

Abstract
Lung cancer is a leading global cause of cancer‐related death, and lung adenocarcinoma (LUAD) accounts for ~ 50% of lung cancer. Here, we screened for novel and specific biomarkers of LUAD by searching for differentially expressed mRNAs (DEmRNAs) and microRNAs (DEmiRNAs) in LUAD patient expression data within The Cancer Genome Atlas (TCGA). The identified optimal diagnostic miRNA biomarkers were used to establish classification models (including support vector machine, decision tree, and random forest) to distinguish between LUAD and adjacent tissues. We then predicted the targets of identified optimal diagnostic miRNA biomarkers, functionally annotated these target genes, and performed receiver operating characteristic curve analysis of the respective DEmiRNA biomarkers, their target DEmRNAs, and combinations of DEmiRNA biomarkers. We validated the expression of selected DEmiRNA biomarkers by quantitative real‐time PCR (qRT‐PCR). In all, we identified a total of 13 DEmiRNAs, 2301 DEmRNAs and 232 DEmiRNA–target DEmRNA pairs between LUAD and adjacent tissues and selected nine DEmiRNAs (hsa‐mir‐486‐1, hsa‐mir‐486‐2, hsa‐mir‐153, hsa‐mir‐210, hsa‐mir‐9‐1, hsa‐mir‐9‐2, hsa‐mir‐9‐3, hsa‐mir‐577, and hsa‐mir‐4732) as optimal LUAD‐specific biomarkers with great diagnostic value. The predicted targets of these nine DEmiRNAs were significantly enriched in transcriptional misregulation in cancer and central carbon metabolism. Our qRT‐PCR results were generally consistent with our integrated analysis. In summary, our study identified nine DEmiRNAs that may serve as potential diagnostic biomarkers of LUAD. Functional annotation of their target DEmRNAs may provide information on their roles in LUAD.
Keywords: biomarker, DEmiRNA, LUAD, lung adenocarcinoma, microRNA, TCGA
Abbreviations
- AUC
area under the curve
- DEmiRNA
differentially expressed microRNA
- DEmRNA
differentially expressed mRNA
- DOR
diagnostic odds ratio
- FDR
false discovery rate
- GO
gene ontology
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LUAD
lung adenocarcinoma
- miRNA
microRNA
- NSCLC
non‐small‐cell lung cancer
- qRT‐PCR
quantitative real‐time PCR
- ROC
receiver operating characteristic
- SVM
support vector machine
- TCGA
The Cancer Genome Atlas
Lung cancer is the most frequently diagnosed cancer and a leading cause of cancer‐related death worldwide 1. Approximately 4/5 diagnosed lung cancers are non‐small‐cell lung cancer (NSCLC), and lung adenocarcinoma (LUAD) is the most common histological subtype of NSCLC, accounting for ~ 50% of lung cancer 2, 3. Due to the lack of effective diagnostic tools, most LUAD patients are diagnosed at the later stages, and the 5‐year survival rate of LUAD has shown no significant improvement over the past few decades 4. Hence, it is essential to identify the novel, non‐invasive, and specific biomarkers for the diagnosis of LUAD.
MicroRNAs (miRNAs) are a class of small non‐coding RNAs of 20–22 nucleotides that regulate gene expression at the post‐transcriptional level and participate in a variety of biological pathways. Aberrantly expressed miRNAs have been found to be involved in various cancers, including LUAD 5, breast cancer 6, gastrointestinal cancer 7, ovarian cancer 8, and esophageal cancer 9, acting as oncogenes or antioncogenes 10, 11. In accumulated studies, aberrantly expressed miRNAs have been found to have potential diagnostic value for LUAD. Some well‐characterized miRNAs function as a diagnostic or prognostic biomarker for LUAD, such as miRNA‐339‐5p, miRNA‐21, miRNA‐383, and miR‐7 3, 12, 13.
The Cancer Genome Atlas (TCGA) is a central bank for multidimensional experimental cancer data that contributes to uncovering the molecular mechanisms of cancer. In the present study, we used TCGA to obtain and further analyze the miRNA and mRNA expression data of LUAD patients. The key miRNAs with great diagnostic value were identified and their roles in LUAD were further revealed by investigating the function of their target genes.
Materials and methods
Integrated profiles in TCGA
We downloaded the clinical data of LUAD patients from TCGA (http://tcga-data.nci.nih.gov/). rsem‐normalized mRNA expression profiles (Level 3‐IlluminaHiseq_RNASeqV2 data) and miRNA expression profile (Level 3‐IlluminaHiSeq‐miRNASeq data) of LUAD and adjacent normal tissues were downloaded from TCGA data portal (http://tcga-data.nci.nih.gov/) as well. Only patients who were diagnosed as LUAD histologically were included in the study.
Identifying miRNAs and mRNAs to distinguish tumor from normal
Based on the read count of each sample, the differentially expressed miRNAs (DEmiRNAs) and mRNAs (DEmRNAs) in LUAD compared to normal tissues were calculated via the R‐bioconductor package deseq2 (http://bioconductor.org/packages/DESeq2/). We performed multiple comparisons by using the Benjamini and Hochberg method to obtain the false discovery rate (FDR). The threshold was FDR < 0.05 and |log2 fold change| > 4 for DEmiRNAs. In order to gain an overview of the characteristics of the miRNA expression profile, a heat‐map was further generated by hierarchical clustering analysis based on the normalized expression values of all DEmiRNAs using the r package (https://www.r-project.org/). For DEmRNAs, the threshold was defined as FDR < 0.05 and |log2 fold change| > 1.5.
Statistics for classification and prediction
To identify optimal diagnostic miRNA biomarkers for LUAD, we performed a feature selection procedures as follows. Firstly, the importance value of each DEmiRNA was ranked according to the mean decrease in accuracy using the random forest analysis. Then, the optimal number of features was found by subsequently adding one DEmiRNA at a time in a top‐down forward‐wrapper approach. The diagnostic odds ratio (DOR) is defined as the ratio of the odds of the positivity of a diagnostic test among a diseased population relative to that in the non‐diseased population. DOR was assessed by using the support vector machine (SVM) at each increment, and the optimal diagnostic miRNA biomarkers for LUAD were identified.
These optimal DEmiRNAs with diagnostic value for LUAD were used to establish classification models including decision tree, the SVM model, and random forest to better distinguish LUAD and adjacent tissues. The decision tree model was established by using the rpart package (https://cran.r-project.org/web/packages/rpart/). The SVM model was established by using the e1071 package in r, and the random forest model was established by using the ‘randomforest’ package (https://cran.r-project.org/web/packages/randomForest/). The three kinds of classification models were compared by the average misjudgment rates of their 10‐fold cross‐validations. Diagnostic ability of classification prediction was evaluated by obtaining the area under a receiver operating characteristic (ROC) curve (AUC) and DOR. A heat‐map of the optimal DEmiRNAs with diagnostic value for LUAD was generated by hierarchical clustering analysis by using the r package.
Identifying mRNA targets of miRNA
To uncover the potential roles of the identified optimal DEmiRNA diagnostic biomarkers, we predicted their targets. Considering the opposite trend in the expression of miRNA and their targets, we screened the significant negatively co‐expressed DEmiRNA–DEmRNA pairs by pairwise Pearson correlation coefficients. DEmiRNA–DEmRNA pairs with P < 0.05 and r < 0 were defined as significant negative co‐expression pairs. The putative targets of DEmiRNAs were predicted by six bioinformatic algorithms (RNA22, miRanda, miRDB, miRWalk, PICTAR2 and Targetscan) of mirwalk2.0 (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/mir-mir-self.html). The targets recorded by more than four algorithms were served as target mRNAs of miRNAs. Moreover, the confirmed targets of miRNAs obtained by mirwalk2.0 were served as target mRNAs of miRNAs as well. Finally, significant negatively co‐expressed DEmiRNA–DEmRNA pairs overlapped with miRNA–target mRNA pairs were used to construct the DEmiRNA–DEmRNA interaction network by using cytoscape software (http://www.cytoscape.org/).
Functional annotation of miRNA targets
To uncover the biological functions and detect the potential pathways of target DEmRNAs of DEmiRNAs, the online software genecodis was used to perform the functional annotation, including gene ontology (GO) classification (molecular functions, biological processes, and cellular component) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment. Statistical significance was defined as FDR < 0.05.
Confirmation of differentially expressed miRNAs and mRNAs
Ten LUAD tumor tissues and 10 normal adjacent tissues were obtained from 10 patients (P1–P10) who were diagnosed as LUAD based on pathological analysis. Patient characteristics are displayed in Table 1. We obtained written informed consent from the patients and approval from the ethics committee of Chinese PLA General Hospital. The study conformed to the Declarations of Helsinki. Total RNA was extracted with the TRIzol reagent (Invitrogen, Shanghai, China). One microgram RNA was used to synthesize cDNA using SuperScript® III Reverse Transcriptase (Invitrogen). Quantitative real‐time PCR (qRT‐PCR) was performed with Power SYBR® Green PCR Master Mix (Applied Biosystems, Carlsbad, CA, USA) in the ABI7500 real‐time PCR system (Applied Biosystems). And the reverse transcriptions of miRNAs were performed using the miScript II RT Kit (Qiagen, Hilden, Germany). qRT‐PCR was performed with miScript SYBR Green PCR Kit (Qiagen). Relative gene expression was analyzed using the method. Statistical significance was assessed by one‐way ANOVA. The human 18S rRNA and U6 were used as endogenous controls for mRNA and miRNA expression in the analysis.
Table 1.
Patient characteristics. P1–P10, patient 1–patient 10 with LUAD
| Index | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Age (years) | 48 | 60 | 59 | 64 | 74 | 69 | 46 | 54 | 66 | 31 |
| Male/female | Male | Female | Female | Female | Male | Male | Male | Male | Female | Male |
| BMI | 24.3 | 18.3 | 25.1 | 26.6 | 21 | 22.3 | 23.7 | 31.1 | 24.4 | 19.7 |
| TNM stage | T1BN0M0 | T1AN0M0 | T1BN0M0 | T2AN0M0 | T2AN0M0 | T2AN2M0 | T1AN0M0 | T2AN0M0 | T1AN0M0 | T1N2M0 |
| Family history | No | No | No | No | No | No | No | No | No | No |
| History of other cancers | No | No | No | No | No | No | No | No | No | No |
Results
Identification of differentially expressed miRNAs and mRNAs between LUAD and adjacent tissues
The mRNA expression profiles of 533 patients with LUAD and 59 normal adjacent tissues were obtained. The median age of these 533 patients was 67 years. Female and male patients account for 53.7% and 46.3% of the population, respectively. The information of the TNM stage was as follows: T1N0M0–T1N2M1 (32.8%), T2N0M0–T2N2M1 (53.8%), T3N0M0–T3N2M1 (9.2%) and T4N0M0–T1N3M1 (3.6%). A total of 19 450 mRNAs were detected. mRNAs with read count value = 0 in more than 20% (n = 107) of tumors or in more than 20% (n = 12) of adjacent tissues were considered to be low/not expressed mRNAs.
The miRNA expression profiles of 519 patients with LUAD and 46 normal adjacent tissues were obtained. The median age of these 519 patients was 67 years. Female and male patients account for 53.6% and 46.4% of the population, respectively. The information of the TNM stage was as follows: T1N0M0–T1N2M1 (33.3%), T2N0M0–T2N2M1 (53.0%), T3N0M0–T3N2M1 (9.4%) and T4N0M0–T1N3M1 (3.7%). A total of 1881 miRNAs were detected. miRNAs with read count value = 0 in more than 60% (n = 311) of tumors or in more than 60% (n = 28) of adjacent tissues were considered to be low/not expressed miRNAs.
After filtering the low/not expressed mRNAs or miRNAs, 16 043 mRNAs and 559 miRNAs were retained for analysis, respectively. A total of 13 DEmiRNAs (three down‐regulated and 10 up‐regulated miRNAs) between LUAD and normal tissues were identified with FDR < 0.05 and |log2 fold change| > 4. A total of 2301 DEmRNAs (933 down‐regulated and 1368 up‐regulated mRNAs) between LUAD and normal tissues were identified with FDR < 0.05 and |log2 fold change| > 1.5. Hierarchical clustering analysis of DEmiRNAs and the top 100 DEmRNAs is displayed in Fig. 1A and B, respectively.
Figure 1.
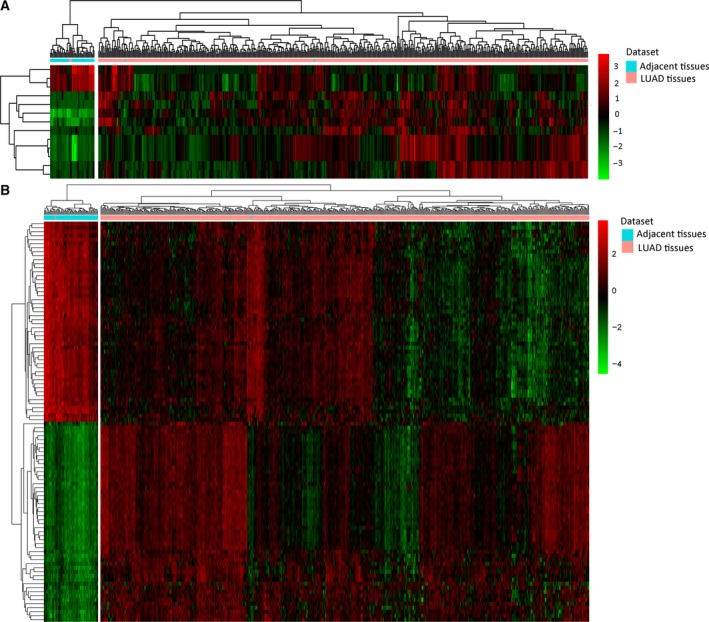
Hierarchical clustering analysis of DEmiRNAs and the top 100 DEmRNAs between LUAD and adjacent tissues. (A) DEmiRNAs; (B) the top 100 DEmRNAs. Rows and columns represent DEmiRNAs/DEmRNAs and tissue samples. The color scale represents the expression levels.
Identification of optimal diagnostic miRNA biomarkers for LUAD
To identify the optimal diagnostic miRNA biomarkers for LUAD, we performed random forest feature selection and classification (SVM, decision tree and random forest) procedures. All DEmiRNAs were ranked according to the standardized drop in prediction accuracy (Fig. 2A). Then, we compared the DOR increment for a specific number of miRNAs by subsequently adding one miRNA at a time in a top‐down forward‐wrapper approach. We found that the DOR of nine DEmiRNAs reached the highest point for the first time (Fig. 2B). Hence, these nine DEmiRNAs were defined as the optimal diagnostic miRNA biomarkers for LUAD (Table 2).
Figure 2.
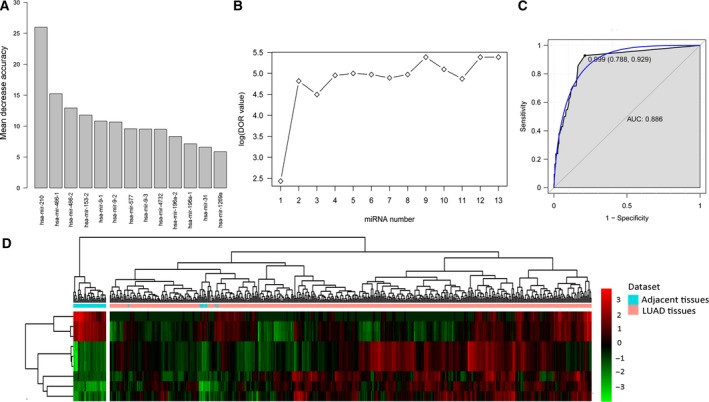
Identification of miRNA biomarkers for LUAD. (A) The variance rate of prediction accuracy for each DEmiRNA. (B) The variance rate of classification performance when increasing the number of the predictive miRNAs. (C) ROC results of combinations of nine miRNA biomarkers based on random forest classification model. (D) Hierarchical clustering analysis of the nine miRNA biomarkers between LUAD and adjacent tissues. Rows and columns represent DEmiRNAs and tissue samples. The color scale represents the expression levels.
Table 2.
The nine optimal diagnostic miRNA biomarkers for LUAD. Data were analyzed by Wald test
| miRNA | Log2 fold change | P‐value | FDR | Regulation |
|---|---|---|---|---|
| hsa‐mir‐210 | 4.536466384 | 1.20E‐97 | 3.36E‐95 | Up |
| hsa‐mir‐486‐1 | −4.277102805 | 3.47E‐84 | 6.47E‐82 | Down |
| hsa‐mir‐486‐2 | −4.268035702 | 2.17E‐83 | 3.03E‐81 | Down |
| hsa‐mir‐153‐2 | 4.204883454 | 1.11E‐52 | 3.26E‐51 | Up |
| hsa‐mir‐9‐1 | 5.296508956 | 9.75E‐63 | 4.19E‐61 | Up |
| hsa‐mir‐9‐2 | 5.303453252 | 7.22E‐63 | 3.67E‐61 | Up |
| hsa‐mir‐577 | 5.026458962 | 1.40E‐39 | 2.11E‐38 | Up |
| hsa‐mir‐9‐3 | 5.303960101 | 2.77E‐63 | 1.55E‐61 | Up |
| hsa‐mir‐4732 | −4.357779935 | 2.26E‐61 | 9.03E‐60 | Down |
These nine optimal DEmiRNAs with diagnostic value for LUAD were used to establish classification models including decision tree, SVM and random forest. The 10‐fold cross‐validation indicated that the misjudgment rate of SVM, decision tree and random forest was 5.12%, 1.59% and 1.42%, respectively. This result suggested that the random forest model, with the smallest average misjudgment rate, could effectively predict LUAD. Based on the classification model of random forest, the DOR of these nine DEmiRNAs was 7735.1; the ROC curves are displayed in Fig. 2C and hierarchical clustering analysis of the nine DEmiRNAs is displayed in Fig. 2D.
miRNA–mRNA interactions in LUAD
To better understand the role of these nine optimal DEmiRNAs in LUAD, their potential target was also analyzed. According to the miRNA–mRNA expression correlation analysis, we obtained 3341 DEmiRNA–DEmRNA pairs that were negatively correlated (P < 0.05, r < 0). Moreover, we obtained 409 miRNA–target mRNA pairs based on miRWalk including 362 miRNA–target mRNA pairs predicted by more than four algorithms and 47 confirmed miRNA–target mRNA pairs. After screening the overlapped miRNA–mRNA pairs among both these 409 miRNA–target mRNA pairs and 3341 negative DEmiRNA–DEmRNA co‐expression pairs, we obtained 232 DEmiRNA–target DEmRNA pairs including 116 DEmRNAs (70 up‐regulated and 46 down‐regulated DEmRNAs) and nine DEmiRNAs (six up‐regulated and three down‐regulated DEmiRNAs). Based on the DEmiRNA–target DEmRNA interaction network, hsa‐mir‐486‐1 (degree = 63) and hsa‐mir‐486‐2 (degree = 63) were two hub DEmiRNAs (Fig. 3).
Figure 3.
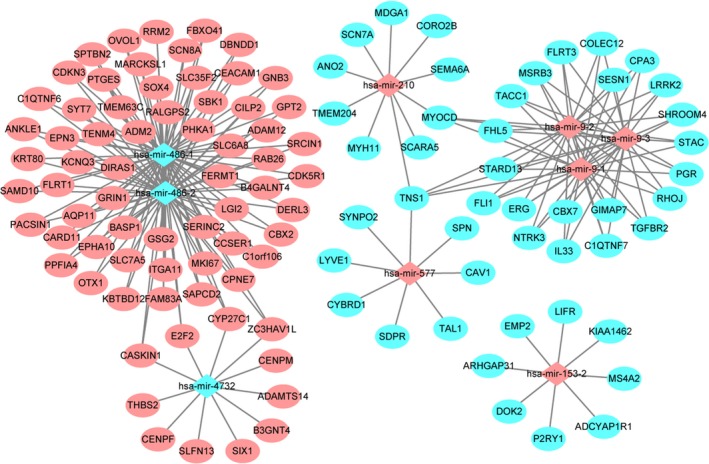
LUAD‐specific DEmiRNA–DEmRNA interaction network. The ellipses and rhombuses represent the DEmRNAs and DEmiRNAs, respectively. Red and blue represent up‐ and down‐regulation, respectively.
Functional enrichment analysis of miRNA targets
The 116 target DEmRNAs of the DEmiRNAs were used to conduct the GO and KEGG enrichment analysis. GO enrichment analysis (Table 3) revealed that the miRNA targets were significantly enriched in circulatory system development (P < 0.05), plasma membrane region (P < 0.05), caveola (P < 0.05) and regulation of synapse assembly (P < 0.05). KEGG pathway enrichment analysis (Table 3) showed that transcriptional misregulation in cancer (P‐value < 0.05) and central carbon metabolism in cancer (P‐value < 0.05) were two significantly enriched pathways.
Table 3.
Functional annotation of targeted DEmRNAs of DEmiRNAs. Data were analyzed using the standard accumulative hypergeometric statistical test. BP, biological processes; CC, cellular components; MF, molecular functions
| Category | ID | Description | Log (P‐value) | Symbols |
|---|---|---|---|---|
| GO (BP) | GO:0072359 | Circulatory system development | −6.703660169 | CEACAM1, CAV1, EMP2, ERG, MYH11, NTRK3, SIX1, SOX4, TAL1, TGFBR2, THBS2, BASP1, FLRT3, TENM4, RHOJ, TMEM204, ADM2, STARD13, MYOCD, GRIN1, CDK5R1, SRCIN1, LRRK2, ADAM12, TNS1, FERMT1, IL33 |
| GO (CC) | GO:0098590 | Plasma membrane region | −5.861204601 | ADCYAP1R1, CEACAM1, CAV1, EMP2, GRIN1, P2RY1, SPTBN2, TGFBR2, SLC7A5, SDPR, SYT7, FLRT3, PACSIN1, FERMT1, SHROOM4, CYBRD1, SRCIN1, LRRK2 |
| GO (CC) | GO:0005901 | Caveola | −5.706471821 | ADCYAP1R1, CAV1, EMP2, TGFBR2, SDPR, LRRK2, CDK5R1, GSG2, SBK1, CEACAM1, DOK2, SRCIN1, FAM83A, DIRAS1, EPN3, CARD11, NTRK3, P2RY1, SPN, SERINC2, PHKA1, EPHA10 |
| GO (BP) | GO:0051963 | Regulation of synapse assembly | −5.440592972 | GRIN1, NTRK3, SIX1, THBS2, FLRT3, FLRT1, CEACAM1, CAV1, TNS1, ITGA11, FERMT1, ARHGAP31, KIAA1462, TMEM204, SRCIN1, SYNPO2, SPTBN2, LRRK2, CDKN3, EMP2, TGFBR2, CDK5R1, MYOCD, DIRAS1, SERINC2, LIFR, DOK2, FAM83A, EPHA10, BASP1, TENM4, SOX4, TAL1, SPN, ADAMTS14, CILP2, P2RY1, PACSIN1 |
| GO (CC) | GO:0043005 | Neuron projection | −5.242350102 | ADCYAP1R1, GNB3, GRIN1, KCNQ3, P2RY1, SCN8A, CDK5R1, SYT7, BASP1, FLRT3, FLRT1, TENM4, PACSIN1, SEMA6A, SRCIN1, LRRK2, AQP11, NTRK3, SPTBN2, DOK2, TAL1, RHOJ, SHROOM4, CEACAM1, CENPF, FERMT1, CYBRD1, OTX1, SIX1, THBS2, TACC1, ADM2, SPN, CAV1 |
| GO (BP) | GO:0007417 | Central nervous system development | −4.874607656 | CENPF, GRIN1, NTRK3, OTX1, SIX1, SOX4, SPTBN2, TACC1, TAL1, TGFBR2, CDK5R1, BASP1, TENM4, SHROOM4, LRRK2, MDGA1, FLRT3 |
| GO (BP) | GO:0001570 | Vasculogenesis | −4.476808317 | CEACAM1, CAV1, EMP2, TGFBR2, MYOCD, MYH11, P2RY1, SCN7A, SLC6A8, STAC, CDKN3, OVOL1, SOX4, SPN, PTGES, ADM2, SIX1, BASP1, TMEM204, LRRK2 |
| GO (CC) | GO:0044306 | Neuron projection terminus | −4.379028359 | GRIN1, SYT7, FLRT3, FLRT1, PACSIN1, LRRK2, PPFIA4, RAB26, P2RY1, CDK5R1, SRCIN1, CEACAM1, CPA3, IL33, SLC6A8, EMP2 |
| GO (BP) | GO:0072001 | Renal system development | −4.023285604 | CENPF, OVOL1, SIX1, SOX4, BASP1, MYOCD, LRRK2, AQP11, ERG, MYH11, NTRK3, TGFBR2, FLRT3, TENM4, CAV1, ADAM12, ITGA11, TMEM204, CDK5R1, STARD13, CEACAM1 |
| GO (CC) | GO:0009986 | Cell surface | −4.021192798 | ADCYAP1R1, CEACAM1, EMP2, MS4A2, GRIN1, KCNQ3, P2RY1, SPN, TGFBR2, TNS1, PPFIA4, AQP11, SCARA5, CAV1, MKI67, NTRK3, PGR, SIX1, PTGES, COLEC12, LRRK2, SCN7A, CYBRD1, SOX4, RRM2, C1QTNF6 |
| GO (MF) | GO:0003779 | Actin binding | −3.668606032 | CEACAM1, MYH11, SPTBN2, TNS1, CORO2B, SHROOM4, MARCKSL1, LRRK2, SYNPO2, CAV1, EMP2, NTRK3, CDK5R1, PACSIN1, RHOJ, CENPF, MS4A2, DOK2, ITGA11, ADM2, GNB3, COLEC12, FERMT1, SRCIN1, ADAMTS14 |
| GO (CC) | GO:0043235 | Receptor complex | −3.62673173 | ADCYAP1R1, CEACAM1, GRIN1, LIFR, NTRK3, TGFBR2, ITGA11, CARD11, SPN, IL33, EMP2, CAV1, SIX1, SOX4, MARCKSL1, MYOCD, FLI1, TAL1, ANKLE1, SLC7A5, DOK2, CDK5R1, SRCIN1 |
| GO (BP) | GO:0042063 | Gliogenesis | −3.613567247 | NTRK3, P2RY1, SOX4, TAL1, CDK5R1, TENM4, IL33, SIX1 |
| GO (MF) | GO:0001077 | Transcriptional activator activity, RNA polymerase II core promoter proximal region sequence‐specific | −3.476014856 | ERG, FLI1, OTX1, PGR, SIX1, SOX4, MYOCD, OVOL1, E2F2, TAL1, BASP1, CEACAM1, CAV1, TGFBR2, SEMA6A, LRRK2, CENPF, CBX2, ANKLE1 |
| GO (MF) | GO:0005516 | Calmodulin binding | −3.378339299 | GRIN1, KCNQ3, MYH11, PHKA1, SYT7, MARCKSL1 |
| GO (BP) | GO:0051051 | Negative regulation of transport | −3.338324909 | CEACAM1, CAV1, P2RY1, SOX4, PACSIN1, SRCIN1, IL33, DERL3, LRRK2, SYT7, RAB26, CARD11 |
| GO (BP) | GO:0003179 | Heart valve morphogenesis | −3.127090085 | ERG, SOX4, TGFBR2, TAL1, MDGA1, SIX1, MYOCD, E2F2, ANKLE1, OTX1, FLRT3, TENM4, CEACAM1, STARD13, MKI67 |
| GO (BP) | GO:1903034 | Regulation of response to wounding | −3.000153384 | CEACAM1, CAV1, NTRK3, TGFBR2, SCARA5, ERG, SYT7, P2RY1, AQP11, LYVE1, FLRT3, PACSIN1, COLEC12, GNB3, SIX1, CDK5R1, SESN1, LRRK2, SOX4, GRIN1, SPN, IL33, RAB26, FLRT1, FLI1, SPTBN2, DIRAS1, EMP2 |
| GO (BP) | GO:0048013 | Ephrin receptor signaling pathway | −2.989818807 | GRIN1, NTRK3, CDK5R1, EPHA10, TGFBR2, ADCYAP1R1, LIFR, MDGA1 |
| GO (BP) | GO:0051899 | Membrane depolarization | −2.798135634 | CAV1, SCN7A, SCN8A, LRRK2, ADCYAP1R1, GRIN1, KCNQ3, P2RY1, STAC, SYT7, ANO2, TMEM63C, CEACAM1, SLC6A8, SLC7A5, SERINC2, SCARA5 |
| KEGG pathway | ko04713 | Circadian entrainment | −1.885359167 | ADCYAP1R1, GNB3, GRIN1 |
| KEGG pathway | hsa05202 | Transcriptional misregulation in cancer | −1.867247226 | ERG, FLI1, SIX1, TGFBR2 |
| KEGG pathway | ko05030 | Cocaine addiction | −1.587657877 | GRIN1, CDK5R1 |
| KEGG pathway | hsa05212 | Pancreatic cancer | −1.374868302 | E2F2, TGFBR2 |
| KEGG pathway | hsa05230 | Central carbon metabolism in cancer | −1.362710373 | NTRK3, SLC7A5 |
| KEGG pathway | hsa04115 | p53 signaling pathway | −1.316099311 | RRM2, SESN1 |
ROC analyses
By using the proc package in the r language, we performed ROC analyses to assess the diagnostic value of DEmiRNAs and their targets. The AUC under binomial exact confidence interval was calculated and a ROC curve was generated.
ROC curve analysis
We performed ROC curve analyses and calculated the AUC to assess the discriminatory ability of these nine DEmiRNAs and six differentially expressed targets between LUAD and adjacent tissues. The expression levels of these nine DEmiRNAs and six differentially expressed targets between LUAD and adjacent tissues are displayed in Fig. 4. The AUC of nine DEmiRNAs and six targets including hsa‐mir‐210 (0.988), hsa‐mir‐9‐1 (0.949), hsa‐mir‐9‐2 (0.949), hsa‐mir‐9‐3 (0.949), hsa‐mir‐153‐2 (0.962), hsa‐mir‐486‐1 (0.944), hsa‐mir‐486‐2 (0.943), hsa‐mir‐577 (0.915), hsa‐mir‐4732 (0.894), FLI1 (0.892), NTRK3 (0.877), SLC7A5 (0.919), TGFBR2 (0.907), ERG (0.924) and SIX1 (0.850) was more than 0.85 (Fig. 5).
Figure 4.
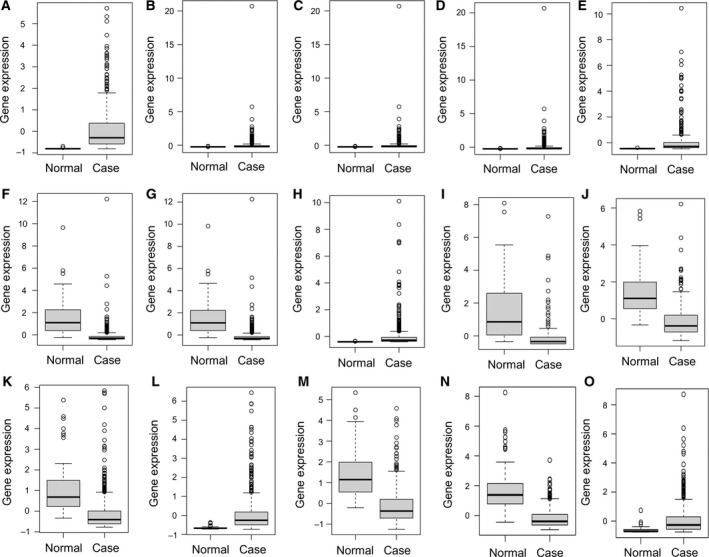
The expression levels of DEmiRNAs and DEmRNAs between LUAD and adjacent tissues. The x‐axes represent normal (LUAD) and control (adjacent tissues) groups. The y‐axes represent gene expression levels. (A) hsa‐mir‐210, (B) hsa‐miR‐9‐1, (C) hsa‐miR‐9‐2, (D) hsa‐miR‐9‐3, (E) hsa‐mir‐153‐2, (F) hsa‐mir‐486‐1, (G) hsa‐mir‐486‐2, (H) hsa‐mir‐577, (I) hsa‐mir‐4732, (J) FLI1, (K) NTRK3, (L) SLC7A5, (M) TGFBR2, (N) ERG and (O) SIX1.
Figure 5.
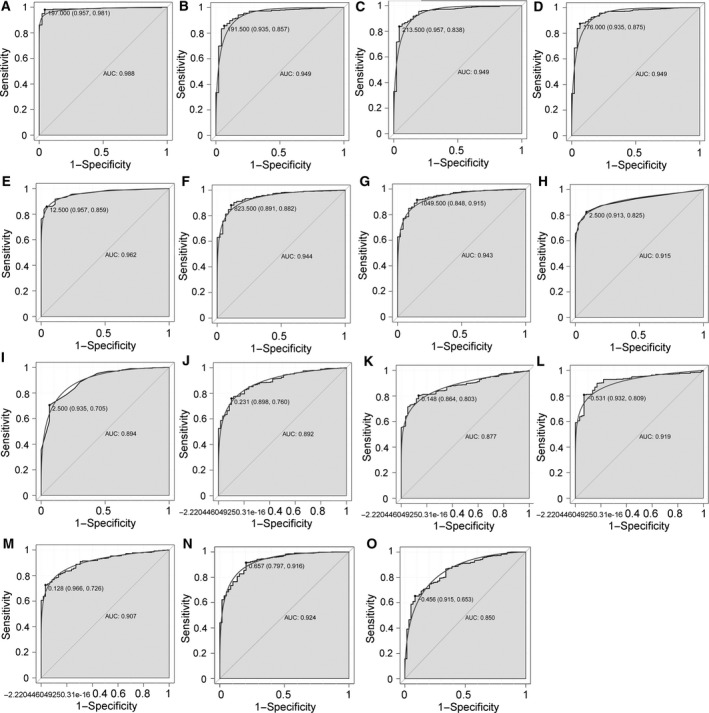
The ROC curves of selected DEmiRNAs and DEmRNAs between LUAD and adjacent tissues. The x‐axes show 1 − specificity (the proportion of false positives) and y‐axes show sensitivity (the proportion of true positives). (A) hsa‐mir‐210, (B) hsa‐miR‐9‐1, (C) hsa‐miR‐9‐2, (D) hsa‐miR‐9‐3, (E) hsa‐mir‐153‐2, (F) hsa‐mir‐486‐1, (G) hsa‐mir‐486‐2, (H) hsa‐mir‐577, (I) hsa‐mir‐4732, (J) FLI1, (K) NTRK3, (L) SLC7A5, (M) TGFBR2, (N) ERG and (O) SIX1.
Confirmation of differentially expressed miRNAs and mRNAs
Quantitative real‐time‐PCR of 10 pairs of LUAD and the adjacent tissues were used to verify the expression of five DEmiRNAs (hsa‐mir‐210‐3p, hsa‐mir‐486‐3p, hsa‐mir‐153‐3p, hsa‐mir‐9‐5p, hsa‐mir‐577). Based on TCGA, hsa‐mir‐486‐3p was down‐regulated while the other four DEmiRNAs were up‐regulated in LUAD compared to adjacent tissues. According to the qRT‐PCR results, all the five DEmiRNAs were up‐regulated, which was consistent with the results for TCGA, generally (Fig. 6).
Figure 6.
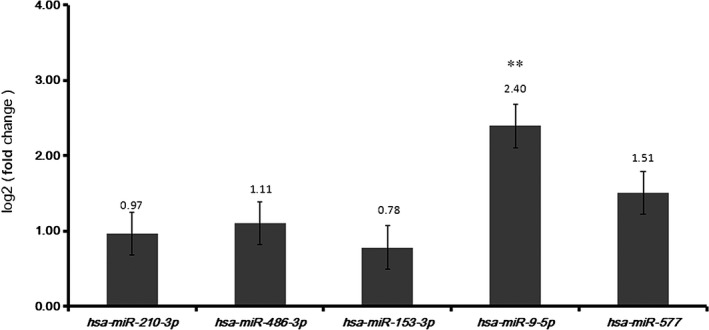
qRT‐PCR results for DEmiRNAs between LUAD and adjacent tissues. Statistical significance was assessed by one‐way ANOVA. The error bars indicate SD. n = 3. The x‐axis and y‐axis represent DEmiRNAs and log fold change, respectively. **P < 0.01.
Discussion
Lung adenocarcinoma is the most frequent subtype of lung cancer, with high global incidence and mortality 14. It is essential to explore accurate and specific biomarkers for it. Accumulated evidence indicates that miRNAs play crucial roles in the progress of LUAD. However, research into the diagnostic and prognostic value of miRNAs is currently in its infancy.
In this study, we performed genome‐wide analysis of miRNAs and mRNAs in a large number of patients with LUAD from TCGA. Altered miRNA expression patterns were found between LUAD and adjacent tissues, suggesting a potential diagnostic role for miRNA for LUAD. To identify the optimal LUAD‐specific miRNA biomarkers, we searched for combinations of miRNAs among the 13 DEmiRNAs whose expression pattern may distinguish LUAD from adjacent tissues with the highest DOR by using random forest feature selection. A nine‐miRNA combination including three down‐regulated miRNAs (hsa‐mir‐486‐1, hsa‐mir‐486‐2, and hsa‐mir‐4732) and six up‐regulated miRNAs (hsa‐mir‐210, hsa‐mir‐153‐2, hsa‐mir‐9‐1, hsa‐mir‐9‐2, hsa‐mir‐577, and hsa‐mir‐9‐3) were served as optimal LUAD‐specific miRNA biomarkers. The 10‐fold cross‐validation of three kinds of models (decision tree, SVM, and random forest) suggested that the random forest model, with the smallest average misjudgment rate of 1.42%, could effectively predict LUAD. Moreover, the AUC of all these nine DEmiRNAs in LUAD was more than 0.85. Taken together, our study has demonstrated the feasibility and potential diagnostic value of these nine miRNA biomarkers in LUAD.
Among them, hsa‐mir‐486‐1 and hsa‐mir‐486‐2 were two hub DEmiRNAs based on the DEmiRNA–DEmRNA interaction network, suggesting their importance in LUAD. Down‐regulation of mir‐486 has been demonstrated to be involved in various cancers including LUAD 15, 16, 17. Moreover, mir‐486 was reported to be cytotoxic in A549 LUAD cells by repressing the expression of bone morphogenetic protein‐2 reporter gene 18. Hsa‐mir‐153 was found to be down‐regulated in NSCLC tissues and cell lines; it inhibits migration and invasion of NSCLC by targeting ADAM19 19 and protein kinase B (AKT) 20. These three miRNAs were speculated to serve as tumor suppressors in LUAD.
The other six DEmiRNAs were all up‐regulated in LUAD compared to adjacent tissue based on our study, which suggested their potential oncogenic roles in LUAD. Hsa‐mir‐210 was the most up‐regulated miRNA in LUAD compared to adjacent tissue based on our study, which was consistent with previous studies 21, 22. Moreover, up‐regulation of hsa‐mir‐210 was found to be closely associated with distant metastases of LUAD 21. Both hsa‐mir‐9 and hsa‐mir‐577 were also found to be up‐regulated in human NSCLC tissues and cell lines compared to normal lung tissues 16, 23, 24. Hsa‐mir‐9 was reported to play an oncogenic role in the proliferation, invasion, and migration of NSCLC cells through mechanisms such as regulating SOX7 23, eukaryotic translation initiation factor 5A2 25, FoxO1 26, epithelial‐to‐mesenchymal transition and the signal transduction pathway 27. Hsa‐mir‐577 could play a role in the cell proliferation of esophageal squamous cell carcinomas and glioblastoma multiforme by regulating a tumor‐associated antigen, testis specific 10 28, and the Wnt signaling pathway 29, respectively. However, the precise role of mir‐577 in LUAD remains unknown.
Although there is no study that reports an association between hsa‐mir‐4732 and LUAD, hsa‐mir‐4732 was indicated to be associated with breast cancer 30. Our study is the first to report the down‐regulation of hsa‐mir‐4732 in LUAD compared to adjacent tissues.
To deeply research the biological functions of these DEmiRNAs in LUAD, we conducted a functional annotation of the target DEmRNAs of these DEmiRNAs. Two significantly enriched pathways in LUAD and their related DEmRNAs, namely transcriptional misregulation in cancer (TGFBR2, FLI1, ERG and SIX1) and central carbon metabolism (NTRK3 and SLC7A5), were speculated to play key roles in LUAD regulated by DEmiRNAs. TGFBR2 was a putative tumor suppressor gene in the TGF‐β signaling pathway. Loss of Tgfbr2 in a K‐ras‐induced LUAD mouse model has been found to induce a highly invasive phenotype associated with lymph node metastasis and reduced survival. In our study, TGFBR2 was down‐regulated in LUAD tissues compared to adjacent tissues, which was in agreement with previous study. Both FLI1 and ERG are members of erythroblast transformation‐specific oncogene family. FLI1 was down‐regulated in squamous lung cancer 31 and could up‐regulate TGFBR2 in LUAD and might be a potential regulator of LUAD 32. Overexpressed ERG variant 2 was found in lung tumors compared to adjacent tissues, which suggested that ERG might exert an oncogenic effect in lung tumors through functions encoded by variant 2 33. Aberrantly methylated NTRK3 has been demonstrated to be involved in various cancers including colorectal cancer and lung cancer 34, 35. Poor survival in lung cancer was closely associated with NTRK3 36. TGFBR2, FLI1, ERG and NTRK3 were shared DEmRNAs of hsa‐mir‐9‐1, hsa‐mir‐9‐2 and hsa‐mir‐9‐3, which suggested that mir‐9 might play a crucial role in LUAD by regulating these four lung cancer‐related genes.
The SIX family was demonstrated to play crucial roles in the tumorigenesis of NSCLC and was associated with the prognosis of patients with NSCLC 37. As a member of the SIX family, Six1 was found to be up‐regulated in LUAD and contributes to preinvasive‐to‐invasive adenocarcinoma progression by inducing epithelial–mesenchymal transition and nuclear atypia 38. The protein SLC7A5 is part of a two‐protein complex with SLC3A2 and the gene was hypermethylated and its expression increased in lung cancer 35. Moreover, SLC7A5 was also reported to be differentially expressed between adenocarcinoma and squamous cell lung cancers 39, 40. Both SLC7A5 and Six1 were target genes of hsa‐mir‐486 and hsa‐mir‐4732, respectively. We speculate that hsa‐mir‐486–SLC7A5 and hsa‐mir‐4732–Six1 interactions might also be involved in the pathogenesis of LUAD. Moreover, the ROC analysis of the present study indicated that all these six targets have great diagnostic value for LUAD and might be potential biomarkers of LUAD as well.
In conclusion, our study identified nine DEmiRNAs that serve as potential biomarkers of LUAD. Functional annotation of their target DEmRNAs in LUAD provided new clues for exploring their precise roles in LUAD. Further validation studies in prospective datasets are needed to test the predictive power for diagnosis before this is applied clinically.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
Z‐PR and X‐BH drafted the manuscript; X‐DT and J‐TG participated in data collection; L‐BZ, Z‐QX and J‐QD carried out the data analysis; S‐WZ, J‐YP, and X‐YC had significant roles in the study design and manuscript review. All authors read and approved the final manuscript.
Acknowledgements
This study was funded by Project of the Beijing Municipal Science and Technology Commission (Z161100000116075) and Wu Jieping Medical Foundation (320.6799.15048).
Zhi‐Peng Ren and Xiao‐Bin Hou contributed equally to this work and should be considered as co‐first authors.
References
- 1. Powrozek T, Mlak R, Dziedzic M, Malecka‐Massalska T and Sagan D (2017) Analysis of primary‐miRNA‐3662 and its mature form may improve detection of the lung adenocarcinoma. J Cancer Res Clin Oncol 143, 1941–1946. [DOI] [PubMed] [Google Scholar]
- 2. Peng Z, Pan L, Niu Z, Li W, Dang X, Wan L, Zhang R and Yang S (2017) Identification of microRNAs as potential biomarkers for lung adenocarcinoma using integrating genomics analysis. Oncotarget 8, 64143–64156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sun Y, Mei H, Xu C, Tang H and Wei W (2018) Circulating microRNA‐339‐5p and ‐21 in plasma as an early detection predictors of lung adenocarcinoma. Pathol Res Pract 214, 119–125. [DOI] [PubMed] [Google Scholar]
- 4. Sui J, Yang RS, Xu SY, Zhang YQ, Li CY, Yang S, Yin LH, Pu YP and Liang GY (2017) Comprehensive analysis of aberrantly expressed microRNA profiles reveals potential biomarkers of human lung adenocarcinoma progression. Oncol Rep 38, 2453–2463. [DOI] [PubMed] [Google Scholar]
- 5. Babu KR and Muckenthaler MU (2016) miR‐20a regulates expression of the iron exporter ferroportin in lung cancer. J Mol Med (Berlin, Germany) 94, 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Asiaf A, Ahmad ST, Arjumand W and Zargar MA (2018) MicroRNAs in breast cancer: diagnostic and therapeutic potential. Methods Mol Biol (Clifton, NJ) 1699, 23–43. [DOI] [PubMed] [Google Scholar]
- 7. Fawzy MS, Toraih EA, Ibrahiem A, Abdeldayem H, Mohamed AO and Abdel‐Daim MM (2017) Evaluation of miRNA‐196a2 and apoptosis‐related target genes: ANXA1, DFFA and PDCD4 expression in gastrointestinal cancer patients: A pilot study. PLoS ONE 12, e0187310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Han X, Zhang Y, Wang D, Fu X, Li M and Wang A (2017) Upregulation of microRNA‐18b induces phosphatase and tensin homolog to accelerate the migration and invasion abilities of ovarian cancer. Oncol Lett 14, 5631–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang X, Li M, Wang Z, Han S, Tang X, Ge Y, Zhou L, Zhou C, Yuan Q and Yang M (2015) Silencing of long noncoding RNA MALAT1 by miR‐101 and miR‐217 inhibits proliferation, migration, and invasion of esophageal squamous cell carcinoma cells. J Biol Chem 290, 3925–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kent OA and Mendell JT (2006) A small piece in the cancer puzzle: microRNAs as tumor suppressors and oncogenes. Oncogene 25, 6188–6196. [DOI] [PubMed] [Google Scholar]
- 11. Carthew RW and Sontheimer EJ (2009) Origins and mechanisms of miRNAs and siRNAs. Cell 136, 642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao S, Gao X, Zang S, Li Y, Feng X and Yuan X (2017) MicroRNA‐383‐5p acts as a prognostic marker and inhibitor of cell proliferation in lung adenocarcinoma by cancerous inhibitor of protein phosphatase 2A. Oncol Lett 14, 3573–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheng MW, Shen ZT, Hu GY and Luo LG (2017) Prognostic significance of microRNA‐7 and its roles in the regulation of cisplatin resistance in lung adenocarcinoma. Cell Physiol Biochem 42, 660–672. [DOI] [PubMed] [Google Scholar]
- 14. Kerr KM (2009) Pulmonary adenocarcinomas: classification and reporting. Histopathology 54, 12–27. [DOI] [PubMed] [Google Scholar]
- 15. Navon R, Wang H, Steinfeld I, Tsalenko A, Ben‐Dor A and Yakhini Z (2009) Novel rank‐based statistical methods reveal microRNAs with differential expression in multiple cancer types. PLoS ONE 4, e8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu L, Todd NW, Xing L, Xie Y, Zhang H, Liu Z, Fang H, Zhang J, Katz RL and Jiang F (2010) Early detection of lung adenocarcinoma in sputum by a panel of microRNA markers. Int J Cancer 127, 2870–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu Z, Chen X, Zhao Y, Tian T, Jin G, Shu Y, Chen Y, Xu L, Zen K, Zhang C et al (2010) Serum microRNA signatures identified in a genome‐wide serum microRNA expression profiling predict survival of non‐small‐cell lung cancer. J Clin Oncol 28, 1721–1726. [DOI] [PubMed] [Google Scholar]
- 18. Fotinos A, Nagarajan N, Martins AS, Fritz DT, Garsetti D, Lee AT, Hong CC and Rogers MB (2014) Bone morphogenetic protein‐focused strategies to induce cytotoxicity in lung cancer cells. Anticancer Res 34, 2095–2104. [PMC free article] [PubMed] [Google Scholar]
- 19. Shan N, Shen L, Wang J, He D and Duan C (2015) MiR‐153 inhibits migration and invasion of human non‐small‐cell lung cancer by targeting ADAM19. Biochem Biophys Res Comm 456, 385–391. [DOI] [PubMed] [Google Scholar]
- 20. Yuan Y, Du W, Wang Y, Xu C, Wang J, Zhang Y, Wang H, Ju J, Zhao L, Wang Z et al (2015) Suppression of AKT expression by miR‐153 produced anti‐tumor activity in lung cancer. Int J Cancer 136, 1333–1340. [DOI] [PubMed] [Google Scholar]
- 21. Daugaard I, Veno MT, Yan Y, Kjeldsen TE, Lamy P, Hager H, Kjems J and Hansen LL (2017) Small RNA sequencing reveals metastasis‐related microRNAs in lung adenocarcinoma. Oncotarget 8, 27047–27061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vosa U, Vooder T, Kolde R, Vilo J, Metspalu A and Annilo T (2013) Meta‐analysis of microRNA expression in lung cancer. Int J Cancer 132, 2884–2893. [DOI] [PubMed] [Google Scholar]
- 23. Han L, Wang W, Ding W and Zhang L (2017) MiR‐9 is involved in TGF‐beta1‐induced lung cancer cell invasion and adhesion by targeting SOX7. J Cell Mol Med 21, 2000–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee HY, Han SS, Rhee H, Park JH, Lee JS, Oh YM, Choi SS, Shin SH and Kim WJ (2015) Differential expression of microRNAs and their target genes in non‐small‐cell lung cancer. Mol Med Rep 11, 2034–2040. [DOI] [PubMed] [Google Scholar]
- 25. Xu G, Shao G, Pan Q, Sun L, Zheng D, Li M, Li N, Shi H and Ni Y (2017) MicroRNA‐9 regulates non‐small cell lung cancer cell invasion and migration by targeting eukaryotic translation initiation factor 5A2. Am J Transl Res 9, 478–488. [PMC free article] [PubMed] [Google Scholar]
- 26. Chen X, Zhu L, Ma Z, Sun G, Luo X, Li M, Zhai S, Li P and Wang X (2015) Oncogenic miR‐9 is a target of erlotinib in NSCLCs. Sci Rep 5, 17031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang R, Chen XF and Shu YQ (2015) Prediction of non‐small cell lung cancer metastasis‐associated microRNAs using bioinformatics. Am J Cancer Res 5, 32–51. [PMC free article] [PubMed] [Google Scholar]
- 28. Yuan X, He J, Sun F and Gu J (2013) Effects and interactions of MiR‐577 and TSGA10 in regulating esophageal squamous cell carcinoma. Int J Clin Exp Pathol 6, 2651–2667. [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang W, Shen C, Li C, Yang G, Liu H, Chen X, Zhu D, Zou H, Zhen Y, Zhang D et al (2016) miR‐577 inhibits glioblastoma tumor growth via the Wnt signaling pathway. Mol Carcinog 55, 575–585. [DOI] [PubMed] [Google Scholar]
- 30. Persson H, Kvist A, Rego N, Staaf J, Vallon‐Christersson J, Luts L, Loman N, Jonsson G, Naya H, Hoglund M et al (2011) Identification of new microRNAs in paired normal and tumor breast tissue suggests a dual role for the ERBB2/Her2 gene. Can Res 71, 78–86. [DOI] [PubMed] [Google Scholar]
- 31. Bai J and Hu S (2012) Transcriptome network analysis reveals potential candidate genes for squamous lung cancer. Int J Mol Med 29, 95–101. [DOI] [PubMed] [Google Scholar]
- 32. Meng X, Lu P, Bai H, Xiao P and Fan Q (2012) Transcriptional regulatory networks in human lung adenocarcinoma. Mol Med Rep 6, 961–966. [DOI] [PubMed] [Google Scholar]
- 33. Xi L, Feber A, Gupta V, Wu M, Bergemann AD, Landreneau RJ, Litle VR, Pennathur A, Luketich JD and Godfrey TE (2008) Whole genome exon arrays identify differential expression of alternatively spliced, cancer‐related genes in lung cancer. Nucleic Acids Res 36, 6535–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Behrouz Sharif S, Hashemzadeh S, Mousavi Ardehaie R, Eftekharsadat A, Ghojazadeh M, Mehrtash AH, Estiar MA, Teimoori‐Toolabi L and Sakhinia E (2016) Detection of aberrant methylated SEPT9 and NTRK3 genes in sporadic colorectal cancer patients as a potential diagnostic biomarker. Oncol Lett 12, 5335–5343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mullapudi N, Ye B, Suzuki M, Fazzari M, Han W, Shi MK, Marquardt G, Lin J, Wang T, Keller S et al (2015) Genome wide methylome alterations in lung cancer. PLoS ONE 10, e0143826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rolfo C and Raez L (2017) New targets bring hope in squamous cell lung cancer: neurotrophic tyrosine kinase gene fusions. Lab Invest 97, 1268–1270. [DOI] [PubMed] [Google Scholar]
- 37. Liu Q, Li A, Tian Y, Liu Y, Li T, Zhang C, Wu JD, Han X and Wu K (2016) The expression profile and clinic significance of the SIX family in non‐small cell lung cancer. J Hematol Oncol 9, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mimae T, Okada M, Hagiyama M, Miyata Y, Tsutani Y, Inoue T, Murakami Y and Ito A (2012) Upregulation of notch2 and six1 is associated with progression of early‐stage lung adenocarcinoma and a more aggressive phenotype at advanced stages. Clin Cancer Res 18, 945–955. [DOI] [PubMed] [Google Scholar]
- 39. Kaira K, Oriuchi N, Imai H, Shimizu K, Yanagitani N, Sunaga N, Hisada T, Tanaka S, Ishizuka T, Kanai Y et al (2008) Prognostic significance of L‐type amino acid transporter 1 expression in resectable stage I‐III nonsmall cell lung cancer. Br J Cancer 98, 742–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kido Y, Tamai I, Uchino H, Suzuki F, Sai Y and Tsuji A (2001) Molecular and functional identification of large neutral amino acid transporters LAT1 and LAT2 and their pharmacological relevance at the blood‐brain barrier. J Pharm Pharmacol 53, 497–503. [DOI] [PubMed] [Google Scholar]