration depth of 110 nm. ECs were fixed, saturated and permeabilized, and then treated with Ab anti‐VEGFR2 (A3) and appropriate Alexa Fluor secondary Ab.
Quantification of immunofluorescence analysis
Immunofluorescence images were acquired on a TCS SPE or TCS SP8 STED confocal laser‐scanning microscopes (Leica Microsystems). Different fields (tissue samples: 15–20; cell samples: 5–8) per sample section were randomly chosen for analysis. When the same molecule was evaluated in different samples, laser power, gain, and offset settings were maintained. Images were quantified using ImageJ software or TCS SP8 quantification software. For each analysis, at least 3 different experiments were performed.
Microarray experiments
RNA extracted using a miRNeasy Mini Kit was amplified and labeled using an Illumina TotalPrep RNA Amplification Kit, and 750 ng of cRNA probe was hybridized to a HumanHT‐12 v4.0 Expression Bead Chip. All experiments were performed in triplicate. Cubic spline‐normalized probe intensity data, together with detection P‐values, were obtained using GenomeStudio software V2011.01. We selected probes with a detection P < 0.05. For each gene, we retained the associated probe with the largest mean expression value across all samples. For each probe, the log2 signal was converted to the log2 ratio against the global average expression of that probe in all samples. Data were clustered using Gene Expression Data Analysis Suite (GEDAS; www://gedas.bizhat.com), and LIMMA (Smyth, 2002) was used to identify the modulated genes. A threshold | log2 FC | of > 0.5 and an adjusted P < 0.1 were used to select differentially expressed genes. Statistical analyses were performed in the R environment (http://www.R-project.org).
GSEA was performed using the public platform at http://www.broadinstitute.org/gsea/msigdb/downloads.jsp. In particular, after gene filtering for all the datasets, probes were collapsed on Gene Symbols, again selecting the probe with the largest mean expression across all the experiments for each gene. GSEA statistics were calculated with the default settings based on a Pearson metric. P‐values and FDRs were calculated by repeating sample permutations 1,000 times. The data were further analyzed for enrichment in biological themes (GO—Biological Processes, Molecular Functions, Cellular Components) by using the DAVID resource (http://david.abcc.ncifcrf.gov).
miRs annotation
We defined a set of miRs associated with angiogenesis on the basis of published literature with evidence based on experimental data (Chamorro‐Jorganes et al, 2013). For each miR, we considered the corresponding pre‐miR based on the annotations provided by the Ensembl database, version Human GRCh37 (GENCODE 19, miRBase 18). In case the miR was intragenic, we identified the host gene and selected only those originating from the same DNA strand. Of these, we further chose only miRs with VEGFR2 as a validated target (Chamorro‐Jorganes et al, 2011; Chan et al, 2013). Finally, for this short list of candidates, we checked significant TFEB binding peak(s) in the putative core promoter region of the host gene.
ChIP‐seq
For genome‐wide analysis of TFEB binding, sequencing libraries were constructed using the NEBNext® ChIP‐seq Library Prep Reagent Set for Illumina and a NextSeq 500 Illumina sequencer. ChIP‐seq reads were aligned to the hg19 genome assembly using Bowtie v0.12.7 with the following parameters: ‐q –max/dev/null ‐v 1 ‐S –sam‐nohead ‐m 1. Data were filtered using the following specifications: Duplicate reads were filtered out. BedGraph files were generated by using MACS tool. Peak calling was performed as described previously (Krepelova et al, 2014) using a P‐value cutoff = 1E‐08. TFEB target genes were defined as those having a peak between −500 and +100 from the annotated transcription start site.
ChIP
Chromatin immunoprecipitation of TFEB was performed as described previously (Krepelova et al, 2014). Briefly, approximately 2 × 107 crosslinked ECs were resuspended in 250 μl of SDS lysis buffer (10 mM EDTA, 1% SDS, 16.7 mM Tris, pH 8) with protease inhibitors and incubated for 10 min on ice. After sonication, the cell lysate was centrifuged at 12,000 g for 10 min at 4°C. The supernatant was diluted ten‐fold with ChIP dilution buffer (16.7 mM Tris–HCl, pH 8.0, 167 mM NaCl, 1.2 mM EDTA, 1% Triton) before immunoprecipitation.
The supernatant was incubated with 5 μg of Ab anti‐TFEB or IgG with rotation at 4°C for 16 h. Samples treated with IgG were used as a negative control. Afterward, previously BSA‐saturated beads (Dynabeads® Protein G) were added for 2 h. Immunoprecipitated complexes were extensively washed before adding SDS elution buffer (50 mM Tris–HCl, pH 8.0, 10 mM EDTA, 1% SDS, DTT 5 mM, NaCl 150 mM) for 30 min at room temperature. After decrosslinking, DNA was purified using a QIAQuick PCR Purification Kit according to the manufacturer's instructions.
Luciferase reporter assay
Cells were seeded in 24‐well plates at a density of 4 × 104 cells per well. After ChIP‐seq analysis, to identified the relative TFEB MACS peak on the different promoter gene, ECs were transfected with CDK4‐luciferase full‐length promoter (from J. Modiano, Addgene plasmid #86656) and CDK4‐luciferase fragment A (from J. Modiano, Addgene plasmid #86658) in which the predicted ChIP‐seq TFEB peak near TSS on CDK4 promoter is deleted. ECs were transfected with pMCS‐CypridiLuc_AC167_MYO1C_promoter (sequences −1,500/+200 from TSS of the longest isoform of MYO1C gene according to Ensembl GRCh37, transcript ID: ENST00000359786), pMCS‐CypridiLuc_AC167_MYO1C_promoter_d1 (in which the putative TFEB binding site has been deleted), pMCS‐CypridiLuc_AC167_MYO1C_promoter_d2 (in which 100 bps around the same putative TFEB binding site has been deleted) synthesized Geneart support (Thermo Fisher Scientific) and with pMCV‐GreenReLuc using Lipofectamine® RNAiMAX Reagent according to the manufacturer's instructions.
Luciferase activities were analyzed with Pierce Cypridina Luciferase Glow Assay Kit and Pierce Renilla Luciferase Glow Assay Kit or The Dual‐Luciferase® Reporter (DLR™) Assay System using a Glomax 20/20 luminometer (Turner Biosystems, Sunnyvale, CA, USA). The relative reporter activity was calculated by normalizing the luciferase activity with Renilla luciferase activity.
Western blot
Western blotting and quantitative analysis using a ChemiDoc Touch Imaging System (Bio‐Rad) and Image Lab software 5.2.1 (Bio‐Rad) were performed as described (Napione et al, 2012a) with the use of specific Abs above indicated. The following Abs were used: anti‐TFEB; anti‐VEGFR2 (55B11), anti‐p‐Tyr‐1175‐VEGFR2 (D5B11), anti‐PLCγ‐1, anti‐p‐Tyr783‐PLCγ‐1, anti‐ERK‐1/2, anti‐p‐ERK‐1/2 (T202/Y204; clone E10), anti‐Src (36D10), anti‐p‐Src (Tyr416; D49G4), anti‐CDK4 (D9G3E), anti‐Rb (4H1), anti‐p‐Rb (Ser780; C84F6) and anti‐p‐Rb (Ser807/811; D20B12), anti‐PCNA (PC10), anti‐ULK1 (D8H5), anti‐ATG9A (D409D); anti‐LC3; anti‐CD31; anti‐MYO1‐C, anti‐LAMP1, and anti‐α‐tubulin (B‐5‐1‐2); anti‐cyclin D1 (SP4), anti‐E2F1, and anti‐E2F2 (TFE‐25).
Biochemical analysis of PM distribution of VEGFR2
ECs were starved in M199 for 3 h and then stimulated with VEGF‐A at 37°C (30 ng/ml) to allow VEGFR2 internalization. Cells were placed at 4°C, and the remaining receptors on PM were labeled with 0.15 mg/ml of impermeant sulfo‐NHS‐SS‐biotin in PBS for 10 min. The unreacted biotin was quenched with TBA (25 mM Tris, pH 8, 137 mM NaCl, 5 mM KCl, 2.3 mM CaCl2, 0.5 mM MgCl2, and 1 mM Na2HPO4), and ECs were solubilized in lysis buffer (20 mM Tris, pH 7.5, 125 mM NaCl, 10% glycerol, 1% NP‐40, protease inhibitor cocktail). This fraction represents the total cellular VEGFR2. PM biotin‐VEGFR2 complexes were separated from total VEGFR2 (biotin‐free) using streptavidin‐agarose beads (Napione et al, 2012a). After protein extraction from the beads, equivalent volumes of the PM and total samples were resolved by SDS–PAGE and blotted with specific Abs. Loading controls were performed with an anti‐CD31 or α‐tubulin Abs.
VEGFR2 internalization assay
VEGFR2 internalization assays were performed as previously described (Valdembri et al, 2009). ECs were PM‐labeled at 4°C with 0.5 mg/ml sulfo‐NHS‐SS‐biotin in PBS for 30 min on ice. Labeled cells were washed with cold MEM 1% FBS and cold PBS, and endocytosis was induced using prewarmed MEM, 1% FBS, containing VEGF‐A (30 ng/ml) and 0.1 M primaquine. At the indicated times, ECs were transferred to ice, and biotin was removed from the PM by incubation with 20 mM sodium 2‐mercaptoethanesulfonate (MesNa) in 50 mM Tris–HCl (pH 8.6), 100 mM NaCl, and 0.015N NaOH for 1 h at 4°C. MesNa was quenched by the addition of 20 mM iodoacetamide for 10 min. Cells were lysed in a specific buffer at 4°C (25 mM Tris–HCl, pH 7.6, 100 mM NaCl, 2 mM MgCl2, 1 mM Na3VO4, 0.5 mM EGTA, 1% Triton X‐100, 5% glycerol, protease inhibitor cocktail). The levels of biotinylated VEGFR2 were determined by Capture ELISA. Briefly, Maxisorp 96‐well plates were coated overnight with 5 μg/ml of anti‐VEGFR2 (89109) Ab at 4°C and were blocked in PBS containing 0.1% Tween‐20 with 5% BSA for 1 h at RT. VEGFR2 was captured by overnight incubation of the cell lysate at 4°C. Unbound material was removed by washing, and the wells were incubated with streptavidin‐conjugated horseradish peroxidase for 1 h at 4°C. Biotinylated VEGFR2 was detected with a chromogenic reaction.
The percentage of internalization of VEGFR2 was calculated as the percent of biotin‐VEGFR2 complexes in cell lysates after VEGF‐A stimulation with respect to all the biotin‐VEGFR2 complexes in unstimulated cells.
“Antibody feeding assay” was performed as previously described (Gourlaouen et al, 2013). ECs were incubated for 3 h in serum‐free M199 medium at 37°C; then, PM VEGFR2 was labeled with anti‐VEGFR2 extracellular domain Ab (R&D System) for 30 min at 4°C; and then, cells were fixed or transferred to prewarmed serum‐free M199 medium containing VEGF‐A (30 ng/ml) at 37°C for 15 min to permit internalization. Cells were then incubated with the anti‐Rab5 Ab and the appropriate Alexa Fluor secondary Abs.
Statistical analysis
Sample sizes were not selected according to a specific power analysis but just in agreement with similar experiments done in other laboratories working in vascular development and quoted in the specific references. No statistical methods were used to predetermine sample size.
We did not randomize sample/animals because our experimental design did not require this type of strategy. The investigators were not blinded to allocation during experiments and outcome assessment. The data are indicated as the mean ± SEM. Statistical analyses were performed using unpaired Student's t‐test (two‐tailed) or one‐way ANOVA, followed by post hoc pairwise analysis tests, as indicated using GraphPad Software (La Jolla, CA, USA, www.graphpad.com). A P < 0.05 was considered significant.
Author contributions
Conceptualization, GD, SO, FB, LP, GC, EA; Methodology, GD, EA, AB, CSp, CSe; Formal Analysis, DC, AP, FN; Investigation, GD, GC, AN, VC, FN; Resources, AB; Writing—original draft, GD, FB; Writing—review & editing, FB; Supervision, FB; Funding acquisition, FB, SO, AB.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC) grants: 12182, 18652, 14284, 11982, 17639, 20240, and IG 2015, Id 17639; Ministry of University and Research RBAP11BYNP, RBFR08F2FS‐002, MIUR FIRB RBAP11Z3YA; European Research Council Advanced Investigator: 694282 (LYSOSOMICS); U.S. National Institutes of Health: R01‐NS078072; the Huffington Foundation; Fondazione Cassa di Risparmio, Italian Telethon Foundation (TGM16CB6), Regione Piemonte Project Deflect and EU‐Trascan (Project TopMeso). The authors thank L. Tamagnone, L. Napione, G. Serini (University of Torino, Italy), S. Sigismund (IFOM, Milano, Italy) for helpful discussions, T. Meerloo (UCSD, San Diego, US), and S. Giove (Candiolo Cancer Institute‐FPO‐IRCCS, Candiolo, Italy) for technical support.
The EMBO Journal (2019) 38: e98250
Contributor Information
Gabriella Doronzo, Email: gabriella.doronzo@unito.it.
Federico Bussolino, Email: federico.bussolino@unito.it.
Data availability
The datasets generated during the current study are available in The Gene Expression Omnibus of the National Center for Biotechnology Information (accession number GSE88896).
DEGs list is available in Dataset EV1.
References
- Baeyens N, Bandyopadhyay C, Coon BG, Yun S, Schwartz MA (2016) Endothelial fluid shear stress sensing in vascular health and disease. J Clin Invest 126: 821–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballmer‐Hofer K, Andersson AE, Ratcliffe LE, Berger P (2011) Neuropilin‐1 promotes VEGFR‐2 trafficking through Rab11 vesicles thereby specifying signal output. Blood 118: 816–826 [DOI] [PubMed] [Google Scholar]
- Bartlett CS, Jeansson M, Quaggin SE (2016) Vascular growth factors and glomerular disease. Annu Rev Physiol 78: 437–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blessing AM, Rajapakshe K, Reddy Bollu L, Shi Y, White MA, Pham AH, Lin C, Jonsson P, Cortes CJ, Cheung E, La Spada AR, Bast RC, Merchant FA, Coarfa C, Frigo DE (2017) Transcriptional regulation of core autophagy and lysosomal genes by the androgen receptor promotes prostate cancer progression. Autophagy 13: 506–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcagnì A, Kors L, Verschuren E, De Cegli R, Zampelli N, Nusco E, Confalonieri S, Bertalot G, Pece S, Settembre C, Malouf GG, Leemans JC, de Heer E, Salvatore M, Peters DJ, Di Fiore PP, Ballabio A (2016) Modelling TFE renal cell carcinoma in mice reveals a critical role of WNT signaling. Elife 5: e17047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK (2011) Molecular mechanisms and clinical applications of angiogenesis. Nature 473: 298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamorro‐Jorganes A, Araldi E, Penalva LO, Sandhu D, Fernández‐Hernando C, Suárez Y (2011) MicroRNA‐16 and microRNA‐424 regulate cell‐autonomous angiogenic functions in endothelial cells via targeting vascular endothelial growth factor receptor‐2 and fibroblast growth factor receptor‐1. Arterioscler Thromb Vasc Biol 31: 2595–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamorro‐Jorganes A, Araldi E, Suárez Y (2013) MicroRNAs as pharmacological targets in endothelial cell function and dysfunction. Pharmacol Res 75: 15–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan LS, Yue PY, Wong YY, Wong RN (2013) MicroRNA‐15b contributes to ginsenoside‐Rg1‐induced angiogenesis through increased expression of VEGFR‐2. Biochem Pharmacol 86: 392–400 [DOI] [PubMed] [Google Scholar]
- Cho CH, Lee CS, Chang M, Jang IH, Kim SJ, Hwang I, Ryu SH, Lee CO, Koh GY (2004) Localization of VEGFR‐2 and PLD2 in endothelial caveolae is involved in VEGF‐induced phosphorylation of MEK and ERK. Am J Physiol Heart Circ Physiol 286: H1881–H1888 [DOI] [PubMed] [Google Scholar]
- Cora’ D, Re A, Caselle M, Bussolino F (2017) MicroRNA‐mediated regulatory circuits: outlook and perspectives. Phys Biol 14: 045001 [DOI] [PubMed] [Google Scholar]
- Dang LT, Lawson ND, Fish JE (2013) MicroRNA control of vascular endothelial growth factor signaling output during vascular development. Arterioscler Thromb Vasc Biol 33: 193–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Malta C, Siciliano D, Calcagni A, Monfregola J, Punzi S, Pastore N, Eastes AN, Davis O, De Cegli R, Zampelli A, Di Giovannantonio LG, Nusco E, Platt N, Guida A, Ogmundsdottir MH, Lanfrancone L, Perera RM, Zoncu R, Pelicci PG, Settembre C et al (2017) Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science 356: 1188–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuel R, Sergin I, Bhattacharya S, Turner J, Epelman S, Settembre C, Diwan A, Ballabio A, Razani B (2014) Induction of lysosomal biogenesis in atherosclerotic macrophages can rescue lipid‐induced lysosomal dysfunction and downstream sequelae. Arterioscler Thromb Vasc Biol 34: 1942–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eremina V, Sood M, Haigh J, Nagy A, Lajoie G, Ferrara N, Gerber HP, Kikkawa Y, Miner JH, Quaggin SE (2003) Glomerular‐specific alterations of VEGF‐A expression lead to distinct congenital and acquired renal diseases. J Clin Invest 111: 707–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson C, Kopp JB, Kabir MG, Backx PH, Gerber HP, Ferrara N, Barisoni L, Alpers CE, Quaggin SE (2008) VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 358: 1129–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser S, Wolburg K, Wolburg H, Breier G, Kurzchalia T, Risau W (1998) Vascular endothelial growth factor induces endothelial fenestrations in vitro . J Cell Biol 140: 947–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Lu H, Liang W, Garcia‐Barrio MT, Guo Y, Zhang J, Zhu T, Hao Y, Chen YE (2018) Endothelial TFEB (transcription factor EB) positively regulates postischemic angiogenesis. Circ Res 122: 945–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrando IM, Chaerkady R, Zhong J, Molina H, Jacob HK, Herbst‐Robinson K, Dancy BM, Katju V, Bose R, Zhang J, Pandey A, Cole PA (2012) Identification of targets of c‐Src tyrosine kinase by chemical complementation and phosphoproteomics. Mol Cell Proteomics 11: 355–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follenzi A, Ailles LE, Bakovic S, Geuna M, Naldini L (2000) Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV‐1 pol sequences. Nat Genet 25: 217–222 [DOI] [PubMed] [Google Scholar]
- Frangi AF, Niessen WJ, Vincken KL, Viergever MA (1998) Multiscale vessel enhancement filtering In Medical Image Computing and Computer‐Assisted Intervention – MICCAI'98, Wells WM, Colchester A, Delp SL. (eds), Lecture Notes in Computer Science Vol. 1496, pp 130–137. Berlin: Springer Verlag; [Google Scholar]
- Gariano RF, Gardner TW (2005) Retinal angiogenesis in development and disease. Nature 438: 960–966 [DOI] [PubMed] [Google Scholar]
- Gourlaouen M, Welti JC, Vasudev NS, Reynolds AR (2013) Essential role for endocytosis in the growth factor‐stimulated activation of ERK1/2 in endothelial cells. J Biol Chem 288: 7467–7480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg MJ, Ostap EM (2013) Regulation and control of myosin‐I by the motor and light chain‐binding domains. Trends Cell Biol 23: 81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarani V, Deflorian G, Franco CA, Krüger M, Phng LK, Bentley K, Toussaint L, Dequiedt F, Mostoslavsky R, Schmidt MH, Zimmermann B, Brandes RP, Mione M, Westphal CH, Braun T, Zeiher AM, Gerhardt H, Dimmeler S, Potente M (2011) Acetylation‐dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature 473: 234–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haq R, Fisher DE (2011) Biology and clinical relevance of the microphthalmia family of transcription factors in human cancer. J Clin Oncol 29: 3474–3482 [DOI] [PubMed] [Google Scholar]
- Hubbi ME, Hu H, Kshitiz Ahmed I, Levchenko A, Semenza GL (2013) Chaperone‐mediated autophagy targets hypoxia‐inducible factor‐1α (HIF‐1α) for lysosomal degradation. J Biol Chem 288: 10703–10714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackstadt R, Hermeking H (2015) MicroRNAs as regulators and mediators of c‐MYC function. Biochim Biophys Acta 1849: 544–553 [DOI] [PubMed] [Google Scholar]
- Jakobsson L, Franco CA, Bentley K, Collins RT, Ponsioen B, Aspalter IM, Rosewell I, Busse M, Thurston G, Medvinsky A, Schulte‐Merker S, Gerhardt H (2010) Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol 12: 943–953 [DOI] [PubMed] [Google Scholar]
- Jopling HM, Odell AF, Pellet‐Many C, Latham AM, Frankel P, Sivaprasadarao A, Walker JH, Zachary IC, Ponnambalam S (2014) Endosome‐to‐plasma membrane recycling of VEGFR2 receptor tyrosine kinase regulates endothelial function and blood vessel formation. Cells 3: 363–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M (2001) Tie2‐Cre transgenic mice: a new model for endothelial cell‐lineage analysis in vivo . Dev Biol 230: 230–242 [DOI] [PubMed] [Google Scholar]
- Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, Ambesi‐Impiombato A, Califano A, Migliazza A, Bhagat G, Dalla‐Favera R (2010) The DLEU2/miR‐15a/16‐1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 17: 28–40 [DOI] [PubMed] [Google Scholar]
- Krepelova A, Neri F, Maldotti M, Rapelli S, Oliviero S (2014) Myc and max genome‐wide binding sites analysis links the Myc regulatory network with the polycomb and the core pluripotency networks in mouse embryonic stem cells. PLoS One 9: e88933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrecque L, Royal I, Surprenant DS, Patterson C, Gingras D, Béliveau R (2003) Regulation of vascular endothelial growth factor receptor‐2 activity by caveolin‐1 and plasma membrane cholesterol. Mol Biol Cell 14: 334–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanahan A, Zhang X, Fantin A, Zhuang Z, Rivera‐Molina F, Speichinger K, Prahst C, Zhang J, Wang Y, Davis G, Toomre D, Ruhrberg C, Simons M (2013) The neuropilin 1 cytoplasmic domain is required for VEGF‐A‐dependent arteriogenesis. Dev Cell 25: 156–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y‐K, Rhodes WT (1990) Nonlinear image processing by a rotating kernel transformation. Optics Letters 15: 1383–1385 [DOI] [PubMed] [Google Scholar]
- Little MH, McMahon AP (2012) Mammalian kidney development: principles, progress, and projections. Cold Spring Harb Perspect Biol 4: a008300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Fan Y, Qiao C, Liang W, Hu W, Zhu T, Zhang J, Chen YE (2017) TFEB inhibits endothelial cell inflammation and reduces atherosclerosis. Sci Signal 10: eaah4214 [DOI] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9: 153–166 [DOI] [PubMed] [Google Scholar]
- Mansueto G, Armani A, Viscomi C, D'Orsi L, De Cegli R, Polishchuk EV, Lamperti C, Di Meo I, Romanello V, Marchet S, Saha PK, Zong H, Blaauw B, Solagna F, Tezze C, Grumati P, Bonaldo P, Pessin JE, Zeviani M, Sandri M et al (2017) Transcription factor EB controls metabolic flexibility during exercise. Cell Metab 25: 182–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Chen Y, Gucek M, Puertollano R (2012) MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8: 903–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathiassen SG, De Zio D, Cecconi F (2017) Autophagy and cell cycle: a complex landscape. Front Oncol 7: 51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, Puri C, Pignata A, Martina JA, Sardiello M, Palmieri M, Polishchuk R, Puertollano R, Ballabio A (2011) Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell 21: 421–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napione L, Pavan S, Veglio A, Picco A, Boffetta G, Celani A, Seano G, Primo L, Gamba A, Bussolino F (2012a) Unraveling the influence of endothelial cell density on VEGF‐A signaling. Blood 119: 5599–5607 [DOI] [PubMed] [Google Scholar]
- Napione L, Strasly M, Meda C, Mitola S, Alvaro M, Doronzo G, Marchiò S, Giraudo E, Primo L, Arese M, Bussolino F (2012b) IL‐12‐dependent innate immunity arrests endothelial cells in G0‐G1 phase by a p21(Cip1/Waf1)‐mediated mechanism. Angiogenesis 15: 713–725 [DOI] [PubMed] [Google Scholar]
- Napolitano G, Ballabio A (2016) TFEB at a glance. J Cell Sci 129: 2475–2481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, Ballabio A (2011) Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet 20: 3852–3866 [DOI] [PubMed] [Google Scholar]
- Park C, Kim TM, Malik AB (2013) Transcriptional regulation of endothelial cell and vascular development. Circ Res 112: 1380–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavel M, Rubinsztein DC (2017) Mammalian autophagy and the plasma membrane. FEBS J 284: 672–679 [DOI] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, Settleman J, Stephanopoulos G, Dyson NJ, Zoncu R, Ramaswamy S, Haas W, Bardeesy N (2015) Transcriptional control of autophagy‐lysosome function drives pancreatic cancer metabolism. Nature 524: 361–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phng LK, Potente M, Leslie JD, Babbage J, Nyqvist D, Lobov I, Ondr JK, Rao S, Lang RA, Thurston G, Gerhardt H (2009) Nrarp coordinates endothelial Notch and Wnt signaling to control vessel density in angiogenesis. Dev Cell 16: 70–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitulescu ME, Schmidt I, Benedito R, Adams RH (2010) Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat Protoc 5: 1518–1534 [DOI] [PubMed] [Google Scholar]
- Polager S, Ofir M, Ginsberg D (2008) E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 27: 4860–4864 [DOI] [PubMed] [Google Scholar]
- Raben N, Puertollano R (2016) TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu Rev Cell Dev Biol 32: 255–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roa S, Avdievich E, Peled JU, Maccarthy T, Werling U, Kuang FL, Kan R, Zhao C, Bergman A, Cohen PE, Edelmann W, Scharff MD (2008) Ubiquitylated PCNA plays a role in somatic hypermutation and class‐switch recombination and is required for meiotic progression. Proc Natl Acad Sci USA 105: 16248–16253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson GS, Ju M, Shih SC, Xu X, McMahon G, Caldwell RB, Smith LE (2001) Nonvascular role for VEGF: VEGFR‐1, 2 activity is critical for neural retinal development. FASEB J 15: 1215–1217 [DOI] [PubMed] [Google Scholar]
- Samarelli AV, Riccitelli E, Bizzozero L, Silveira TN, Seano G, Pergolizzi M, Vitagliano G, Cascone I, Carpentier G, Bottos A, Primo L, Bussolino F, Arese M (2014) Neuroligin 1 induces blood vessel maturation by cooperating with the α6 integrin. J Biol Chem 289: 19466–19476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A (2009) A gene network regulating lysosomal biogenesis and function. Science 325: 473–477 [DOI] [PubMed] [Google Scholar]
- Sawamiphak S, Seidel S, Essmann CL, Wilkinson GA, Pitulescu ME, Acker T, Acker‐Palmer A (2010) Ephrin‐B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature 465: 487–491 [DOI] [PubMed] [Google Scholar]
- Schoors S, Bruning U, Missiaen R, Queiroz KC, Borgers G, Elia I, Zecchin A, Cantelmo AR, Christen S, Goveia J, Heggermont W, Goddé L, Vinckier S, Van Veldhoven PP, Eelen G, Schoonjans L, Gerhardt H, Dewerchin M, Baes M, De Bock K et al (2015) Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 520: 192–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serini G, Ambrosi D, Giraudo E, Gamba A, Preziosi L, Bussolino F (2003) Modeling the early stages of vascular network assembly. EMBO J 22: 1771–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A (2011) TFEB links autophagy to lysosomal biogenesis. Science 332: 1429–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A (2012) A lysosome‐to‐nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31: 1095–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, Wollenberg AC, Di Bernardo D, Chan L, Irazoqui JE, Ballabio A (2013a) TFEB controls cellular lipid metabolism through a starvation‐induced autoregulatory loop. Nat Cell Biol 15: 647–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Medina DL, Ballabio A (2013b) Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 14: 283–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Gordon E, Claesson‐Welsh L (2016) Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol 17: 611–625 [DOI] [PubMed] [Google Scholar]
- Sison K, Eremina V, Baelde H, Min W, Hirashima M, Fantus IG, Quaggin SE (2010) Glomerular structure and function require paracrine, not autocrine, VEGF‐VEGFR‐2 signaling. J Am Soc Nephrol 21: 1691–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK (2002) Limma: linear models for microarray data. Bioinformatics. New York: Springer; [Google Scholar]
- Steingrímsson E, Tessarollo L, Reid SW, Jenkins NA, Copeland NG (1998) The bHLH‐Zip transcription factor Tfeb is essential for placental vascularization. Development 125: 4607–4616 [DOI] [PubMed] [Google Scholar]
- Sun CY, She XM, Qin Y, Chu ZB, Chen L, Ai LS, Zhang L, Hu Y (2013) miR‐15a and miR‐16 affect the angiogenesis of multiple myeloma by targeting VEGF. Carcinogenesis 34: 426–435 [DOI] [PubMed] [Google Scholar]
- Takahashi T, Yamaguchi S, Chida K, Shibuya M (2001) A single autophosphorylation site on KDR/Flk‐1 is essential for VEGF‐A‐dependent activation of PLC‐gamma and DNA synthesis in vascular endothelial cells. EMBO J 20: 2768–2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura N, Huang XL, Naruse T, Hamaguchi I, Dumont DJ, Yancopoulos GD, Suda T (1998) Critical role of the TIE2 endothelial cell receptor in the development of definitive hematopoiesis. Immunity 9: 677–686 [DOI] [PubMed] [Google Scholar]
- Tiwari A, Jung JJ, Inamdar SM, Nihalani D, Choudhury A (2013) The myosin motor Myo1c is required for VEGFR2 delivery to the cell surface and for angiogenic signaling. Am J Physiol Heart Circ Physiol 304: H687–H696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdembri D, Caswell PT, Anderson KI, Schwarz JP, Konig I, Astanina E, Caccavari F, Norman JC, Humphries MJ, Bussolino F, Serini G (2009) Neuropilin‐1/GIPC1 signaling regulates alpha5beta1 integrin traffic and function in endothelial cells. PLoS Biol 7: e25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Lüthi U, Barberis A, Benjamin LE, Mäkinen T, Nobes CD, Adams RH (2010) Ephrin‐B2 controls VEGF‐induced angiogenesis and lymphangiogenesis. Nature 465: 483–486 [DOI] [PubMed] [Google Scholar]
- Xu H, Ren D (2015) Lysosomal physiology. Annu Rev Physiol 77: 57–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokomizo T, Yamada‐Inagawa T, Yzaguirre AD, Chen MJ, Speck NA, Dzierzak E (2012) Whole‐mount three‐dimensional imaging of internally localized immunostained cells within mouse embryos. Nat Protoc 7: 421–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue J, Tigyi G (2010) Conservation of miR‐15a/16‐1 and miR‐15b/16‐2 clusters. Mamm Genome 21: 88–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarkada G, Heinolainen K, Makinen T, Kubota Y, Alitalo K (2015) VEGFR3 does not sustain retinal angiogenesis without VEGFR2. Proc Natl Acad Sci USA 112: 761–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Data Availability Statement
The datasets generated during the current study are available in The Gene Expression Omnibus of the National Center for Biotechnology Information (accession number GSE88896).
DEGs list is available in Dataset EV1.