

Abstract
Methylation at Arg198 and Arg200 residues of the EGFR extracellular domain by PRMT1 have been demonstrated to enhance EGFR activation by the canonical ligands, EGF and TGFα. On the other hand, RNase 5 was identified as a new ligand of EGFR recently. However, the interplay between EGFR methylation and RNase 5 in EGFR activation is still unclear. Here, we showed that RNase 5 activated EGFR and enhanced cell proliferation in colorectal cancer cells. PRMT1 positively regulated EGFR signaling activation by RNase 5. Inhibition of EGFR methylation by methylation-site mutagenesis reduced the binding affinity of RNase 5 to EGFR and abrogated RNase 5-mediated EGFR activation, suggesting that PRMT1-mediated EGFR methylation is critical for EGFR activation by RNase 5. Notably, RNase 5 diminished the inhibitory activity of cetuximab on colorectal cancer cells, implying RNase 5 is a potential biomarker to predict cetuximab response in colorectal cancer.
Keywords: EGFR, arginine methylation, PRMT1, cetuximab, RNase, angiogenin
Introduction
Aberrant activation of the epidermal growth factor receptor (EGFR) and its downstream signaling have been associated with the initiation and progression of a number of cancer types, such as lung cancer, glioblastoma, head and neck cancer, and colorectal cancer [1]. Several anticancer therapeutics have been developed to target EGFR, including tyrosine kinase inhibitors (TKIs) for lung cancer, and cetuximab for colorectal cancer [1]. The biological functions of EGFR are largely regulated by post-translational modifications [2]. Arginine (Arg) methylation is an emerging post-translational modification of EGFR [3]. Methylation at Arg198 and Arg200 residues within the dimer interface of the EGFR extracellular domain by protein arginine methyltransferase 1 (PRMT1) augments the binding affinity of ligands EGF and TGFα to EGFR [4], thereby enhancing ligand-induced receptor activation and EGFR-dependent cell proliferation in colorectal cancer [4] and triple-negative breast cancer cells [5]. Moreover, EGFR arginine methylation confers resistance to cetuximab through induction and stabilization of the active EGFR dimer conformation [4]. Therefore, arginine methylation of EGFR have been proposed as a biomarker for predicting resistance to cetuximab in colorectal cancer [6].
Recently, human ribonuclease 5 (RNase 5, also known as angiogenin) has been identified as a non-canonical EGFR ligand in an RNase catalytic-independent manner [7]. Activation of EGFR signaling by RNase 5 confers sensitivity to erlotinib, an EGFR TKI, in pancreatic ductal adenocarcinoma [7]. Elevated plasma RNase 5 level is a potential predictive marker for erlotinib sensitivity [7]. However, the functional impact of arginine methylation on EGFR activation by RNase 5 is not clear. Thus, there is a need to characterize the precise interplay between EGFR arginine methylation and RNase 5-mediated EGFR activation in colorectal cancer.
Materials and methods
Cell culture
Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/F12 supplemented with 10% fetal bovine serum and antibiotics. In this series of experiments, cells were treated with RNase 5 in serum-starved conditions for 24 hr.
shRNA construct and transfection
Lentiviral-based pLKO.1 PRMT1 shRNA vector was indicated in our previously study [4]. For lentiviral production, PLKO.1 PRMT1 shRNA vector, packaging (pCMV-dr8.Z dvpr), and envelope (pCMV-VSV-G) plasmids were cotransfected into 293T cells using PEI Reagent (Polysciences). After 48-hour transfection, HT29 colon cancer cells were infected with viral particles. Stable knockdown clones were selected by culturing medium with 4 μg/ml puromycin for 2 weeks.
Constructs, reagents, peptides and antibodies
EGFR (GI: 2811086), PRMT1 (GI: 359338974) and EGFR (R198/200K) mutagenesis constructs were prepared as described previously [3,4]. Anti-EGFR antibody (catalog 06-847, Millipore) was used to detect full-length EGFR. Antibodies against phospho-Tyr 1086 (catalog ab5650, Abcam) was used for detection of EGFR activation. Antibodies to phosphor-AKT (Ser 473) (catalog 9271, Cell Signaling) was used to detect the EGFR downstream signaling activation. Anti-RN-ase 5 (catalog AF265, R&D Systems) was used to detect RNase 5 level after overexpression. Anti-tubulin antibody was purchased from Sigma-Aldrich (catalog T5168).
Cell viability assay
Cell viability was analyzed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma) assay. Cells were seeded at 5×103 cells/100 μl/well in 96-well plates and treated with or without cetuximab (50 µg/ml). After incubation at 37°C for 72 hr, 10 μl of MTT (5 mg/ml in PBS) was added to each well and cells were incubated at 37°C for 3 hr. The water-insoluble purple precipitate was then solubilized in 100 μl of DMSO per well. Absorbance of the wells was measured at 595 nm with a reference wavelength of 650 nm using a BioTek Synergy Neo multi-mode reader.
Cell migration and invasion assays
Cell migration and invasion were performed by using BioCoat Control Cell Culture Insertsin and BioCoat Growth Factor Reduced Matrigel Invasion Chambers (BD Biosciences), respectively. Culture medium containing 10% fetal bovine serum as a chemoattractant was added to the plate, and cells (1×105 cells/24-well chamber) in serum-free culture medium with the indicated treatments were added to the chamber. The chamber was then incubated at 37°C for 72 hr (for HT29 cells) to allow cells to penetrate an 8 mm pore size, uncoated membrane or a Matrigel-coated membrane. Cells remained on the upper surface of the membrane were removed with a cotton swab, and those on the underside of the membrane were fixed with 4% paraformaldehyde, stained with 0.5% crystal violet, and microscopically counted from three random fields of each membrane. The values of cell migration and invasion are shown as numbers of migrated cells and invaded cells per field, respectively. The relative fold at empty vector without treatment or the first bar was defined as [4].
Detection of RNase 5-EGFR binding affinity by ELISA
ELISA 96-well plates were captured with 3 μg/ml anti-EGFR antibody (Abcam) in 0.2 M sodium phosphate buffer (pH 6.5) at 100 μl/well overnight at room temperature. The plates were then rinsed three times with phosphate-buffered saline with 0.05% Tween-20 (PBST) and blocked with 200 μl/well of 1% BSA solution containing 0.05% Tween-20 at 37°C for 2 hr. After rinsing three times with PBST, 100 μl/well of A431-RIPA lysates or RIPA buffer only as a negative control were added and incubated at 37°C for 1.5 hr. The plates were then washed with 400 μl/well of 1 M sucrose three times, shaking 1 min per time, followed by the addition of recombinant RNase 5 at a series of diluted concentrations in RIPA buffer. After incubation at 37°C for 1.5 hr, wells were washed with 400 μl/well of 1 M sucrose three times, shaking 1 min per time, and 100 μl/well of biotin-conjugated detection antibodies in blocking buffer was added for incubation at 37°C for 1 hr. The plates were washed with 1 M sucrose three more times with shaking, and 100 μl/well of streptavidin-conjugated HRP (1:2,000 in blocking buffer) was added and incubated for 30 min at room temperature. The wells were washed again with 1 M sucrose three times with shaking, and 100 μl/well of TMB as a peroxidase substrate was added and incubated for 30 min at room temperature. The reaction was terminated by the addition of 50 μl/well of stop solution. The optical density was determined at 450 nm, corrected by subtraction of readings at 570 nm, with use of a BioTek Synergy Neo multi-mode reader. The dissociation constant (KD) and Bmax were estimated by the above binding data and then transformed to create a Scatchard plot with the GraphPad Prism program (version 6; Prism Software Inc., San Diego, USA).
Results
RNase 5 has been demonstrated to act as an EGFR ligand in pancreatic ductal adenocarcinoma [7]. However, its activity on other EGFR-driven cancer types is unclear. We found that exogenous RNase 5 treatment induced EGFR phosphorylation and downstream signaling activation in colorectal cancer cells (Figure 1A). Ectopic expression of RNase 5 in colorectal cancer cells enhanced cell proliferation (Figure 1B). Together, these results suggest that RNase 5 activates EGFR in colorectal cancer cells.
Figure 1.
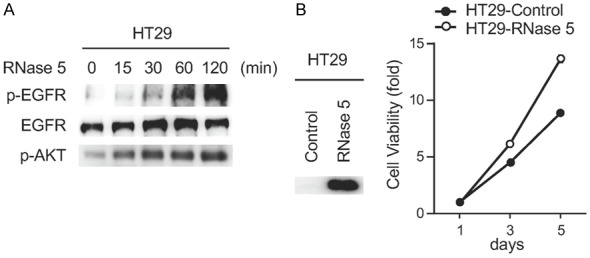
RNase 5 activates EGFR and enhances cell proliferation in colorectal cancer cells. A. Immunoblot of HT29 cells treated with RNase 5 for indicated time. B. The viability of HT29 cells with ectopic RNase 5 expression were tested at the indicated time points. Each experiment was performed twice in triplicate.
PRMT1-mediated EGFR methylation at Arg198 and Arg200 residues enhances EGFR activation by EGF and TGFα [4]. Since whole-transcriptome analysis reveals that RNase 5 and EGF elicit similar signaling events to regulate gene transcription [7], we sought to investigate whether EGFR Arg198 and Arg200 methylation is involved in RNase 5-mediated EGFR activation. Results showed that ectopic PRMT1 expression enhanced RNase 5-induced EGFR signaling activation (Figure 2A). In contrast, knockdown of endogenous PRMT1 abolished EGFR activation by RNase 5 (Figure 2B). Moreover, inhibition of EGFR methylation by methylation-site mutagenesis (R198/200K) reduced the binding affinity of RNase 5 to EGFR (Figure 2C) and suppressed RNase 5-mediated EGFR activation (Figure 2D). Collectively, these results suggest that EGFR methylation by PRMT1 is required for RNase 5-mediated EGFR activation.
Figure 2.
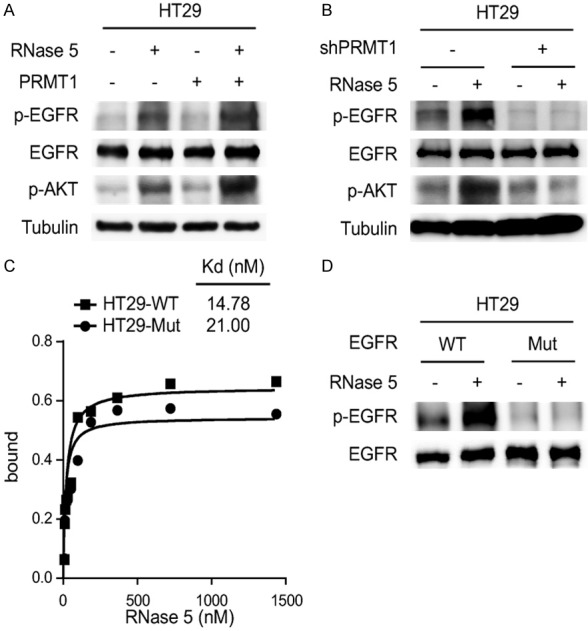
PRMT1-mediated EGFR methylation is required for EGFR activation by RNase 5. A. HT29 control and PRMT1 overexpressing cells were treated with RNase 5, and EGFR and AKT activation were evaluated by Western blotting with the specific phospho-antibodies. B. HT29 control (WT) and PRMT1 knockdown (shPRMT1) cells were treated with RNase 5, and EGFR and AKT activation were evaluated by Western blotting with the specific phospho-antibodies. C. Binding curves measuring the EGFR-RNase 5 binding affinity of HT29 cells expressing EGFR wild type (WT) or EGFR R198/200K mutant (Mut). D. HT29 cancer cells expressing either EGFR wild type (WT) or EGFR R198/200K mutant (Mut) were treated with RNase 5, and EGFR activation was evaluated by Western blotting with phospho-EGFR (Y1068) antibody.
High RNase 5 enhances the response to erlotinib treatment in pancreatic ductal adenocarcinoma [7]. However, whether RNase 5 also regulates the inhibitory effect of cetuximab on colorectal cancer cells is still unknown. Results showed that RNase 5 inhibited cetuximab-mediated suppression on cell proliferation and migration of colorectal cancer cells (Figure 3A and 3B), implying that RNase 5 may serve as a potential biomarker to predict cetuximab response in colorectal cancer.
Figure 3.
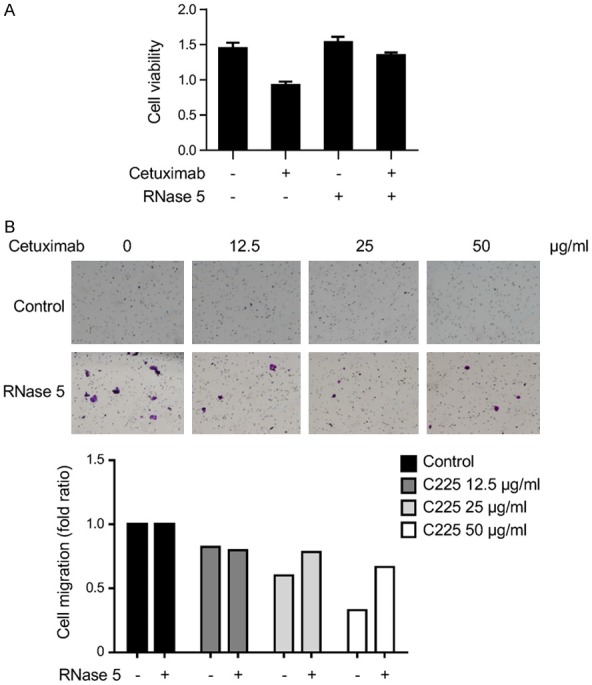
RNase 5 attenuates cetuximab-mediated suppression on colon cancer cell growth and migration. A. HT29 cells were treated with cetuximab (50 μg/ml) in the presence or absence of RNase 5 protein (0.5 μg/ml) and subjected to MTT assay. B. HT29 cells were subjected to cell migration assay in the presence or absence of cetuximab (C225) and RNase 5.
Discussion
Previous study has shown that methylation at Arg198 and Arg200 residues of the EGFR extracellular domain positively regulates the binding affinity of EGF to EGFR [4]. Arg198 and Arg200 are located at the hinge region between domain 1 and domain 2. Methylation of these sites is predicted to favor the active conformation of EGFR, leading to an enhanced ligand-stimulated EGFR signaling [4]. Our current study demonstrated that Arg198 and Arg200 methylation is also involved in RNase 5-mediated EGFR activation, suggesting that RNase 5 may induce EGFR activation through a similar mechanism as EGF. X-ray crystallography reveals that EGF binds to the EGFR extracellular domain I through Met21, Ile23 and Leu26 of EGF, and to the EGFR extracellular domain III through Tyr13, Leu15, Arg41, Gln43, Arg45 and Leu47 of EGF [8]. EGF-EGFR binding results in the exposure of EGFR dimerization surface and subsequent EGFR dimerization [8]. Similar to EGF, RNase 5 also induces EGFR dimerization, and EGFR domains I and III are involved in RNase 5-EGFR binding [7]. RNase 5 binds to EGFR through the C-terminal region of RNase 5 (amino acids 74-123). However, sequence alignment analysis reveals that, among the conserved residues between RNase 5 C-terminal region and EGF, only one residue is involved in the EGF-EGFR binding (RNase 5 Gln93 vs. EGF Gln43) [7]. Therefore, it is still unclear how RNase 5 induces EGFR dimerization and activation. A co-crystal structure is needed to understand how Arg198 and Arg200 methylation regulates RNase 5-mediated EGFR activation.
Two classes of EGFR inhibitors have been developed to target EGFR for cancer therapy, including EGFR TKIs that target the receptor kinase domain, such as erlotinib, and monoclonal antibodies that target the receptor extracellular domain, such as cetuximab [1]. Previous study demonstrated that RNase 5 knockdown desensitizes pancreatic cancer cells to erlotinib treatment [7]. Moreover, elevated plasma RNase 5 level of pancreatic adenocarcinoma patients positively correlates with response to erlotinib [7]. These findings collectively suggest that RNase 5 plays a positively regulatory role in erlotinib sensitivity. Interestingly, our current results showed that RNase 5 suppressed the inhibitory activity of cetuximab on colorectal cancer cells (Figure 3A and 3B). Does RNase 5 exhibit differential regulatory activities on two classes of EGFR inhibitors? Further studies are required to evaluate the predictive value of RNase 5 as a biomarker in cetuximab response.
Acknowledgements
This work was funded in part by the NIH/NCI under award number P30CA016672, R01 CA211615, U01 CA201777, and AI116722; the Cancer Prevention & Research Institute of Texas (RP160710 and RP150245); The University of Texas MD Anderson Cancer Center-China Medical University and Hospital Sister Institution Fund (to M.-C.H.); the Ministry of Health and Welfare, China Medical University Hospital Cancer Research Center of Excellence (MOHW107-TDU-B-212-114024; MOHW107-TDU-B-212-112015); a fellowship under the Ministry of Science and Technology (MOST, Taiwan) New Partnership Program award for the Connection to the Top Labs in the World (MOST-106-2911-I-006-524); the Center for Biological Pathways; and an NIH T32 Training Grant in Cancer Biology (5T32CA186892; to H.-H.L.).
Disclosure of conflict of interest
None.
References
- 1.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 2.Linggi B, Carpenter G. ErbB receptors: new insights on mechanisms and biology. Trends Cell Biol. 2006;16:649–656. doi: 10.1016/j.tcb.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Hsu JM, Chen CT, Chou CK, Kuo HP, Li LY, Lin CY, Lee HJ, Wang YN, Liu M, Liao HW, Shi B, Lai CC, Bedford MT, Tsai CH, Hung MC. Crosstalk between Arg1175 methylation and Tyr1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat Cell Biol. 2011;13:174–181. doi: 10.1038/ncb2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liao HW, Hsu JM, Xia W, Wang HL, Wang YN, Chang WC, Arold ST, Chou CK, Tsou PH, Yamaguchi H, Fang YF, Lee HJ, Lee HH, Tai SK, Yang MH, Morelli MP, Sen M, Ladbury JE, Chen CH, Grandis JR, Kopetz S, Hung MC. PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. J Clin Invest. 2015;125:4529–4543. doi: 10.1172/JCI82826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakai K, Xia W, Liao HW, Saito M, Hung MC, Yamaguchi H. The role of PRMT1 in EGFR methylation and signaling in MDA-MB-468 triple-negative breast cancer cells. Breast Cancer. 2018;25:74–80. doi: 10.1007/s12282-017-0790-z. [DOI] [PubMed] [Google Scholar]
- 6.Korphaisarn K, Chou CK, Xia WY, Clarke CN, Katkhuda R, Davis JS, Raghav KP, Liao HW, Wu JY, Menter DG, Maru DM, Hung MC, Kopetz S. Arginine methylation of EGFR: a new biomarker for predicting resistance to anti-EGFR treatment. Am J Cancer Res. 2017;7:2587–2599. [PMC free article] [PubMed] [Google Scholar]
- 7.Wang YN, Lee HH, Chou CK, Yang WH, Wei Y, Chen CT, Yao J, Hsu JL, Zhu C, Ying H, Ye Y, Wang WJ, Lim SO, Xia W, Ko HW, Liu X, Liu CG, Wu X, Wang H, Li D, Prakash LR, Katz MH, Kang Y, Kim M, Fleming JB, Fogelman D, Javle M, Maitra A, Hung MC. Angiogenin/ribonuclease 5 is an EGFR ligand and a serum biomarker for erlotinib sensitivity in pancreatic cancer. Cancer Cell. 2018;33:752–769. e8. doi: 10.1016/j.ccell.2018.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, Kim JH, Saito K, Sakamoto A, Inoue M, Shirouzu M, Yokoyama S. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110:775–787. doi: 10.1016/s0092-8674(02)00963-7. [DOI] [PubMed] [Google Scholar]