

Abstract
Isocitrate dehydrogenases (IDHs) are enzymes involved in the production of α-ketoglutarate (αkg) in normal cellular metabolism. Cells with IDH mutations reduce αkg to 2-hydroxyglutarate (2HG), an oncometabolite, and 2HG directly transforms normal cells to malignant cells through histone demethylation and epigenetic dysregulation. However, whether IDH mutations affect cancer stromal cells is elusive, and little is known whether 2HG may impact the tumor microenvironment. We hypothesized that the IDH mutant cancer secretome and metabolites would stimulate primitive vascular-endothelial genesis. The secretome of IDH1 mutant human fibrosarcoma cells was harvested following medium starvation and was used to treat vascular-endothelial cells using a tube formation assay. GSK864, an allosteric IDH1 inhibitor, was supplemented to the fibrosarcoma secretome to determine its effects on vascular-endothelial tube formation. Exogenous 2HG or as supplemented in the GSK864-treated secretome was applied to further induce vascular-endothelial perturbation. Total vascular-endothelial tube lengths were quantified using NIH/Image J. Two-sided Student’s t-tests and Mann-Whitney U tests were used for statistical analysis. The IDH1 mutant fibrosarcoma secretome stimulated vascular-endothelial tube formation by ~138% relative to control. Remarkably, GSK864 attenuated vascular-endothelial tube formation by ~36%, but 2HG not only reversed GSK864 attenuation of tube formation, but also significantly stimulated vascular-endothelial tubes in the GSK864-treated fibrosarcoma secretome. Importantly, 2HG alone augmented vascular-endothelial tube formation that was equivalent to the fibrosarcoma secretome. Thus, 2HG stimulates vascular-endothelial genesis in conjunction with the fibrosarcoma secretome, despite pre-emptive inhibition of IDH1 mutation with GSK864, suggesting that 2HG enables oncogenic angiogenesis via paracrine signaling. Stimulation of vascular-endothelial genesis by 2HG alone, independent of the cancer secretome, suggests that 2HG also activates oncogenic angiogenic pathways in cancer stromal cells. Thus, the IDH mutant cancer secretome stimulates primitive oncogenic angiogenesis through 2HG and/or paracrine pathways. Taken together, these findings suggest novel mechanisms by which the IDH mutant cancer secretome and/or metabolite, specifically 2HG, interacts with the tumor microenvironment by inducing oncogenic angiogenesis in favor of metastasis.
Keywords: IDH, isocitrate dehydrogenase, 2HG, 2-hydroxyglutarate, histone demethylation, angiogenesis, tumor microenvironment, sarcoma, glioma, leukemia
Introduction
Isocitrate dehydrogenases (IDHs) are vital enzymes for the metabolism and homeostasis of normal cells. IDHs catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate (αKG) via the tricarboxylic acid cycle [6,44]. IDH genes may undergo missense mutations at arginine residues of their active site [6]. IDH mutations were first identified in clinical glioma samples [6,44], and have been subsequently shown to cause multiple malignancies including acute myeloid leukemia, sarcomas, melanomas, and liver, breast, prostate, colorectal and thyroid cancers [7,43].
IDH mutant oncogenesis provides direct evidence that normal cells can be transformed into malignant cells by altered cellular metabolism [7,33,43]. IDH mutations represent heterozygous point mutations that cause both a loss of normal IDH enzyme activity and a gain of de novo function by reducing αKG to 2-hydroxyglutarate (2HG) [6,17,21,33]. The wild-type IDH continues to produce αKG that is required for normal cellular metabolism, whereas the mutant IDH gains the ability to convert αKG to 2HG [17,21,33]. Whereas IDH1 mutations occur primarily in the cytosol, IDH2 mutations take place in mitochondria [6,17].
Excessive 2HG directly transforms normal cells to malignant cells through histone demethylation and epigenetic dysregulation [22], and blocks cell differentiation by competitive inhibition of αKG-dependent dioxygenases [21,22,35,43]. 2HG further promotes aerobic glycolysis and glutamate metabolism through the HIF1α pathway [7,43]. Quantitatively, 2HG in millimolar concentrations in IDH-mutant tumors was sufficient to mediate histone demethylases and methylcytosine dioxygenases of the ten eleven translocation (TET) family [21,22,33,45]. Thus, 2HG, as an oncometabolite resulting from IDH mutations, is the central culprit for direct transformation towards oncogenesis [43]. In addition to their considerable significance in cancer biology and metabolism, IDH mutations have been key targets for therapeutics. Several small-molecule IDH inhibitors have been approved by the Food and Drug Administration (FDA) for the treatment of acute myeloid leukemia and gliomas [11,43].
The tumor microenvironment represents a highly complex milieu of the oncogenic extracellular matrix and cancer stromal cells including blood vessel cells, immune cells, fibroblasts, and bone marrow-derived inflammatory cells [1,10,13,19,31]. Immune cells include lymphocytes and dendritic cells [19,46]. Inflammatory cells include leukocytes, monocytes and granulocytes [31,34]. Blood vessel cells include vascular-endothelial cells, smooth muscle cells, myofibroblasts and pericytes [13,19]. Classic cancer biology has primarily focused on cancer cells of different tissue origins. The roles of cancer stromal cells in the pro-survival, proliferation, immune surveillance, invasion and metastasis of cancer cells have been increasingly recognized [13,19,20,36]. In contrast to cancer cells that are highly heterogeneous and may undergo repetitive mutations, cancer stromal cells are relatively stable in their genome and may not be as susceptible to drug resistance, thus representing understudied and attractive therapeutic targets [4,19,32]. Metastasis is the primary cause of cancer mortality. It is estimated that metastasis is responsible for about 90% of cancer deaths [20,38]. Metastasis occurs when cancer cells invade surrounding tissues and enter blood vessels [5,20,32,38]. Thus, one of the most significant steps of cancer metastasis is oncogenic angiogenesis without which malignant cells cannot enter the blood stream [12,14,19].
Little is known in literature regarding the tumor microenvironment of IDH mutant cancers, in sharp contrast to emerging knowledge of the tumor microenvironment of other malignancies [13,19,31,46]. Nonetheless, there is indirect but conflicting evidence on whether IDH mutations may have a positive or negative effect on oncogenic angiogenesis. It was speculated that IDH1 mutations may disrupt blood vessels and the brain barrier [39]. Also, IDH1 mutation was associated with a distinct hypoxic and angiogenic transcriptome [15]. However, 2HG overexpression led to defective collagen type IV and disrupted angiogenic gene expression profiles [23]. Accordingly, we hypothesized that IDH1 mutant cancer cells and/or their oncometabolite stimulate vascular-endothelial cells to form primitive vascular tubes as a surrogate model for in vivo oncogenic angiogenesis.
Materials and methods
Cell culture
Human fibrosarcoma cells (HT-1080) were acquired from ATCC (Cat. #CCL-121; Manassas, VA, USA). We selected HT-1080 because: 1) HT-1080 harbors IDH1 mutations [2,8], 2) no effective biological therapy has been developed for human fibrosarcoma [2,8] and 3) fibroblasts are a ubiquitous component among stromal cells of numerous cancers [2,8,19]. Vials of frozen fibrosarcoma cells were removed from -80°C freezer and thawed. The cells were then transferred to a 15-mL tube, and 10-mL of RPMI with 1% Antibiotic-Antimycotic (Gibco, Fair Lawn, NJ, USA) added to the 15-mL tube, and centrifuged for 7 min at 1,000 rpm.
Following supernatant aspiration, cells were resuspended in 12 mL of RPMI (Gibco, Fair Lawn, NJ, USA) and transferred to a T75 flask. The flask was then incubated at 37°C, 5% CO2 and 100% humidity. For trypsinization, all medium was removed from the flask. Cells were rinsed with 1 mL of 5-fold diluted trypsin in a T75 flask. A total of 3 mL Trypsin EDTA 0.05% was added to a T75 flask. The flask was then incubated for 3 min. Cells were examined under the microscope to confirm detachment. Cells were split accordingly, and the flask was replenished with 12 mL of medium. All cultures were maintained and assayed in at least biological triplicates.
Human dermal fibroblasts were acquired from ATCC (HFF-1, Cat. #SSRC-1041; Manassas, VA, USA). We selected human dermal fibroblasts because fibroblasts are a ubiquitous component of cancer stroma [2,8,19]. Fibroblasts were expanded in L-DMEM with 10% fetal bovine serum, 2 mM L-glutamine, 1% Antibiotic-Antimycotic (Gibco, Fair Lawn, NJ, USA) at 37°C with 5% CO2. For tripsinization, a total of 3 mL Trypsin EDTA 0.05% was added to a T75 flask. Cells were examined under the microscope to confirm detachment. Cells were split accordingly, and the medium was replenished. All cultures were maintained and assayed in at least biological triplicates.
Cell proliferation
The proliferation of human fibrosarcoma cells (HT-1080) and human dermal fibroblasts (HFF-1) (ATCC, Manassas, VA, USA) in at least biological triplicates was assayed. An aliquot of Cell-Titer 96® Aqueous One Solution Reagent was thawed at room temperature or in a water bath at 37°C. A total of 15 μL of the Cell-Titer 96® Aqueous One Solution Reagent (Promega, Madison, WI, USA) was pipetted into each well at a density of 3×103 cells per well of the 96-well plate with 100 μL of culture medium. The plate was incubated for 1-4 hrs at 37°C and 5% CO2. The absorbance was read at 490 nm using a 96-well plate reader.
2-hydroxyglutarate (2HG)
We treated vascular-endothelial cells with 2-hydroxyglutarate (2HG) for multiple reasons. First, 2HG is a pivotal oncometabolite and a direct product of IDH mutant cells [6,7]. In literature, 2HG has been primarily studied in the context of IDH mutant malignant cells, but rarely in cancer stromal cells [43]. Thus, our experiment addressing 2HG’s effects on vascular-endothelial cells was designed to demonstrate whether 2HG impacts vascular tube formation. Second, whether 2HG mediates other oncogenic angiogenesis pathways such as VEGF, which is a potent angiogenic factor, has been speculated but lacks specific experimental evidence in IDH mutant cancers. 2HG was acquired from Sigma (St. Louis, MO, USA) and prepared by diluting in ddH2O and per additional manufacturer’s guidelines. 2HG at concentrations of 3, 6, 9, 12, 15 and 17 μmol was pipetted to 96 well plates containing cultures of human fibrosarcoma cells (HT-1080). This concentration range covered those that have been previously reported on the IDH mutant cancer cells in literature [21,22,35,43]. There were a total of 5 biological samples.
GSK864
GSK864 was acquired from Sigma (St. Louis, MO, USA) and prepared by diluting in DMSO and per additional manufacturer’s guidelines. We selected GSK864 for multiple reasons. GSK864 is a direct inhibitor of IDH1 mutation in fibrosarcoma cells [2,8]. GSK864 is a small molecule and selective allosteric IDH1 inhibitor [2,8,24,28]. GSK864 readily penetrates cells and is a potent inhibitor of intracellular 2HG production [24,28]. GSK864 binds to an allosteric site and inactivates both wild-type IDH1 and mutant IDH1 to a catalytic conformation [24,28]. Here, GSK864 at concentrations of 0.2, 0.4, 0.8, 1.2 and 2.0 μmol was pipetted to a 96-well plate containing cultures of either human fibrosarcoma cells (HT-1080) or human dermal fibroblasts (HFF-1). This concentration range covered those previously reported [24,28]. There were a total of 5 biological samples.
IDH mutant cancer secretome
Human fibrosarcoma cells (HT-1080) were plated in T25 flasks. Cells were first treated with the Roswell Park Memorial Institute medium (RPMI) (Thermo Fisher Scientific, Grand Island, New York, USA) or 0.8 μmol GSK864 supplemented RPMI medium for 24 hrs. After the medium was discarded from the flasks, cells were exposed to serum-free medium for starvation over 24 hrs. Then, the cell supernatant secretome was collected, centrifuged at 1200 rpm for 5 min to remove debris, further filtered with a 0.22-μm membrane. The fibrosarcoma secretome was collected for GSK864 treatment at 0.8 μmol for 24 hrs, followed by addition of 50 μL of 12 μmol 2HG into 5 mL of endothelial basal medium for treating vascular-endothelial cells. All cultures were maintained and assayed with at least biological triplicates.
Vascular-endothelial tube formation assay
Vascular-endothelial cells were acquired from ATCC (EA.hy926; CRL-2922; Manassas, VA, USA). We selected EA.hy926 cells for multiple reasons. EA.hy926 cells are among the most commonly used endothelial cells in experimental investigations [27,29]. Specifically, EA.hy926 cells have been broadly used for oncogenic angiogenesis [27,29]. EA.hy926 cells were cultured in endothelial basal medium (ATCC), at 1×106 cells per T25 tissue culture flask.
Basement Membrane Extract (BME) with reduced growth factors (Cultrex; Trevigen, Gaithersburg, MD, USA) was thawed in an ice bath and placed at 2-4°C overnight. Following 24-hr thawing, the BME solution was aliquoted into a 96-well plate at 50 μL per well, followed by visual assessment to ensure even gel distribution in the entire plate. The plate was then centrifuged at 250×g for 5 min at 4°C to remove any bubbles that may have been trapped in the BME, followed by incubation for 60 min. A total of 5 μL of 2 μmol of Calcein AM solution (2 mg/ml, Trevigen) was added to 5 mL of Basal Medium (Sigma). Endothelial cells were washed with sterile 5-mL PBS. Then the medium was added to a T25 flask and incubated for 30 min at 37°C and 5% CO2. After 30 min, cells were washed twice with 5 mL of sterile PBS at room temperature.
Cells were then harvested by adding 1 mL of Trypsin EDTA 0.25% and incubated for 3 min, and transferred to a 15 mL conical tube. Following addition of 2-mL Basal Medium, cells were counted and centrifuged at 200×g for 3 min. Following removal of the supernatant, cells were resuspended. A total of 4-mL Basal Medium was added, followed by splitting cells into 4 tubes per group. The tubes were centrifuged again at 200×g for 3 min.
A total of 4 different cell suspensions at a density of 1×106 cells per mL were prepared for cell count. A total of 100 μL cell suspension with 10,000 cells was carefully added in 96-well plates atop the gelled BME. The plate was incubated at 37°C and 5% CO2 for 24 hrs. Vascular-endothelial tube formation was visualized with phase contrast and fluorescence microscopy at 485 nm excitation/520 nm emission. The total vascular-endothelial tube lengths were measured from a total of 9-12 randomly selected image fields per sample per group using NIH/Image J.
Statistical analysis
All quantitative data were subjected to statistical analysis. Means and standard deviation or standard errors were used in all charts. Two-sided Student’s t-tests were used for paired comparisons with normal data distribution. For samples with skewed distribution, Mann-Whitney U tests were performed. A p value < 0.05 was considered as statistically significant. All statistical analyses were performed with SPSS 1.0.0.1126 (64-bit Edition).
Results
The fibrosarcoma secretome significantly stimulated vascular-endothelial genesis
Vascular-endothelial cells, such as those found in the tumor microenvironment, are pivotal for oncogenic angiogenesis and metastasis [19]. Using basal medium treated vascular-endothelial cells as a control, we found that the human fibrosarcoma secretome substantially stimulated vascular-endothelial tube formation (Figure 1B), in comparison with sparse vascular-endothelial tubes in the control (Figure 1A). Quantitatively, the average length of vascular-endothelial tubes treated with the human fibrosarcoma secretome was approximately 138% greater than that treated with basal medium (P < 0.001) (Figure 1C), suggesting that IDH-mutant fibrosarcoma cells produce a secretome highly capable of stimulating in vivo angiogenesis, potentially leading to metastasis. These findings are novel and suggest that the secretome of IDH mutant cancer cells harbors factor(s) with oncogenic angiogenic capacity in a paracrine manner.
Figure 1.
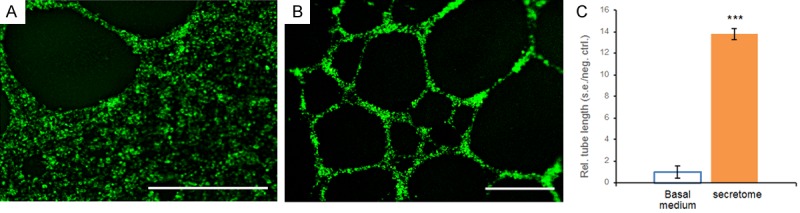
Representative images and quantification of vascular-endothelial tube formation in basal medium and the human fibrosarcoma secretome. A: Vascular-endothelial cells treated with basal medium showing sparse tube formation. B: The fibrosarcoma secretome stimulated abundant vascular-endothelial tube formation. Scale bars: 400 μm. C: Quantification of vascular-endothelial tube lengths in basal medium and the fibrosarcoma secretome. Rel.: relative; s.e.: standard error; neg.: negative; ctrl.: control; n=3 independent biological samples per group; ***: P < 0.001.
GSK864 significantly reversed fibrosarcoma’s ability to stimulate vascular-endothelial genesis
We then postulated that IDH1 attenuation by a small-molecule inhibitor may reduce cancer cells’ ability to stimulate oncogenic angiogenesis. Although GSK864 is known to attenuate 2HG production of the IDH1 mutant cancer cells [2,8,24,28], little is known whether 2HG mediates cancer stromal cells. We collected the GSK864-treated fibrosarcoma secretome, and then treated vascular-endothelial cells. Remarkably, GSK864 substantially reduced vascular-endothelial tube formation relative to the untreated fibrosarcoma secretome (Figure 2A, 2B). Quantitatively, the average total tube length induced by the GSK864-treated fibrosarcoma secretome was reduced by ~36% of that of the untreated fibrosarcoma secretome (P < 0.001) (Figure 2C). These findings suggest that GSK864, known to inhibit IDH1 mutations, substantially alters the cancer secretome by reducing its capacity for oncogenic angiogenesis and metastasis. This leads to our next postulate that 2HG, a pivotal culprit of IDH mutations, may mediate oncogenic angiogenesis.
Figure 2.
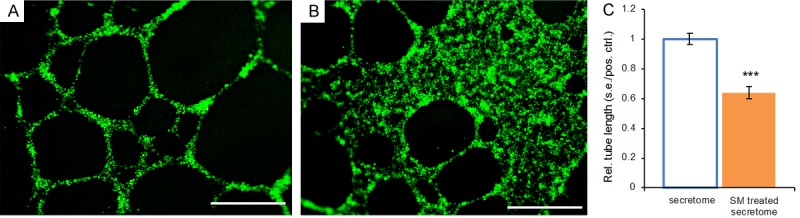
Representative images and quantification of vascular-endothelial tube formation in the cancer secretome and a small molecule (SM)-treated cancer secretome. A: Abundant vascular-endothelial tubes induced by the fibrosarcoma secretome. B: Substantial reduction in vascular-endothelial tubes induced by the GSK864-treated fibrosarcoma secretome. SM: small molecule, GSK864; Scale bars: 400 μm. C: Quantification of vascular-endothelial tube lengths in the fibrosarcoma secretome and a small molecule (SM)-treated fibrosarcoma secretome. SM: GSK864. Rel.: relative; s.e.: standard error; pos.: positive; ctrl.: control; n=3 independent biological samples per group; ***: P < 0.001.
2HG reversed GSK864’s inhibitory effects on cancer-induced vascular-endothelial genesis
To follow up on GSK864’s inhibitory effects on cancer-induced angiogenesis, we supplemented 2HG to the GSK864-treated fibrosarcoma secretome. Indeed, 2HG reversed GSK864’s inhibitory effects on vascular-endothelial tube formation (Figure 3C), in comparison to that induced by the untreated fibrosarcoma secretome and GSK864-treated fibrosarcoma secretome (Figure 3A, 3B). Quantitatively, the average length of vascular-endothelial tubes formed upon treatment with the 2HG-supplemented, GSK864-treated fibrosarcoma secretome approximately doubled over that treated with the GSK864-treated fibrosarcoma secretome and was even ~38% more than the fibrosarcoma secretome (P < 0.001) (Figure 3D). These surprising findings suggest that 2HG, as an oncometabolite of IDH-mutant malignancies, stimulates oncogenic angiogenesis and potentially metastasis in a paracrine manner by activating multiple angiogenic factors.
Figure 3.

Representative images and quantification of vascular-endothelial tube formation in the fibrosarcoma secretome, a small molecule (SM)-treated fibrosarcoma secretome, and 2HG supplemented and SM-treated fibrosarcoma secretome. A: The fibrosarcoma secretome induced abundant vascular-endothelial tube formation. B: The GSK864-treated fibrosarcoma secretome reduced tube formation. C: 2HG supplemented in the SM-treated fibrosarcoma secretome enhanced tube formation. Scale bars: 400 μm. D: Quantification of vascular-endothelial tube lengths in the fibrosarcoma secretome, a small molecule (SM)-treated fibrosarcoma secretome (by GSK864) and the 2HG supplemented, GSK864-treated fibrosarcoma secretome. Rel.: relative; s.e.: standard error; pos.: positive; ctrl.: control; n=6 independent biological samples per group; ***: P < 0.001.
2HG alone stimulated vascular-endothelial genesis on a par with the fibrosarcoma secretome
To follow up on the surprising finding of 2HG reversal of GSK864 attenuation on cancer secretome-induced vascular-endothelial tube formation, and to address another postulate of whether 2HG alone may stimulate oncogenic angiogenesis, we added 2HG to endothelial basal medium, devoid of any influence from the fibrosarcoma secretome. Strikingly, 2HG alone directly stimulated vascular-endothelial tube formation (Figure 4B), on a par with the fibrosarcoma secretome (Figure 4A). The 2HG dosage at 12 μmol was exactly the same as in the experiment with exogenous 2HG added to the GSK864-treated fibrosarcoma secretome in the experiment as described in Figure 3. Quantitatively, the average total length of vascular-endothelial tubes induced by 2HG alone in basal medium showed a lack of statistically significant differences from that induced by the fibrosarcoma secretome (Figure 4C). These novel findings suggest that 2HG alone, independent of IDH-mutant malignant cells from which 2HG is produced, enhances oncogenic angiogenesis by acting directly on vascular-endothelial cells in cancer stroma.
Figure 4.

Representative images and quantification of vascular-endothelial tube formation in the fibrosarcoma secretome and 2HG in endothelial basal medium. A: Abundant vascular-endothelial tube formation induced by the fibrosarcoma secretome. B: 2HG in endothelial basal medium induced similar vascular-endothelial tube formation. Scale bars: 400 μm. C: Quantification of vascular-endothelial tube lengths in the fibrosarcoma secretome and 2HG in endothelial basal medium. Rel.: relative; s.e.: standard error; pos.: positive; ctrl.: control; n=3 independent biological samples per group; n.s.: no (statistical) significance.
Proliferation of human fibrosarcoma cells was affected by 2HG and GSK864
Given 2HG’s effects to stimulate oncogenic angiogenesis, we conducted another experiment to determine to what degree 2HG may further stimulate the proliferation of human fibrosarcoma cells beyond its proliferative baseline [8]. Indeed, 2HG significantly augmented the proliferation of human fibrosarcoma cells in a dose dependent manner, with an average increase in proliferation rates by 50% to 70% (Figure 5A). Although 2HG concentration at 3 μmol began to show pro-proliferative effects on human fibrosarcoma cells, we selected 12 μmol to determine its effects on vascular-endothelial tube formation in our experiments above (Figures 3 and 4), because 12 μmol represented the mid-range plateau. As elucidated in Figure 3, 2HG indeed stimulated vascular-endothelial tube formation and even reversed GSK864’s inhibitory effects on primitive oncogenic angiogenesis.
Figure 5.
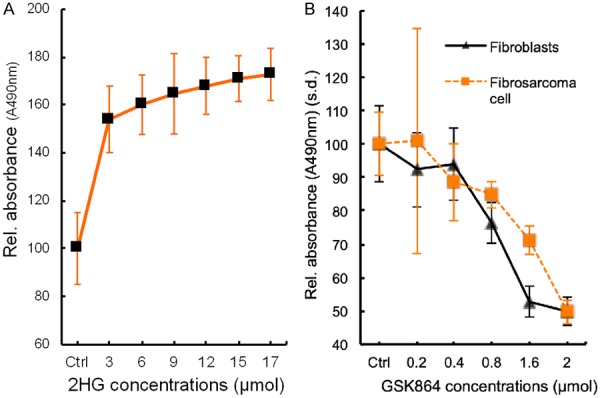
Cell proliferation. A: 2-hydroxyglutarate (2HG) stimulated human fibrosarcoma cell (HT-1080) proliferation beyond its baseline in a dose-dependent manner. s.d.: standard deviation. n=5 independent biological samples. B: GSK864 inhibited the proliferation of both human fibroblasts and human fibrosarcoma cells (HT-1080) in a dose-dependent manner. s.d.: standard deviation. Proliferation of human fibrosarcoma cells (HT-1080) and fibroblasts at 0.8, 1.6 and 2 μmol GSK864 concentrations were significantly lower than control (ctrl) and 0.2 and 0.4 μmol (P < 0.001; n=5 independent biological samples per group).
GSK864 is a potent small-molecule allosteric inhibitor of IDH1 mutant, intracellular 2HG production [24,28]. Given our observation that GSK864 reduced vascular-endothelial tube formation (Figure 3), we followed up with another experiment to determine whether GSK864 would attenuate the proliferation of stromal fibroblasts, such as those found in cancer stroma, along with fibrosarcoma cells. Indeed, GSK864 attenuated the proliferation of both fibrosarcoma cells and normal fibroblasts with a similar trend (Figure 5B). Despite their different proliferation rates, GSK864 significantly reduced the proliferation of fibrosarcoma cells and normal fibroblasts at doses of 0.8, 1.6 and 2 μmol, relative to control and at doses of 0.2 and 0.4 μmol (Figure 5B). These findings suggest that GSK864 not only attenuated the proliferation of IDH1 mutant cancer cells, but also fibroblasts such as those found in cancer stroma. Thus, GSK864 attenuation of vascular-endothelial tube formation (Figure 3), reversable by IDH mutant 2HG to which GSK864 inhibits, may represent a novel me-chanism by which GSK864 or other small molecules may be harnessed to target cancer stromal cells.
Discussion
The present data, as summarized in Figure 6, are novel and absent in literature. Our findings provide experimental evidence, for the first time, for how IDH mutant cancer secretome and/or oncometabolite impacts cancer stromal cells. An IDH mutant oncometabolite, 2-hydroxyglutarate (2HG), is shown to stimulate vascular-endothelial genesis in conjunction with the fibrosarcoma secretome, despite preemptive inhibition of IDH1 mutation with GSK864, a small-molecule IDH1 inhibitor, suggesting that 2HG may augment oncogenic angiogenesis via paracrine signaling (Figure 6). In parallel, 2HG alone stimulates vascular-endothelial tube formation, and may directly activate angiogenic pathways in cancer stromal cells (Figure 6). These findings enrich previous work that established the concept of the tumor microenvironment and metastasis via vascular invasion [5,32,38], and provide clues that the IDH mutant tumors may undergo metastasis by augmenting oncogenic angiogenesis.
Figure 6.
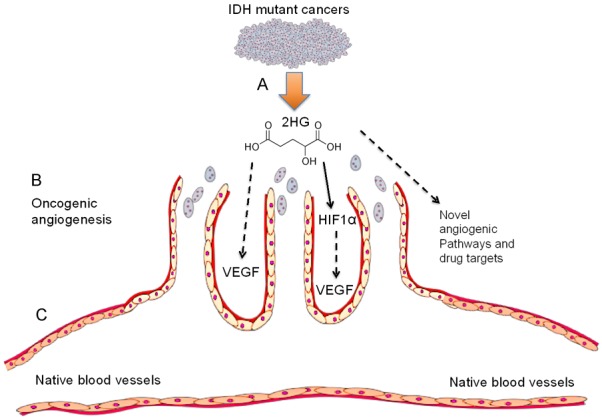
Summary of the present data and future studies. A: IDH mutant cancers produce 2HG, an oncometabolite that transforms normal cells into malignant cells via histone demethylation and epigenetic dysregulation. However, whether 2HG or IDH mutations impact cancer stroma is elusive. Our data show that 2HG alone or in the cancer secretome stimulates vascular-endothelial tube formation, as a surrogate model for in vivo oncogenic angiogenesis and metastasis. B: Vascular sprouts: vertically oriented cancer blood vessels via oncogenic angiogenesis. C: Horizontal blood vessel: native host vasculature. 2HG may activate HIF1α and/or VEGF signaling.
These findings suggest that 2HG’s paracrine effects to stimulate oncogenic angiogenesis and its direct effects are not mutually exclusive. The greater capacity of vascular-endothelial genesis by 2HG supplemented in the GSK864-treated fibrosarcoma secretome than 2HG alone in basal medium is likely due to 1) greater 2HG concentration that is derived from both exogenous 2HG added (12 μmol) plus endogenous 2HG produced by fibrosarcoma cells. Equivalent vascular-endothelial tube formation induced by the cancer secretome and 2HG alone in basal medium provides experimental evidence that 2HG may stimulate oncogenic angiogenesis by directly acting on vascular-endothelial cells and/or through multiple angiogenic pathways that may include HIF1α and/or VEGF. HIF1α signaling was proposed to promote angiogenesis by IDH mutant cancer cells [30], and recently 2HG was shown to activate aerobic glycolysis and glutamate metabolism through HIF1α [26]. VEGF is a pivotal angiogenic factor in tissue development and multiple malignancies [5,32,38], but its roles in IDH mutant tumors are elusive [30]. Whether 2HG directly activates VEGF signaling, via HIF1α or other paracrine factors secreted from stromal cells warrants new investigations (Figure 6). Additionally, the chemokine receptor CXCR7 was immuno-localized to vascular endothelium of clinical IDH-mutant glioma samples, implicating CXCR7 as another potential pathway for IDH mutation induced oncogenic angiogenesis [3].
Cancer cells are genetically unstable and undergo repetitive mutations [33,40,43], which underscores the difficulty in developing novel therapeutics against malignant cells, and accounts for some of cancer treatment relapse and tumor recurrence. For example, IDH mutant tumors have begun to show resistance to some of the novel small-molecule inhibitors developed such as ivosidenib (AG-120) [11]. Contrastingly, cancer stromal cells possess relative genetic stability [33,40,43], and therefore are attractive targets for developing therapeutics that, for example, targets vascular-endothelial cells. Our finding of GSK864 inhibition of vascular-endothelial genesis provides additional clues for targeting cancer stromal cells. GSK864 inhibits both wild-type and mutant IDH1 isoforms of cancer cells [24,28]. IDH mutations are heterozygous and involve arginine substitution to histidine, yielding a wild-type and mutant heterodimer [17,21,33,45]. GSK864 is an allosteric IDH1 inhibitor that attenuates intracellular 2HG production by inactivating wild-type and mutant IDH1s [24,28]. GSK864’s half life in human and mouse liver cells is in the range of 20-70 min [24,42]. Hence, most of the supplemented GSK864 in the fibrosarcoma secretome must have lost its bioactivity during the time course of vascular-endothelial tube formation for 24 hrs. Thus, 2HG’s efficacy in stimulating vascular-endothelial genesis in the GSK864-treated cancer secretome is likely due to its own merit, and probably not substantially affected by GSK864. However, the mechanisms by which 2HG or GSK864 affects vascular-endothelial cells or other cancer stromal cells are elusive and warrant investigations.
Several anti-2HG small molecules have been FDA-approved for the treatment of gliomas and acute myeloid leukemia [43]. However, there is no effective biological treatment for sarcomas including fibrosarcoma that is the current model system. The insight unveiled by our findings regarding 2HG stimulation and GSK864 inhibition of vascular-endothelial tube formation in the fibrosarcoma secretome serves as a model system for investigating molecular pathways and for developing novel sarcoma therapeutics via small molecules and/or vaccines. Further understanding of how T cells respond to IDH mutant fibrosarcoma may also aid in the development of enhanced tumor surveillance and novel therapeutics. For example, 2HG accumulation in the IDH mutant glioma activated CD8+ T lymphocytes via the HIF1α pathway, and enhanced in vivo cancer cell proliferation [41]. In immortalized human astrocytes and syngeneic mouse glioma models, IDH1 mutation or 2HG impaired CD8+ T cell accumulation and CXCL10 chemotaxis [16,41]. Novel peptides activated IDH mutation-specific CD4(+) T-helper-1 cell responses in patient samples of IDH1-mutant gliomas [36]. Little is known in literature regarding biological therapies for sarcomas.
The present study has a number of limitations. A total of three well-established cell lines were utilized. Although these cell lines are broadly adopted in cancer biology, patients’ primary tumor cells would be of interest for devising patient-specific therapeutics via, for example, CRISPR/Cas9 or other gene editing tools. Stromal cells utilized in the present study are dermal fibroblasts and vascular-endothelial cells that have been commonly adopted in cancer biology, rather than from the patients’ primary cancer stroma cells. Our future work will profile patients’ cancer stromal cells via RNASeq and single-cell RNASeq, followed by utilization of patient’s cancer stromal cells for the development of patient-specific molecular therapeutics. Our future experiment will also profile the proteome of the fibrosarcoma secretome in complement to RNASeq analysis. Within these constraints, our findings have unveiled several novel features of vascular-endothelial response to the cancer secretome and to 2HG, an IDH mutant oncometabolite. These data serve as an in vitro surrogate model for manipulating oncogenic angiogenesis for the benefit of understanding cancer biology and developing novel therapeutics, especially by targeting the cancer microenvironment.
Disclosure of conflict of interest
None.
References
- 1.Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M, Choi K, Fromme RM, Dao P, McKenney PT, Wasti RC, Kadaveru K, Mazutis L, Rudensky AY, Pe’er D. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. 2018;174:1293–1308. e36. doi: 10.1016/j.cell.2018.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badur MG, Muthusamy T, Parker SJ, Ma S, McBrayer SK, Cordes T, Magana JH, Guan KL, Metallo CM. Oncogenic R132 IDH1 mutations limit NADPH for de novo lipogenesis through (D)2-Hydroxyglutarate production in fibrosarcoma cells. Cell Rep. 2018;25:1680. doi: 10.1016/j.celrep.2018.10.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Birner P, Tchorbanov A, Natchev S, Tuettenberg J, Guentchev M. The chemokine receptor CXCR7 influences prognosis in human glioma in an IDH1-dependent manner. J Clin Pathol. 2015;68:830–4. doi: 10.1136/jclinpath-2015-202886. [DOI] [PubMed] [Google Scholar]
- 4.Cheng CJ, Bahal R, Babar IA, Pincus Z, Barrera F, Liu C, Svoronos A, Braddock DT, Glazer PM, Engelman DM, Saltzman WM, Slack FJ. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature. 2015;518:107–10. doi: 10.1038/nature13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clark AG, Vignjevic DM. Modes of cancer cell invasion and the role of the microenvironment. Curr Opin Cell Biol. 2015;36:13–22. doi: 10.1016/j.ceb.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 6.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–44. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dang L, Yen K, Attar EC. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol. 2016;27:599–608. doi: 10.1093/annonc/mdw013. [DOI] [PubMed] [Google Scholar]
- 8.Ding Y, Boguslawski EA, Berghuis BD, Young JJ, Zhang Z, Hardy K, Furge K, Kort E, Frankel AE, Hay RV, Resau JH, Duesbery NS. Mitogen-activated protein kinase kinase signaling promotes growth and vascularization of fibrosarcoma. Mol Cancer Ther. 2008;7:648–58. doi: 10.1158/1535-7163.MCT-07-2229. [DOI] [PubMed] [Google Scholar]
- 9.Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suvà ML, Bernstein BE. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529:110–4. doi: 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Intlekofer AM, Shih AH, Wang B, Nazir A, Rustenburg AS, Albanese SK, Patel M, Famulare C, Correa FM, Takemoto N, Durani V, Liu H, Taylor J, Farnoud N, Papaemmanuil E, Cross JR, Tallman MS, Arcila ME, Roshal M, Petsko GA, Wu B, Choe S, Konteatis ZD, Biller SA, Chodera JD, Thompson CB, Levine RL, Stein EM. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature. 2018;559:125–129. doi: 10.1038/s41586-018-0251-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Cell. 2005;7:513–20. doi: 10.1016/j.ccr.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 13.Katheder NS, Khezri R, O’Farrell F, Schultz SW, Jain A, Rahman MM, Schink KO, Theodossiou TA, Johansen T, Juhász G, Bilder D, Brech A, Stenmark H, Rusten TE. Microenvironmental autophagy promotes tumour growth. Nature. 2017;541:417–420. doi: 10.1038/nature20815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaur A, Webster MR, Marchbank K, Behera R, Ndoye A, Kugel CH 3rd, Dang VM, Appleton J, O’Connell MP, Cheng P, Valiga AA, Morissette R, McDonnell NB, Ferrucci L, Kossenkov AV, Meeth K, Tang HY, Yin X, Wood WH 3rd, Lehrmann E, Becker KG, Flaherty KT, Frederick DT, Wargo JA, Cooper ZA, Tetzlaff MT, Hudgens C, Aird KM, Zhang R, Xu X, Liu Q, Bartlett E, Karakousis G, Eroglu Z, Lo RS, Chan M, Menzies AM, Long GV, Johnson DB, Sosman J, Schilling B, Schadendorf D, Speicher DW, Bosenberg M, Ribas A, Weeraratna AT. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature. 2016;532:250–4. doi: 10.1038/nature17392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kickingereder P, Sahm F, Radbruch A, Wick W, Heiland S, Deimling Av, Bendszus M, Wiestler B. IDH mutation status is associated with a distinct hypoxia/angiogenesis transcriptome signature which is non-invasively predictable with rCBV imaging in human glioma. Sci Rep. 2015;5:16238. doi: 10.1038/srep16238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kohanbash G, Carrera DA, Shrivastav S, Ahn BJ, Jahan N, Mazor T, Chheda ZS, Downey KM, Watchmaker PB, Beppler C, Warta R, Amankulor NA, Herold-Mende C, Costello JF, Okada H. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J Clin Invest. 2017;127:1425–1437. doi: 10.1172/JCI90644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, Travins J, Weiss S, Looper R, Ligon KL, Verhaak RG, Yan H, Kaelin WG Jr. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484–8. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JH, Lee JE, Kahng JY, Kim SH, Park JS, Yoon SJ, Um JY, Kim WK, Lee JK, Park J, Kim EH, Lee JH, Lee JH, Chung WS, Ju YS, Park SH, Chang JH, Kang SG, Lee JH. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature. 2018;560:243–247. doi: 10.1038/s41586-018-0389-3. [DOI] [PubMed] [Google Scholar]
- 19.Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375–9. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 20.Liu Q, Zhang H, Jiang X, Qian C, Liu Z, Luo D. Factors involved in cancer metastasis: a better understanding to “seed and soil” hypothesis. Mol Cancer. 2017;16:176. doi: 10.1186/s12943-017-0742-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, Kaelin WG Jr. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621–5. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O’Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–8. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma S, Jiang B, Deng W, Gu ZK, Wu FZ, Li T, Xia Y, Yang H, Ye D, Xiong Y, Guan KL. D-2-hydroxyglutarate is essential for maintaining oncogenic property of mutant IDH-containing cancer cells but dispensable for cell growth. Oncotarget. 2015;6:8606–20. doi: 10.18632/oncotarget.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma T, Zou F, Pusch S, Xu Y, von Deimling A, Zha X. Inhibitors of mutant isocitrate dehydrogenases 1 and 2 (mIDH1/2): an update and perspective. J Med Chem. 2018;61:8981–9003. doi: 10.1021/acs.jmedchem.8b00159. [DOI] [PubMed] [Google Scholar]
- 25.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O, Stephanopoulos G. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481:380–4. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011;481:385–8. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okamoto T, Akita N, Kawamoto E, Hayashi T, Suzuki K, Shimaoka M. Endothelial connexin32 enhances angiogenesis by positively regulating tube formation and cell migration. Exp Cell Res. 2014;321:133–41. doi: 10.1016/j.yexcr.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 28.Okoye-Okafor UC, Bartholdy B, Cartier J, Gao EN, Pietrak B, Rendina AR, Rominger C, Quinn C, Smallwood A, Wiggall KJ, Reif AJ, Schmidt SJ, Qi H, Zhao H, Joberty G, Faelth-Savitski M, Bantscheff M, Drewes G, Duraiswami C, Brady P, Groy A, Narayanagari SR, Antony-Debre I, Mitchell K, Wang HR, Kao YR, Christopeit M, Carvajal L, Barreyro L, Paietta E, Makishima H, Will B, Concha N, Adams ND, Schwartz B, McCabe MT, Maciejewski J, Verma A, Steidl U. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat Chem Biol. 2015;11:878–886. doi: 10.1038/nchembio.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pedersen AK, Mendes Lopes de Melo J, Mørup N, Tritsaris K, Pedersen SF. Tumor microenvironment conditions alter Akt and Na+/H+ exchanger NHE1 expression in endothelial cells more than hypoxia alone: implications for endothelial cell function in cancer. BMC Cancer. 2017;17:542. doi: 10.1186/s12885-017-3532-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prensner JR, Chinnaiyan AM. Metabolism unhinged: IDH mutations in cancer. Nat Med. 2011;17:291–3. doi: 10.1038/nm0311-291. [DOI] [PubMed] [Google Scholar]
- 31.Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, Holland EC, Sutton JC, Joyce JA. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science. 2016;352:aad3018. doi: 10.1126/science.aad3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–37. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raffel S, Falcone M, Kneisel N, Hansson J, Wang W, Lutz C, Bullinger L, Poschet G, Nonnenmacher Y, Barnert A, Bahr C, Zeisberger P, Przybylla A, Sohn M, Tönjes M, Erez A, Adler L, Jensen P, Scholl C, Fröhling S, Cocciardi S, Wuchter P, Thiede C, Flörcken A, Westermann J, Ehninger G, Lichter P, Hiller K, Hell R, Herrmann C, Ho AD, Krijgsveld J, Radlwimmer B, Trumpp A. BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature. 2017;551:384–388. doi: 10.1038/nature24294. [DOI] [PubMed] [Google Scholar]
- 34.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martín-Algarra S, Mandal R, Sharfman WH, Bhatia S, Hwu WJ, Gajewski TF, Slingluff CL Jr, Chowell D, Kendall SM, Chang H, Shah R, Kuo F, Morris LGT, Sidhom JW, Schneck JP, Horak CE, Weinhold N, Chan TA. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171:934–949. e16. doi: 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brüstle A, Harris IS, Holmes R, Wakeham A, Haight J, You-Ten A, Li WY, Schalm S, Su SM, Virtanen C, Reifenberger G, Ohashi PS, Barber DL, Figueroa ME, Melnick A, Zúñiga-Pflücker JC, Mak TW. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488:656–9. doi: 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, Menn O, Osswald M, Oezen I, Ott M, Keil M, Bal J, Rauschenbach K, Grabowska AK, Vogler I, Diekmann J, Trautwein N, Eichmüller SB, Okun J, Stevanović S, Riemer AB, Sahin U, Friese MA, Beckhove P, von Deimling A, Wick W, Platten M. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. 2014;512:324–7. doi: 10.1038/nature13387. [DOI] [PubMed] [Google Scholar]
- 37.Seehawer M, Heinzmann F, D’Artista L, Harbig J, Roux PF, Hoenicke L, Dang H, Klotz S, Robinson L, Doré G, Rozenblum N, Kang TW, Chawla R, Buch T, Vucur M, Roth M, Zuber J, Luedde T, Sipos B, Longerich T, Heikenwälder M, Wang XW, Bischof O, Zender L. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature. 2018;562:69–75. doi: 10.1038/s41586-018-0519-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seton-Rogers S. Ageing matrix promotes metastasis. Nat Rev Cancer. 2018;18:721. doi: 10.1038/s41568-018-0079-3. [DOI] [PubMed] [Google Scholar]
- 39.Shih AH, Levine RL. IDH1 mutations disrupt blood, brain, and barriers. Cancer Cell. 2012;22:285–7. doi: 10.1016/j.ccr.2012.08.022. [DOI] [PubMed] [Google Scholar]
- 40.Trujillo JA, Sweis RF, Bao R, Luke JJ. T cell-inflamed versus Non-T cell-inflamed tumors: a conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol Res. 2018;6:990–1000. doi: 10.1158/2326-6066.CIR-18-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Veliça P, You J, Chia GS, Sim J, Doedens A, Abelanet A, Evans CE, Griffiths JR, Poellinger L, Goldrath AW, Johnson RS. S-2-hydroxyglutarate regulates CD8+ T-lymphocyte fate. Nature. 2016;540:236–241. doi: 10.1038/nature20165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Urban DJ, Martinez NJ, Davis MI, Brimacombe KR, Cheff DM, Lee TD, Henderson MJ, Titus SA, Pragani R, Rohde JM, Liu L, Fang Y, Karavadhi S, Shah P, Lee OW, Wang A, McIver A, Zheng H, Wang X, Xu X, Jadhav A, Simeonov A, Shen M, Boxer MB, Hall MD. Assessing inhibitors of mutant isocitrate dehydrogenase using a suite of pre-clinical discovery assays. Sci Rep. 2017;7:12758. doi: 10.1038/s41598-017-12630-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waitkus MS, Diplas BH, Yan H. Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell. 2018;34:186–195. doi: 10.1016/j.ccell.2018.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–73. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yusuf RZ, Wang YH, Scadden DT. The secrets of the bone marrow niche: metabolic priming for AML. Nat Med. 2012;18:865–867. doi: 10.1038/nm.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou P, Shaffer DR, Alvarez Arias DA, Nakazaki Y, Pos W, Torres AJ, Cremasco V, Dougan SK, Cowley GS, Elpek K, Brogdon J, Lamb J, Turley SJ, Ploegh HL, Root DE, Love JC, Dranoff G, Hacohen N, Cantor H, Wucherpfennig KW. In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature. 2014;506:52–57. doi: 10.1038/nature12988. [DOI] [PMC free article] [PubMed] [Google Scholar]