

Abstract
Chimeric antigen receptor (CAR) immunotherapy has recently shown promise in clinical trials for B-cell malignancies; however, designing CARs for T-cell based diseases remain a challenge since most target antigens are shared between normal and malignant cells, leading to CAR-T cell fratricide. CD7 is highly expressed in T-cell acute lymphoblastic leukemia (T-ALL), but it is not expressed in one small group of normal T lymphocytes. Here, we constructed monovalent CD7-CAR-NK-92MI and bivalent dCD7-CAR-NK-92MI cells using the CD7 nanobody VHH6 sequences from our laboratory. Both CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI cells consistently showed specific and potent anti-tumor activity against T-cell leukemia cell lines and primary tumor cells. We observed robust cytotoxicity of the bivalent mdCD7-CAR-NK-92MI monoclonal cells against primary T-ALL samples. In agreement with the enhanced cytotoxicity of mdCD7-CAR-NK-92MI cells, significant elevations in the secretion of Granzyme B and interferon γ (IFN-γ) were also found in mdCD7-CAR-NK-92MI cells in response to CD7-positive primary T-ALL cells compared with NK-92MI-mock cells. Furthermore, we also demonstrated that mdCD7-CAR-NK-92MI cells significantly inhibited disease progression in xenograft mouse models of T-ALL primary tumor cells. Our data suggest that CD7-CAR-NK-92MI cells can be used as a new method or a complementary therapy for treating T-cell acute lymphocytic leukemia.
Keywords: T-cell acute lymphoblastic leukemia, chimeric antigen receptor, CD7, NK-92MI, American type culture collection
Introduction
T-cell malignancies represent a class of hematologic cancers with high rates of relapse and mortality in both children and adults, for which there are currently no effective or targeted therapies [1,2]. T-cell acute lymphoblastic leukemia (T-ALL) is a highly heterogeneous hematological malignancy that accounts for 25% of adult acute lymphocyte leukemia cases and 15% of pediatric acute lymphocyte leukemia cases [3]. Currently, treatment strategies for T-ALL include intensive chemotherapy [4], allogeneic hematopoietic stem cell transplantation (allo-HSCT) [5], antiviral therapy [6], and molecular targeted therapy [7]. However, intensive chemotherapy and allo-HSCT often do not prevent treatment-refractory relapse. For those who relapse after initial therapy, salvage chemotherapy regimens induce remissions in 20-40% of cases. Although allo-HSCT is the only curative therapy, it has been associated with a risk of treatment related mortality [8].
In recent years, chimeric antigen receptor T cell (CAR-T) therapy has shown promise results, as a powerful, new adoptive-immunotherapy technique in treating several solid and hematological cancers, most notably B-cell lymphocytic leukemia and lymphoma [9-11]. CAR-T therapy utilizes modified patient T lymphocytes to target and eliminate malignancies in a major histocompatibility complex-independent manner. The key to effectively applying this technology is identifying a suitable target for the CAR. The optimal target antigen should be highly expressed by tumor cells and absent on normal cells, or be expressed only by normal cells whose temporary absence is clinically manageable [12]. Thus, leukemias and lymphomas of B-cell origin can be targeted with CARs directed against CD19 or CD22, which are normally expressed only by B-lymphoid cells [13,14]. Infusion of autologous T cells expressing anti-CD19 CARs into patients with refractory B-cell leukemia and lymphoma resulted in significant clinical responses [15-17]. These results have provided indisputable evidence supporting the power of this technology in clinical practice.
CD7 is a lineage-specific antigen that is highly expressed on the blasts of T-ALL and 30% of acute myeloid leukemia (AML) [18-21]. CD7 is commonly expressed in T-ALL and normal T lymphocytes, but at least absent on a small group of normal T lymphocytes [22]. Besides, CD7 appears not to be pivotal contribution to T-cell development or function, since disrupting the CD7 gene in murine T-cell progenitor cells permits normal T-cell development and homeostasis, with only minor alterations in T-cell effector functions [23,24]. Therefore, CD7 may be a particularly appropriate target for treating T-ALL. However, several challenges have limited the development of CD7-CAR-T cells. First, the shared expression of CD7 antigens between effector cells and malignancies results in fratricide in clinical practice. Second, harvesting adequate numbers of autologous T cells from the patients with refractory disease, without contamination by malignant cells, is technically challenging.
Natural killer (NK) cells play an essential role in innate immune defenses against malignant cells, making them potentially effector cells for adoptive immunotherapy [25]. The NK-92 cell line derived from the peripheral blood mononuclear cells of a non-Hodgkin’s lymphoma patient is the only NK cell line tested in clinical trials for immunotherapy, and its safety and expansion feasibility have been validated in a phase I trial for renal cell cancer and melanoma [26,27]. NK-92 cells lack almost all inhibitory killer cell immunoglobulin-like receptors (KIRs) except for KIR2DL4, which inhibits NK cell activation by binding to human leukocyte antigen molecules on target cells [28]. NK-92MI cells were derived from the NK-92 cell line by stably transfecting an interleukin-2 (IL-2) gene, making them IL-2-independent [27,29], conferring the same characteristics to activated NK cells as the parental NK-92 cells [30]. CAR-modified NK cells are expected to be exhausted shortly after tumor cell lysis [31]. This transient effect may preclude the need for an inducible safety switch [32,33]. In addition, NK cells have been observed to mediate antitumor effects with little risk for graft-versus-host disease and have been validated in CAR applications [34], as has the efficacy in several clinical trials [35-39].
In this study, we constructed two CD7-CAR-NK-92MI cell lines (monovalent CD7-CAR-NK-92MI and bivalent dCD7-CAR-NK-92MI). Our results show that both CD7-CAR NK-92MI cells could specifically eliminate CD7+ T-ALL cell lines and CD7+ T-ALL primary tumor cells in vitro. We also demonstrate that bivalent dCD7-CAR-NK-92MI monoclonal cells (mdCD7-CAR-NK-92MI cells) have potent tumor-directed cytotoxicity in a mouse xenograft model of T-ALL primary tumor cells, significantly improving overall survival of mice compared to controls.
Materials and methods
Cell lines and primary tumor cells
The CD7-positive Jurkat and CCRF-CEM leukemia cell lines, and the CD7-negative Raji lymphoblastic cell line were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). The NK-92MI cell line, which expresses human IL-2, was also purchased from the ATCC. T-ALL primary tumor cells were provided by the Department of Hematology and approved by the Ethics Committee at the Jiangsu Province Hospital of Traditional Chinese Medicine. Jurkat, CCRF-CEM, Raji, and T-ALL primary cells were cultured in RPMI-1640 medium (Hyclone) supplemented with 10% fetal bovine serum (Gibco). NK-92MI cells were incubated in an alpha modification of Eagle’s minimum essential medium (MEM-α, Invitrogen, Carlsbad, CA) containing 2 mM l-glutamine and 1.5 g L-1 sodium bicarbonate. All cells used in this study were cultured in the presence of 0.1 mg/mL streptomycin (Gibco) and 100 U/mL penicillin (Gibco) at 37°C in a humidified atmosphere containing 5% CO2.
Construction of the CD7 specific-CAR vector
We constructed two CD7-CAR vectors using the monovalent and bivalent CD7 nanobody sequence (VHH6) identified in our laboratory by screening [40]. The monovalent CD7 nanobody sequence VHH6 or bivalent CD7 nanobody sequence VHH6-VHH6 was joined to the signal peptide, Fc hinge, CD28 transmembrane and intracellular domain, and intracellular domain of 4-1BB and CD3ζ, resulting in the CD7-CAR or dCD7-CAR cassette, respectively. The CD7-CAR and dCD7-CAR cassettes were individually sub-cloned into XbaI and XhoI sites of the pHULK PiggyBac Mammalian Expression Vector and designated as the CD7-CAR plasmid and dCD7-CAR plasmids, respectively.
Generation of CD7-specific CAR-modified NK-92MI cells
To construct the CD7-CAR-NK-92MI and dCD7-CAR-NK92-MI stable cell lines, NK-92MI cells were suspended in 100 µl electroporation buffer (catalog #VCA-1001, Lonza) containing 5 “µg of the CD7-CAR plasmid or the dCD7-CAR plasmid, and electroporated with an Amaxa Nucleofector 2b device (Lonza, Germany) using program U14. NK-92MI cells electroporated without CAR plasmid were used as a negative control. After electroporation, NK-92MI cells were placed in a 6-well plate (Labserv, Fisher Scientific, USA) and cultured in MEM-α. CD7-CAR-positive and dCD7-CAR-positive NK-92MI cells were sorted with a FACSAriaTM III Cell Sorter (BD Biosciences, NJ) and were cultured in medium with puromycin (1 μg/ml) (Thermo Fisher Scientific, MA).
Flow cytometry
To detect CD7-CAR or dCD7-CAR expression on the surface of NK-92MI cells, we stained NK-92MI, CD7-CAR-NK-92MI, and dCD7-CAR-NK-92MI cells with APC-conjugated anti-human IgG Fc antibodies (Jackson ImmunoResearch). To detect expression of the CD7 antigen on the cell surface, we stained Jurkat, CCRF-CEM, Raji, T-ALL primary tumor, NK-92MI, CD7-CAR-NK-92MI, and dCD7-CAR-NK-92MI cells with APC-conjugated anti-CD7 antibodies (Becton Dickinson). Specifically, the cells were incubated with antibodies at 37°C for 15 min, washed three times with phosphate-buffered saline (PBS), and then analyzed using a flow cytometer (BD Biosciences).
Western blotting
CD7-NK-92MI, dCD7-NK-92MI, and NK-92MI cells were prepared separately. NK-92MI cells served as a control group. The lysates were separated by 8-12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore). The membranes were blocked with 5% skim milk for 1 h and incubated overnight with a mouse anti-human CD3ζ antibody (BD) at 4°C. Immune complexes were detected by incubating the PVDF membranes with a horseradish peroxidase (HRP)-conjugated anti-mouse IgG1-Fc antibody (Solarbio) at 37°C for 1 h and developing the membranes with enhanced chemiluminescence reagents (Millipore).
Cytotoxicity assays
We used the CFSE/7-AAD flow cytometry assay method to test the cytotoxicity of CD7-CAR-NK-92MI or dCD7-CAR-NK-92MI cells to T-ALL tumor cell lines or primary tumor cells. The target cells (Jurkat, CCRF-CEM, Raji, T-ALL primary tumor cell sample 1) were resuspended in PBS and treated with 1 μL carboxyfluorescein succinimidyl ester (CFSE) at 37°C for 30 min. Raji cells served as a negative target cell control group. The effector cells were added at various effector:target (E:T) ratios, and co-cultured with target cells for 4 or 24 h in a 24-well plate, after which the cells were collected, re-suspended in an equal volume of PBS, and 7-AAD (7-aminoactinomycin D; BD) was added. The percentage of specific lysis (CFSE-positive and 7-AAD-positive) was determined by flow cytometry [41]. CFSE-positive cells were the target cells, and the percentage of 7-AAD-positive cells reflected the death rate of the target cells, while the percentage of 7-AAD-negative cells reflected the percentages of leftover target cells.
CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI monoclonal screening
Mouse trophoblasts (5 × 103) were added to each well of a 96-well plate, with each well containing 200 µl of MEM-α medium. Monoclonal CD7-CAR-NK-92MI or dCD7-CAR-NK-92MI cells was sorted using a FACSAriaTM III Cell Sorter (BD Biosciences, NJ) and APC-conjugated anti-human IgG Fc antibodies (Jackson ImmunoResearch). Then, the monoclonal cells were added to a 96-well plate containing mouse trophoblast cells for co-culture. Colonies observed under a microscope after about two weeks were expanded in a 48-well plate. Finally, the CAR-positive rates of monoclonal cells were tested using a flow cytometer (BD Biosciences).
Cytotoxicity analysis of different monoclonal cells
CCRF-CEM cells (CD7-positive tumor cells) were selected as target cells for killing experiments, and various monovalent or bivalent CD7-CAR-NK-92MI monoclonal cells served as effector cells. The experimental method used was the same as that described above (2.6. Cytotoxicity assays). After co-culture for 24 h at an E:T ratio of 1:1, flow cytometric analysis was performed to screen for the most active monoclonal cell line.
Cytokine secretion of different monoclonal cells
The ability of CD7-NK-92MI and dCD7-NK-92MI cells to produce interferon γ (IFN-γ) and Granzyme B in response to CCRF-CEM cells (CD7-positive tumor cells) was analyzed using a cytometric bead array (CBA) system. The effector cells were co-cultured with a constant number of target cells (2 × 105) at an E:T ratio of 1:1 in 24-well microplates in a final volume of 1 ml RPMI 1640 complete medium. After incubation for 24 h, the supernatant was collected and used for CBA assays. The Human Granzyme B CBA Flex Set D7 Kit (catalog #560304) and the Human IFN-γ CBA Flex Set E7 Kit (catalog #558269) were obtained from BD.
Cytotoxicity of bivalent mdCD7-CAR-NK-92MI monoclonal cells to T-ALL primary tumor cells
We used the most active bivalent CD7-CAR-NK-92MI monoclonal cells (mdCD7-CAR-NK-92MI) to kill T-ALL primary tumor cells in vitro. T-ALL primary tumor cells were labeled with CFSE for 30 min at 37°C and seeded into round-bottomed 24-well plates, after which mdCD7-CAR-NK92MI cells were added at various E:T ratios and co-cultured with target cells for 24 h. After 24 h, the supernatant was collected for analysis of IFN-γ and granzyme B secretion using the CBA kit, and the cells were resuspended in PBS containing 1 μL 7-aminoactinomycin D (7-AAD; BD). The cytotoxicity rates were then analyzed using a FACSCalibur flow cytometer (BD).
Patient-derived xenograft (PDX) model
Six to seven-week-old, female NOD-PrkdcscidIl2rgtm1/Bcgen (B-NSG) mice (Biocytogen) were engrafted with T-ALL primary tumor cells (T-ALL sample 1) by tail vein injection. T-ALL primary tumor cells were provided by the Department of Hematology at Jiangsu Hospital of Traditional Chinese Medicine. Three B-NSG mice were injected with T-ALL primary tumor cells (1 × 107 cells per mouse), and the moribund mice were euthanized. After a red cell-lysis procedure, the spleen T-ALL cells collected from the mice were immunotyped by flow cytometry. We transplanted those T-ALL cells into 30 mice, which were divided into two groups (i.e., the low-tumor load group and the high-tumor load group). Each mouse in the low-tumor load group was injected with 2 × 106 T-ALL cells, and each mouse in the high tumor load group was injected with 1 × 107 T-ALL cells. The low-tumor load group and high-tumor load group mice were divided into three cohorts: (i) mice subjected to PBS (n = 5), (ii) mice subjected to intravenous (i.v.) administration of 1 × 107 60Co-irradiated (10 Gy) NK-92MI cells (n = 5), and (iii) mice subjected to i.v. administration of 1 × 107 60Co-irradiated (10 Gy) mdCD7-NK-92MI cells (n = 5). Three days after the mice were injected with the tumor cells, they were further treated by injection with NK-92MI/md-CD7-CAR-NK-92MI cells, once every 2-4 days for a total of five injections according to the state of the mice. Three days after the last administration, the tumor burden and NK-92MI/mdCD7-CAR-NK-92MI of the mice were measured by blood sampling from the eyelids. All in vivo mouse experiments were conducted in accordance with the experimental animal management regulations of Soochow University.
Statistical analysis
Statistical analyses were performed using GraphPad Prism Software (version 5.0). Paired t-tests were used to compare differences between groups. The brackets in the figures indicate which groups were compared, and *, **, and ***indicate P < 0.05, < 0.01, and < 0.001, respectively.
Results
Characterization of the CD7-CAR/dCD7-CAR construct and generation of CAR-NK-92MI cells
To target CD7, we constructed two CD7-CARs using the monovalent and bivalent nanobody VHH6 sequences developed in our laboratory. The monovalent CD7-CAR was comprised of a signal peptide, an anti-CD7 nanobody sequence (VHH6), a human Fc hinge region, a CD28 transmembrane domain, and CD28 and 4-1BB intracellular signaling domains in tandem with the CD3ζ signaling domain. The bivalent dCD7-CAR contained a signal peptide, anti-CD7 nanobody repeat sequence (VHH6-VHH6), a human Fc hinge region, a CD28 transmembrane domain, and CD28 and 4-1BB intracellular signaling domains in tandem with the CD3ζ signaling domain. A schematic representation of the resulting CAR constructs is shown in Figure 1A. The CD7-CAR and dCD7-CAR cassettes were sub-cloned into the pHULK PiggyBac Mammalian Expression Vector and designated as the CD7-CAR plasmid and dCD7-CAR plasmids, respectively.
Figure 1.
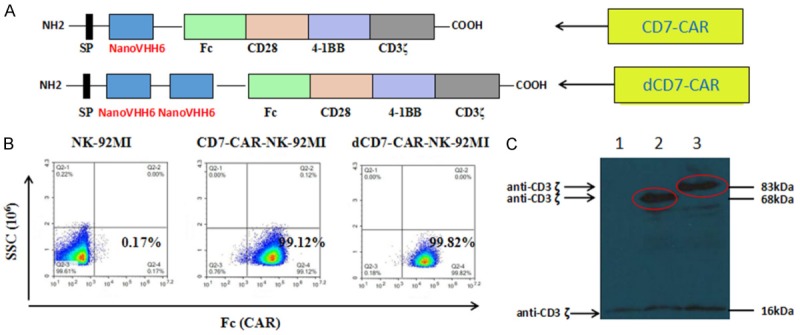
Organization of the CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI construct and its expression. A. Schematic representation of the CD7-specific CAR vector. The CD7-CAR construct contained a signal peptide sequence, a monovalent nanobody VHH6 sequence (with dCD7-CAR containing a bivalent VHH6 sequence), a hinge domain (Fc), two co-stimulatory domains (CD28 and 4-1BB), and the intracellular signaling domain CD3ζ. B. Flow cytometry analysis of cell-surface expression of CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI on NK-92MI cells. C. Western blot analysis of CD7-CAR expression in NK-92MI cells. Lysates of NK-92MI (lane 1), CD7-NK-92MI (lane 2), and dCD7-NK-92MI (lane 3) cells were separated by SDS-PAGE under reducing conditions. Immunoblot analysis was performed with a CD3ζ chain-specific mAb followed by exposure to an HRP-conjugated anti-mouse antibody and chemiluminescent detection. The positions of endogenous and chimeric CD3ζ fusion proteins and of molecular weight standards (kDa) are indicated.
After electroporation, CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI cells were sorted by flow cytometry using an anti-Fc antibody. Following sorting, CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI cells were selected in vitro for 3-4 months in medium containing puromycin (1 μg/ml), resulting in stable transgene expression. CAR protein expression on the cell surface of CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI cells was demonstrated by flow cytometry, which revealed that approximately 99% of both CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI were CAR+ (Figure 1B).
Expression of the CD7-CAR and dCD7-CAR fusion proteins was also examined by western blotting (Figure 1C). Under reducing conditions, the endogenous CD3ζ chain was detected as a 16-kDa band in NK-92MI (lane 1), CD7-CAR-NK-92MI (lane 2), and dCD7-CAR-NK-92MI (lane 3) cell lysates. Two additional bands were only observed in CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI cells, and matched the calculated size (68-kDa or 83-kDa) of the monovalent CD7-CAR fusion protein or the bivalent dCD7-CAR fusion protein, respectively (Figure 1C).
Expression of CD7 in NK-92MI and T-ALL cells
CD7 expression was examined in NK-92MI, CD7-CAR-NK-92MI, and dCD7-CAR-NK-92MI cells by flow cytometry. The results showed that 8.42% of NK-92MI cells expressed cell-surface CD7, while only below 1% of NK-92MI cells transfected with CD7-CAR (CD7-CAR-NK-92MI or dCD7-CAR-NK-92MI) were CD7+ (Figure 2A). These findings indicate that CD7-CAR-NK-92MI or dCD7-CAR-NK-92MI cells can specifically kill CD7+ NK-92MI cells. We also examined CD7 expression on the surface of leukemia cell lines (Jurkat and CCRF-CEM), a lymphoblastic cell line (Raji), and cells from T-ALL primary tumor sample 1. The results showed that CD7-positive rates in CCRF-CEM and Jurkat cells were nearly 100% (Figure 2B) and 93% for T-ALL sample 1 (Figure 2C, the control group was T-ALL cells that were not incubated with CD7 antibody.), whereas Raji cells were CD7-negative (Figure 2B).
Figure 2.
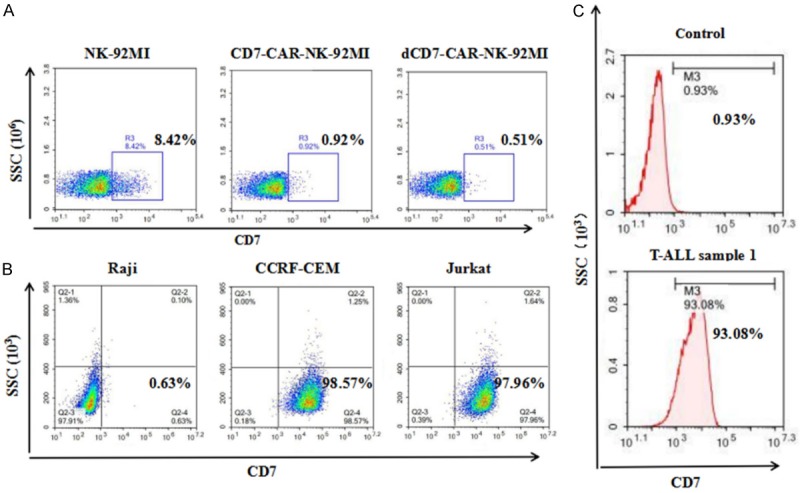
The expression level of CD7 in NK-92MI cells and T-ALL tumor cells. A. Expression of CD7 in NK-92MI cells and changes of CD7 expression on NK-92MI cells after transfection with the CD7-CAR construct. B. CD7 expression levels in T-ALL tumor cell lines (CCRF-CEM and Jurkat), or Raji cells (a CD7-negative target cell line). C. CD7 expression levels in primary T-ALL tumor cells (sample 1).
CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI cells eliminated T-ALL cell lines in vitro
We evaluated the anti-tumor activity of CD7-CAR-NK92-MI and dCD7-CAR-NK92-MI cells using CD7+ T-ALL cell lines (CCRF-CEM and Jurkat cells). The CD7-negative lymphoblastic cell line, Raji, was used as a negative cell line. CD7-CAR-NK92-MI and dCD7-CAR-NK92-MI cells were tested after co-culturing for 4 or 24 h in vitro with CCRF-CEM cells at an E:T cell ratio of 1:1. Compared to control NK-92MI cells, we observed that CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI cells consistently and robustly eliminated CD7+ CCRF-CEM cells. At an E:T cell ratio of 1:1, CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI cells showed significant specific cytotoxicity against CCRF-CEM cells compared to control NK92-MI cells, regardless of the co-culture time (4 or 24 h; Figure 3A). We also evaluated the ability of CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI cells to kill CD7+ Jurkat cells at E:T cell ratios of 1:1 and 5:1. After 24 hours of incubation with Jurkat cells, the results showed that the percentage of leftover tumor cells in the CD7-CAR-NK-92MI group and the dCD7-CAR-NK-92MI group were significantly lower than the control NK-92MI group (Figure 3B). CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI exerted significant cytotoxicity against Jurkat cells compared to control NK-92MI cells. However, no difference in cytotoxicity was detected between NK-92MI and either CD7-CAR-NK-92MI or dCD7-CAR-NK92-MI versus Raji cells at an E:T cell ratio of 5:1 (Figure S1). These results indicate that CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI were cytotoxic specifically against CD7-positive tumor cells, but not CD7-negative cells.
Figure 3.

CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI cells specifically eliminated CD7-expressing T-ALL cell lines and primary tumor cells in vitro. A. At a 1:1 E:T cell ratio, CD7-CARNK-92MI and dCD7-CAR-NK-92MI cells targeted and lysed the CD7+ T-ALL cell line CCRF-CEM. B. CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI cells showed specific lysis of CD7+ Jurkat cells at an E:T cell ratio of 1:1 or 5:1, the 7-AAD negative cell population represents the percentage of leftover Jurkat cells. C. At a 1:1 or 5:1 E:T cell ratio, the CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI cells eliminated primary T-ALL tumor cells.
The antitumor effects of CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI against T-ALL primary tumor cells
To assess their anti-leukemia potentials, we tested the efficiencies of CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI cells in recognizing and killing primary T-ALL tumor cells. Co-culture experiments were conducted using T-ALL primary sample 1 from a leukemia patient unresponsive to standard chemotherapy. The CD7-positive rate in T-ALL sample 1 was 93% (Figure 2C). The result showed that CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI cells had significant specific cytotoxicity against primary T-ALL tumor cells compared to control NK-92MI cells with E:T cell ratios of 1:1 and 5:1 (Figure 3C). These finding indicate that CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI are specifically toxic to T-ALL cell lines and primary T-ALL tumor cells in vitro.
Comparison of the activity of different monovalent CD7-CAR-NK-92MI and bivalent dCD7-CAR-NK-92MI monoclonal cells
To maintain the stable expression of CAR on CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI cells in long-term culture, we further performed monoclonal cell screening. We finally screened eight monovalent CD7-CAR-NK-92MI monoclonal cell lines, and the CD7-CAR-positive rates of eight monoclonal cell lines were close to 100% (Figure 4A). We used CCRF-CEM cells as target cells and compared the killing activity of eight monoclonal cells against CCRF-CEM cells in vitro (Figure 4B). The results indicate that eight monovalent CD7-CAR-NK-92MI monoclonal cell lines showed significant cytotoxicity to CCRF-CEM cells at an E:T cell ratio of 1:1, but there was no significant difference in cytotoxicity. Then we analyzed the secretion of IFN-γ and Granzyme B after incubation of eight monovalent CD7-CAR-NK-92MI monoclonal cell lines with CCRF-CEM cells (Figure 5A, 5B). The results showed that the release of IFN-γ was significantly different in different monoclonal cell lines, but the release of granzyme B was not significantly different. In addition, we screened six bivalent dCD7-CAR-NK-92MI monoclonal cell lines, and the CD7-CAR positive rates of six monoclonal cell lines were also close to 100% (Figure 4C). Six bivalent dCD7-CAR-NK-92MI monoclonal cell lines also showed significant cytotoxicity to CCRF-CEM cells at an E:T cell ratio of 1:1 (Figure 4D). We also analyzed IFN-γ and Granzyme B secretion after incubating six bivalent dCD7-CAR-NK-92MI monoclonal cell lines with CCRF-CEM cells (Figure 5C, 5D). The results showed that the levels of IFN-γ and Granzyme B released were significantly different among the different monoclonal cell lines.
Figure 4.

Comparison of the cytotoxicity of different monovalent CD7-CAR-NK92-MI and bivalent dCD7-CAR-NK-92MI monoclonal cell lines against CCRF-CEM cells. A. Detection of the CAR-positive rate of eight monovalent CD7-CAR-NK-92MI monoclonal cell lines screened. B. Cytotoxicity of eight monovalent CD7-CAR-NK-92MI monoclonal cell lines to CCRF-CEM cells. The cytotoxicities of different monoclonal cell lines against CCRF-CEM cells after a 24-h co-culture at an E:T cell ratio of 1:1 are shown, using eight CD7-CAR-NK-92MI monoclonal cell lines. C. Detection of the CAR-positive rate of six bivalent dCD7-CAR-NK-92MI monoclonal cell lines screened. D. Cytotoxicity of six bivalent dCD7-CAR-NK-92MI monoclonal cell lines to CCRF-CEM cells. The cytotoxicities of different monoclonal cell lines against CCRF-CEM cells after a 24-h co-culture at an E:T cell ratio of 1:1 were compared, using six dCD7-CAR-NK-92MI monoclonal cell lines.
Figure 5.
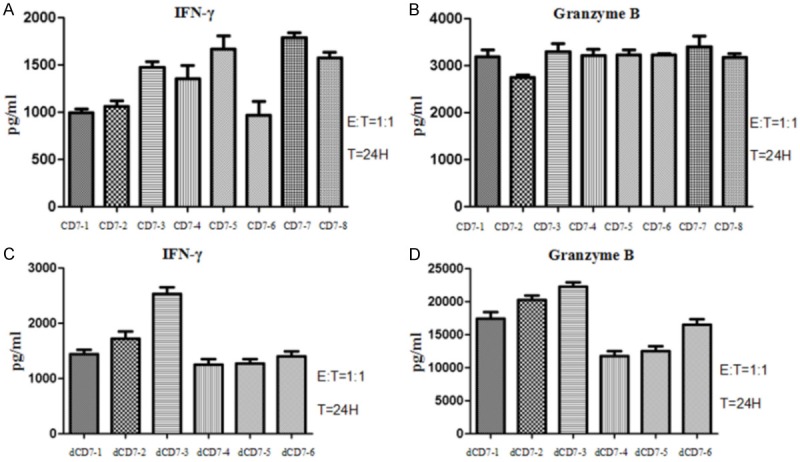
Comparison of cytokine secretions in different monoclonal cell lines. A. IFN-γ secretion after 24 h in co-cultures with an E:T cell ratio of 1:1, using eight CD7-CAR-NK-92MI monoclonal cell lines against CCRF-CEM cells. B. Granzyme B secretion after 24 h in co-cultures with an E:T ratio of 1:1, using eight CD7-CAR-NK-92MI monoclonal cell lines against CCRF-CEM cells. C. IFN-γ secretion after 24 h in co-culture with an E:T cell ratio of 1:1, using six dCD7-CAR-NK-92MI monoclonal cell lines against CCRF-CEM cells. D. Granzyme B secretion after 24 h in co-cultures with an E:T ratio of 1:1, using six dCD7-CAR-NK-92MI monoclonal cell lines against CCRF-CEM cells.
These results demonstrated that, although the cytotoxicities of the monovalent CD7-CAR-NK-92MI and the bivalent dCD7-CAR-NK-92MI cells to CCRF-CEM cells were very close, the dCD7-CAR-NK-92MI cells was slightly stronger (Figure 4B, 4D). IFN-γ and Granzyme B release was variable among the different monoclonal cell lines (Figure 5). The ability of secreting Granzyme B by bivalent dCD7-CAR-NK-92MI monoclonal cells was significantly higher than the monovalent CD7-CAR-NK-92MI monoclonal cells (Figure 5B, 5D). The dCD7-3 clone derived from the dCD7-CAR-NK-92MI monoclonal cell line showed the strongest cytokine secretion capacity among all monoclonal cell lines tested. We named the bivalent dCD7-3 monoclonal cell line as the mdCD7-CAR-NK-92MI cell line.
mdCD7-CAR-NK-92MI cells recognized and eliminated primary T-ALL tumor cells
To further evaluate the capacity of mdCD7-CAR-NK-92MI cells to respond to primary tumors, we tested their ability to kill T-ALL primary tumor samples from T-ALL patient 2 (T-ALL sample 2) in vitro, and measured cytokine production upon co-culture with primary T-ALL tumor cells. CD7 was highly expressed in T-ALL sample 2 (Figure S2A). After 24 hours of incubation with primary T-ALL cells, we observed the percentage of leftover tumor cells in the mdCD7-CAR-NK-92MI group was significantly lower than the control NK-92MI group at an E:T cell ratio of 1:1 or 5:1 (Figure S2B). The results showed robust cytotoxicity of the mdCD7-CAR-NK-92MI cells against primary T-ALL tumor cells at an E:T cell ratio of 1:1 or 5:1, after 24 hours of co-culture. We also observed that the mdCD7-CAR-NK-92MI cells produced increased levels of IFN-γ and Granzyme B (Figure S2C, S2D). These results indicate that the mdCD7-CAR-NK-92MI cells efficiently eradicated primary T-ALL tumor cells in vitro.
mdCD7-CAR-NK-92MI cells demonstrated potent anti-leukemic activity in vivo
To evaluate the antitumor activity of mdCD7-CAR-NK-92MI cells in vivo, we used mouse xenograft model of patient-derived T-ALL. Three B-NSG mice were injected with primary T-ALL tumor cells (1 × 107 cells per mouse), and the moribund mice were euthanized (Figure S3A). After a red cells lysis proceed, the spleen T-ALL cells collected from one mouse were immunotyped by flow cytometry as being comprised of nearly 90% T-ALL tumor cells (Figure S3B). To test the mdCD7-CAR-NK-92MI cells effect on the PDX model, we transplanted those T-ALL cells to 30 mice. The experimental scheme of the animal experiment is shown in Figure 6A. We administered 2.0 × 106 T-ALL cells (T-ALL dose 1) or 1.0 × 107 T-ALL cells (T-ALL dose 2) to B-NSG mice (15 mice per group) via i.v. injection. After 3 days, NK-92MI and md-CD7-CAR-NK-92MI cells were administrated once every 2-4 days for a total of five injections. Three days after the last administration, the tumor burden of the mice was measured by blood sampling from the eyelids. The results showed that mdCD7-CAR-NK-92MI cells significantly prolonged mouse survival compared with the PBS-control group and the NK-92MI group (Figure 6C, 6E). Flow cytometric analysis also demonstrated mdCD7-CAR-NK-92MI showed robust control and suppression of primary T-ALL tumor growth in xenogeneic mouse models (Figure 6F, 6G). We also detected the proportion of NK-92MI/CD7-CAR-NK-92MI cells in peripheral blood of mice by flow cytometry, the result showed that NK-92MI/CD7-CAR-NK-92MI cells were not detected in mice after three days of NK-92MI cell reinfusion (Figure S4), this indicates that the duration of NK-92MI cells in mice is very short. In addition, slight weight loss was transiently observed for most mice, which fully recovered after the last injection of mdCD7-CAR-NK-92MI cells (Figure 6B, 6D).
Figure 6.

mdCD7-CAR-NK-92MI cells demonstrated potent antitumor activity in a PDX model. A. Schematic representation of a primary T-ALL xenograft model. B-NSG mice (15 mice per group) were injected i.v. with 2.0 × 106 T-ALL cells (T-ALL dose 1) or 1.0 × 107 T-ALL cells (T-ALL dose 2). After 3 days, NK-92MI and md-CD7-CAR-NK-92MI cells were administrated once every 2-4 days for a total of five injections. B. The average body weights of mice in the low-tumor load group (T-ALL dose 1). C. Survival curves of mice in the low-tumor load group (T-ALL dose 1). n = 5. Statistical analysis was performed using the log-rank test. **P < 0.01. D. Average body weights of mice in the high-tumor load group (T-ALL dose 2). E. Survival curves of mice in the high-tumor load group (T-ALL dose 2). n = 5. Statistical analysis was performed using the log-rank test. **P < 0.01. F. Flow cytometric analysis of the peripheral blood tumor burden after 17 days of mdCD7-CAR-NK-92MI cell therapy. G. Statistical analysis of the peripheral blood tumor burden after 17 days of mdCD7-CAR-NK-92MI cell therapy. **P < 0.01, n = 3.
In summary, in PDX mouse models, mdCD7-CAR-NK-92MI cells robustly reduced tumor burdens, controlled tumor growth, and significantly prolonged survival in the B-NSG mice injected with primary T-ALL tumor cells when compared to those treated with NK-92MI cells as a control.
Discussion
Durable remissions in patients with refractory B-cell leukemia and lymphoma can be achieved with CAR-T cells, but effective options are lacking for patients with T-cell malignancies. We chose to target CD7 due to its high expression in most T-cell malignancies [42-44] and negative expression on approximately 9% of normal peripheral T cells [22]. In addition to T-cell malignancies, CD7 is expressed in approximately 24% of AML cases and is thought to be a marker of leukemic stem cells [20,45]. Furthermore, we wanted to target an antigen that could be deleted in T cells without impacting immune function. A CD7-deficient mouse model showed a normal lymphocyte population and maintained T cell functions [23]. Thus, CD7 may be a proper target for both T cell malignancies and AML with CD7 expression on the blasts.
Although CD7 is an attractive therapeutic target for T-cell malignancies, effector T cells modified with CD7-specific CARs fail to substantially downregulate CD7 expression, resulting in extensive fratricide and precluded T-cell expansion. Indeed, CD7 CAR-T cells have been generated by several groups and exhibited great potential in treating T cell malignancies. Among of them, gene editing to knock out CD7 molecule is a attractive approach to avoid the fratricide effect. However, in clinical practice, it is impossible to perform CD7 CAR-T trial if using autologous T cells from the patients because non-malignant T cells are hard to be selected by current cell sorting technique from leukapheresis sample. Accordingly, this will lead to no normal T cells for preparing CAR-T cells. Allogeneic T cells from healthy donor may be a solution for CD7 CAR-T cell preparation. It has been recently reported that T-ALL cells can be eliminated by allogeneic CD7-CAR-T cells that are simultaneously knock out of CD7 and T cell receptors alpha chain by CRISPR/Cas9 gene editing [46]. But some challenges still remain to be solved, including gene editing efficiency and potential off target effect which may result in GVHD (graft-versus-host disease), poor persistence, and even serious side effect due to off-targeting. Therefore, our strategy provide another option for T-ALL treatment because NK-92 and NK-92MI cells have been used in multiple clinical trials and did not show risk of GVHD. In addition, the persistence of NK-92 and NK-92MI cells may be worse, however, since NK-92 or NK-92MI cells are off-the-shelf cell products, repeated administration of NK-92 or NK-92MI cells to the patients could be a solution.
In this study, we constructed monovalent CD7-CAR-NK-92MI and bivalent dCD7-CAR-NK-92MI cells using the CD7 nanobody VHH6 sequences identified in our laboratory. Due to a small molecular weight [47], rapid tissue penetration [48], high solubility and stability [49], high specificity of antigen binding, and weak immunogenicity [50], nanobodies are often used in cancer treatment. Nanobody is an antibody fragment consisting of a single monomeric variable antibody domain derived from camelidae heavy-chain antibodies. In our previous studies [40], we demonstrated that our CD7 nanobody exhibited high affinity and specificity to CD7 molecule which lays a solid foundation for successful CAR-NK-92MI therapy. Nanobody may lead to the induction of an immune response when injected into non-camelid hosts. However, it has been thought that nanobody is expected to produce lower immunogenicity due to high sequence homology (more than 80%) between the human VH framework and nanabody framework [51].
We found that CD7-CAR-NK-92MI and dCD7-CAR-NK-92MI cells possessed potent and specific target-directed anti-tumor effects in vitro. Both of the cells lysed malignant target cell lines during co-culture with CCRF-CEM and Jurkat cells. In addition, these cells produced potent and specific anti-tumor effects on primary T-ALL tumor samples. We screened eight monovalent CD7-CAR-NK-92MI and six bivalent dCD7-CAR-NK-92MI monoclonal cell lines, and compared the activities of the monoclonal cells. The mdCD7-CAR-NK-92MI monoclonal cell line was the most active cell line of all monoclonal cell lines. In agreement with the enhanced cytotoxicity of mdCD7-CAR-NK-92MI cells, significant elevations in the secretion of Granzyme B and interferon γ (IFN-γ) were also found in mdCD7-CAR-NK-92MI cells in response to CD7-positive primary T-ALL cells compared with NK-92MI-mock cells. We also tested the secretion of IL-6 and TNF-α in mdCD7-CAR-NK cells using CBA system (Human IL-6 CBA Flex Set A7 Kit, catalog #558276; Human TNF CBA Flex Set D9 Kit, catalog #558273 ), the results showed that mdCD7-CAR-NK-92MI released very little TNF-α and IL-6 after incubation with CCRF-CEM cells (Figure S5). In addition, although CD7-CAR-NK-92MI cells could release high amount of IFNγ during activation in vitro, due to the short duration of NK-92MI cells in vivo, it should not cause a serious CRS response. Our mouse experiments also confirmed that there was no significant toxicity after transfusion of mdCD7-CAR-NK-92MI cells. Only slight weight loss was transiently observed for most mice, which fully recovered after the last injection of mdCD7-CAR-NK-92MI cells (Figure 6B, 6D). Furthermore, mdCD7-CAR-NK-92MI cells robustly reduced tumor burdens, controlled tumor growth, and significantly prolonged survival in B-NSG mice injected with primary T-ALL tumors.
Unlike CAR-T, CAR-NK-92-based therapies will have to balance the persistence of CAR-NK-92 cells with tumor-lytic efficacy. CAR-NK-92-based therapies may serve as a quick-acting transient treatments for depleting target tumor cells bridging transplantation. Unlike CAR-modified autologous T cells, CAR-modified NK-92 or NK-92MI cells have several advantages: (1) they can kill tumor cells directly via the release of toxic granules, (2) they may release low amounts of cytokines and can reduce the risk of cytokine storms, and (3) they can be expanded on a large scale and developed as “off-the-shelf” products. The potential disadvantages of using NK-92 or NK-92MI cells in CAR therapy include a lack of persistence, which may be overcome by multiple administrations of CAR-NK92 cells.
In conclusion, to our knowledge, this is the first report of CD7-CAR-NK-92MI constructed based on anti-CD7 nanobody sequences, that showed a therapeutic potential on T-ALL. We not only demonstrated the specific cytotoxicity of CD7-CAR-transduced NK-92MI cells on T-ALL in vitro, but also showed that CD7-CAR-NK-92MI has an inhibitory effect on tumor cells in PDX mice model, whose survival were increased compared with the control group. This study suggests that CD7-CAR-NK-92-MI cells can be used alone temporarily, or as a bridge for standard treatment procedures.
Acknowledgements
This work was supported by the National Key R&D Program of China (2016YFC1303403), Priority Academic Program Development of Jiangsu Higher Education Institutions, the National Natural Science Foundation of China (Grant No. 31471283), the Collaborative Innovation Major Project (Grant No. XYXT2015304), the Six Talent Peaks Project in Jiangsu Province (No. SWYY-CXTD-010), the Science and Technology Development Program of Jiangsu Province (grant Nos. BE2016809), and the Nanjing Science and Technology Development Program (grant Nos. 201503011).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Karrman K, Johansson B. Pediatric T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2017;56:89–116. doi: 10.1002/gcc.22416. [DOI] [PubMed] [Google Scholar]
- 2.Ma H, Abdul-Hay M. T-cell lymphomas, a challenging disease: types, treatments, and future. Int J Clin Oncol. 2017;22:18–51. doi: 10.1007/s10147-016-1045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Durinck K, Goossens S, Peirs S, Wallaert A, Loocke WV, Matthijssens F, Pieters T, Milani G, Lammens T, Rondou P, Roy NV, Moerloose BD, Benoit Y, Haigh J, Speleman F, Poppe B, Van Vlierberghe P. Novel biological insights in T-cell acute lymphoblastic leukemia. Exp Hematol. 2015;43:625–639. doi: 10.1016/j.exphem.2015.05.017. [DOI] [PubMed] [Google Scholar]
- 4.Utsunomiya A, Choi I, Chihara D, Seto M. Recent advances in the treat-ment of adult T-cell leukemia-lymphomas. Cancer Sci. 2015;106:344–351. doi: 10.1111/cas.12617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin HN, Liu CY, Pai JT, Chang FP, Yang CF, Yu YB, Hsiao LT, Chiou TJ, Liu JH, Gau JP, Tzeng CH, Cheng PM, Hong YC. How to predict the outcome in mature T and NK cell lymphoma by currently used prognostic models? Blood Cancer J. 2012;2:e93. doi: 10.1038/bcj.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kchour G, Tarhini M, Kooshyar MM, El Hajj H, Wattel E, Mahmoudi M, Hatoum H, Rahimi H, Maleki M, Rafatpanah H, Rezaee SA, Yazdi MT, Shirdel A, de Thé H, Hermine O, Farid R, Bazarbachi A. Phase 2 study of the efficacy and safety of the combination of arsenic trioxide, interferon alpha, and zidovudine in newly diagnosed chronic adult T-cell leukemia/lymphoma (ATL) Blood. 2009;113:6528–6532. doi: 10.1182/blood-2009-03-211821. [DOI] [PubMed] [Google Scholar]
- 7.Zain JM, O’Connor O. Targeted treatment and new agents in peripheral T-cell lymphoma. Int J Hematol. 2010;92:33–44. doi: 10.1007/s12185-010-0614-9. [DOI] [PubMed] [Google Scholar]
- 8.Litzow MR, Ferrando AA. How I treat T-cell acute lymphoblastic leukemia in adults. Blood. 2015;126:833–841. doi: 10.1182/blood-2014-10-551895. [DOI] [PubMed] [Google Scholar]
- 9.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, Borquez-Ojeda O, Qu J, Wasielewska T, He Q, Bernal Y, Rijo IV, Hedvat C, Kobos R, Curran K, Steinherz P, Jurcic J, Rosenblat T, Maslak P, Frattini M, Sadelain M. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K, Quintás-Cardama A, Larson SM, Sadelain M. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007;13:5426–5435. doi: 10.1158/1078-0432.CCR-07-0674. [DOI] [PubMed] [Google Scholar]
- 11.Firor AE, Jares A, Ma Y. From humble beginnings to success in the clinic: chimeric antigen receptor-modified T-cells and implications for immunotherapy. Exp Biol Med (Maywood) 2015;240:1087–1098. doi: 10.1177/1535370215584936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campana D, Janossy G, Bofill M, Trejdosiewicz LK, Ma D, Hoffbrand AV, Mason DY, Lebacq AM, Forster HK. Human B cell development. I. Phenotypic differences of B lymphocytes in the bone marrow and peripheral lymphoid tissue. J Immunol. 1985;134:1524–1530. [PubMed] [Google Scholar]
- 14.Nadler LM, Anderson KC, Marti G, Bates M, Park E, Daley JF, Schlossman SF. B4, a human B lymphocyte-associated antigen expressed on normal, mitogen-activated, and malignant B lymphocytes. J Immunol. 1983;131:244–250. [PubMed] [Google Scholar]
- 15.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, Lindgren CG, Lin Y, Pagel JM, Budde LE, Raubitschek A, Forman SJ, Greenberg PD, Riddell SR, Press OW. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012;119:3940–3950. doi: 10.1182/blood-2011-10-387969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Del Poeta G, Stasi R, Venditti A, Cox C, Aronica G, Masi M, Bruno A, Simone MD, Buccisano F, Papa G. CD7 expression in acute myeloid leukemia. Leuk Lymphoma. 1995;17:111–119. doi: 10.3109/10428199509051710. [DOI] [PubMed] [Google Scholar]
- 19.Janossy G, Coustan-Smith E, Campana D. The reliability of cytoplasmic CD3 and CD22 antigen expression in the immunodiagnosis of acute leukemia: a study of 500 cases. Leukemia. 1989;3:170–181. [PubMed] [Google Scholar]
- 20.Miwa H, Nakase K, Kita K. Biological characteristics of CD7(+) acute leukemia. Leuk Lymphoma. 1996;21:239–244. [PubMed] [Google Scholar]
- 21.Shimamoto T, Ohyashiki JH, Ohyashiki K, Kawakubo K, Inatomi Y, Fujieda H, Nakazawa S, Kimura N, Miyauchi J, Toyama K. Clinical and biologic characteristics of CD7 acute myeloid leukemia: our experience and literature review. Cancer Genet Cytogenet. 1994;73:69–74. doi: 10.1016/0165-4608(94)90185-6. [DOI] [PubMed] [Google Scholar]
- 22.Reinhold U, Abken H, Kukel S, Moll M, Müller R, Oltermann I, Kreysel HW. CD7-T cells represent a sub-set of normal human blood lymphocytes. J Immunol. 1993;150:2081–2089. [PubMed] [Google Scholar]
- 23.Bonilla FA, Kokron CM, Swinton P, Geha RS. Targeted gene disruption of murine CD7. Int Immunol. 1997;9:1875–1883. doi: 10.1093/intimm/9.12.1875. [DOI] [PubMed] [Google Scholar]
- 24.Lee DM, Staats HF, Sundy JS, Patel DD, Sempowski GD, Scearce RM, Jones DM, Haynes BF. Immunologic characterization of CD7-deficient mice. J Immunol (Baltim Md 1950) 1998;160:5749–5756. [PubMed] [Google Scholar]
- 25.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, Klingemann H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy. 2008;10:625–632. doi: 10.1080/14653240802301872. [DOI] [PubMed] [Google Scholar]
- 27.Tam YK, Maki G, Miyagawa B, Hennemann B, Tonn T, Klingemann HG. Characterization of genetically altered, interleukin 2-independent natural killer cell lines suitable for adoptive cellular immunotherapy. Hum Gene Ther. 1999;10:1359–1373. doi: 10.1089/10430349950018030. [DOI] [PubMed] [Google Scholar]
- 28.Tonn T, Becker S, Esser R, Schwabe D, Seifried E. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J Hematother Stem Cell Res. 2001;10:535–44. doi: 10.1089/15258160152509145. [DOI] [PubMed] [Google Scholar]
- 29.Tam YK, Miyagawa B, Ho VC, Klingemann HG. Immunotherapy of malignant melanoma in a SCID mouse model using the highly cytotoxic natural killer cell line NK-92. J Hematother. 1999;8:281–290. doi: 10.1089/106161299320316. [DOI] [PubMed] [Google Scholar]
- 30.Favors SE, Curd LM, Gregg RK. Use of the antiinflammatory cytokine interleukin-11 to reverse HIV-1gp120 repression of a natural killer cell line. Cell Immunol. 2012;276:1–5. doi: 10.1016/j.cellimm.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 31.Glienke W, Esser R, Priesner C, Suerth JD, Schambach A, Wels WS, Grez M, Kloess S, Arseniev L, Koehl U. Advantages and applications of CAR-expressing natural killer cells. Front Pharmacol. 2015;6:21. doi: 10.3389/fphar.2015.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, Heslop HE, Rooney CM, Brenner MK, Dotti G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24:1160–1170. doi: 10.1038/leu.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu C, Hong SG, Winkler T, Spencer DM, Jares A, Ichwan B, Nicolae A, Guo V, Larochelle A, Dunbar CE. Development of an inducible caspase-9 safety switch for pluripotent stem cell-based therapies. Mol Ther Methods Clin Dev. 2014;1:14053. doi: 10.1038/mtm.2014.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suck G, Odendahl M, Nowakowska P, Seidl C, Wels WS, Klingemann HG, Tonn T. NK-92: an ‘off-the-shelf therapeutic’ for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol Immunother. 2016;65:485–492. doi: 10.1007/s00262-015-1761-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boissel L, Betancur-Boissel M, Lu W, Krause DS, Van Etten RA, Wels WS, Klingemann H. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. Oncoimmunology. 2013;2:e26527. doi: 10.4161/onci.26527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, Peng Y, Mao H, Yi L, Ghoshal K, He X, Devine SM, Zhang X, Caligiuri MA, Hofmeister CC, Yu J. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28:917–927. doi: 10.1038/leu.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang H, Zhang W, Shang P, Zhang H, Fu W, Ye F, Zeng T, Huang H, Zhang X, Sun W, Man-Yuen Sze D, Yi Q, Hou J. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol Oncol. 2014;8:297–310. doi: 10.1016/j.molonc.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schonfeld K, Sahm C, Zhang C, Naundorf S, Brendel C, Odendahl M, Nowakowska P, Bönig H, Köhl U, Kloess S, Köhler S, Holtgreve-Grez H, Jauch A, Schmidt M, Schubert R, Kühlcke K, Seifried E, Klingemann HG, Rieger MA, Tonn T, Grez M, Wels WS. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther. 2015;23:330–338. doi: 10.1038/mt.2014.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang C, Burger MC, Jennewein L, Genssler S, Schonfeld K, Zeiner P, Hattingen E, Harter PN, Mittelbronn M, Tonn T, Steinbach JP, Wels WS. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst. 2015;108 doi: 10.1093/jnci/djv375. [DOI] [PubMed] [Google Scholar]
- 40.Tang J, Li J, Zhu X, Yu Y, Chen D, Yuan L, Gu Z, Zhang X, Qi L, Gong Z, Jiang P, Yu J, Meng H, An G, Zheng H, Yang L. Novel CD7-specific nanobody-based immunotoxins potently enhanced apoptosis of CD7-positive malignant cells. Oncotarget. 2016;7:34070–34083. doi: 10.18632/oncotarget.8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.You FT, Jiang LC, Zhang BZ, Lu Q, Zhou Q, Liao XY, Wu H, Du KQ, Zhu YC, Meng HH, Gong ZS, Zong YH, Huang L, Lu M, Tang JR, Li YF, Zhai XC, Wang XL, Ye SS, Chen D, Yuan L, Qi L, Yang L. Phase 1 clinical trial demonstrated that MUC1 positive metastatic seminal vesicle cancer can be effectively eradicated by modified anti-MUC1 chimeric antigen receptor transduced T cells. Sci China Life Sci. 2016;59:386–97. doi: 10.1007/s11427-016-5024-7. [DOI] [PubMed] [Google Scholar]
- 42.Campana D, Behm FG. Immunophenotyping of leukemia. J Immunol Methods. 2000;243:59–75. doi: 10.1016/s0022-1759(00)00228-3. [DOI] [PubMed] [Google Scholar]
- 43.Khalidi HS, Chang KL, Medeiros LJ, Brynes RK, Slovak ML, Murata-Collins JL, Arber DA. Acute lymphoblastic leukemia. Survey of immunophenotype, french-american-british classification, frequency of myeloid antigen expression, and karyotypic abnormalities in 210 pediatric and adult cases. Am J Clin Pathol. 1999;111:467–476. doi: 10.1093/ajcp/111.4.467. [DOI] [PubMed] [Google Scholar]
- 44.Patel JL, Smith LM, Anderson J, Abromowitch M, Campana D, Jacobsen J, Lones MA, Gross TG, Cairo MS, Perkins SL. The immunophenotype of T-lymphoblastic lymphoma in children and adolescents: a children’s oncology group report. Br J Haematol. 2012;159:454–461. doi: 10.1111/bjh.12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tiftik N, Bolaman Z, Batun S, Ayyildiz O, Isikdogan A, Kadikoylu G, Muftuoglu E. The importance of CD7 and CD56 antigens in acute leukaemias. Int J Clin Pract. 2004;58:149–152. doi: 10.1111/j.1368-5031.2004.0018.x. [DOI] [PubMed] [Google Scholar]
- 46.Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, Rettig MP, Wang B, Eissenberg LG, Ghobadi A, Gehrs LN, Prior JL, Achilefu S, Miller CA, Fronick CC, O’Neal J, Gao F, Weinstock DM, Gutierrez A, Fulton RS, Dipersio JF. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. 2018;32:1970–1983. doi: 10.1038/s41375-018-0065-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. 2013;82:775–797. doi: 10.1146/annurev-biochem-063011-092449. [DOI] [PubMed] [Google Scholar]
- 48.Vaneycken I, Govaert J, Vincke C, Caveliers V, Lahoutte T, Baetselier PD, Raes G, Bossuyt A, Muyldermans S, Devoogdt N. In vitro analysis and in vivo tumor targeting of a humanized, grafted nanobody in mice using pinhole SPECT/micro-CT. J Nucl Med. 2010;51:1099–1106. doi: 10.2967/jnumed.109.069823. [DOI] [PubMed] [Google Scholar]
- 49.Dumoulin M, Conrath K, Van Meirhaeghe A, Meersman F, Heremans K, Frenken LG, Muycdermans S, Wyns L, Matagne A. Single-domain anti-body fragments with high conformational stability. Protein Sci. 2002;11:500–515. doi: 10.1110/ps.34602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Unciti-Broceta JD, Del Castillo T, Soriano M, Magez S, Garcia-Salcedo JA. Novel therapy based on camelid nanobodies. Ther Deliv. 2013;4:1321–1336. doi: 10.4155/tde.13.87. [DOI] [PubMed] [Google Scholar]
- 51.Muyldermans S, Cambillau C, Wyns L. Recognition of antigens by single-domain antibody fragments: the superfluous luxury of paired domains. Trends Biochem Sci. 2001;26:230–235. doi: 10.1016/s0968-0004(01)01790-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.