

Abstract
Manganese‐zinc ferrite nanoparticles were synthesized by using a hydrothermal treatment, coated with silica, and then tested as efficient cellular labels for cell tracking, using magnetic resonance imaging (MRI) in vivo. A toxicity study was performed on rat mesenchymal stem cells and C6 glioblastoma cells. Adverse effects on viability and cell proliferation were observed at the highest concentration (0.55 mM) only; cell viability was not compromised at lower concentrations. Nanoparticle internalization was confirmed by transmission electron microscopy. The particles were found in membranous vesicles inside the cytoplasm. Although the metal content (0.42 pg Fe/cell) was lower compared to commercially available iron oxide nanoparticles, labeled cells reached a comparable relaxation rate R 2, owing to higher nanoparticle relaxivity. Cells from transgenic luciferase‐positive rats were used for in vivo experiments. Labeled cells were transplanted into the muscles of non‐bioluminescent rats and visualized by MRI. The cells produced a distinct hypointense signal in T2‐ or T2*‐weighted MR images in vivo. Cell viability in vivo was verified by bioluminescence.
Keywords: nanoparticles, doping, cell labeling, cell transplantation, magnetic resonance imaging
1. Introduction
Regenerative medicine has undergone rapid developments in recent decades.1 Although the high expectations for cell transplantations have yet to be fully met,2 many experimental methods have already found their way into clinical practice.3 The experimental development and preclinical/clinical utilization of both cell transplantations and tissue engineering have also benefitted from progress in nanotechnology. Monitoring of cell implants often requires that cells are appropriately labeled, thus enabling tracking by optical methods, positron emission tomography (PET) or magnetic resonance imaging (MRI).4 MRI occupies a special position among these imaging methods. The method is non‐invasive and requires no ionizing radiation. MRI is not limited by the depth of the implant in the tissue and delivers reasonably high spatial resolution, while preclinical experiments are easily transferable to clinical practice.5 Although its sensitivity is low compared to PET, it can be enhanced using suitably designed labels such as nanoparticle labels, which can be also utilized as drug delivery or therapy systems.6
Several types of contrast agents are used for tracking by MRI. T1 contrast agents produce positive contrast, however, their usage is limited to bigger implants, as their signal corresponds to the actual size. Single cells or implants with subvoxel size may not be detectable.7 Promising are also paramagnetic chemical exchange saturation transfer (paraCEST) agents,8 which may enable to distinguish various implants labeled by modified agents with different saturation frequency.9 However, their sensitivity is rather low. Among non‐proton MRI, fluorinated tracers are explored.10 19F nuclei have similar resonance frequency and similar gyromagnetic ratio, therefore, they produce similar signal as protons. However, concentration of the tracer is usually very low compared to proton concentration, and so is the fluorine signal. Magnetic nanoparticles produce strong T2 and T2* contrast. Due to the so called “blooming effect”, they influence proton signal in much bigger area than is their size. Therefore, even single cells may be detectable.11 Currently, the most commonly used nanoparticles for imaging by MRI are based on coated superparamagnetic iron oxides (maghemite γ‐Fe2O3, magnetite Fe3O4 and intermediate phases).12 Coating ensures the colloidal stability and biocompatibility of such nanoparticles and can be further functionalized for special purposes.13 Some iron oxide nanoparticles are commercially available.
Although maghemite and magnetite particles have high magnetization, which yields high relaxivity, other materials possess even better properties. For example, a relaxivity of up to three times higher has been achieved using ferromagnetic particles based on lanthanum‐strontium‐manganese perovskites.14 Doping of ferrites using metallic ions may substantially increase magnetization and relaxivity.15 Moreover, ferrites can be modified by doping using other metal ions to change their physical properties according to specific requirements. Cobalt ferrites,16 cobalt‐zinc ferrites17 and manganese‐zinc ferrites18 have been previously studied with a view to their use in biomedicine.
Besides relaxivity, the main issues with nanoparticles are biocompatibility and toxicity. Nanoparticle composition, size, coating and possible functionalization can all negatively influence interactions in vivo.19 Toxic effects are particularly associated with the generation of reactive oxygen species, leading to oxidative damage of biological macromolecules and, in turn, cellular dysfunction.20 Even standard iron oxide nanoparticles have been proven to increase the risk of oxidative injury to lipids, proteins and DNA,21 while the introduction of other metallic ions may cause further problems.
Suitable and inert coating is therefore necessary not only to ensure the stability of nanoparticle suspension but also to create a barrier between the potentially toxic metallic core and its surroundings.22 Polysaccharides are widely used,23 as they are biocompatible and can ease cellular uptake.24 Although in some applications polysaccharide biodegradability is preferential, in the case of diagnostic nanoparticles it is questionable. As polysaccharides can be metabolized,25 they do not offer a stable coating, thus leaving the bare iron oxide (or other magnetic core) exposed to the cellular environment. Silica shell, on the other hand, is an inert layer that better separates the particle core from its surroundings. Silica and silica‐coated nanoparticles have been integrated into a broad range of medical applications and their different methods of preparation have been discussed.26 Silica coating improves colloidal stability and reduces toxic effects in the case of both iron oxide nanoparticles26 and various ferrites.27 This type of coating thus improves nanoparticle dispersion in water, protects the core from leaching in acidic environments and ensures a chemically inert surface.
In this study, we synthesized and tested manganese‐zinc ferrites (Mn−Zn ferrites) coated with silica as a cell label for in vivo applications.
2. Results
2.1. Magnetic Nanoparticles and Their Characterization
According to powder X‐ray diffraction (for the diffraction pattern see the Supporting Information, Figure ES1), the MZF nanoparticles were prepared as a single‐phase product with the spinel structure of symmetry and the mean size of crystallites d XRD=11 nm. The chemical composition of bare particles was refined to Mn0.61Zn0.42Fe1.97O4 based on the X‐ray fluorescence spectroscopy (data not shown).
Figure 1A shows a representative transmission electron micrograph of silica‐coated MZF particles. Their magnetic cores were formed by small clusters of ferrite crystallites covered, as a whole, by a smooth and continuous shell of amorphous nature. The image analysis revealed that the mean size of individual ferrite crystallites was 10 nm, the mean size of whole magnetic cores was 30 nm, and the mean silica coating thickness was 17 nm.
Figure 1.
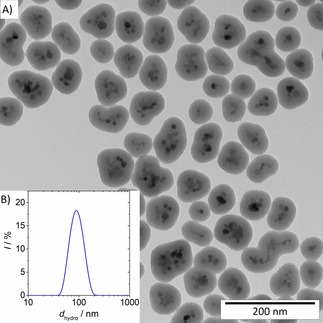
Representative transmission electron microscopy image of silica‐coated MZF nanoparticles (A) and distribution of their hydrodynamic size in an aqueous suspension (B).
The DLS measurements evidenced colloidal stability of the silica‐coated product in water. The hydrodynamic size distribution depicted in Figure 1B was characterized by Z‐average of 87 nm and polydispersity index of 0.065 nm. The zeta potential at neutral pH was determined to −40 mV.
The hysteresis loops of bare MZF particles measured at low and room temperatures are presented in Figure 2A. Specific magnetization reached M=107.1 and 53.3 Am2/kg in magnetic field of 1 T at 5 K and 300 K, respectively. The hysteresis loop at room temperature seems to be either anhysteretic or the coercivity of the sample is below the experimental limit given by remnant fields in the superconducting winding of magnetometer.
Figure 2.
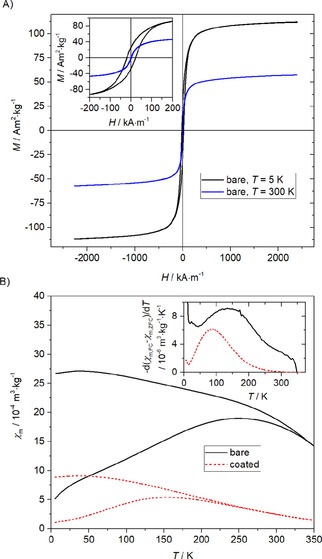
Magnetic properties: hysteresis loops of bare MZF particles at low and room temperatures (A) and ZFC/FC susceptibility measurements for the bare and silica‐coated products (B). The inset show the temperature derivative of the ZFC‐FC susceptibility difference.
The zero‐field‐cooled (ZFC) and field‐cooled (FC) susceptibility measurements are shown for both the bare and silica‐coated products in Figure 2B together with the temperature derivative of the FC‐ZFC susceptibility difference. The irreversibility temperature, the ZFC maximum and the maximum of the temperature derivative are shifted to lower temperatures for the silica‐coated product.
2.2. In Vitro Tests
The high relaxivity of the silica‐coated Mn−Zn ferrite nanoparticles predetermined the nanoparticles as a superb T2 contrast agent. A decrease of r2 relaxivity with increasing temperature was observed, see the Supporting Information, Figure ES2; however, relaxivity at body temperature reached 295±6 s−1 mM−1(Mn0.61Zn0.42Fe1.97O4) at 0.5 T, which is approximately two times higher than relaxivity of a commercially available carboxydextran‐coated iron oxide nanoparticles (analogue of Resovist).
2.3. Cell Viability
The coated nanoparticles were tested using: rat mesenchymal stem cells (MSCs) isolated from bone marrow (BM‐MSCs); rat glioblastoma cells (cell line C6); and rat mesenchymal stem cells from the adipose tissue (AT‐MSCs). The latter were isolated from genetically modified Lewis rats with ubiquitous expression of a gene for the luciferase enzyme, which enabled their visualization by bioluminescence. The trypan blue exclusion test revealed comparable viability of cells cultured in the presence of nanoparticles and unlabeled cells in the control sample in both bone marrow mBM‐MSCs and C6 cells at 0.05 and 0.11 mM(Mn0.61Zn0.42Fe1.97O4) concentrations. The highest nanoparticle concentration in the medium (0.55 mM(Mn0.61Zn0.42Fe1.97O4)) caused a substantial decrease in viability (see Table 1). Therefore, we used a modified protocol for AT‐MSCs (used in the in vivo experiments) with shorter labeling time (24 hours) and lower concentrations (up to 0.2 mM(Mn0.61Zn0.42Fe1.97O4)). Cell viability did not differ from that of the unlabeled cells for any of the used concentrations of nanoparticles according to the modified protocol (Table 1). Also, number of harvested viable cells (gain) did not significantly differ.
Table 1.
Cell viability and gain (average value±standard deviation) after labeling by silica‐coated Mn−Zn ferrite nanoparticles.
| Labeling conc. [mM] | BM‐MSCs[a] | C6[a] | AT‐MSCs[b] | |||
|---|---|---|---|---|---|---|
| Viability [%] | Gain [%] | Viability [%] | Gain [%] | Viability [%] | Gain [%] | |
| 0.05 | 95±6 | 95±6 | 89±6 | 94±54 | 88±9 | 72±12 |
| 0.1 | 90±10 | 88±10 | 88±10 | 79±34 | 94±2 | 103±3 |
| 0.2 | – | – | – | – | 95±1 | 106±8 |
| 0.55 | 51±24[c] | 66±8[c] | 61±33 | 34±9 | – | – |
| unlabeled cells | 96±4 | 100±14 | 88±17 | 100±52 | 92±2 | 100±2 |
[a] 48‐hour labeling. [b] 24‐hour labeling. [c] Significantly differs from unlabeled cells (control).
2.4. Apoptosis Detection
Terminal deoxynucleotidyl transferase dUTP nick end labeling confirmed no apoptosis increase in either type of tested cells even at the highest concentration (Table 2 and Figure ES3). Flow cytometry charts (data shown in Figure ES4) revealed apoptosis in positive control samples only.
Table 2.
Apoptosis results for cells labeled with different concentrations of silica‐coated MZF nanoparticles. Cells were incubated for 48 h and then labeled with FITC‐dUTP; the proportion of FITC‐positive apoptotic events within nucleated singlet cells was determined. The negative control represents unlabeled stained cells, while the positive cells are apoptosis‐induced cells included in the kit.
| Sample | TUNEL [%] | Sample | TUNEL [%] |
|---|---|---|---|
| C6 negative control | 0.10 | BM‐MSCs negative control | 0.34 |
| C6 labeled at 0.05 mM | 0.04 | BM‐MSCs labeled at 0.05 mM | 0.98 |
| C6 labeled at 0.11 mM | 0.04 | BM‐MSCs labeled at 0.11 mM | 0.96 |
| C6 labeled at 0.55 mM | 0.25 | BM‐MSCs labeled at 0.55 mM | 1.39 |
| Positive control | 34.22 | Positive control | 26.37 |
2.5. Real‐Time Cell Proliferation
The influence of nanoparticles was observed in all three concentrations of nanoparticles during real‐time cell proliferation measurement; however, only the highest concentration (0.55 mM(Mn0.61Zn0.42Fe1.97O4)) substantially slowed down the proliferation of BM‐MSCs (Figure 3).
Figure 3.
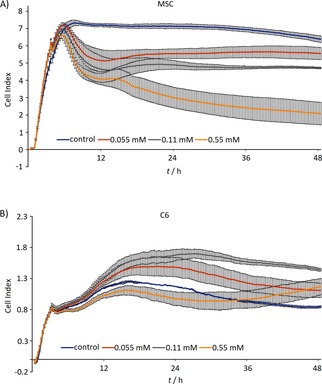
Real‐time proliferation of cells after labeling assessed by impedance measurement in microplates seeded with BM‐MSCs (A) and C6 cells (B).
2.6. Reactive Oxygen Species Production in Labeled BM‐MSCs
All cells stained with CellROX® Deep Red reagent revealed slight production of ROS. The production increase was not proportional to the time of exposition to the nanoparticles. The proportion of dead cells was low and independent of ROS production (Table 3 and Figure ES5). Flow cytometry charts documenting the results are shown in Figure 4.
Table 3.
Percentage of intact cells (BM‐MSCs) and ROS‐positive cells in the sample; middle fluorescent intensity of the CellROX® Deep Red reagent (MFI). Cells were exposed to 0.11 mM(Mn0.61Zn0.42Fe1.97O4).
| Sample | Intact cells in the sample [%] | ROS‐positive cells [%] | ROS production (MFI) |
|---|---|---|---|
| Positive control (TBHP) | 87.0 | 98.9 | 366.4 |
| 3 h labeling | 91.1 | 56.1 | 51.9 |
| 6 h labeling | 91.9 | 49. 7 | 51.1 |
| 12 h labeling | 92.5 | 48.4 | 48.5 |
| 24 h labeling | 90.4 | 48.9 | 49.4 |
Figure 4.
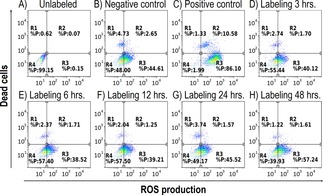
FACS analysis of ROS production in unlabeled cells (A), negative control (B), positive control (C), cells labeled at 0.11 mM(Mn0.61Zn0.42Fe1.97O4) concentration 3 hours (D), 6 hours (E), 12 hours (F), 24 hours (G) and 48 hours (H). Dot plots show multiparametric analysis of ROS production and cell death. Horizontal axis: CELLRox DeepRed Reagent, vertical axis: SYTOXBlue. Quadrant gate: region R1: dead cells negative for ROS, R2: dead cells positive for ROS, R3: ROS production in living cells, R4: live intact cells without ROS production. No significant difference between negative cells and NPs‐treated cells was found. As depicted on negative control, BM‐MSCs have basal ROS production.
2.7. Transmission Electron Microscopy
The method confirmed that labeling cells with silica‐coated nanoparticles and endocytosis without further transfection agents was successful. The nanoparticles were found inside the cytoplasm; they formed clusters in membranous vesicles (endosomes/lysosomes), whereas the nucleus and other important organelles (mitochondria) were free of the particles; see Figure 5.
Figure 5.
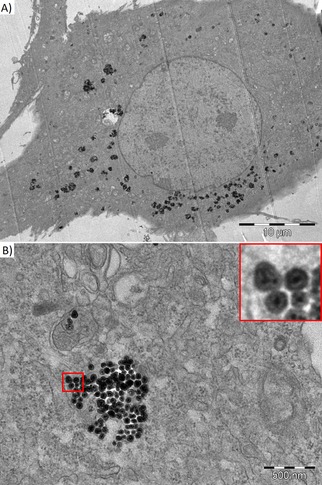
Transmission electron microscopy of BM‐MSC labeled by silica‐coated MZF nanoparticles at 0.11 mM(Mn0.61Zn0.42Fe1.97O4) concentration. The nanoparticles were found in the cytoplasm outside the nucleus (A) and in composing clusters in membranous vesicles (B). The insert (a detailed view of the red‐bordered area) shows single particles formed by small clusters of Mn−Zn ferrite crystallites coated by an intact silica layer.
2.8. In Vitro MR Relaxometry of Cell Suspensions and Cellular Metal Content
Dependence of the R2 relaxation rate of the labeled cells related to 1 million of cells per 1 mL on magnetic field strength is shown in Figure ES6. Cells labeled at a 0.2 mM(Mn0.61Zn0.42Fe1.97O4) concentration in the culture medium showed a relaxation rate of 6.1 s−1/(106 cells/mL) at 4.7 T field strength, even with low cellular metal content (Table 4).
Table 4.
Metal content in the cells after labeling by silica‐coated Mn−Zn ferrite nanoparticles.
| Labeling concentration [mM] (Mn0.61Zn0.42Fe1.97O4) | Mn content [pg/cell] | Fe content [pg/cell] |
|---|---|---|
| 0.05 | 0.033±0.015 | 0.075±0.023 |
| 0.1 | 0.043±0.013 | 0.109±0.025 |
| 0.2 | 0.126±0.005 | 0.420±0.016 |
| Control | 0.003±0.002 | 0.031±0.016 |
2.9. In Vitro Bioluminescence Imaging (BLI)
Viability of AT‐MSCs was also confirmed by bioluminescence. The bioluminescence signal from labeled AT‐MSCs (observed after adding D‐luciferin) decreased with increasing nanoparticle concentration used for labeling (Figure 6).
Figure 6.
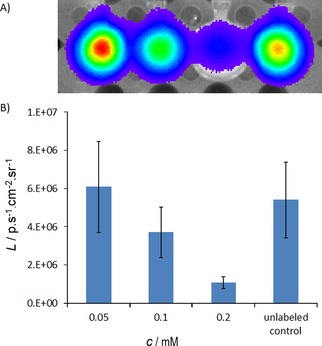
In vitro bioluminescence of cells (from left to right: cells labeled at concentrations c=0.05, 0.1, 0.2 mM(Mn0.61Zn0.42Fe1.97O4) and unlabeled cells) after the addition of luciferin to the medium (A). Quantified average radiance L plotted for different concentrations shows quenching of the signal by high nanoparticle concentrations in the cell pellet (B).
2.10. In Vivo Cell Transplantation
The bioluminescent cells from the adipose tissue (each graft contained 5 million cells) were successfully transplanted into the rat muscle; the transplant was monitored by optical and MR imaging in vivo.
Transplanted cells (both labeled and unlabeled) produced a bioluminescent signal after intravenous application of D‐luciferin on Day 1 (Figure 7A, B), which confirmed that the grafts were viable.
Figure 7.
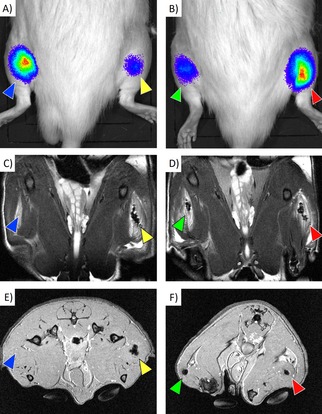
In vivo imaging of the engrafted cells: Bioluminescence images (A, B), coronal T2‐weighted MR images (C, D), and transversal T2*‐weighted MR images (E, F) of rats with transplanted cells. Both labeled and unlabeled cells were detectable by bioluminescence imaging. Unlabeled cells (blue arrows) provided no detectable MR signal, whereas cells labeled at 0.2 mM (yellow arrows), 0.1 mM (green arrows) and 0.05 mM (red arrows) were detectable as distinct hypointense areas.
The signal gradually decreased to 10 % within one week after cell transplantation in all grafts including the graft with unlabeled cells (see Figure ES7).
The labeled cells were trackable on both T2‐weighted (Figure 7C, D) and T2*‐weighted (Figure 7E, F) MR images in vivo. The hypointense signal caused by the nanoparticles was detectable from the beginning (Day 1) until the end of the experiment (Day 28) without visible changes.
3. Discussion
Mn−Zn ferrite nanoparticles were successfully prepared by a hydrothermal procedure, and no admixtures were evidenced by powder X‐ray diffraction. The substoichiometric amount of iron, (Mn+Zn) : Fe=1 : 1.8, was intentionally employed in the reaction mixture to suppress possible formation of hematite as a minor admixture.27b Importantly, the chemical composition of the product tightly reflected the ratio of Mn : Zn used for the synthesis. The composition close to Mn0.6Zn0.4Fe2O4 was selected based on the previous studies18b,27b of hydrothermally prepared ≈10 nm Mn1‐xZnxFe2O4 nanoparticles to achieve magnetic particles with magnetization as high as possible and superparamagnetic behaviour at room/body temperature at the same time. Particles with lower zinc content showed even higher magnetization but were characterized by a considerable fraction of blocked particles at room temperature18b (see the details for x=0.21 and 0.31). By all means, room‐temperature magnetization of the present Mn0.61Zn0.42Fe1.97O4 sample was high enough for the intended application, and the bare sample was almost completely in the superparamagnetic state according to the ZFC/FC susceptibility studies (see Figure 2). Furthermore, the distribution of blocking temperatures, described by the temperature derivative of the FC‐ZFC susceptibility difference (the insert in Figure 2B),28 indicated that the blocking of silica‐coated particles was shifted to lower temperatures, i. e. the coated particles were definitely in the superparamagnetic state.
The inspection of the coated product by transmission electron microscopy evidenced that the ferrites crystallites formed roughly 30 nm clusters exhibiting higher relaxivity than well dispersed ≈10 nm nanoparticles.29 Finally, the colloidal stability of the coated product, which can be explained by the efficient Coulombic repulsion of particles (see the highly negative zeta potential), and the reasonable hydrodynamic size were crucial conditions to consider the particles for the subsequent biological study.
Both in vitro and in vivo tests of the novel nanoparticles revealed that they can be used as a suitable and efficient tool for cell tracking by MRI. The relaxivity of the silica‐coated Mn−Zn ferrite nanoparticles, despite the strong temperature dependence of r2, is (at body temperature) approximately double that of commercially available iron oxide‐based particles30 due to their higher magnetization. High relaxivity is important for detection sensitivity. Actually, the study of monodisperse 15 nm Mn1‐xZnxFe2O4 nanoparticles by Jang et al. already showed extraordinarily high transverse relaxivities of Mn−Zn ferrite particles, and the maximum values of r 2 if expressed per amount of magnetic ions was observed for the composition x=0.4, specifically 860 s−1 mM(Fe+Mn)−1 in magnetic field of 4.7 T.31 However, the comparison with our data is not so straightforward due to the much higher magnetic field and absence of thick silica coating (particles were stabilized by small molecules of 2,3‐dimercaptosuccinic acid) in the previous study.
Testing of viability and cell proliferation confirmed the safety of the nanoparticles. Adverse effects (lower viability and slower proliferation) were observed at the highest concentration only (0.55 mM), which led us to modify the labeling protocol for in vivo applications.
The slight production of ROS in all samples of stained cells did not increase the ratio of dead cells. The proportion of dead cells was comparable to the unlabeled control. Even in the positive control sample, the dead cell count was not as high as expected, which documents the superior resistance of MSCs to ROS.
Successful labeling and the presence of nanoparticles inside the cells were confirmed by TEM. The particles found in the cytoplasm had intact coating and did not enter the nucleus or other important organelles.
Viability of the labeled AT‐MSCs was also verified by in vitro bioluminescence imaging. Interestingly, we observed a decreasing bioluminescence signal in line with increasing nanoparticle concentration. As the independent viability test showed no significant effect of the nanoparticles on viability, we attributed the decrease in bioluminescence more to signal quenching by the nanoparticles than to any change in viability.
The relaxation rate R2 of water in the cell suspension sample, which reflects both the relaxivity of the nanoparticles and their amount inside the cells, determined the sensitivity of cell detection using MRI. Due to higher nanoparticle relaxivity, cells labeled at a 0.2 mM(Mn0.61Zn0.42Fe1.97O4) nanoparticle concentration in the culture medium exceeded the relaxation rates of cells labeled by commercially available iron oxide nanoparticles, even with low metal content (0.13 pg Mn+0.42 pg Fe per cell in the case of Mn‐Zn ferrite nanoparticles, 14.6 pg Fe per cell for Endorem‐labeled cells).28 We also hypothesize that the stability of the silica coating may prevent the formation of larger nanoparticle clusters (causing a decrease in relaxivity), which cannot be excluded in the case of dissolving coating based on polysaccharides.
A higher relaxation rate can be attained using iron oxide‐based nanoparticles with modified coating or using transfection agents,32 which improve their uptake.24 However, a substantially higher iron content is also generated, which threatens viability by oxidative stress.33 Saccharide coating in particular may be metabolized, thus exposing the core to the cell environment.21 Silica coating, contrarily, represents an inert insoluble cover (see also TEM images in Figure 5B showing nanoparticles with intact coating inside the cells). Silica does not intensify the uptake, but represents an effective barrier between the core and the cellular environment.
The cells were successfully transplanted into the rat muscle, observed by both bioluminescence and MR imaging. The disappearance of bioluminescence over time probably corresponded to graft rejection, as no immunosuppression was used in the experiment. The signal disappeared in all implants including the unlabeled cells, which supports our hypothesis, that nanoparticle toxicity, which was also ruled out during extensive in vitro experiments, is not responsible for this effect.
Similar to the in vitro experiment, we observed lower bioluminescence in the case of cells labeled by higher concentrations of nanoparticles in the culture medium, which we explained by partial quenching of the signal by nanoparticles. Nevertheless, we also observed some variations in the bioluminescent signal of a rather stochastic nature during the experiment. We suppose that they reflect the different absorption rates of light in tissue due to the different depths of the implantation site. The signal intensity therefore may strongly depend on the actual position of the animal in the imager, which does not allow exact quantification of the surviving transplanted cells using the bioluminescence signal (see Figure ES8).
In vivo MR imaging detected grafts of the labeled cells in both T2‐weighted and T2*‐weighted MR images. Since the unlabeled control sample provided a similar signal to the target tissue, no contrast was observed in the area of the control graft.
The hypointense signal caused by the nanoparticles persisted throughout the whole experiment (28 days). This finding seemingly contradicts the optical imaging results, which proved that the grafts were rejected within one week after transplantation (see Figure ES7). Nevertheless, it should be acknowledged that MRI does not detect the transplanted cells themselves, but the magnetic label they carry. Therefore, MRI cannot respond to cell viability.34 We presume that the nanoparticles remained in the poorly vascularized muscle tissue (probably inside macrophages) even in the case of cell death.
4. Conclusions
Magnetic nanoparticles based on manganese‐zinc ferrites with silica coating can be used as safe cell labels and possess suitable physical and biochemical properties. Their high transverse relaxivity increases detection sensitivity and enables the decrease of nanoparticle concentrations during labeling, which may minimize possible adverse effects on cell viability. Both in vitro and in vivo experiments confirmed the safety and biocompatibility of the tested nanoparticles as well as their ease of use for cell labeling and cell tracking.
Experimental Section
Preparation of Magnetic Nanoparticles
Mn−Zn ferrite (MZF) nanoparticles were synthesized by hydrothermal procedure and subsequent encapsulation into silica18 Briefly, stock solutions of manganese(II) nitrate, zinc nitrate and iron(III) nitrate with chemically determined metal contents were used as starting materials. Appropriate amounts were combined in the molar ratio Mn : Zn : Fe=0.6 : 0.4 : 1.8 to reach a total metal content of 5.6 mmol. The solution was deoxygenated and the pH of the solution was adjusted to 10.0 by addition of ammonia under inert atmosphere, which led to the formation of brown precipitates. The mixture was transferred into a Berghof DAB‐2 pressure vessel and subjected to hydrothermal treatment under autogenous pressure at 180 °C for 12 h. The product was washed several times with water and then dried.
The MZF nanoparticles (150 mg) were treated with ice‐cold 1 M nitric acid in an ultrasound bath for 15 min and centrifuged before similar treatment with ice‐cold 0.1 M citric acid. After washing with water, the particles were redispersed in water (20 mL) alkalized with ammonia. The citrate‐stabilized particles were transferred to the mixture of ethanol (340 mL), water (70 mL) and ammonia (22 mL). Tetraethoxysilane (4.33 mL) was added under ultrasound agitation and mechanical stirring; the mixture was then stirred only mechanically at 60 °C for 4 h; then stirred without heating at room temperature overnight. The product was separated via centrifugation and washed with ethanol and water. Finally, size fractionation was carried out by repeated differential centrifugation 1,860 rcf for 15 minutes.
Fundamental Characterization of Magnetic Nanoparticles
The phase composition and crystal structure of bare nanoparticles and mean size of crystallites were studied by powder X‐ray diffraction with CuKα radiation. Bruker D8 diffractometer was employed and the diffraction pattern was analyzed by Rietveld method in the FullProf program. The mean size of crystallites was determined based on the analysis of peak broadening, for which the Thompson‐Cox‐Hastings pseudo‐Voigt profile was applied to separate the size and strain contribution. The instrumental contribution to the broadening was determined on a strain‐free tungsten powder with crystallite size of 9.4 μm.
The accurate ratio of metals in the bare sample was determined by X‐ray fluorescence spectroscopy on an Axios spectrometer.
Magnetic properties were measured by SQUID magnetometry in DC fields by using a Quantum Design MPMS XL system. The ZFC/FC studies were carried out in magnetic field of 1.6 kA/m.
The morphology and size of nanoparticles were analyzed by transmission electron microscopy on a Philips CM 120 system.
The colloidal stability of silica‐coated particles in aqueous suspension and their hydrodynamic size were probed by dynamic light scattering (DLS) on a Malvern Zetasizer Nano S. In addition, the zeta potential of the particles was measured on the same instrumentation by the laser Doppler electrophoresis.
Nanoparticle Relaxometry
Magnetic resonance T2 relaxation time measurements were carried out on a 0.5 T Bruker Minispec MQ20 relaxometer (Bruker BioSpin, Rheinstetten, Germany) in order to characterize the particles and their impact on the contrast of MR images. A standard Carr‐Purcell‐Meiboom‐Gill (CPMG) sequence (echospacing TE=2 ms, number of echoes varied between 500 and 2000, recovery time TR=5000 ms, 8 acquisitions, 2 dummy scans) was used for measuring the diluted nanoparticle suspension (concentration c=0.094 mM(Mn0.61Zn0.42Fe1.97O4)) within a temperature range of 5–71 °C.
Relaxivities r2 were calculated as reciprocal values of the relaxation times T2 related to molar concentration (c) of formula units after deducting the solvent contribution (T2 c°ntr°l) according to Equation (1):
| (1) |
Cells
The coated nanoparticles were tested using: rat mesenchymal stem cells (MSCs) isolated from bone marrow (BM‐MSCs); rat glioblastoma cells (cell line C6); and rat mesenchymal stem cells from the adipose tissue (AT‐MSCs) of genetically modified Lewis rats with ubiquitous expression of a gene for the luciferase enzyme (Lew‐Tg(Gt(ROSA)26Sor‐luc)11Jmsk, National BioResource Project – Rat, Kyoto, Japan).
BM‐MSCs from Wistar rats were isolated from rat bone marrow as described previously.30 Briefly, bone marrow was washed out from the femurs and tibias of 14‐day‐old rats and placed on a 10 cm2 Petri dish. The cells were cultivated in low‐glucose DMEM supplemented with 10 % of FBS, penicillin/streptomycin and Primocin. Nonadherent cells were removed after 72 hours by replacing the medium. After forming large colonies (>1 mm), cells were harvested and placed into the 75 cm2 cultivation flasks.
Rat glioma cell line C6 (ATCC® CCL‐107™) was obtained from Mr. Čestmír Altaner of the Institute of Experimental Oncology SA, Bratislava, Slovakia. Rat glioblastoma cells (cell line C6) were cultivated in high‐glucose DMEM supplemented with 10 % of FBS, non‐essential amino‐acids (NEAA) and 4 mM L‐Glutamin, penicillin/streptomycin and Primocin.
AT‐MSCs were isolated from visceral adipose tissue according to a standard protocol.34 Briefly, the tissue was digested by collagenase, filtered, over‐layered on 5 mL of Ficoll 1.077 g/mL (Ficoll‐Paque Premium, GE Healthcare Bio Sciences AB, Uppsala, Sweden) and spun. The cells in the interphase layer were collected by Pasteur pipette and seeded in a tissue culture flask, then grown to reach a sufficient amount. The cells were cultivated in DMEM‐low glucose media supplemented with 10 % FBS and 1 % penicillin/streptomycin/glutamine (all Thermo Fisher Scientific).
All cell cultures were maintained under standard conditions (37° C, 5 % CO2). Medium was replaced twice per week. When reaching 70–90 % confluence, cells were harvested using Trypsin/EDTA and seeded into the new cultivation flasks (split ratio 1 : 2).
Due to limited access to luciferase‐positive AT‐MSCs for the in vivo experiments, toxicity analysis was performed on BM‐MSCs and C6 cells. AT‐MSCs underwent viability tests and relaxometry only prior to transplantation.
Cells were routinely tested for Mycoplasma contamination.
Labeling
BM‐MSCs and C6 cells were redistributed into flasks (5×106 cells per 45 mL/180 cm2 flask) and labeled by adding an aqueous suspension of the nanoparticles at three concentrations: 0.05, 0.1, and 0.55 mM (Mn0.61Zn0.42Fe1.97O4). A control sample of cells cultured in the medium without the contrast agent was prepared simultaneously. If not stated otherwise, the cells were incubated for 48 hours (37 °C, 5 % CO2 atmosphere), after which the medium was poured out and the cells washed three times with phosphate buffered saline (PBS, formulation: Monopotassium phosphate 0.125 g, Sodium phosphate dibasic dodecahydrate 0.72 g, Sodium chloride 5 g and Potassium chloride 0,125 g in 500 g of purified water. pH=7.2) using 20 mL per flask. The cells were detached from the flask by adding 12.5 mL of 0.05 % trypsin (Sigma‐Aldrich, St. Louis, MO, USA). After incubation for 5 min, trypsinization was stopped by adding 25 mL of 10 % fetal bovine serum (Sigma‐Aldrich). The cells were collected and centrifuged at 1500 rpm for 5 min. The pellet was resuspended in PBS (20 mL) and centrifuged once more to remove residual contrast agent from the cultured medium. The cell pellet was subsequently resuspended in PBS.
AT‐MSCs used for the in vivo experiment were labeled similarly, with labeling concentrations of 0.05, 0.1 and 0.2 mM(Mn0.61Zn0.42Fe1.97O4). The labeling time was shortened to 24 hours to minimize the possible adverse effects. Modified formulation of PBS was used (Potassium dihydrogen phosphate 0.10 g, Sodium hydrogen phosphate dodecahydrate 1.45 g, Potassium chloride 0.10 g, Sodium chloride 4.00 g in 500 g of purified water, pH=7.4).
Cell Growth and Viability
BM‐MSCs and C6 cells were seeded in a 75 cm2 cultivation flask in definite amounts, after which nanoparticles in appropriate concentrations were added. The cells were subsequently cultivated for 48 hours, harvested and cell viability was determined using the trypan blue (Sigma‐Aldrich) exclusion test35 in a Burker chamber under a light microscope.
Cell viability was expressed as a ratio of the viable (unstained) cells to all cells (by percentage). To consider also possible loss of cells due to lower adherence, gain (percentage of harvested viable cells; harvested viable unlabeled cells represent 100 %) was calculated for all types of cells.
Real‐Time Cell Proliferation
Cell proliferation curves were obtained using the xCELLigence® RTCA DP instrument (ACEA Biosciences, San Diego, USA). The method is based on the measurement of electrical impedance, which increases during cell growth through the use of a special 16‐well microplate (E‐Plate) equipped with gold electrodes at the bottom. The presence of adherent cells impedes the flow of charge carriers in the electrolyte (cultivation media or saline). The actual impedance depends on cell shape and size, the number of cells and the attachment quality (https://www.aceabio.com/products/rtca‐dp/). Impedance is expressed as the Cell Index (dimensionless parameter). Briefly, 50 μL of cultivation media was added to each well and the background impedance measured. Next, cells (BM‐MSCs and C6) at a density of 20 000 per well were seeded and left to attach for 30 min. During the log phase (approximately 2 h after the beginning of the experiment), the appropriate amount of nanoparticles were added to 50 μL of the cultivation media to achieve a final concentration in the well (0.05, 0.11 and 0.55 mM(Mn0.61Zn0.42Fe1.97O4)). Cells were then cultivated under standard cultivation conditions. Impedance was recorded every 15 minutes for 48 hours, with each experiment performed in doublets.36
Apoptosis Detection
To determine the level of apoptosis, cells (both C6 and BM‐MSCs) were labeled with the appropriate amount of nanoparticles and incubated for 48 h. They were then harvested, fixed with 4 % formaldehyde, kept overnight at 4 °C and stained for TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) (APO‐DIRECT™ Kit, BD Biosciences, San Jose, CA, USA). dUTP was stained with FITC. Cells were stained according to the manufacturer's recommendations, including the positive cell control provided in the kit. All samples were then run on an Apogee A50‐Micro flow cytometer (Apogee Flow Systems, Hertfordshire, UK), equipped with blue, red and violet lasers. They were then compared to unlabeled cells (negative control) and to the positive control using FlowLogic™ Software (Inivai Technologies, Mentone, Australia).
Reactive Oxygen Species (ROS) Production in Labeled Cells
A time‐programmed experiment was designed to detect the production of reactive oxygen species in labeled cells. ROS manifestation was analyzed using the CellROX™ Deep Red Flow Cytometry Assay Kit (Life Technologies, Carlsbad, CA, USA). Cell‐permeable CellROX® Deep Red reagent is non‐fluorescent while in a reduced state, but exhibits strong fluorescence emission when oxidized in the presence of reactive radicals. The kit also includes the common inducer of ROS tert‐Butyl hydroperoxide (TBHP), the antioxidant N acetylcysteine (NAC) and the SYTOX® Blue Dead Cell stain.
Six flasks were seeded by BM‐MSCs in freshly prepared media and exposed to 0.11 mM(Mn0.61Zn0.42Fe1.97O4) for 3, 6, 12, 24, and 48 hours. A control contained untreated cells. All flasks were then harvested. Unlabeled cells were divided into an unstained cytometer setup sample, a stained negative control and a positive control with TBHP. With the exception of the cytometer setup sample, all samples were then incubated with 500 nM CellROX® Deep Red reagent. During the last 15 min, 1 μL (1 μM final concentration) of the SYTOX® Blue Dead Cell stain was added. All samples were immediately analyzed using the Apogee A50 Micro flow cytometer. The dead cell count and red fluorescence of the CellROX® Deep Red reagent were evaluated using FlowLogic™ software.
Transmission Electron Microscopy (TEM)
Internalization of silica‐coated MZF nanoparticles inside the cells was verified by TEM as described previously.21 The BM‐MSCs grown on coverslips were incubated with the nanoparticles for 48 h, fixed in 2.5 % glutaraldehyde in 0.1 M Sörensen's buffer for 48 h at 4 °C and stained with 1 % osmium tetraoxide in 0.1 M Sörensen's buffer for 2 h. The cells were then dehydrated in ethanol, immersed in propylene oxide and flat‐embedded in Epon 812 using gelatin capsules. After polymerization for 72 h at 60 °C, the coverslips were removed from cells embedded in polymer blocks by soaking in liquid nitrogen. Ultra‐thin sections of 60 nm were examined with a Philips Morgagni 268D transmission electron microscope (FEI Inc., Hillsboro, OR, USA).
In Vitro Bioluminescence Imaging (BLI)
Viability of the luciferase‐positive‐labeled cells (AT‐MSCs) later used for in vivo cell transplantation was also verified by bioluminescence imaging. Viable cells emit light in the presence of luciferin due to a photochemical reaction catalyzed by firefly luciferase. Optical images were acquired on an IVIS Lumina XR imager (PerkinElmer, USA). Test tubes containing 250×103 cells in 0.5 mL of media were scanned before and after the addition of 5 μl of D‐luciferin (30 mg/mL, MEDESA, Polička, Czech Republic). The exposure time of 1 min, an open aperture and an open emission filter were applied.
Metal Content Analysis
The content of metal ions in AT‐MSCs was determined by inductively coupled plasma mass spectrometry (ICP‐MS). Samples were transferred by concentrated nitric acid into teflon vessels for microwave decomposition (Uniclever BMI−Z, Plazmatronics, Poland). A mixture of 3 mL nitric acid and 1 mL hydrofluoric acid (both Suprapur, Merck, Darmstadt, Germany) was used for sample decomposition. The decomposed samples were transferred to a 50 mL volumetric flask and after appropriate dilution spiked with an internal standard solution (100Rh). Both the calibration solutions and the IS solution were prepared from solutions at a concentration of 1.000±0.002 g/L (Merck). Distilled and demineralized water (conductivity <0.1 μS/cm) (Millipore, Bedford, MA, USA) was used to prepare all solutions.
ICP‐MS measurements were performed using an Elan DRC‐e system (PerkinElmer, Concord, Canada) equipped with a concentric PTFE nebulizer, a cyclonic spray chamber, a high‐efficiency quartz torch, a dynamic reaction cell (DRC) for the elimination of spectral interferences and the Gilson 212 peristaltic pump. Monitored nuclides included 57Fe, 66Zn and 55Mn.
Measurement conditions were: Plasma power 1 100 W, sample uptake 1.2 mL/min, nebulizer Air Flow 0.75 L/min, plasma Air Flow 11 L/min, auxiliary Air Flow 1 L/min, dwell time 50 ms per nuclide, sweeps/reading 10, total acquisition time 20 s.
In Vitro MR Relaxometry
Labeled AT‐MSCs for in vitro MR relaxometry were fixed by 4 % formaldehyde and resuspended in 4 % porcine gelatin (Sigma‐Aldrich) to avoid sample sedimentation. Samples containing 50×103, 100×103, 200×103, 400×103 and 800×103 cells were prepared. T2 relaxation time measurements were carried out on a 0.5 T Bruker Minispec MQ20 relaxometer, a 1.0 T Bruker Minispec MQ40 relaxometer and a 4.7 T Bruker Biospec imager (all instruments manufactured by Bruker BioSpin, Rheinstetten, Germany) at room temperature. A standard CPMG sequence (TE=2 ms, number of echoes varied between 500 and 2000, TR=5000 ms, 8 acquisitions, 2 dummy scans) was used at 0.5 and 1 T. A similar CPMG sequence with different timing (TE=7.2 ms or 14.4 ms, TR=5000 ms, number of echoes 256) was used at 4.7 T due to hardware limitations.
Relaxation rates R2 of the labeled cells related to cell concentrations (s−1/(106 cells/mL)) were calculated as reciprocal values of the relaxation times T2 divided by the cell concentration (c cells) after deducting the contribution of the unlabeled cells (T2 c°ntr°l) according to Equation (2):
| (2) |
As the control unlabeled cells were fixed and mounted in the same way, deduction of 1/T2 c°ntr°l thus ensures elimination of the contribution of the unlabeled cells as well as of the gelatin with formaldehyde used for cell fixation.
In Vivo Cell Transplantation
As a proof of principle, the labeled cells were transplanted to two rats and imaged in vivo. Luciferase‐positive AT‐MSCs for in vivo transplantation were isolated and labeled as described previously. The suspension of 5×106 cells for each tested concentration of nanoparticles was concentrated to 200 μL before transplantation. Luciferase‐negative inbred Lewis rats (Velaz, Prague, Czech Republic) were used for the in vivo experiment. Hind legs were shaved and suspensions of unlabeled cells and cells labeled by nanoparticles at 0.05, 0.1 and 0.2 mM(Mn0.61Zn0.42Fe1.97O4) concentrations were injected using a syringe into the calf muscle under anesthesia induced by passive inhalation of isoflurane (Isofluran; Torrex Chiesi Pharma, Vienna, Austria).
Optical Imaging
Rats with transplanted bioluminescent cells were measured 1, 3 and 7 days after transplantation. The animals were anesthetized using isoflurane: 5 % concentration in air for induction, 1.5–3 % for maintenance. A cannula was inserted into the tail vein for intravenous administration of D‐luciferin dissolved in sterile PBS (a dose of 15 mg). Optical images were acquired on an IVIS Lumina XR imager (PerkinElmer, USA) with an exposure time of 1 min, an open aperture and an open emission filter. A photographic image was acquired for anatomical co‐registration of the signal. The rats were examined before administration of D‐luciferin, immediately after administration and then every 2 minutes for 10 min. The bioluminescent color‐coded images were superimposed on the photographic images and analyzed using a Living Image software package (PerkinElmer, USA). Signal intensity was assessed as photons per second per square centimeter per steradian (p/s/cm2/sr) from the area containing the transplant.
Magnetic Resonance Imaging
The animals were scanned using magnetic resonance imaging 1, 3, 7, 14, 21 and 28 days after transplantation.
The animals were anesthetized using isoflurane: 5 % concentration in air for induction, 1.5–3 % for maintenance. Passive inhalation was sufficient for the experiment. The animals were placed on a heated holder and fixed, and then scanned using a Bruker Biospec 4.7 T MR imager (Bruker BioSpin, Rheinstetten, Germany) equipped with a 7 cm resonator coil. The scanning protocol was as follows:
Gradient echo localizer for proper positioning of the subsequent imaging sequences.
T2‐weighted turbo spin echo sequence with fat saturation, effective TE=36 ms, TR=3200 ms, number of acquisitions NA=4, slice thickness 1 mm, field of view FOV=75×60 mm, matrix 256×256, transversal and coronal orientations.
T2*‐weighted gradient echo sequence, flip angle FA=30°, TE=4.6 ms, TR=80 ms, NA=32, slice thickness 1 mm, field of view FOV=75×60 mm, matrix 512×256, transversal orientation.
The whole protocol lasted 40 minutes.
All animal experimental protocols were approved by the Ethics Committee of the Institute for Clinical and Experimental Medicine and the Ministry of Health of the Czech Republic (No. 54/2017) in accordance with European Communities Council Directive 86/609/EEC. The laboratory was accredited to usage of experimental animals by the Ministry of Agriculture of the Czech Republic (No. 55386/2016‐MZE‐17214).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We are grateful to Petra Veselá from the Department of Microscopy (Head: Jan Malínský, PhD), Institute of Experimental Medicine, CAS, for her help with sample processing and TEM scan performance. The financial support of the Czech Science Foundation (project No. 16‐04340S), the Ministry of Health of the Czech Republic (Institute for Clinical and Experimental Medicine – IKEM, Project IN 00023001), the Ministry of Industry and Trade of the Czech Republic (grant project TIP FR‐TI3/521), the Ministry of Education and Youth of the Czech Republic (National Sustainability Project LO1309 and Czech‐BioImaging – LM2015062) is gratefully acknowledged. This paper was presented, in part, at the World Molecular Imaging Congress (September 13–16, 2017, Philadelphia, PA, USA) as an oral presentation with interim findings (Herynek V et al: Manganese‐zinc ferrites for cell labeling and imaging in vivo, control ID: 2723909). The abstract of the presentation was published in Mol. Imaging Biol. 2017, 19(Suppl 1): DOI: https://doi.org/10.1007/s11307‐017‐1138‐y. The actual paper, however, has never been published.
V. Herynek, K. Turnovcová, A. Gálisová, O. Kaman, D. Mareková, J. Koktan, M. Vosmanská, L. Kosinová, P. Jendelová, ChemistryOpen 2019, 8, 155.
References
- 1. Mahla R. S., Int J Cell Biol 2016, 6940283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liew C. G., Moore H., Ruban L., Shah N., Cosgrove K., Dunne M., Andrews P., Ann. Med. 2005, 37, 521–532. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Berebichez-Fridman R., Gomez-Garcia R., Granados-Montiel J., Berebichez-Fastlicht E., Olivos-Meza A., Granados J., Velasquillo C., Ibarra C., Stem Cells Int 2017, 2638305; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Chang C. A., Lawrence M. C., Naziruddin B., Curr Opin Organ Transplant 2017, 22, 437–443; [DOI] [PubMed] [Google Scholar]
- 3c. Yoshida Y., Yamanaka S., Circ. Res. 2017, 120, 1958–1968. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Connell J. J., Patrick P. S., Yu Y., Lythgoe M. F., Kalber T. L., Regener. Med. 2015, 10, 757–772; [DOI] [PubMed] [Google Scholar]
- 4b. Fu Y., Kedziorek D., Kraitchman D. L., J. Cardiovasc. Pharmacol. 2010, 3, 24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Bulte J. W., AJR Am. J. Roentgenol. 2009, 193, 314–325; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Toso C., Vallee J. P., Morel P., Ris F., Demuylder-Mischler S., Lepetit-Coiffe M., Marangon N., Saudek F., Shapiro A. M. James, Bosco D., Berney T., Am. J. Transplant. 2008, 8, 701–706. [DOI] [PubMed] [Google Scholar]
- 6. McNamara K., Tofail S. A. M., Adv. Phys. 2017, 2, 54–88. [Google Scholar]
- 7. Henning T. D., Saborowski O., Golovko D., Boddington S., Bauer J. S., Fu Y. J., Meier R., Pietsch H., Sennino B., McDonald D. M., Daldrup-Link H. E., Eur Radiol 2007, 17, 1226–1234. [DOI] [PubMed] [Google Scholar]
- 8. Woods M., Woessner D. E., Sherry A. D., Chem. Soc. Rev. 2006, 35, 500–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nicholls F. J., Ling W., Ferrauto G., Aime S., Modo M., Sci. Rep. 2015, 5, 14597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ruiz-Cabello J., Barnett B. P., Bottomley P. A., Bulte J. W., NMR Biomed. 2011, 24, 114–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shapiro E. M., Sharer K., Skrtic S., Koretsky A. P., Magn. Reson. Med. 2006, 55, 242–249. [DOI] [PubMed] [Google Scholar]
- 12. Stephen Z. R., Kievit F. M., Zhang M., Mater Today (Kidlington) 2011, 14, 330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu W., Wu Z., Yu T., Jiang C., Kim W. S., Sci. Technol. Adv. Mater. 2015, 16, 023501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pollert E., Kaman O., Veverka P., Veverka M., Marysko M., Zaveta K., Kacenka M., Lukes I., Jendelova P., Kaspar P., Burian M., Herynek V., Philos. Trans. R. Soc. London 2010, 368, 4389–4405. [DOI] [PubMed] [Google Scholar]
- 15. Joshi H. M., J. Nanopart. Res. 2013, 15, 1–19. [Google Scholar]
- 16. Veverka M., Veverka P., Kaman O., Lancok A., Zaveta K., Pollert E., Knizek K., Bohacek J., Benes M., Kaspar P., Duguet E., Vasseur S., Nanotechnology 2007, 18, 345704. [Google Scholar]
- 17. Veverka M., Zaveta K., Kaman O., Veverka P., Knizek K., Pollert E., Burian M., Kaspar P., J Phys D Appl Phys 2014, 47, 065503. [Google Scholar]
- 18.
- 18a. Kaman O., Kulickova J., Herynek V., Koktan J., Marysko M., Dedourkova T., Knizek K., Jirak Z., J. Magn. Magn. Mater. 2017, 427, 251–257. [Google Scholar]
- 18b.J. Stergar, Z. Jirák, P. Veverka, L. Kubíčková, T. Vrba, J. Kuličková, K. Knížek, F. Porcher, J. Kohout, O. Kaman, J. Magn. Magn. Mater 2018, in press, DOI: https://doi.org/10.1016/j.jmmm.2018.11.020.
- 19. Yildirimer L., Thanh N. T., Loizidou M., Seifalian A. M., Nano Today 2011, 6, 585–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Manke A., Wang L., Rojanasakul Y., BioMed Res. Int. 2013, 2013, 942916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Novotna B., Jendelova P., Kapcalova M., P. Rossner, Jr. , Turnovcova K., Bagryantseva Y., Babic M., Horak D., Sykova E., Toxicol. Lett. 2012, 210, 53–63. [DOI] [PubMed] [Google Scholar]
- 22. Yu M., Huang S. H., Yu K. J., Clyne A. M., Int. J. Mol. Sci. 2012, 13, 5554–5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang Y., Kohler N., Zhang M., Biomaterials 2002, 23, 1553–1561. [DOI] [PubMed] [Google Scholar]
- 24. Salatin S., Khosroushahi A. Yari, J. Cell. Mol. Med. 2017, 21, 1668–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Arbab A. S., Wilson L. B., Ashari P., Jordan E. K., Lewis B. K., Frank J. A., NMR Biomed. 2005, 18, 383–389. [DOI] [PubMed] [Google Scholar]
- 26. Malvindi M. A., De Matteis V., Galeone A., Brunetti V., Anyfantis G. C., Athanassiou A., Cingolani R., Pompa P. P., PLoS One 2014, 9, e85835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.
- 27a. Girgis E., Wahsh M. M. S., Othman A. G. M., Bandhu L., Rao K. V., Nanoscale Res. Lett. 2011, 6, 460; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27b. Kaman O., Dedourkova T., Koktan J., Kulickova J., Marysko M., Veverka P., Havelek R., Kralovec K., Turnovcova K., Jendelova P., Schrofel A., Svoboda L., J. Nanopart. Res. 2016, 18, 100; [Google Scholar]
- 27c. Novotna B., Turnovcova K., Veverka P., P. Rossner, Jr. , Bagryantseva Y., Herynek V., Zvatora P., Vosmanska M., Klementova M., Sykova E., Jendelova P., Nanotoxicology 2016, 10, 662–670. [DOI] [PubMed] [Google Scholar]
- 28. Lu J. J., Deng H. Y., Huang H. L., J. Magn. Magn. Mater. 2000, 209, 37–41. [Google Scholar]
- 29. Dedourkova T., Kaman O., Veverka P., Koktan J., Veverka M., Kulickova J., Jirak Z., Herynek V., IEEE T Magn 2015, 51, 5300804. [Google Scholar]
- 30. Jendelova P., Herynek V., De Croos J., Glogarova K., Andersson B., Hajek M., Sykova E., Magn. Reson. Med. 2003, 50, 767–776. [DOI] [PubMed] [Google Scholar]
- 31. Jang J. T., Nah H., Lee J. H., Moon S. H., Kim M. G., Cheon J., Angew. Chem. Int. Ed. 2009, 48, 1234–1238; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 1260–1264. [Google Scholar]
- 32.
- 32a. Babic M., Horak D., Trchova M., Jendelova P., Glogarova K., Lesny P., Herynek V., Hajek M., Sykova E., Bioconjugate Chem. 2008, 19, 740–750; [DOI] [PubMed] [Google Scholar]
- 32b. Horak D., Babic M., Jendelova P., Herynek V., Trchova M., Pientka Z., Pollert E., Hajek M., Sykova E., Bioconjugate Chem. 2007, 18, 635–644; [DOI] [PubMed] [Google Scholar]
- 32c. Kedziorek D. A., Kraitchman D. L., Methods Mol. Biol. 2010, 660, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pongrac I. M., Pavicic I., Milic M., Ahmed L. Brkic, Babic M., Horak D., Vrcek I. Vinkovic, Gajovic S., Int. J. Nanomed. 2016, 11, 1701–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fabryova E., Jirak D., Girman P., Zacharovova K., Galisova A., Saudek F., Kriz J., Transplant. Proc. 2014, 46, 1963–1966; [DOI] [PubMed] [Google Scholar]
- 35. Carvalho K. A., Cury C. C., Oliveira L., Cattaned R. I., Malvezzi M., Francisco J. C., Pachalok A., Olandoski M., Faria-Neto J. R., Guarita-Souza L. C., Transplant. Proc. 2008, 40, 839–841. [DOI] [PubMed] [Google Scholar]
- 36. Jirakova K., Seneklova M., Jirak D., Turnovcova K., Vosmanska M., Babic M., Horak D., Veverka P., Jendelova P., Int. J. Nanomed. 2016, 11, 6267–6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary